

Summary
Proteasomes are heterogeneous in forms and functions, but how the equilibrium among the 20S, 26S, and 30S proteasomes is achieved and altered is elusive. Here, we present a protocol for purifying and characterizing proteasome species. We describe steps for generating stable cell lines; affinity purifying the proteasome species; and characterizing them through native PAGE, activity assay, size-exclusion chromatography, and mass spectrometry. These standardized methods may contribute to biochemical studies of cellular proteasomes under both physiological and pathological conditions.
For complete details on the use and execution of this protocol, please refer to Choi et al. (2023).1
Subject areas: Cell Biology, Mass Cytometry, Protein Biochemistry, Protein expression and purification
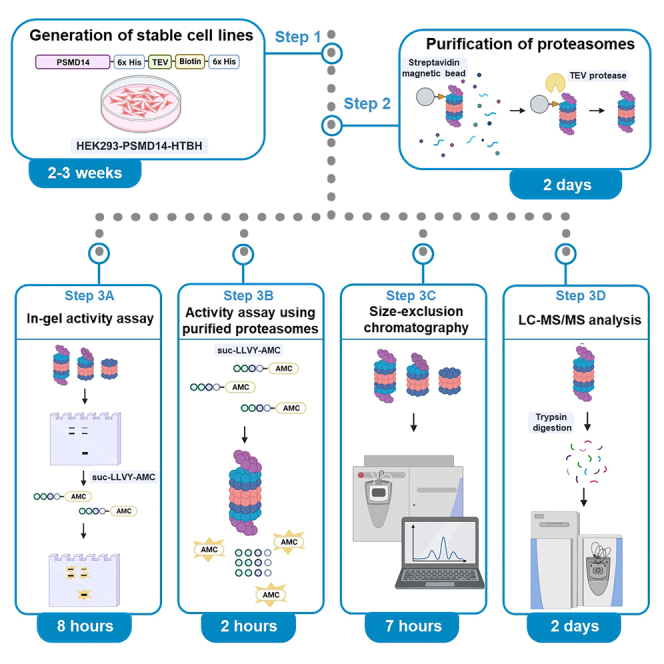
Graphical abstract
Highlights
-
•
Protocol for generating cell lines stably expressing tagged proteasome subunits
-
•
Detailed protocol for tandem affinity purification of human proteasomes
-
•
Biochemical validation of proteasome composition, integrity, and activity
-
•
Mass spectrometric analysis of purified proteasomes and associating proteins
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Proteasomes are heterogeneous in forms and functions, but how the equilibrium among the 20S, 26S, and 30S proteasomes is achieved and altered is elusive. Here, we present a protocol for purifying and characterizing proteasome species. We describe steps for generating stable cell lines; affinity purifying the proteasome species; and characterizing them through native PAGE, activity assay, size-exclusion chromatography, and mass spectrometry. These standardized methods may contribute to biochemical studies of cellular proteasomes under both physiological and pathological conditions.
Before you begin
In the ubiquitin-proteasome system, the proteasome is responsible for the irreversible degradation of polyubiquitinated substrates. The proteasome holoenzyme consists of a 20S core particle (CP; molecular weight ∼730 kDa) and one or two 19S regulatory particle (RP; ∼930 kDa) that recognizes, unfolds, and translocates the substrates to the catalytic chamber.2,3 The singly-capped (RP-CP configuration) and doubly-capped (RP2-CP) holoenzymes are referred to as the 26S and 30S proteasomes. They exist in cells with stand-alone 20S proteasomes; the ratio of these proteasome species (20S, 26S, and 30S) appears to dynamically alter to maintain cellular proteostasis upon metabolic needs and environmental changes.4,5 Their structural similarity makes it difficult to directly analyze the equilibrium changes in proteasome species at the cellular level.
The protocol below describes a series of methods for generating an A549-based stable cell line for proteasome purification (Figure 1), affinity-purifying the proteasome species (Figure 2), and their analyses using non-denaturing (native) polyacrylamide gel electrophoresis (PAGE), size-exclusion chromatography (SEC), activity assay using fluorogenic peptide substrate (Figures 3 and 4), and mass spectrometry (MS). Please note that, in this protocol, we use the official names of proteasome subunits, determined by the HUGO gene nomenclature committee at the European Bioinformatics Institute (https://www.genenames.org/).
Figure 1.

Workflow for generation of A549-PSMD14-HTBH cells
pQCXIP-PSMD14-HTBH and pVSV-G plasmids are transfected to 293-GP cells. A549 cells are treated with the retrovirus-containing media from HEK293-GP cells, followed by puromycin selection to remove untransduced cells.
Figure 2.

Strategy to purify proteasome species
A549-PSMD14-HTBH cells are lysed and bound to streptavidin magnetic beads to affinity-purify the proteasome. After enrichment of the 26S proteasome holoenzyme complex using the biotin-streptavidin interaction, the proteasome can be obtained by treating the TEV protease.
Figure 3.

Confirmation of purified proteasome species using SDS-PAGE
(A) To confirm whether PSMD14-HTBH transduction was successful, SDS-PAGE analysis followed by immunoblotting was performed using both the 6× His tag and PSMD14 antibodies.
(B) The purified proteasome species from A549-PSMD14-HTBH cells were confirmed through CBB staining.
(C) The purified proteasomes were also confirmed through immunoblotting.
Figure 4.

Characterization of diverse proteasome species
(A) The proteasome purified using HTBH-tagged PSMD14 (a 19S subunit) and the proteasome purified in the same way using HTBH-tagged PSMB5 (a 20S subunit) were separated using Native Page, and in-gel activity assay was performed using fluorogenic peptide substrates, followed by subsequent immunoblotting; Exp., Exposure.
(B) The activity of proteasome can be quantitatively measured by fluorometer. The proteasomal activity significantly increased when ATPγS, which induces the 26S and 30S to their active states, was used than when normal ATP was used. Each point represents the mean (± SD) of three independent experiments (N=3).
(C) Purified proteasomes from A549-PSMB5-HTBH cells were subjected to SEC. The immunoblotting using separated fractions reveals the 20S and 26S/30S proteasomes based on the reactivity towards 19S and 20S subunit antibodies.
Generation of A549-PSMD14-HTBH cell line
Timing: 2–3 weeks
The procedure below describes the generation of a stable cell line that stably expresses PSMD14-HTBH (Figure 1).
-
1.
DAY 1 (30 min).
Seed 8 × 105 HEK293-GP cells in a 6-cm2 culture dish in 5 mL of complete DMEM media (DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 2 mM L-glutamine) and incubate overnight at 37°C.
Note: HEK293-GP cell line is based on HEK293 and stably expresses the viral gag and pol proteins.
-
2.
DAY 2 (1 h).
The following day or when cells were at ∼60% confluency, transfect HEK293-GP cells with retroviral plasmids expressing PSMD14-HTBH using Lipofectamine 3000.-
a.Make tube 1 by mixing plasmid DNA (pQCXIP-PSMD14-HTBH and pVSV-G), P3000 transfection reagents, and Opti-MEM media in a sterilized 1.5 mL microcentrifuge tube according to the recipe below.
Reagent Amount pQCXIP-PSMD14-HTB (500 ng/μL) 4 μL pVSVG (500 ng/μL) 4 μL P3000 8 μL Opti-MEM 184 μL Total 200 μL -
b.Make tube 2 by mixing 6 μL of Lipofectamine 3000 with 194 μL of Opti-MEM in a sterilized 1.5 mL microcentrifuge tube.
-
c.After carefully putting all the mixture in tube 1 into tube 2, incubate them at room temperature (RT) for 10 min.
-
d.Add 400 μL of the mixture made in step c into HEK293-GP cells, gently shake the culture dish to mix well, and incubate for 6 h.
-
e.Change the media to remove the transfection reagents.
-
i.Wash the cells once with Dulbecco’s phosphate-buffered saline (DPBS).
-
ii.Replace the media with 2.5 mL of complete DMEM.
-
iii.Incubate the cells at 37°C for additional 42 h.Note: Despite some claims of Lipofectamine 3000 being noncytotoxic, we recommend replacing the transfection media with fresh media after 6 h of transfection
-
i.
-
a.
-
3.
DAY 3 (30 min).
Seed 2 × 105 A549 cells (or your cells-of-interests) in 2 mL of complete RPMI media per well into 6-well plates and culture for a day for the cells to reach a 50%–60% confluency before retroviral infection.
Note: The same protocol can be applied to other cells to generate cell lines stably overexpressing HTBH tagged PSMD14 (or other subunits).
Note: Similar to complete DMEM media, complete RPMI media is supplemented with 10% FBS, 1% penicillin-streptomycin, and 2 mM L-glutamine.
-
4.
DAY 4 (30 min).
Transduction of A549 cells with retrovirus-containing media from the HEK293-GP cells.-
a.After 48 h post-transfection, collect the whole media of HEK293-GP cells containing retrovirus expressing PSMD14-HTBH in a 15-mL conical tube.
-
b.Centrifuge the tube at 400× g for 5 min at RT for complete removal of HEK293-GP cells.
-
c.Collect supernatants and prepare a virus mixture according to the recipe below.
Reagent Final concentration Amount DMEM media from HEK293-GP cells (containing retrovirus) N/A 2 mL RPMI (complete media) 10% (v/v) 2 mL Polybrene (10 mg/mL in DMSO) 5 μg/mL 2 μL Total 4.0 mL -
d.Remove the cell media of each well and add the virus mixture at 2 mL/well to A549 or target cells for transduction.
-
e.Incubate cells for additional 48 h at 37°C.
 Pause point: If you are not ready to transduce the cells right away (for example, inadequate cell confluency of target cells), store the DMEM media with retrovirus at ‒80°C for later use.Note: We typically do not use media containing retroviruses if they have been stored for more than a month. Also, do not repeat freezing and thawing of this media.
Pause point: If you are not ready to transduce the cells right away (for example, inadequate cell confluency of target cells), store the DMEM media with retrovirus at ‒80°C for later use.Note: We typically do not use media containing retroviruses if they have been stored for more than a month. Also, do not repeat freezing and thawing of this media.
-
a.
-
5.
DAY 6 (12 h).
Selection of A549-PSMD14-HTBH stable cell lines.-
a.Culture retrovirus-treated A549 cells in a 10-cm2 dish.
-
b.After cells are completely attached to the culture dish (more than 10 h), treat puromycin (final 1 μg/mL)-containing RPMI media to retrovirus-treated A549 cells.
-
c.Incubate A549 cells for more than 24 h for positive selection of A549-PSMD14-HTBH.Note: Although only for 24 h puromycin treatment works well for selection, for more effective selection, we recommend 1 μg/mL of puromycin for the starting concentration and a gradual increase of the concentration up to 2.5 μg/mL of puromycin for further selection.(every 24 h increase the puromycin concentration by 0.5 μg/mL) If you plan to use other cell lines, the effective concentration of puromycin selection should be determined in advance through dose-dependent treatment of puromycin followed by cell viability assays (e.g., MTT, XTT, and Cell Titer Glo).6
-
a.
-
6.
DAY 7 (20 min).
After puromycin selection for 24 h (or more if necessary), wash your cells with PBS stored in RT and change your media with complete RPMI.
CRITICAL: After selection, confirm the HTBH-tagged PSMD14 expression through Western blotting using anti‒6×His or PSMD14 antibodies or HRP-conjugated streptavidin.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PSMA4 (1:1,000) | Enzo Life Sciences | Cat# PW8115; RRID: AB_2171569 |
| PSMB5 (1:1,000) | Thermo Fisher Scientific | Cat# PA1-977; RRID: AB_2172052 |
| PSMC2 (1:1,000) | Santa Cruz Biotechnology | Cat# sc-166972; RRID: AB_10611067 |
| PSMB6 (1:1,000) | Thermo Fisher Scientific | Cat# PA1-978; RRID: AB_2172197 |
| PSMD1 (1:1,000) | Santa Cruz Biotechnology | Cat# sc-514809 |
| Anti-β-actin antibody (1:5,000) | Merck | Cat# A1978 RRID: AB_476692 |
| 6x-His tag monoclonal antibody (1:3,000) | Thermo Fisher Scientific | Cat# MA1-21315 RRID: AB_557403 |
| PSMD4 (1:1,000) | Thermo Fisher Scientific | Cat# PA5-17259 RRID: AB_10978153 |
| Goat anti-rabbit IgG (H + L) secondary antibody, HRP (1:10,000) | Thermo Fisher Scientific | Cat# 31460 RRID: AB_228341 |
| Goat anti-mouse IgG, peroxidase conjugated, H + L (1:10,000) | Merck | Cat# AP124P RRID: AB_90456 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Welgene | Cat# LM001-05 |
| RPMI-1640 | Welgene | Cat# LM011-01 |
| l-glutamine | Welgene | Cat# LS 002-01 |
| Gibco fetal bovine serum | Thermo Fisher Scientific | Cat# 12483020 |
| Gibco Opti-MEM | Thermo Fisher Scientific | Cat# 31985070 |
| Lipofectamine 3000 transfection reagent | Thermo Fisher Scientific | Cat# L3000075 |
| Penicillin-streptomycin | Welgene | Cat# LS 202-02 |
| Dulbecco’s phosphate-buffered saline (D-PBS) | Welgene | Cat# LB001-02 |
| Puromycin | Cayman Chemical | Cat# 13884 |
| suc-LLVY-AMC | Bachem | Cat# I-1395 |
| 1.5 mL microcentrifuge tube | Tarsons | Cat# T500010-N |
| Tris base | Merck | Cat# 77-86-1 |
| Hydrochloric acid | Daejung Chemicals | Cat# 4090-4400 |
| Glycerine | Daejung Chemicals | Cat# 4066-4400 |
| Magnesium chloride anhydrous | Daejung Chemicals | Cat# 5504-4405 |
| Adenosine 5′-triphosphate (ATP) disodium salt | Bio Basic | Cat# AB0020 |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | Cat# R8061 |
| Tobacco etch virus (TEV) protease | Thermo Fisher Scientific | Cat# 12575015 |
| Bovine serum albumin | Bovogen | Cat# BSAS0.05 |
| Aprotinin | AG Scientific | Cat# A-1420 |
| Leupeptin hemisulfate | AG Scientific | Cat# L-1165 |
| Pepstatin A | AG Scientific | Cat# P-1519 |
| Sodium chloride | Daejung Chemicals | Cat# 7548-4105 |
| Potassium chloride | Daejung Chemicals | Cat# 6566-4400 |
| EDTA | Daejung Chemicals | Cat# 4000-4405 |
| Tween 20 | Duksan General Science | Cat# 4892 |
| ATPγS (adenosine-5'-(γ-thio)-triphosphate, tetralithium salt) | Jena Bioscience | Cat# NU-406-25 |
| Polybrene | Merck | Cat# TR-1003-G |
| Sodium dodecyl sulfate (SDS) | Kanto | Cat# 37203-11 |
| Urea | Merck | Cat# U1250-1KG |
| Iodoacetamide | Merck | Cat# I1149-5G |
| Sequencing grade modified trypsin | Promega | Cat# V5111 |
| Sodium chloride solution | Merck | Cat# S5150-1L |
| Ammonium bicarbonate | Merck | Cat# 09830-500G |
| Formic acid (LCMS grade) | Thermo Fisher Scientific | Cat# AAB-A117-50 |
| Acetonitrile (LCMS grade) | Thermo Fisher Scientific | Cat# A955212 |
| Tris-HCl, pH 8.5 | Nippon Gene | Cat# 316-90405 |
| Water (HPLC grade) | Thermo Fisher Scientific | Cat# W54 |
| Water (LCMS grade) | Thermo Fisher Scientific | Cat# W64 |
| UltraPure SDS solution, | Thermo Fisher Scientific | Cat# 24730020 |
| Trifluoroacetic acid | Thermo Fisher Scientific | Cat# 85183 |
| indexed Retention Time (iRT) | Biognosys | Cat# 43891 |
| Ammonium hydroxide | Merck | Cat# 5438300250 |
| Protein LoBind tubes | Eppendorf | Cat# 0030108116 |
| Empore SDB-RPS extraction disks | Thermo Fisher Scientific | Cat# 13-110-022 |
| QSP low-retention pipette tips | Thermo Fisher Scientific | Cat# TLR070-Q |
| Methanol (LCMS grade) | Thermo Fisher Scientific | Cat# A456-4 |
| Minisart NML surfactant-free cellulose acetate standard syringe filter | Sartorius | Cat# S6534-FMOSK |
| LCMS certified clear glass screw neck total recovery vial | Waters | Cat# 600000671CV |
| Critical commercial assays | ||
| NuPAGE 3%–8% Tris-acetate protein gels | Thermo Fisher Scientific | Cat# EA0375BOX |
| Novex Tris-glycine native running buffer (10X) | Thermo Fisher Scientific | Cat# LC2672 |
| Novex Tris-glycine native sample buffer (2X) | Thermo Fisher Scientific | Cat# LC2673 |
| Amicon Ultra-0.5 centrifugal filter unit | Merck | Cat# UFC5010BK |
| BioMag streptavidin | Bangs Laboratories | Cat# BM551 |
| Pierce BCA protein assay kit | Thermo Fisher Scientific | Cat# 23221 |
| Experimental models: Cell lines | ||
| Human: A549 | KCLB | Cat# 10185 |
| Human: A549-PSMB5-HB | Choi et al. | N/A |
| Human: A549-PSMD14-HB | Choi et al. | N/A |
| Human: HEK293-GP2 | Takara Bio | Cat# 631458 |
| Recombinant DNA | ||
| Plasmid: pVSV-G | Addgene | Cat# 14888 |
| Plasmid: pQCXIP-PSMB5-HB | Choi et al. | N/A |
| Plasmid: pQCXIP-PSMD14-HB | Choi et al. | N/A |
| Software and algorithms | ||
| MaxQuant (version 1.6.1.0 and above) | Max Planck Institute of Biochemistry | https://www.maxquant.org/ |
| Perseus (version 1.6.15.0 and above) | Max Planck Institute of Biochemistry | https://maxquant.net/perseus/ |
| Xcalibur (version 4.5) | Thermo Fisher Scientific | N/A |
| BioRender | BioRender.com | N/A |
| Other | ||
| Dounce tissue grinder, 15 mL | Wheaton | Cat# 357544 |
| MagJET separation rack | Thermo Fisher Scientific | Cat# MR02 |
| Cell scraper | SPL Life Sciences | Cat# 90031 |
| Cooling microcentrifuge | LaboGene | Cat# 1730R |
| Tube rotator | SeouLin Bioscience | Cat# SLRM-3 |
| Water bath | JSR | Cat# JSWB-11T |
| Mini gel tank | Thermo Fisher Scientific | Cat# A25977 |
| 5 mL syringe | Koreavaccine | Cat# S15-27-167 |
| ChemiDoc MP imaging system | Bio-Rad | Cat# 12003154 |
| Power supply | Major Science | Cat# MP-310 |
| PVDF membrane | Merck | Cat# IPVH00010 |
| 15 mL tube | SPL Life Sciences | Cat# 50215 |
| 96-well black plate | SPL Life Sciences | Cat# 30296 |
| Tecan infinite m200 fluorometer | Tecan | |
| Whatman membrane filters nylon (pore size 0.2 μm, diam. 47 mm) | Whatman | Cat# SL.MCE02047A |
| Size-exclusion chromatography columns (Superose 6 Increase 10/300 GL) | Cytiva | Cat# 29-0915-96 |
| Orbitrap Exploris 480 mass spectrometer | Thermo Fisher Scientific | Cat# BRE725539 |
| UltiMate 3000 RSLCnano system | Thermo Fisher Scientific | Cat# ULTIM3000RSLCNANO |
| Acclaim PepMap C18 reversed phase HPLC column | Thermo Fisher Scientific | Cat# 160454 |
| Analytical EASY-Spray column | Thermo Fisher Scientific | Cat# ES903 |
| Ultrasonic bath | Branson | Cat# CPX5800 |
| Thermomixer compact | Eppendorf | N/A |
| CentriVap benchtop centrifugal vacuum concentrator with glass lid | Labconco | Cat# 7810015 |
Materials and equipment
Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 25 mM | 0.25 mL |
| Glycerol | 10% (v/v) | 1 mL |
| MgCl2 (1 M) | 50 mM | 50 μL |
| ATP (250 mM) | 1 mM | 40 μL |
| Dithiothreitol (DTT) (1 M) | 1 mM | 10 μL |
| PIC (100 ×) | 1 X | 0.1 mL |
| ddH2O | N/A | 8.55 mL |
| Total | 10 mL |
Note: Lysis buffer without ATP, DTT, and PIC can be stored for 3 months at 4°C. Add ATP, DTT, and PIC to the buffer right before you use it.
Protease inhibitor cocktail (PIC; 100×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Aprotinin (in ddH2O) | 80 μM | 2.6 mg |
| Leupeptin (in ddH2O) | 2 mM | 4.8 mg |
| Pepstatin A (in DMSO) | 1 mM | 3.43 mg |
| DMSO | N/A | Up to 5 mL |
| Total | 5 mL |
Note: Protease inhibitor cocktail can be stored for more than 1 year at ‒20°C. For long-term storage, we usually store it in 500 μL aliquots.
ATP (250 mM)
| Reagent | Final concentration | Amount |
|---|---|---|
| Adenosine 5′-triphosphate (ATP) disodium salt | 250 mM | 2,067 mg |
| Tris-base (2 M) | 500 mM | 3.75 mL |
| MgCl2 (1 M) | 250 mM | 3.75 mL |
| ddH2O | N/A | Up to 15 mL |
| Total | 15 mL |
Note: The pH of Tris-base (2 M) does not need to be adjusted. ATP (250 mM) can be stored for more than 6 months at ‒20°C. We usually store it in 300 μL aliquots.
Washing buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 25 mM | 0.75 mL |
| Glycerol | 10% (v/v) | 3 mL |
| MgCl2 (1 M) | 50 mM | 0.15 mL |
| ATP (250 mM) | 1 mM | 0.12 mL |
| Dithiothreitol (DTT; 1 M) | 1 mM | 30 μL |
| ddH2O | N/A | 25.95 mL |
| Total | 30 mL |
Note: Washing buffer without ATP and DTT can be stored for 3 months at 4°C. Add ATP and DTT to the buffer right before you use it.
TEV washing buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 50 mM | 0.5 mL |
| Glycerol | 10% (v/v) | 1 mL |
| ATP (250 mM) | 1 mM | 40 μL |
| ddH2O | N/A | 8.46 mL |
| Total | 10 mL |
Note: TEV washing buffer without ATP can be stored for 3 months at 4°C. Add ATP to the buffer right before you use it.
TEV protease buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 50 mM | 0.25 mL |
| Glycerol | 10% (v/v) | 0.5 mL |
| ATP (250 mM) | 2 mM | 40 μL |
| AcTEV Protease | 15 unit / mL | 7.5 μL |
| ddH2O | N/A | 4 mL |
| Total | 5 mL |
Note: TEV protease buffer should be freshly prepared and used. Make the buffer according to the sample number (approximately 500 μL of TEV protease buffer required per tube).
Native gel running buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Novex Tris-Glycine Native Running Buffer (10×) | 1× | 20 mL |
| EDTA (0.5 M) | 0.1 mM | 40 μL |
| ATP (250 mM) | 0.5 mM | 0.4 mL |
| MgCl2 (1 M) | 5 mM | 1 mL |
| DTT (1 M) | 0.5 mM | 0.1 mL |
| ddH2O | N/A | 178.46 mL |
| Total | 200 mL |
Note: We recommend preparing native gel running buffer freshly right before we use it.
In-gel activity assay buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 50 mM | 0.75 mL |
| ATP (250 mM) | 2 mM | 60 μL |
| MgCl2 (1 M) | 5 mM | 75 μL |
| suc-LLVY-AMC (20 mM) | 12.5 μM | 0.15 mL |
| ddH2O | N/A | 13.965 mL |
| Total | 15 mL |
Note: In-gel activity assay buffer without ATP and suc-LLVY-AMC can be stored for 3 months at 4°C. Add ATP and suc-LLVY-AMC to the buffer right before you use it and keep the buffer from light.
Proteasome activity assay buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 50 mM | 10 mL |
| ATP or ATPγS (250 mM) | 1 mM | 0.8 mL |
| EDTA (0.5 M) | 1 mM | 0.4 mL |
| Bovine serum albumin | 1 mg/mL | 0.2 g |
| Dithiothreitol (DTT) (1 M) | 1 mM | 0.2 mL |
| ddH2O | N/A | 188.6 mL |
| Total | 200 mL |
Note: Proteasome activity assay buffer without ATP and DTT can be stored for 2 months at 4°C. Add ATP and DTT to the proteasome activity assay buffer right before you use it.
Size-exclusion chromatography buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl, pH 7.5 (1 M) | 25 mM | 6.25 mL |
| MgCl2 (1 M) | 5 mM | 1.25 mL |
| ATP (250 mM) | 1 mM | 1 mL |
| DTT (1 M) | 1 mM | 0.25 mL |
| ddH2O | N/A | 241.25 mL |
| Total | 250 mL |
Note: Size-exclusion chromatography buffer without ATP and DTT can be stored for 3 months at 4°C. Add ATP and DTT to the buffer right before you use it. The buffer should be filtered through a membrane filter with 0.2-μm pore size before use.
Denaturation buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DTT | 0.1 M | 0.01543 g |
| Tris-HCl pH 8.5 (1 M) | 0.1 M | 100 μL |
| 10% Sodium dodecyl sulfate (SDS) | 2% | 200 μL |
| water (HPLC grade) | N/A | 700 μL |
| Total | 1 mL |
Note: Denaturation buffer should be freshly prepared and used.
Urea buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Urea | 8 M | 4.8048 g |
| Tris-HCl, pH 8.5 (1 M) | 0.1 M | 1 mL |
| water (HPLC grade) | N/A | Up to 10 mL |
| Total | 10 mL |
Note: Urea buffer can be stored for 1 year at RT.
E1 buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% Trifluoroacetic acid (TFA) | 0.2% | 8 μL |
| ACN (LCMS grade) | 60% | 600 μL |
| water (LCMS grade) | N/A | 392 μL |
| Total | 1 mL |
Note: E1 buffer should be freshly prepared and used.
Sample injection buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Formic acid (LCMS grade) | 0.1% | 1 μL |
| ACN (LCMS grade) | 2% | 20 μL |
| water (LCMS grade) | N/A | 959 μL |
| indexed Retention Time (iRT) | 50× | 20 μL |
| Total | 1 mL |
Note: Sample injection buffer can be stored for 1 year at RT.
-
•
Ammonium bicarbonate (ABC) buffer: water (HPLC grade) with 40 mM ABC, pH 8.0.
Note: ABC buffer can be stored at RT for up to 12 months.
-
•
Alkylation buffer: Urea buffer with 50 mM Iodoacetamide (IAA).
Note: Alkylation buffer should be freshly prepared and used. IAA should be stored in the dark as it is sensitive to light.
-
•
NaCl buffer: water (HPLC grade) with 0.5 M NaCl.
Note: NaCl buffer can be stored at RT for up to 12 months.
-
•
Trypsin buffer: 0.1 μg/μL Trypsin in 200 μL ABC buffer.
Note: Trypsin buffer can be stored at ‒20°C for up to 6 months.
-
•
Acidification buffer: water (LCMS grade) with 10% TFA.
-
•
Washing buffer (MS): water (LCMS grade) with 0.2% TFA.
Note: Acidification buffer and Washing buffer (MS) should be freshly prepared and used.
-
•
E2 buffer: 80% ACN (LCMS grade) with 5% ammonium hydroxide.
Note: E2 buffer should be freshly prepared and used.
-
•
LC Solvent A: water (LCMS grade) with 0.1% formic acid (LCMS grade).
-
•
LC Solvent B: 80% ACN (LCMS grade) with 0.1% formic acid (LCMS grade).
Note: We recommend to de-gas the LC solvent through sonication before use. The presence of bubbles in the solvent can lead to issues with pressure and peaks in analysis. The LC Solvents A and B can be stored for 6 months at RT.
LC-MS/MS setup
-
•
This protocol uses a high-resolution Orbitrap-Exploris 480 MS coupled with an UltiMate 3000 RSLCnano system. Other LC-MS systems with similar capabilities can be applied.
-
•
A two-column system consisting of an Acclaim PepMap u-precolumn cartridge (C18, 5 μm, 100 Å, 300 μm × 5 mm) and an analytical EASY-Spray column (C18, 2 μm, 100 Å, 75 μm × 50 cm) was used.
-
•
Peptides were separated using an 160-min segmented gradient as follows: 6%–8% solvent B for 7 min, 8%–30% solvent B for 113 min, 30%–95% solvent B for 5 min, 95% solvent B for 10 min, 95%–6% solvent B for 5 min, 6% solvent B for 20 min. Refer to tables below: the LC gradient profile and LC parameter settings below (Tables 1 and 2).
-
•
The MS is operated in a DDA mode, employing full MS scans with 1 Microscan at a resolution of 120,000 over a mass range of m/z 350–1,800, with detection in the Orbitrap. After each full MS scan, the top twenty intense ions with multiple charged ions are selected for the MS/MS scan. Fragment ion spectra through higher-energy collisional dissociation (HCD) are acquired in the orbitrap. The data was acquired using Xcalibur software v4.5. Refer to the table of Parameters used for the MS operation below (Table 3).
Table 1.
LC gradient profile
| Time | Flow rate (nL/min) | Percent of solvent B |
|---|---|---|
| 0 | 300 | 6 |
| 7 | 300 | 8 |
| 120 | 300 | 30 |
| 125 | 300 | 95 |
| 135 | 300 | 95 |
| 140 | 300 | 6 |
| 160 | 300 | 6 |
Table 2.
LC parameter settings
| Injection volume | 3 μL |
|---|---|
| Column temperature | Control off |
| Sampler temperature | 4°C |
| Max pressure limit | 900 Bar |
Table 3.
Parameters used for the operation of the mass spectrometer
| Parameter | Value |
|---|---|
| Polarity | positive |
| Global settings | |
| Methods Duration (min) | 160 |
| Ion Source Type | NSI |
| Spray Voltage: Positive Ion (V) | 1800 |
| Ion Transfer Tube Temp (°C) | 290 |
| Default Charge State | 2 |
| Full MS Scans | |
| Mass analyzer | Orbitrap |
| Automatic Gain Control (AGC) target | 300% |
| Microscans | 1 |
| Orbitrap resolution | 120,000 |
| Scan range (m/z) | 350–1,800 |
| Max Injection Time (ms) | 25 |
| AGC Target | Custom |
| DataType | Profile |
| Filter Settings | |
| Include Charge State (s) | 2–6 |
| Exclusion Duration (s) | 35 |
| Mass tolerance (ppm) | 10 |
| Dependent MS/MS Scans | |
| Mass analyzer | Orbitrap |
| Automatic Gain Control (AGC) target | 100,000 |
| Microscans | 1 |
| Activation type | HCD |
| Normalized collision energy (%) | 30 |
| Isolation Window | 1.2 |
| First Mass | 110 |
| Number of dependent scans | 20 (from most intense precursors) |
| Scan rate | Normal |
| Mass range | Normal |
| Charge exclusion | Exclude +1 charge state |
| Isolation Mode | Quadrupole |
| Ion trap scan rate | Rapid |
| Max Injection Time (ms) | Custom |
| Resolution | 15000 |
| Maximum ion time | 25 ms |
| Loop count | 10 |
| Isolation offset | 0 |
Step-by-step method details
Purification of proteasomes
The workflows below describe the purification of mammalian proteasomes in two steps: (1) lysis of A549-PSMD14-HTBH cells and binding proteasomes to streptavidin magnetic beads, (2) eluting the proteasomes bound to the beads by cleaving them using TEV proteases (Figure 2).
-
1.
Grow and expand A549-PSMD14-HTBH cells that stably express proteasome subunit, PSMD14 which has a tandem 6× his and biotin tag.
Note: Typically, we can get about 30 μg of purified proteasome from a 90%-confluent 150-mm culture dish. We recommend preparing proteasomes from 6 to 8 plates for one-time purification.
-
2.
Prepare 10 mL of Lysis buffer and 40 mL of washing buffer.
-
3.Harvest cells.
-
a.Remove all of the cell culture media from the 150-mm cell culture plates.
-
b.Add ∼10 mL of PBS onto the plate to completely remove media.
-
c.Remove PBS from the plates.
-
d.Add 1 mL of ice-cold Lysis buffer per plate and immediately scrape the cells of the plates.
-
e.Transfer detached cells to a 15-mL Dounce tissue grinder.
-
a.
Note: Cells collected with Lysis buffer from multiple culture dishes can be loaded in a grinder. But do not load more than ∼5 mL of buffer to avoid overflows.
-
4.
Lyse the cells by gently moving the Dounce tissue grinder up and down about 15 times.
-
5.
Transfer the buffer which has lysed cells into multiple 1.5-mL microcentrifuge tubes, maximum 1 mL at a time.
-
6.
Centrifuge the tubes at 16,000× g for 15 min at 4°C.
-
7.During centrifugation, wash streptavidin magnetic beads (BioMag Streptavidin).
-
a.Insert the same number of new 1.5-mL microcentrifuge tubes into a magnetic rack.
-
b.Transfer 100 μL of streptavidin magnetic beads (50% slurry) to each tube.
-
c.Add 1 mL of ice-cold Washing buffer to each tube.
-
d.Resuspend the magnetic beads after removing the tubes from the magnetic rack.
-
e.Insert the tubes into the magnetic rack. After the beads are completely gathered onto the magnet, remove all the buffer.
-
f.Repeat steps (c) – (e) two times.
-
a.
Note: Add the Washing buffer again as quickly as possible to prevent the beads from drying out, when the Washing buffer is removed during bead washing.
Note: Streptavidin-agarose beads can also be used at proteasome purification (e.g. Streptavidin, agarose conjugate, Cat# 16–126, Merck). But for rapid and convenient purification, we recommend using streptavidin magnetic beads.
-
8.
Add the supernatants from whole A549-PSMD14-HTBH cell lysates to the washed beads.
-
9.
Incubate tubes in tube rotator for overnight at 4°C.
-
10.
Prepare 40 mL of washing buffer, 10 mL of TEV washing buffer, and 5 mL of TEV protease buffer.
-
11.Remove all the supernatant in the tubes and wash the streptavidin magnetic beads.
-
a.Add 1 mL of ice-cold Washing buffer to each tube.
-
b.Resuspend the magnetic beads after removing the tubes from the magnetic rack.
-
c.Insert the tubes into the magnetic rack. After the beads are completely gathered onto the magnet, remove all the buffer.
-
d.Repeat steps (a) – (c) two times.
-
a.
-
12.
Briefly wash the streptavidin magnetic beads once with TEV washing buffer.
-
13.
Add 500 μL of TEV protease buffer to the tube.
-
14.
Incubate the tubes for 2 h at 30°C in a water bath.
Note: Gently invert the tubes every 30 minutes for resuspension. Prepare the same number of Amicon Ultra-0.5 centrifugal filter unit (top) that are placed into Amicon microcentrifuge tubes (bottom).
-
15.
Insert the tubes into the magnetic rack, and after beads are completely gathered onto the magnet, transfer the supernatants to Amicon filter units.
-
16.
Centrifuge the Amicon tubes with samples at 14,000× g for 15 min at 4°C.
-
17.
To collect the samples, place the Amicon filter device upside down in a clean microcentrifuge tube.
-
18.
Centrifuge the tubes at 1,000× g for 2 min at 4°C.
-
19.
Measure the concentration of collected purified proteasomes, for example, through BCA assay.
Note: Concentration of purified proteasomes can be measured by a Bradford assay (e.g. Pierce Bradford Plus Protein Assay Reagent)
Note: In assessing the purity and integrity of purified proteasomes, we commonly perform Coomassie Brilliant Blue (CBB) staining in addition to Native PAGE analysis.
In-gel activity assay and immunoblotting of proteasome species
This process is to characterize purified mammalian proteasomes using native PAGE, which not only maintains the native structure and activity of proteasome species but also separates them based on size, such as the 20S proteasome alone, singly-capped RP-CP (26S proteasome), and doubly-capped RP2-CP (30S proteasome). These different proteasome species can be visualized by using fluorogenic peptide substrates, such as suc-LLVY-AMC as well as by subsequent immunoblotting.
-
20.
Prepare 200 mL of ice-cold Native gel running buffer.
-
21.
Assemble native gel (NuPAGE 3–8% Tris-Acetate Protein Gels) into an Invitrogen mini-gel tank.
-
22.
Make the samples by mixing purified proteasomes with 2× Native sample buffer (Novex Tris-Glycine Native Sample Buffer).
Note: Do not boil the sample subjected to native PAGE.
-
23.
Pour Native gel running buffer into the mini-gel tank. And wash each well of native gel using a 5-mL syringe.
Note: Be sure to clean the wells of native gel to remove gel residues which may interrupt proper sample loading and gel running.
-
24.
Load the 1–2 μg sample of purified proteasomes to a well.
-
25.
Run the samples for 4 h at 150 V at 4°C.
-
26.
Prepare 15 mL of In-gel activity assay buffer.
-
27.
Take the gel from the pre-cast cassette and place it in 15 mL of In-gel activity assay buffer.
Note: When you separate the pre-cast gel, carefully open it since native gels are more easily torn than conventional denaturing gels.
-
28.
Incubate the gel in the In-gel activity assay buffer for 10 min at 37°C, protecting it from the light.
-
29.After incubation, place the gel on a ChemiDoc imaging system (ChemiDoc MP Imaging System) and detect the AMC signal.
-
a.To prevent the gel from drying out, spray ddH2O before placing the gel on the ChemiDoc.
-
b.Place the gel on the ChemiDoc.
-
c.Spray 3 mL of In-gel activity assay buffer evenly over the gel.
-
d.The AMC signal can be detected using the EtBr channel.
-
e.Adjust the exposure time between 1 – 10 s to find the optimal images of 26S and 30S proteasomes.
-
a.
-
30.
Add 24 μL of 10% SDS (w/v, in ddH2O) to the remaining 12 mL of In-gel activity assay buffer.
-
31.
Incubate the gel for 10 min at 37°C, protecting it from the light.
-
32.After incubation, place the gel on ChemiDoc (ChemiDoc MP Imaging System) and detect AMC signal again.
-
a.To prevent the gel from drying out, spray ddH2O before placing the gel on the ChemiDoc.
-
b.Place the gel on the ChemiDoc.
-
c.Spray 3 mL of In-gel activity assay buffer containing 0.02% SDS evenly over the gel.
-
d.The AMC signal can be detected using the EtBr channel.
-
e.Adjust the exposure time between 1 – 10 s to find the optimal image of 20S proteasomes.
-
a.
Note: For additional confirmation, we routinely perform subsequent immunoblotting with the native gel following the activity assay.
-
33.
After detecting the AMC signals, carefully wash the native gel with ddH2O.
-
34.
Activate the PVDF membrane (Millipore) by wetting it in absolute methanol for 10 min.
-
35.
Assemble the transfer cassette and transfer it in a mini-gel tank for 70 min at 100 V.
-
36.
Block the PVDF membrane in 2% BSA in TBS-T solution.
-
37.
Make an antibody solution by diluting the antibody (for example, anti-PSMB6 and -PSMD4 antibodies) with a 2% BSA in TBS-T solution at 1:1000.
Note: Other antibodies against the 20S and 19S subunits can be used for immunoblotting.
-
38.
Bind the membrane with antibody cocktails and incubate it on a rocking shaker overnight at 4°C.
-
39.
Wash the membrane with TBS-T solution for 10 min. Repeat this three times.
-
40.
Make an antibody solution by diluting the secondary antibody conjugated with HRP with a 2% BSA in TBS-T solution at 1:10000.
-
41.
Bind the membrane with secondary antibody cocktails and incubate it on a rocking shaker for 1 h at RT.
-
42.
Wash the membrane with TBS-T solution for 10 min. Repeat this three times.
-
43.
After incubation, place the membrane on a gel imaging system and spray ECL solution. Detect the chemiluminescence signal.
Proteasome activity assay using fluorogenic peptide substrates
This process measures the activity of proteasomes using fluorogenic peptide substrates such as suc-LLVY-AMC. Because a microplate-based fluorometer is used, proteasome activity can be measured more precisely and quantitatively than in-gel activity assay. However, the suc-LLVY-AMC hydrolysis activity reflects both 20S and 26S/30S proteasomes. Their ratio in the purified proteasomes can be calculated by using a substance such as ATPrS that binds to the 26S and 30S proteasome and maintains its active state. To the contrary, high concentration of SDS (∼ 1%) inhibits the cleavage between the peptides and the AMC group, instead of activating the 20S proteasome.
-
44.
Prepare 15 mL of Proteasome activity assay buffer.
Note: 15 mL of Proteasome activity assay buffer is enough for assaying one 96-well microplate.
-
45.
Set the fluorometer machine (Tecan infinite m200 fluorometer) in advance. We use Tecan Magellan 7.0 software according to the parameters below (Table 4).
Note: We kinetically monitor proteasome activity by measuring the AMC signal for about an hour. Monitoring for a more extended period will not matter, but a shorter period measurement of AMC signals may result in insufficient observation.
Note: This is a general protocol for measuring proteasome activity using Tecan Infinite m200 fluorometer. If you use different fluorometer, please refer to the manufacturer’s guideline and empirically optimize the measurement parameters in the table.
-
46.
Add 8.33 μL of suc-LLVY-AMC (20 mM) to 12 mL of Proteasome activity assay buffer.
Note: The concentration of suc-LLVY-AMC in Proteasome activity assay buffer can vary according to the samples, such as purified proteasomes and whole-cell lysates. Always prevent suc-LLVY-AMC from exposure to light until using it.
-
47.
Prepare the proteasome samples containing 20S, 26S, and 30S proteasomes by mixing it with Proteasome activity assay buffer which doesn’t have suc-LLVY-AMC at a final concentration of 100 nM.
-
48.
First, add 10 μL of proteasome samples to each well of the 96-well black bottom plate.
Note: We usually put one sample into three individual wells. In addition, we use 10 μL of Proteasome activity assay buffer lacking suc-LLVY-AMC for normalization.
-
49.
Dispense 90 μL of Proteasome activity assay buffer with suc-LLVY-AMC into wells containing samples. The concentration of LLVY-AMC becomes 12.5 μM.
-
50.
Place the 96-well plate after loading on the fluorometer machine and start measuring proteasome activity.
-
51.
After the activity measurement is finished, save the file in an Excel format.
Table 4.
Tecan measurement parameters
| Measurement parameter | Value |
|---|---|
| Plate | SPL flat black plate |
| Temperature | 30°C |
| Wait for Temperature | |
| Minimum | 29.5°C |
| Maximum | 30.5°C |
| Kinetic Cycle | |
| Number of cycles | 21 |
| Kinetic interval | 00:03:00 |
| Shaking | |
| Duration | 5 s |
| Mode | Orbital |
| Amplitude | 4 mm |
| Frequency | 37 rpm |
| Wait (Timer) | |
| Wait time | 00:00:02 |
| Fluorescence Intensity | |
| Wavelength (Excitation) | 380 (20) nm |
| Wavelength (Emission) | 465 (45) nm |
| Mode | Top |
| Mirror (Mirror) | Automatic |
| Read (Number of flashes) | 25 |
| Read (Settle time) | 0 ms |
| Gain (Manual) | 65 |
| Integration (Lag time) | 0 μs |
| Integration (Integration time) | 20 μs |
| Label (Name) | Label1 |
| Move Plate | |
| Move plate | Out |
Analyze proteasomes through size-exclusion chromatography
These steps are to separate and analyze proteasomes using size-exclusion chromatography (SEC). We can confirm changes in the 20S and 19S sub-complexes by immunoblotting with the fractions separated by the ÄKTA liquid chromatography machine.
-
52.
Prepare Size-exclusion chromatography buffer.
-
53.
Before beginning the SEC, make sure the ÄKTA machine has been fully cleaned and the bubbles were removed from the pump, lines, and capillary loop.
Note: All the solutions including ddH2O used in the ÄKTA machine should be filtered through a 0.2 μM membrane filter.
-
54.
Connect the Superose 6 Increase 10/300 GL columns to the ÄKTA properly. In order to equilibrate the connected column with ddH2O followed by Size-exclusion chromatography buffer, put inlet A into filtered ddH2O and inlet B into the filtered Size-exclusion chromatography buffer.
Note: Other size-exclusion chromatography columns that can separate the proteasome species from the 20S core particle (∼730 kDa) to the doubly-capped proteasome (∼2.6 MDa) can also be used.
-
55.
Before injecting the sample, turn on Unicorn7 software and enter the protocol below (Table 5).
Note: 1 column volume (CV) of the Superose 6 Increase 10/300 GL column is 23.562 mL.
-
56.
Using a 1 mL syringe, draw up the purified proteasome samples up to 1 mL. Invert the syringe and tap the syringe gently to remove bubbles and to prevent introducing gases into the ÄKTA.
Note: Prepare more than 500 μg of purified proteasomes for SEC and subsequent biochemical analyses.
Note: Make sure no bubbles remain inside the solution prior to sample injection.
-
57.
Inject the sample into the capillary loop through the injection port.
-
58.
Prepare approximately 100 of 1.5-mL tubes and put them on the fractionation collector.
-
59.
After the fractionation starts, move the collected protein eluates to ice every 5 min.
Note: If you do not analyze the fractionated samples immediately, snap-freeze them quickly with liquid nitrogen. Samples can be stored indefinitely at ‒80°C. We found thawing-refreezing cycles did not significantly impact proteasome integrity but decreased the number of interacting proteins.
-
60.
The fractionated samples are subjected to SDS-PAGE, followed by immunoblotting using various proteasome subunit antibodies.
Table 5.
Unicorn7 software settings
| Parameter | Value |
|---|---|
| Equilibration | |
| Flow rate | 0.2 mL/min |
| Inlet | A |
| Total volume | 2.0 CV |
| Reset UV monitor | |
| Sample application | |
| Flow rate | 0.2 mL/min |
| Loop type | Capillary loop |
| Empty loop | 1 mL |
| Fractionate state | In waste (do not collect) |
| Elution | |
| Flow rate | 0.2 mL/min |
| Inlet | A |
| Isocratic elution volume | 1.50 CV |
| Fractionate state | fraction collector |
| Fraction type | fixed volume fractionation |
| Fixed fractionation volume | 0.25 mL |
Sample preparation for mass spectrometry
This step describes proteasome digestion with trypsin and sample clean-up for mass spectrometric analysis. Filter-aided sample preparation (FASP) is a method for successful bottom-up type proteomic analysis. Following the FASP step, peptides are cleaned up through a desalting process. The desalting step before MS is crucial because excess salt can interfere with peptide ionization and add the background signals in the mass spectra.
-
61.Proteasome digestion with trypsin.
-
a.Add Denaturation buffer to proteasome samples. Vortex and spin down the tubes.
-
b.Sonicate the samples using a water bath sonicator. Vortex and spin down the tubes again.
-
c.Boil the samples for 15 min at 95°C. After cooling at RT, spin down the tubes.Note: To avoid any loss during the heating process, ensure that the tubes are tightly capped and allow sufficient cooling before proceeding to the next step.
-
d.Add 300 μL Urea buffer to the denatured samples.
-
e.Vortex and spin down the tubes.
-
f.Repeat step (b).
-
g.Transfer the samples to Amicon Ultra-0.5 centrifugal filter units.
-
h.Centrifuge at 14,000× g for 15 min at RT. Discard the flow-through from the collection tubes.
-
i.Add 400 μL Urea buffer to the filter. Centrifuge at 14,000× g for 15 min at RT. Discard the flow-through from the collection tubes. Repeat twice.
-
j.Add 200 μL Alkylation buffer to the filters. Incubate for 30 min in a dark place.
-
k.Centrifuge at 14,000× g for 15 min at RT. Discard the flow-through from the collection tubes.
-
l.Add 400 μL Urea buffer to filter. Centrifuge at 14,000× g for 15 min at RT. Discard the flow-through from the collection tubes. Repeat twice.
-
m.Add 400 μL ABC buffer to the filter. Centrifuge at 14,000× g for 15 min at RT. Discard the flow-through from the collection tubes. Repeat twice.
-
n.Add 100 μL ABC buffer and 10 μL Trypsin buffer. Incubate for 16 h on Thermomixer at 650 rpm at 37°C.Note: We recommend to use 1 μg trypsin and an enzyme-to-protein ratio of 1:100.
-
o.Elute the digested peptides in the collection tube from the filter unit.
-
i.Centrifuge at 14,000× g for 15 min.
-
ii.Add 100 μL ABC buffer to the filter. Centrifuge at 14,000× g for 15 min at RT.
-
iii.Add 50 μL ABC buffer to the filter. Centrifuge at 14,000× g for 15 min at RT.
-
iv.Add 50 μL NaCl buffer to the filter. Centrifuge at 14,000× g for 15 min at RT.
-
i.
-
p.Transfer the digested peptides from the collection tubes to low-retention microcentrifuge tubes.Pause point: Digested proteins can be stored at ‒20°C or ‒80°C until use.
-
a.
-
62.Desalt peptide mixtures before MS analysis.Note: The desalting process was performed using the StageTip desalting method using Empore Styrenedivinylbenzene-Reverse Phase Sulfonate (SDB-RPS).7Note: Different types of disks, such as C18 or C8 disks, can be used based on the characteristics of the samples. It is important to ensure that the disk does not dry out at each step.
-
a.Prepare StageTip with a triple-layered SDB-RPS disk plug using 200 μL low-retention tips.
-
b.Column conditioning.
-
i.Add 150 μL methanol to StageTip. Centrifuge at 850× g for 30 s. Discard the flow-through. Repeat twice.
-
ii.Add 150 μL ACN to StageTip. Centrifuge at 850× g for 30 s. Discard the flow-through. Repeat twice.
-
i.
-
c.Equilibrate the column by adding 100 μL Washing buffer (MS). Centrifuge at 850× g for 30 s. Discard the flow-through. Repeat three times.
-
d.Acidify the samples by adding Acidification buffer (Acidification buffer to sample volume ratio 1:10). Vortex and spin down.
-
e.Using pH test paper, check whether the sample pH is in the pH 2 to 3 range.
-
f.Transfer the samples to StageTip. Centrifuge at 850× g for 30 s.
-
g.Wash the column by adding 100 μL Washing buffer (MS). Centrifuge at 850× g for 30 s. Discard the flow-through. Repeat five times.
-
h.Elute the desalted peptides in the Protein LoBind microcentrifuge tubes from StageTip.
-
i.Add 40 μL E1 buffer to StageTip. Elute into a tube using a syringe that fits the tip diameter.
-
ii.Add 40 μL E2 buffer to StageTip. Elute into a tube using a syringe that fits the tip diameter. Repeat twice.Pause point: Desalted peptide can be stored at ‒20°C or ‒80°C until use.
-
iii.Dry the desalted samples in a CentriVap Benchtop Centrifugal Vacuum Concentrator.
-
i.
-
a.
Proteomic analysis by LC-MS/MS
This step describes sample data acquisition of the digested samples by LC-MS/MS using a high-resolution Orbitrap-Exploris 480 mass spectrometry coupled with an Ultimate 3000 RSLCnano system. The analytical parameters are stated previously in the Materials and equipment part (LC-MS/MS setup).
-
63.Resuspend dried samples with Sample injection buffer for injection onto the liquid chromatography system.
-
a.Add 8 μL of Sample injection buffer to the dried sample.
-
b.Vortex sample and spin down the tubes.
-
c.Sonicate the samples, using a water bath sonicator for 1 min.
-
d.Vortex sample again.
-
e.Centrifuge at 21,000× g for 1 h at 4°C.
-
f.Transfer the supernatant to LCMS Certified vials.
-
a.
-
64.
Inject 3 μL of samples onto LC-MS/MS system for analysis.
MS data analysis
This step describes computational analysis to identify the levels of proteasome subunits and proteasome-associated proteins from raw mass spectrometric data using MaxQuant, which is one of the most frequently used software packages for analyzing large mass-spectrometric data sets.
-
65.Analyze the acquired MS data with the MaxQuant software package (version_1.6.1.0) using the parameter described in Parameters used to operate the MaxQuant software (Table 6).
-
a.Download the human UniProt FASTA database (2014.12, 88,657 entries), common contaminant database, and iRT database.
-
b.In the Raw data tab, upload the raw file. Set the experiments for each sample using the ‘Set Experiment’ function.
-
c.In the Group-specific parameters tab, click “Label-free quantification”. Change “None” to “LFQ”.
-
d.In the Global parameters tab, click “Sequence”. Upload the FASTA file. Check the identifier rule corresponding to the UniProt identifier.
-
e.Set the Min. peptide length to 6.
-
f.Click “Label-free quantification”. Check the iBAQ parameter to conduct data analysis using iBAQ values.Note: Given that the default parameters of MQ software are generally optimized to use, there should be no need for additional modifications except for the parameters mentioned here.
-
g.Adjust the “Number of threads” based on your computer’s capabilities. Start the MQ analysis.
-
a.
-
66.
Export the results in the text format (proteingroup.txt) or as a spreadsheet for further processing using Perseus software (version_1.6.15.0).
Table 6.
Parameters used for the operation of the MaxQuant software
| Parameter | Value |
|---|---|
| Variable modifications | Oxidation (M) |
| Acetyl (protein N-term) | |
| Fixed modifications | Carbamidomethyl (C) |
| Maximum number of modifications per peptide | 5 |
| Enzyme | Trypsin |
| Maximum missed cleavages | 2 |
| Minimum peptide length | 6 |
Expected outcomes
Strong expression of C-terminally HTBH-tagged PSMD14 can be confirmed through immunoblotting against PSMD14, biotin, or His×6 tag (Figure 3A). PSMD14-HTBH is integrated into the intact proteasome, enabling proteasome purification through biotin-streptavidin interactions. Affinity-purified proteasome species can be confirmed by gel staining, such as Coomassie Brilliant Blue (CBB) and silver staining, or immunoblotting of proteasome subunits (Figures 3B and 3C). The population changes in the cellular proteasome species can be characterized by various methods. For example, comparing the Native PAGE results using the proteasomes purified from A549-PSMD14-HTBH cells (enriching 19S subcomplexes) and from A549-PSMB5-HTBH cells (enriching 20S proteasomes) will provide useful information. After separating 20S, 26S, and 30S proteasomes, an in-gel activity assay using fluorogenic peptide substrates is performed to identify the proteasome species and measure their activity with lesser sensitivity. Through subsequent immunoblotting, the compositions of 20S, 26S, and 30S proteasomes can be further confirmed (Figure 4A). The proteasome peptidase activity can be quantitatively measured with higher sensitivity when the assay is performed with a fluorometer (Figure 4B). In addition, proteasome species can be separated into free 19S and 20S or 26 and 30S holoenzymes through SEC (Figure 4C). Through MS analysis, we successfully recognized all established proteasome subunits, interacting proteins, and subunits of the proteasome activator complex, ranging from PSME1 to PSME4. We expect that the tandem affinity purification techniques demonstrated here will aid in uncovering further biochemical roles and physiological functions of proteins interacting with the proteasome.
Limitations
Conventionally, proteasome biologists have evaluated their structural and biochemical properties after purifying them through various methods, which include classical enrichment using sequential fractionation and chromatography, affinity purification using ubiquitin (Ub)-like domains, and purification through affinity tags (e.g., 3×FLAG, histidine-biotin with a TEV cleavage site [HTBH], or protein A) fused to various proteasome subunit.8,9 This protocol demonstrates the efficiency of the tandem affinity purification techniques to purify various proteasome species present within cells. Compared to other proteasome enrichment methods, such as 1) classical enrichment using sequential fractionation and chromatography, 2) employing the N-terminal ubiquitin-like (Ubl) domain of human RAD23B as an affinity bait and 3) multiple differential centrifugation,8,10 our tandem affinity ensures the high purity and yield of the purified proteasomes. Overexpression of proteasome subunits with tandem affinity tag for purification may require genetic manipulation and act as a small artifact on intracellular proteasomes, but since the proteasome is the key cellular machinery responsible for the turnover of intracellular proteins, these impacts are expected to be small. Moreover, it allows the identification of diverse proteasome species, which are dynamically altered under varying cellular environments.
In this protocol we used fluorogenic peptide substrates to measure proteasome activity. Activity measured through the use of the substrates has traditionally been thought and used to represent overall proteasome activity. Although not covered in this protocol, in addition to using fluorogenic peptide substrates, there is also a method of measuring proteasome activity by measuring degradation of protein substrates.11 If the method is used together, proteasome activity can be shown more clearly. Another challenge lies in the limited sensitivity of gel- and chromatography-based analyses in detecting the 19S subcomplex. The identification of intact free 19S complexes proves to be particularly challenging when obtained through the purification of 20S subunit-fused affinity tags (e.g., A549-PSMB5-HTBH cells). This stands in contrast to the clear presence of the 20S, 26S, and 30S proteasomes. There is a possibility that a portion of the free 19S subcomplex might be sequestered within liquid droplets or other membrane-less organelles. These locations are not easily accessible through protocols designed for soluble proteins. Consequently, it becomes imperative to explore a range of biochemical conditions to investigate the intracellular assembly/disassembly between the 19S and 20S subcomplexes. This approach will shed light on proteasome dynamics by either revealing the differential susceptibility of these two subcomplexes to biochemical conditions or providing definitive evidence of their interplay.
Troubleshooting
Problem 1
Appropriate proteasome concentrations may not be obtained after proteasome purification (purification of proteasomes step 19).
Potential solution
-
•
When detaching the A549-PSMD14-HTBH cell, try to scrape the cells as gently as possible.
-
•
Loss occurs mostly when the proteasome is less efficiently cleaved from the streptavidin magnetic beads. TEV protease should be used without a repeated freezing and thawing cycle.
Problem 2
When the proteasome is purified and confirmed by CBB staining, protein bands are shown in the size other than the proteasome subunit sizes (purification of proteasomes step 19; Figure 3B).
Potential solution
-
•
The purified proteasome may have many interacting proteins (e.g., ECPAS, VCP, USP14). If you do not want these proteins, increase the salt-concentration in the binding buffer.
Problem 3
When performing In-gel activity assay following Native-PAGE, fluorescent signals of 30S and 26S are not distinguishable (in-gel activity assay and immunoblotting of proteasome species step 10; Figure 4A).
Potential solution
-
•
Gel running should be long enough (at least 150 V for more than 6 h) to separate 30S and 26S proteasomes.
Problem 4
Upon immunoblotting with proteasome subunits after native-PAGE, no specific proteasome population was detected (in-gel activity assay and immunoblotting of proteasome species step 24; Figure 4A).
Potential solution
-
•
This might be a technical (antibody) issue. When native PAGE is performed, the proteasome on the gel maintains its integral structure. If the primary antibody’s epitope (in the PSMA ring) is located near the contact point with the PSMC ring, it can be masked when the 19S is bound to the 20S. Please test other antibodies.
Problem 5
Regarding proteasome activity assay, it is doubtful whether the increase in AMC signal reflects proteasome activity (proteasome activity assay using fluorogenic peptide substrates step 8; Figure 4B).
Potential solution
Problem 6
Protein bands are not visible when performing immunoblotting with protein fractions obtained from size-exclusion chromatography (analyze proteasomes through size-exclusion chromatography step 9; Figure 4C).
Potential solution
-
•
Double check the setting of size-exclusion chromatography machine before running.
-
•
500 μg of purified proteasome is enough for size-exclusion chromatography followed by immunoblotting. But when proteasome subunits are not detectable in immunoblotting, try to inject more amount of purified protein.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Min Jae Lee (minjlee@snu.ac.kr).
Materials availability
This study did not generate new unique materials.
Data and code availability
This paper does not report original code.
Acknowledgments
This work was supported by grants from the National Research Foundation of Korea (2021R1A2C2008023 and RS-2023-00261784 to M.J.L., 2022R1F1A1068852 to D.H., and 2020R1A5A1019023 to D.H. and M.J.L.).
Author contributions
Conceptualization, I.B. and M.J.L.; methodology and formal analysis, I.B. and H.S.; resources, J.K. and D.J.; data curation, D.H. and M.J.L.; writing – original draft, I.B. and H.S.; writing – review and editing, D.H. and M.J.L.; funding acquisition, D.H. and M.J.L.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Dohyun Han, Email: hdh03@snu.ac.kr.
Min Jae Lee, Email: minjlee@snu.ac.kr.
References
- 1.Choi W.H., Yun Y., Byun I., Kim S., Lee S., Sim J., Levi S., Park S.H., Jun J., Kleifeld O., et al. ECPAS/Ecm29-mediated 26S proteasome disassembly is an adaptive response to glucose starvation. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112701. [DOI] [PubMed] [Google Scholar]
- 2.Marshall R.S., Vierstra R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019;6:40. doi: 10.3389/fmolb.2019.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi W.H., Yun Y., Park S., Jeon J.H., Lee J., Lee J.H., Yang S.A., Kim N.K., Jung C.H., Kwon Y.T., et al. Aggresomal sequestration and STUB1-mediated ubiquitylation during mammalian proteaphagy of inhibited proteasomes. Proc. Natl. Acad. Sci. USA. 2020;117:19190–19200. doi: 10.1073/pnas.1920327117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanaka K. The proteasome: overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009;85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahu I., Mali S.M., Sulkshane P., Xu C., Rozenberg A., Morag R., Sahoo M.P., Singh S.K., Ding Z., Wang Y., et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021;12:6173. doi: 10.1038/s41467-021-26427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adan A., Kiraz Y., Baran Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016;17:1213–1221. doi: 10.2174/1389201017666160808160513. [DOI] [PubMed] [Google Scholar]
- 7.Kim S.I., Hwangbo S., Dan K., Kim H.S., Chung H.H., Kim J.W., Park N.H., Song Y.S., Han D., Lee M. Proteomic Discovery of Plasma Protein Biomarkers and Development of Models Predicting Prognosis of High-Grade Serous Ovarian Carcinoma. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2023.100502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo C.L., Collins G.A., Goldberg A.L. Methods to Rapidly Prepare Mammalian 26S Proteasomes for Biochemical Analysis. Methods Mol. Biol. 2018;1844:277–288. doi: 10.1007/978-1-4939-8706-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Negi H., Osei-Amponsa V., Ibrahim B., Evans C.N., Sullenberger C., Loncarek J., Chari R., Walters K.J. An engineered cell line with a hRpn1-attached handle to isolate proteasomes. J. Biol. Chem. 2023;299 doi: 10.1016/j.jbc.2023.104948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besche H.C., Haas W., Gygi S.P., Goldberg A.L. Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry. 2009;48:2538–2549. doi: 10.1021/bi802198q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi W.H., de Poot S.A.H., Lee J.H., Kim J.H., Han D.H., Kim Y.K., Finley D., Lee M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016;7 doi: 10.1038/ncomms10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S., Park S.H., Choi W.H., Lee M.J. Evaluation of Immunoproteasome-Specific Proteolytic Activity Using Fluorogenic Peptide Substrates. Immune Netw. 2022;22:e28. doi: 10.4110/in.2022.22.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi W.H., Kim S., Park S., Lee M.J. Concept and application of circulating proteasomes. Exp. Mol. Med. 2021;53:1539–1546. doi: 10.1038/s12276-021-00692-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This paper does not report original code.