

Abstract
Focusing on advances from the last decade, this review will describe oxidative phenol coupling as applied in the total synthesis of natural products. This review covers catalytic and electrochemical methods with a brief comparison to stoichiometric and enzymatic systems assessing their practicality, atom economy, and other measures. Natural products forged by C–C and C–O oxidative phenol couplings as well as from alkenyl phenol couplings will be addressed. Additionally, exploration into catalytic oxidative coupling of phenols and other related species (carbazoles, indoles, aryl ethers, etc.) will be surveyed. Future directions of this particular area of research will also be assessed.
Graphical Abstract

This review highlights modern uses of oxidative phenol coupling in the total synthesis of natural products, spanning catalytic, electrochemical, stoichiometric and enzymatic approaches.
1. Introduction
The field of natural product chemistry has been significantly impacted by the emergence of oxidative coupling in the early 20th century.1, 2 Discovering new ways to couple aryl fragments continues to aid organic chemists in their journey to construct the most intricate molecules found in nature. Phenols and phenolic-type compounds are a common feature seen in natural products. These moieties offer unique redox chemistry which provides access to further derivatization (cyclization, dearomatization, etc.).3–5 A plethora of biosynthetic pathways involve phenol reactivity, especially oxidative coupling of these species.6–9 As a result, developing oxidative phenolic coupling methods has remained an important challenge in organic chemistry.10, 11 Fig. 1 illustrates the citation and publication count from 1961 to present day on oxidative phenol coupling.
Fig. 1.

Graph of publications/citations on “oxidative phenol coupling” from 1961 to 2023 from a Web of Science topic search. (Citation Report graphic is derived from Clarivate Web of Science, Copyright Clarivate 2023. All rights reserved.)
This review highlights recent synthetic uses of catalytic and electrochemical oxidative phenol couplings in the total synthesis of natural products with an additional motivation in providing a general outlook for the future. Stoichiometric and enzymatic syntheses are canvassed as a means of comparison to catalytic routes. Key factors and general considerations in oxidative coupling are discussed. The natural products are divided based on their respective chemo- and regioselectivity. The natural products to be discussed are shown in Fig. 2.
Fig. 2.

Representative natural products forged by oxidative phenolic couplings categorized by approach used.
2. Fundamentals of oxidative phenol coupling
Oxidative coupling serves as a powerful synthetic technique in constructing new phenolic connections. It offers good functional group tolerance and operates under mild reaction conditions as phenol oxidation is facile. In comparison to other cross-coupling methods, no prefunctionalization (aryl bromides, boronic acids, etc.) is required as the inherent reactivity of each ring is utilized. However, controlling multiple reactive sites remains a challenge and can lead to undesired coupling products. Recent advances in the field have improved upon these selectivity issues by biasing the substrate and developing catalyst features, those of which will be later discussed.
Various products from a general phenol oxidation are illustrated in Scheme 1. If multiple sites are available for reaction, coupling at the para position tends to dominate. One gambit to predispose a substrate for ortho coupling is the installation of blocking substituents at the para position. Pummerer ketone products, such as usnic acid, arise from an important subclass of phenol couplings that involve an additional intramolecular cyclization.12–14 When these coupling products were first introduced in the literature in 1925 by Pummerer et al., the authors postulated that the reaction proceeded via a C-O coupling at the para position of p-cresol followed by a radical cyclization.15 Dismayed by the lack of mechanistic evidence provided in the prior work, Barton et al. published a full follow-up paper refuting the mechanism and structure of the Pummerer ketone.14, 16 The authors argued that the post-coupling radical intermediate proposed by Pummerer et al. would not undergo cyclization, but rather oxidation to a para-phenoxy quinone. Rather, Barton et al. proposed that a C-C ortho-para coupling of p-cresol would afford a neutral quinone intermediate that would undergo β-addition of the hydroxyl to the quinone to furnish the Pummerer ketone. Their revised mechanism and structure remains accepted in the literature today. However, Pummerer ketone products will not be discussed here as there are no recent advances.
Scheme 1.

Potential products from one-electron/proton transfer of phenol.
Sir Derek Barton was one of the early pioneers of oxidative phenol couplings in the 1950s, establishing many of the precedents in which modern applications rely upon. A 1957 perspective from Barton et al. intricately details the various phenol coupling mechanisms at the time and their biosynthetic application in the synthesis of alkaloids, fungal metabolites, and other plant-derived natural products.17 Kozlowski and co-workers recently disclosed a perspective on oxidative phenol coupling methods18, while the current review focuses on applications to natural product synthesis. Oxidative coupling and its role in asymmetric biaryl formation for natural product synthesis is also outlined in a review by Kozlowski et al., which serves as a main repository for pre-2008 work.19 Several research groups have conducted thoughtful analyses of the chemo- and regio-selectivity of oxidative phenol coupling for iron20–22, chromium23, 24, copper25, 26, and vanadium27, 28 systems. In these works, oxidation potentials, global/site nucleophilicities, Sterimol values, and pKa values are utilized to gain control of homo- versus heterocoupling of phenols and other related structures. In one report, the authors discuss how over-oxidation is a common issue as dimeric products are often more oxidizable than their monomeric forms so various oligomeric products can be obtained.29 Takizawa and co-workers have presented a perspective on the use of vanadium catalysts in the atroposelective synthesis of axially chiral compounds via oxidative coupling.30
Oxidative coupling of phenols can be accomplished through chemical, electrochemical, photochemical, and biosynthetic approaches. The groups of Kozlowski23, 24, Pappo20–22, and Lumb25, 31 have established several precedents for these chemical transformations utilizing metal catalysts and molecular oxygen or peroxides as terminal oxidants. The field of electrochemical oxidative couplings have been pioneered by Waldvogel32–36 and Opatz35–37. By utilizing biosynthetic pathways, the Narayan38–41 and Müller42–44 groups have developed enzymatic approaches to tackle these couplings. Photochemistry has also recently emerged as a useful technique to achieve phenolic transformations including dearomatizations and oxidative couplings.29, 45–48
3. Stoichiometric approaches
An abundance of stoichiometric oxidative couplings have been reported in the synthesis of biologically active compounds. The use of stoichiometric oxidants and metals raises concerns over functional group tolerance as well as scalability. Typical procedures utilize stoichiometric loadings of first-row transition metal reagents or hypervalent iodine complexes. Using these approaches, many research groups have succeeded in synthesizing natural products via key oxidative coupling steps. Selected examples of intermolecular couplings by the Vincent group are outlined in Scheme 2. In the first case, one substrate undergoes a discrete oxidation to an iodinated intermediate generating an electrophile in situ that then reacts with the second nucleophilic partner (Scheme 2a). Specifically, 1b reacts with stoichiometric oxidant NIS to generate an intermediate that can engage phenol 1a in a Friedel-Crafts reaction and cyclization to furnish the voacalgine A core (1) in 26% yield.49
Scheme 2.

Stoichiometric couplings of phenols and indoles.
However, further synthetic endeavors by Vincent and co-workers discovered that their originally assigned framework was incorrect. A stoichiometric Ag2O coupling of phenol 2a and pleoiocarpamine (2b) formate salt afforded the revised structure of voacalgine A (2) in 21% yield (Scheme 2b).50 This method proceeds via an ortho-quinone intermediate which undergoes 1,4-addition with the indole. Subsequent trapping of the resultant iminium by the carboxylate then provides voacalgine A (2). The dimeric natural product bipleiophylline (not shown) was also formed albeit in very low yield of 3%. The authors use 2D-NMR analysis to support the assignment of this isochromenoindoline skeleton. Notably, the yields in these stoichiometric processes are not high, highlighting the difficulty in performing these complex oxidative couplings. The products of oxidative phenol couplings are prone to overoxidation as they can often be more oxidizable than the starting materials. Employing harsh oxidative conditions can therefore further degrade potential product formation.18, 51
Intramolecular versions of oxidative couplings have also been employed in total synthesis. In 2014, the Gaunt group reported an enantioselective formal synthesis of (−)-morphine (3).52 An ortho-para oxidative phenol coupling provided a key early-stage intermediate on a multigram scale (Scheme 3). This intramolecular oxidative coupling of 3a was accomplished with PhI(OAc)2 to form the biaryl bond (highlighted in green) and DBU-promoted desymmetrization aided in the production of intermediate 3b with 48% yield over two steps (10 gram scale) resulting in a formal synthesis of (−)-morphine (3). Literature precedent exists for the completion of (−)-morphine (3) from 3b in 16 additional steps.53, 54
Scheme 3.

Stoichiometric ortho-para coupling in formal synthesis of morphine.
Often, late-stage intramolecular oxidative couplings are employed in total synthesis, typically through biomimetic routes. (−)-Vescalin (4), a unique polyphenolic natural product, was synthesized utilizing a sequence of oxidative couplings as shown in Scheme 4.55 The C1-C2 connection in 4a was first assembled with a CuCl2-nBuNH2 oxidative system in a 58% yield with 5 and 20 equivalences, respectively. In this process only the (S)-atropisomer was obtained. The core of (−)-vescalin (4) was furnished with a final atroposelective oxidative coupling to connect C3-C4 in 41% yield with CuCl2 (5 equiv) and bispidine (20 equiv). Hydrogenation and acidic workup completed the sequence to (−)-vescalin (4) which was comprised of 16 steps with an overall yield of around 2%. Yamada and co-workers also disclosed an atroposelective CuCl2-nBuNH2 method which was used in the total synthesis of (−)-strictinin (not shown), a gallate-derived natural product, in a related intramolecular coupling.56 The axial chirality of the hexahydroxydiphenoyl (HHDP) moieties found in compounds like (−)-vescalin (4) was recently elucidated by Wakamori and co-workers. Notably, the (S)-atropisomer formed initially and then isomerized to the natural (R)-configuration.57
Scheme 4.

Biomimetic total synthesis of (−)-vescalin.
The Baran group accessed the arylomycin core (5) with an oxidative phenol coupling via a Cu(III)-TMEDA system (Scheme 5).58 Their approach involved a biomimetic ortho-ortho intramolecular cyclization of 5a to form the arylomycin core (5) in 51–70% yields. The method scaled well with a five gram scale reaction proceeding in 60% yield. Molecular oxygen proved to be the optimal terminal oxidant in this chemistry as stronger oxidants such as oxone and H2O2 led to lower yields. With access to the core via this early-stage approach, the authors concisely synthesized an array of derivatives which allowed for the study of their antibacterial properties.
Scheme 5.

Baran’s stoichiometric synthesis of arylomycin core.
4. Enzymatic approaches
Oxidative biaryl bond formation is ubiquitous across many classes of natural products and synthetic chemists have made tremendous strides in completing these transformations using innovative chemical approaches. However, the unique environments provided by enzymes in biological systems have proven difficult to replicate for these processes. These selective biocatalytic transformations offer impressive functional group tolerance and overall efficiency. Within the last two decades, directed evolution has allowed for the optimization of different enzymes such that these transformations can be performed on non-natural substrates with efficiencies approaching or rivaling those of conventional organic reactions. Enzymes such as horseradish peroxidase (HRP) have found utility in oxidative phenol coupling in the presence of sensitive functional groups.59, 60 Müller and co-workers have summarized many of the enzymatic approaches towards intermolecular phenol oxidative coupling.44 Cytochrome P450 enzymes have proven to be useful platforms as shown elegantly by the Narayan group.41
As previously demonstrated, arylomycins are an important class of antibiotic compounds with phenolic biaryl backbones and present a considerable synthetic challenge. Prior to the stoichiometric oxidative coupling of the arylomycin core (5) by Baran and co-workers (see Scheme 5 above), Dorrestein et al. discovered the gene cluster responsible for production of the arylomycins in Streptomyces roseosporus.61 This work paved way for chemists to bioengineer these clusters to make different arylomycin analogues. In a recent report, an engineered cytochrome P450 enzyme was utilized to accomplish the oxidative cyclization of two phenol units (ortho-ortho) to form the arylomycin core (5) on gram-scale in 84% assay yield.62
Intramolecular oxidative couplings are difficult to achieve chemically, especially for strained ring systems, but enzymes can facilely accomplish these reactions. Mycocyclosin (6), a diketopiperazine metabolite isolated from Mycobacterium tuberculosis, contains a highly strained dityrosine framework. Martí and co-workers used the gene rv2276 which encodes for a P450 enzyme, CYP121, for the oxidative C–C bond formation of 6a to form mycocyclosin (6) (Scheme 6).63 This enzyme is found exclusively in M. tuberculosis and this is its only known biocatalytic activity. Work by the Hutton64 and Schindler65 groups have leveraged Pd(II)-catalyzed Suzuki-Miyaura and Ullman-type couplings in their respective total syntheses of mycocyclosin (6), but no catalytic oxidative coupling methods have been successful.
Scheme 6.

Biogenetic oxidative coupling of mycocyclosin.
There has been lots of work on enzymatic intramolecular oxidative couplings, but biomolecular intermolecular couplings are much less common. The Müller group developed cytochrome P450 enzymes for the stereo- and regioselective synthesis of axially chiral phenolic natural products.43 Using P450 enzymes KtnC and DesC (expressed in Saccharomyces cerevisiae fungi), the authors were able to synthesize P-orlandin (8i) and M-desertorin A (9i), respectively, from the same starting material 7 (Scheme 7). This divergent approach highlights the regio- and stereoselectivity of P450 enzymes in fungi. Additionally, there are literature precedents for the synthesis of P-kotanin (8ii) from P-orlandin (8i).66 While the synthesis of M-desertorin A (9i) to M-desertorin C (9ii) has not been reported, it should also be feasible. Recently, the fungal P450 KtnC was also expressed in yeast to accomplish the in vivo biosynthesis of P-orlandin (8i) by Narayan and co-workers.67
Scheme 7.

Regioselective biocatalytic synthesis of p-orlandin and m-desertorin A.
5. Catalytic and electrochemical C–C oxidative couplings
The formation of C–C bonds via oxidative coupling methods are an especially useful transformation. Without the need for prefunctionalization, new connections between aryl fragments can be forged in fewer synthetic manipulations. Catalytic and electrochemical methods for these reactions allow for more efficient processes that minimize waste and improve upon atom economy while maintaining synthetic efficacy. Within C–C oxidative phenol coupling, a variety of regioselective outcomes can be observed as previously described in Scheme 1. The following section will discuss these transformations based on the different coupling patterns (ortho-ortho, para-para, ortho-para, and others).
5.1. ortho-ortho couplings
Axially chiral phenolic natural products have been common targets amongst those working in the oxidative coupling community. In 2012, the Kozlowski group disclosed an asymmetric naphthol oxidative coupling with a catalytic Cu-cyclic amine system in the total synthesis of (S)-bisoranjidiol (10).68 With molecular oxygen as the oxidant, this approach allowed for the homocoupling of 10a to afford (S)-10b in 62% yield and 87% ee (Scheme 8). An additional nine synthetic steps led to the completion of (S)-bisoranjidiol (10). Attempts to couple the anthracene or anthraquinone units (biomimetic approaches) failed with the former oxidizing to the corresponding anthraquinones and the latter proving resistant to oxidation. As such, the authors coupled the naphthol fragments and then constructed the remaining ring of the anthraquinone via Diels-Alder reactions. In a follow-up publication, divergent syntheses of this framework were achieved to furnish bisanthraquinone analogues (not shown).69
Scheme 8.

Oxidative coupling in the total synthesis of (S)-bisoranjidiol.
In 2007, a research group from the University of Mississippi isolated a marine natural product from the Indonesian sponge Lendenfeldia sp. and determined its structure to be (S)-11 via NMR analysis.70 The Kozlowski group set out in 2012 to synthesize this natural product via an asymmetric oxidative coupling.71 From naphthol 11a, a chiral dimeric vanadium catalyst gave rise to (S)-11b in 82% yield and 81% ee which was confirmed by spectroscopy and crystallography (Scheme 9). An additional four steps led to the synthesis of (S)-11 in 70% yield. When the authors compared their spectra and physical properties (solubility, air/silica stability, etc.) with those reported from the isolation, they discovered that they did not match. With further analysis, Kozlowski and co-workers determined that the isolated material from the plant was actually a tetrabrominated diphenyl ether compound.
Scheme 9.

Atroposelective oxidative coupling in marine metabolite structural revision.
Pappo and co-workers implemented an intermolecular catalytic oxidative cross-coupling method to assemble the arylomycin core and analogues thereof.72 As discussed before, the intramolecular cyclizations proved difficult. In addition, direct oxidative coupling of tyrosine derivatives is challenging due to over oxidation to trimers, etc.9 As such, substrates 12a and 12b with tert-butyl groups, which both blocked formation of higher order adducts and improved reactivity, were employed. Intermolecular cross-coupling using an iron porphyrin catalyst produced 12c in 48% yield (Scheme 10). To obtain optimal yields, the slow addition of the oxidant (m-CPBA) and 12b in small portions was needed. Protecting group removal/macrolactamization and tert-butyl cleavage afforded the (S)-arylomycin core (12) in 60% yield over three steps. Pappo and co-workers were also able to access the core of polycyclic peptide RP 66453 (not shown).
Scheme 10.

Intermolecular catalytic cross-coupling approach to arylomycin core.
Oligomers of guaiacol, a phenolic natural product, are commonly found in the degradation of lignin and other biosynthetic pathways.73, 74 These materials also have potential as sustainable aromatic feedstocks. The Waldvogel group applied boron-doped diamond (BDD) anodes in the anodic coupling of guaiacol and analogues as an entry to unsymmetrical bisphenol products.33 Scheme 11 highlights an interesting shift in observed regioselectivity by varying the R group at the para position of 13a. The ortho-meta product 13i is obtained in a 33% yield when 4-methyl 13a is used and ortho-ortho 13iv is formed in a 18% yield with 4-tert-butyl 13a. The isopropyl variant of 13a led to equal quantities of the ortho-ortho (13ii) and ortho-meta (13iii). As the steric bulk increases, the product distribution shifts to favor ortho-ortho versus ortho-meta homocoupling. It is appears that both the hydroxy and methoxy groups can activate different positions for oxidation and the overall reactivity is dictated by steric effects. The even lower conversion for the isopropyl variant was attributed to its propensity to undergo alternate oxidation reactions including at the benzylic position.
Scheme 11.

Regioselectivity in electrochemical oxidative coupling of guaiacol derivatives.
(−)-Viriditoxin (14), a fungal natural product with antibacterial properties isolated from Aspergillus viridinutans, contains an atropoisomeric biaryl backbone.75 In 2011, the Shaw group reported the total synthesis of (−)-viriditoxin (14) via an asymmetric oxidative naphthol coupling (Scheme 12a).76 At first glance, the backbone appears to be constructed via a para-para connection, but it is actually achieved via an ortho-ortho coupling with post-functionalization to adjust the phenol alkylation states. The authors accessed 14a in an eight step sequence, although yields were variable and scale-up was difficult. They then screened a variety of vanadium catalysts to optimize the diastereoselective homocoupling. With VO(acac)2, a 67% yield of 14b was achieved with moderate diastereoselectivity (76:24 dr). A dimeric (S,R)-vanadium catalyst led to the best results with 87% yield and 89:11 dr and led to (−)-viriditoxin (14) after five additional synthetic steps. Use of a (S,S)-vanadium dimeric catalyst (Scheme 12b) in oxidative coupling of analog 14c resulted in a superior diastereoselectivity (>95:5) albeit with slightly lower yield (85%).77 An additional synthetic step was required to synthesize (−)-viriditoxin (14) in comparison to the former synthesis, but the oxidative coupling precursor 14c was easier to synthesize on scale. Scheme 12c illustrates the proposed asymmetric induction in the coupling process which is governed by the configuration of the pyranone ring and the chiral vanadium catalyst. The diastereomeric transition state B is favored as the equatorial configuration of the R group minimizes steric interactions in comparison to the axial hydrogen clash in transition state A.
Scheme 12.

Oxidative couplings in the total syntheses of (−)-viriditoxin.
An abundance of steroidal compounds contain phenolic frameworks, but their competence in oxidative coupling is not well studied. In 2013, Yu and co-workers disclosed an oxidative coupling of deoxyestrone derivatives with a catalytic copper system.78 Using CuCl2/TMEDA as a catalyst and air as the terminal oxidant, the authors coupled (S)-15a to synthesize a variety of 2-substituted deoxyestrone dimers (S,S)-15 in 22–75% yields and 1:1 dr (Scheme 13). In order to avoid regioselectivity issues, the authors blocked the 2-position of (S)-15a. The success of the reaction was highly affected by the steric and electronic nature of the 2-substituent. Large substituents (tert-butyl, naphthalene) and electron-rich aryls led to lower yields or even no reactivity.
Scheme 13.

Oxidative homocoupling of deoxyestrone derivatives.
5.2. para-para couplings
Couplings at the para-para positions have been reported in a number of systems. However, reaction selectivity remains a complicating factor when both ortho and para sites are available. One common tactic to address regioselection in this context is to employ blocking groups. The Porco group utilized this strategy in oxidative couplings to generate a dimeric lactone natural product (Scheme 14).79 Specifically, a bromine was strategically installed at the ortho position of (±)-gonytolide C generating (+)-16a via a kinetic resolution. An asymmetric conjugate reduction strategy using Cu(OAc)2 and Josiphos SL-J001-2 achieved the kinetic resolution (97:3 er post-recrystallization with 49% yield), which yielded (+)-16a after bromination. Subsequent oxidative coupling with VOF3/TFA/TFAA yielded (+)-gonytolide A (16) in a 12% yield after dehalogenation. Its unnatural atropisomer, (−)-atrop-gonytolide A (not shown), was also obtained in a 35% yield. The bromo group both stabilized radical cation intermediates and aided in the diastereoselectivity of the coupling. The need for such blocking groups to control regioselectivity in oxidative couplings highlights a limitation of current small molecule reagents and catalysts.
Scheme 14.

Atroposelective oxidative coupling in synthesis of (+)-gonytolide A.
In 2011, the Waldvogel group presented a electrochemical para-para coupling of guaiacol derivatives which did not require such a blocking approach in order to achieve a selective para-para coupling.33 Using their BDD anodic approach, 17a was coupled selectively to form the para-para dimer 17 in an 83% yield (Scheme 15). Methoxy substituents have a tendency to promote ortho-quinone formation, so the lack of overoxidation products was surprising to the authors. The oxidation of phenols with BDD anodes offers unique regioselective outcomes, typically unattainable by traditional methods that utilize the steric and electronic bias of the substituents. In addition, the use of 1,1,1,3,3,3-hexfluoro-2-propanol (HFIP) solvent was key and has seen continued use in in both chemical, photochemical, and electrochemical oxidative couplings. Its high polarity and intricate hydrogen-bonding network with aggregating fluorine atoms that creates a unique “solvate cage” stabilizes radical intermediates.80
Scheme 15.

Electrochemical para-para coupling of a guaiacol derivative.
The Pappo group disclosed an oxidative phenol coupling toward the synthesis of fucol-derived natural products in which a blocking group was not employed.81 Oxidative coupling of 18a using their FeCl3/t-BuOOt-Bu system formed para-para product sym-Me4-difucol (18i) along with ortho-para unsym-Me4-difucol (18ii) in 33% and 22% yields, respectively (Scheme 16). With both ortho and para sites open, the method suffered from poor regioselectivity with only a slight preference for para-para over ortho-para (1.5:1). This outcome highlights the need in the field for development of more selective couplings in the presence of multiple reactive sites.
Scheme 16.

Iron-catalyzed para-para and ortho-para oxidative coupling of fucol natural products.
Intermolecular oxidative couplings have been well investigated, but intramolecular approaches have been much more elusive. The Kozlowski lab reported an efficient and mild method for furnishing spirohexadienones via vanadium-catalyzed intramolecular para-para oxidative coupling of phenols.82 Typically, this transformation has been performed with stoichiometric oxidants (VOCl3)83, 84 or via oxidation of the aryl ether products85, 86, which follows a different mechanism. Despite there being multiple sites for potential functionalization, only the para-para coupled product was seen in optimization of this reaction. This bond formation creates a quaternary carbon center arising from dearomatization of the para phenol.
With this method, Kozlowski and co-workers completed the first total synthesis of pulchelstyrene D (19). The key step was intramolecular coupling of 19a with vanadium catalyst V2 in the presence of O2 to afford 19b in 82% yield (Scheme 17a). Three additional steps (34% overall) were needed to introduce the alkene backbone which was not tolerated during the oxidative cyclization. The spirocyclic dracaenone natural product, (±)-10-hydroxy-11-methoxydracaenone (20), was synthesized in a 78% yield from 20a using V2 and molecular oxygen as terminal oxidant (Scheme 17b). This compound has been synthesized previously with protected aryl ether approaches87, 88, but this new method eliminates the need for protecting groups. Scheme 17c describes the oxidative coupling of 21a with a V2/O2 system to produce 21b in an 88% yield. Subsequent selective deprotection of the isopropyl ether in the presence of the methyl ether led to the formation of (±)-spirolouveline (21).
Scheme 17.

Intramolecular oxidative coupling to form phenol-dienones.
5.3. ortho-para couplings
The bark of magnolia trees, specifically Magnolia grandiflora, has a long history for its use in Chinese herbal medicine with anti-inflammatory, anti-oxidant, and a plethora of other desirable biological properties. Honokiol (22), a phenolic natural product, was identified as a major active component of these extracts.89–91 Recently, honokiol (22) has found use in potential treatments for cancer and heart disease as well as commercial products like toothpaste. In 2017, the Kozlowski group reported several efficient methods for the total synthesis of honokiol (22) utilizing an ortho-para oxidative coupling early in the synthesis.92 Scheme 18 illustrates one of the approaches in which 22a and 22b are cross-coupled with commercially available Cr-Salen-Cy and then de-tert-buylated to form 22c in 85% yield. Five subsequent steps led to the total synthesis of honokiol (22). Again, the tert-butyl groups serve to block potential reactive positions to limit the regioisomeric outcomes. Overall, this route minimized the use of toxic reagents and long reaction times while providing a lower step count and higher yields.93–95 Building off of this work, Kozlowski and co-workers also utilized Cr-Salen catalysts to synthesize honokiol-type derivatives leading to analogs that are far more potent against commensal oral bacteria.91, 96
Scheme 18.

Oxidative coupling as first step in improved synthesis of honokiol.
5.4. Other C–C couplings of phenols
Some approaches in the literature have accessed unique regioselective products using oxidative coupling or contain highly-oxygenated species in which one specific regioselective outcome cannot be assigned. Continuing with the work of Waldvogel on the coupling of guaiacol derivatives with BDD anodes, the authors were able to assemble some notable regioisomers.33 Scheme 19a shows the coupling of 23a to form meta-meta product 23 in a 45% yield as well as the oxidative coupling of syringol (24a) to access a meta-para dimer 24 in a 44% yield (Scheme 19b). In these cases, the directing effects of the methoxy groups likely compete with those of the hydroxy groups.
Scheme 19.

meta-meta and meta-para electrochemical oxidative couplings.
Polyketide natural products are abundant across fungal and plant species and possess a wealth of biological activities. A oxygenated axially chiral polyketide, chaetoglobin A (25), was recently found to have potent inhibitory effects on the growth of human breast and colon cancer.97 Kozlowski et al. completed the first total synthesis of chaetoglobin A (25) in 12 steps from commercially available 2,6-dimethoxytoluene.98 Direct coupling of the highly oxidized azaphilone heterocycle was unsuccessful. Thus, a vanadium(V)-catalyzed atroposelective oxidative phenol coupling was employed using 25a to establish the key biaryl bond of 25b in 58% yield and 94:6 dr (Scheme 20). Six subsequent synthetic steps led to chaetoglobin A (25). While the catalyst used appears monomeric, the authors postulate that coordination of acetic acid leads to a pseudo-dimer active species.
Scheme 20.

Atroposelective oxidative couplings in total synthesis of chaetoglobin A.
Morphinandienone alkaloids, isolated from the opium poppy Papaver somniferum, have found extensive use in the medical field, often as analgesics.99 Chemists have been trying to tackle these compounds since the elucidation of their biosynthesis, which involves an intramolecular oxidative coupling of reticuline (not shown).60, 100–103 Prior attempts to accomplish these couplings have required the use of stoichiometric oxidants while suffering from low yields and regioselectivities.104–106 Use of a symmetrical reticuline derivative 26a (Scheme 21) reduces complexity, but there are still two regioisomeric products possible via either 8-2′/6′ or 4a-2′/6′ coupling.
Scheme 21.

Intramolecular oxidative synthesis for the synthesis of (±)-salutaridine derivative.
In 2020, the Kozlowski group explored the selective oxidative coupling of a reticuline derivative using their method to construct phenol-dienones.82 With catalyst V2 (as seen in Scheme 17a), oxidative coupling of reticuline analogue 26a formed (±)-salutaridine derivative (26) in a 71% yield via union of 4a-2′/6′ (Scheme 21). The alternate 8-2′/6′ coupling adduct only formed in trace quantities. These catalytic results rival the initial stoichiometric attempts from Schwartz and co-workers in the 1980s.84
5.5. C–C couplings involving aryl ethers
Phenol-aryl ether oxidative couplings have been extensively reported in the literature and are often achieved with the use of hypervalent iodine oxidation chemistry. Several reviews and book chapters have outlined the use of hypervalent iodine in organic chemistry, especially in oxidative C–C bond formation of aryl ethers.107–110 The examples shown below are not comprehensive but seek to show that these couplings can be broadly utilized, from phenol oligomers to some of the most difficult intramolecular cases. These examples also highlight some of the key advances in catalysis; namely, the use of first-row transition metals, hypervalent iodine complexes, nitrite organocatalysts, and electrochemistry. While not specifically discussed, the Wirth group recently disclosed the total synthesis of (−)-galanthamine (not shown) utilizing an electrochemical phenol-aryl ether oxidative coupling.111
Phlorotannins are a class of natural products typically found in algae and other aquatic plants. These compounds are oligomers of phloroglucinol and play an integral role in the structure of cell walls.112 While their biosynthesis has yet to be elucidated, synthetic chemists have used oxidative coupling strategies to connect these compounds. The Pappo group employed sequential iron-catalyzed oxidative couplings to synthesize a variety of fucol natural products.81 A catalytic FeCl3/t-BuOOt-Bu system of the corresponding monomer provided sym-Me4-difucol (18i, see Scheme 16). Using the same reaction system but with a more reactive solvent, HFIP, further cross-coupling with different equivalents of aryl ether 27a generated Me7-fucol (27i) and Me10-tetrafucol A (27ii) in 30% and 36% yields, respectively (Scheme 22). This method furnishes complex natural and unnatural phlorotannin products that are typically inaccessible via prior approaches. By tuning the catalyst loading, solvent, reaction time, and oxidants, control over an oxidative coupling sequence was achieved.
Scheme 22.

Iron-catalyzed method reported by Pappo toward oligomeric natural products.
Identification of mild terminal oxidants that will generate iodine(III) in-situ is the key barrier to development of hypervalent iodine catalysts in oxidative coupling. Kita and co-workers found one solution to this problem in their efforts to form new C–C bonds in the oxidative coupling of phenols and aryl ethers.113 The authors discovered that m-CPBA was a poor oxidant for these conditions, but in-situ generated bis(trifluoroacetyl) peroxide was optimal. Specifically, urea•H2O2 with excess TFAA led to exclusive formation of this desired peroxide which in turn generated the active iodine(III) species. Screening of other iodoarenes revealed that 4-fluoro-1-iodobenzene provided the best overall yield of 76% in the intramolecular coupling 28a to amaryllidaceae alkaloid core 28 (Scheme 23).
Scheme 23.

Iodoarene-catalyzed oxidative coupling in accessing amaryllidaceae alkaloids.
Aporphine and morphine alkaloids have also been synthesized from aryl ether oxidative coupling approaches. Wang and co-workers reported an efficient intramolecular coupling of phenols with aryl ethers using an organocatalytic approach.114 With a sodium nitrite catalyst and molecular oxygen as the terminal oxidant, Wang et al. forged new C–C bonds in 29a derivatives to afford various aporphine alkaloid precursors (29) in 18–99% yields at room temperature (Scheme 24). NO+ serves as the active species in the phenol single-electron oxidation, producing an NO radical which is oxidized by O2 to complete the cycle after deprotonation. Screening of the different tethers revealed that nitrogen and amide-based linkers also performed well. The authors utilized these backbones in various Lewis acid-mediated rearrangement strategies, demonstrating promise for potential access to the aporphine alkaloid core (29i).
Scheme 24.

Intramolecular oxidative phenol coupling with sodium nitrite to access aporphine alkaloid precursors.
(−)-Thebaine (30), a minor component of the opium poppy, is oft used as a synthetic precursor in the industrial-scale synthesis of other morphinandienone derivatives. Opatz and Waldvogel employed electrochemistry to access (−)-thebaine (30) with a regio- and diastereoselective anodic oxidative coupling.35 As shown in Scheme 25a, (R)-30a was coupled using the anodic coupling conditions to furnish (+)-salutaridine analogue (30b) in a 59% yield. (−)-Thebaine (30) was achieved after six additional steps.
Scheme 25.

Electrochemical oxidations in the syntheses of (−)-thebaine and (−)-oxycodone.
The following year, the Opatz and Waldvogel disclosed the total synthesis of another morphinandienone, (−)-oxycodone (31), with their anodic coupling system.36 With their streamlined synthesis to (R)-30a, the authors rapidly synthesized (+)-salutaridine analogue (30b) with slightly modified conditions in a higher yield (69%) as seen in Scheme 25b. A sequence of seven subsequent steps involving a 1O2 cycloaddition, hydrogenation, and removal of protecting groups led to (−)-oxycodone (31). Notably, the phenol groups of (R)-30a need to be protected for this strategy to succeed (see above for comparison with the unprotected phenol Scheme 21).
6. Catalytic C–O oxidative couplings
The formation of C–O bonds via oxidative coupling methods have proved to be much more elusive than their C–C bond counterparts. In a screening study by the Kozlowski group on aerobic oxidative coupling of phenols, a number of metal catalysts (Mn, Cr, Cu) were found to facilitate C–O bond formation from para-substituted phenols to provide the Pummerer ketone adducts.23 Several reviews have focused on diaryl ether formation by means of non-oxidative couplings including many examples in natural products synthesis.115, 116 The following examples focus on oxidative coupling to form diaryl ethers in the synthesis of natural products. Interestingly, copper catalysis has been the most successful in this context to date.
Lumb and co-workers described a series of foundational studies in which semiquinone radical intermediates obtained from copper-catalyzed oxidations were utilized. This method enabled mild ortho-oxygenation of phenols with molecular oxygen followed by formation of new C–O, C–N, and C–S bonds.117 Application to intramolecular couplings allowed the formation of oxindoles via C–N bond formation.118 In further work, Lumb et al. disclosed copper-catalyzed oxidative cross-couplings of ortho-quinones and phenols.119 Reductive work-up accesses highly oxygenated species that are difficult to obtain by other approaches due poor regioselectivity. The authors highlighted the utility of this approach by synthesizing a xanthone natural product with known biological properties, norathyriol (32). Using catalytic CuCl and N,N′-dibenzylethylenediamine (DBED) under aerobic conditions, 32a and 32b are cross-coupled in a 76% yield after reductive work-up and acetylation to form 32c on a gram-scale (Scheme 26). A series of four additional steps led to the total synthesis of norathyriol (32) in only six steps.
Scheme 26.

Copper catalyzed cross-coupling of ortho-quinones with phenols.
Expanding upon this approach, Lumb and co-workers examined more complex systems including the enantioselective total synthesis of (S,S)-tetramethylmagnolamine (33).120 Previous syntheses had relied upon Ullman-type couplings to forge the C–O bond and these methods required high temperatures and often a stoichiometric amount of base.121 Instead, the authors used a catalytic CuPF6/DBED system to oxidatively couple 33a in a 75% yield (Scheme 27). This process first begins with an ortho oxidation of 33a to form the Cu(II)-semiquinone radical shown in Scheme 27, which then undergoes an oxidative C–O coupling. Two additional steps furnished (S,S)-tetramethylmagnolamine (33) resulting in an overall yield of 21% across seven steps. This route provides a more concise access to tetrahydroisoquinoline alkaloid dimers.
Scheme 27.

Dimerization of tetrahydroisoquinoline alkaloids with Cu catalyst.
The Lumb lab also developed a copper-catalyzed oxidative coupling of phenols and catechols which enabled the total synthesis of another tetrahydroisoquinoline alkaloid (S,S)-thalicarpine (34).122 As shown in Scheme 28, substrates 34a and 34b were oxidatively cross-coupled with CuCl and 4-MeO-Py in 76% yield after reductive work-up. (S,S)-Thalicarpine (34) was obtained after two further steps (deprotection/methylation). This key C–O bond formation served to unite two different alkaloids in a selective fashion that avoided homo-coupling. Previous work towards the synthesis of (S,S)-thalicarpine (34) from Kupchan and co-workers employed Ullman-type coupling conditions (required 140 °C) in which a Br had to be pre-installed for C–O bond formation.123
Scheme 28.

Key phenol-catechol oxidative coupling step in total synthesis of (S,S)-thalicarpine.
While many phenolic oligomers are connected via sequential C–C bonds, a variety of phenolic trimers are connected by both C–C and C–O bonds. Pyrolaside B (35), a phenolic trimer natural product isolated from Pyrola rotundifoliai/calliantha and possessing potent anti-bacterial properties, is such a compound.124 This natural product is composed of a A-A-A trimeric system in which all three subunits are identical but are connected via one C–C and one C–O bond. In 2020, the Kozlowski group successfully synthesized pyrolaside B (35) via an oxidative trimerization cascade (Scheme 29).125 CuCl/pyridine and molecular oxygen oxidatively trimerize monomer 35a to afford spiroketal 35b in a 65% yield. Subsequent reduction and deprotection led to the first synthesis of pyrolaside B (35) in five steps and a 16% overall yield. This method forgoes traditional tactics (Ullman and Suzuki couplings) for assembling these types of structures that require prefunctionalization and multiple protection/deprotection steps. The authors also synthesized a variety of analogues and designed an entry to unsymmetrical trimers.
Scheme 29.
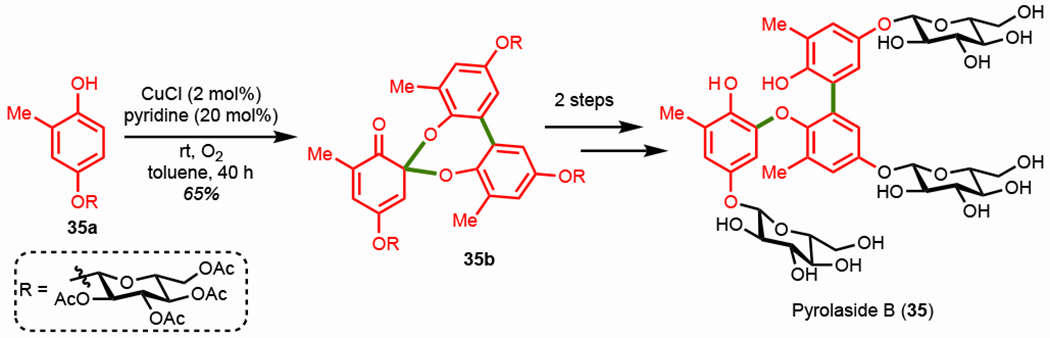
Oxidative coupling trimerization cascade in the synthesis of pyrolaside B.
7. Catalytic and electrochemical alkenyl phenol oxidative couplings
Alkenyl phenols moieties exhibit rich reactivity in natural product biosynthesis and are key components of lignin, a biopolymer that provides structural support for cell walls. The large lignin waste stream from the paper industry has motivated research groups to examine lignin for multiple applications.126, 127 Nature has devised many ways to oxidatively couple these structures to achieve a variety of diverse natural products. Upon single-electron oxidation of alkenyl phenols, radical character can delocalize to the exocyclic alkene portion allowing for a range of different coupling modalities. The Kozlowski group has highlighted the intricacies of alkenyl phenol oxidative coupling and some associated natural products in a recent review.128 These transformations have been difficult to control; stoichiometric oxidants or enzymes result in low yields and/or poor selectivity.129–131
Expanding upon their repertoire of oxidative phenol couplings, Kozlowski and co-workers discovered selective methods for the oxidative homocoupling of alkenyl phenols.132 Monomeric and dimeric vanadium catalysts were studied to bias reactivity toward specific isomers. A dimeric catalyst (similar to one from Scheme 9) was selective for C–O bond formation in the β-O coupling whereas a monomeric catalyst (seen in Scheme 30) led to the β-β coupling. Concentration and solvent also played a critical role in the selectivity where β-β coupling was promoted only in dilute acetonitrile conditions. The β-β couplings arises from C–C oxidative bond formation followed by two nucleophilic additions an XH2 nucleophile such as H2O or RNH2 while β-O results from a C–O oxidative coupling and one nucleophilic addition of water.
Scheme 30.

Oxidative coupling of alkenyl phenols to form the galbelgin core.
With the optimized β-β coupling, Kozlowski and co-workers synthesized derivatives of the galbelgin core (36). These lignin natural products have recently emerged as key targets due to their anti-inflammatory, anti-oxidant, and anti-cancer physiological properties.133 Scheme 30 shows their approach where 36a is homocoupled with a monomeric vanadium catalyst in the presence of H2O/O2 to furnish the galbelgin core (36). The method displayed good functional group tolerance with yields ranging from 17–72%, but poor diastereoselectivity (1.2-1.8:1 dr). Mechanistic experiments and high-throughput experimentation led to a rationale for why this β-β coupling is favored vs the β-O pathway. Solvents with small dipole moments (e.g. 1,4-dioxane) favored the β-O coupling, while large dipole moment solvents (e.g. acetonitrile) generated β-β coupling products. Lower concentrations led to more β-β products while higher concentrations favored the β-O coupling. Increased catalyst loadings also led to a greater selectivity for the β-O pathway over β-β. From this information, β-O coupling likely arises from a metal-bound pathway, while the β-β coupling likely occurs from a homocoupling with a free phenoxyl radical. Additionally, by utilizing other nucleophiles (e.g. anilines) instead of water, the authors were able to complete the synthesis of pyrrolidine analogues via similar β-β coupling.
Liu and co-workers reported the biomimetic oxidative cross-coupling of alkenyl phenols and phenols with an aerobic copper approach.134 An overview of methods to construct neolignan backbones with phenol oxidative couplings has been summarized by Papaioannou et al.135 Biosynthetically, these couplings are often accomplished by iron- and copper-based enzymes where homocoupling is typically observed. Scheme 31a displays the general strategy of Liu where catalytic Cu(OAc)2/air allowed for the synthesis of natural product licarin A (37) in a 60% yield and >20:1 dr from commercially available 37a in one step (Scheme 31a). This approach could also be used to enable cross-couplings (Scheme 31b). Specifically, 38a and 38b can be selectively cross-coupled to form 8-O-4′ neolignan analogues (38i) when EtOAc was used as solvent. With HFIP/PhCF3, 8-5′ neolignan analogues (38ii) were obtained. However, the scope is limited to 4-amino variants of 38b as phenols bearing 4-OMe or 3-NMe2 were unsuccessful.
Scheme 31.

Cross-coupling of phenols and alkenyl phenols in synthesis of neolignan analogues.
Alkenyl phenols can lead to a diverse array of natural products depending on the location of the alkenyl substituent relative to the phenol. Carpanone (39), extracted from carpano trees (Cinnamonmum sp.) in the south Pacific, highlights one of these alternate pathways. Biosynthetically, the assembly of carpanone (39) is proposed to occur in a single step, setting all five stereocenters at once.136 In the 1970s, the Chapman group published the first biomimetic synthesis of carpanone (39) where they used a palladium(II) catalyst to furnish the compound in a single step with a 46% yield as a single diastereomer.137 The observed diastereoselectivity arises from selective endo facial approach during an inverse-electron demand Diels-Alder reaction. This classic total synthesis highlights a rapid assembly of complexity and serves as an inspiration for highly efficient total syntheses.
Due to the very facile oxidation of the alkenyl phenol precursor to carpanone (39) (which even occurs slowly even under ambient conditions), many groups have achieved the oxidative synthesis of carpanone (39) using a variety of first-row transition metal and photochemical approaches.138, 139 For example, Lindsley and co-workers developed a catalytic CuCl2/(−)-sparteine oxidative coupling of benzoxanthenone scaffolds to afford carpanone (39).140 As illustrated in Scheme 32a, the authors homocoupled alkenyl phenol 39a with their optimized conditions to form 39b which then underwent a rapid Diels-Alder [4+2] cycloaddition to form carpanone (39) in 91% yield as a single diastereomer. They were also able to synthesize other benzoxanthenone derivatives in high yields with this one-pot diastereoselective approach. Despite screening a range of chiral amine ligands, the authors saw little to no enantioselectivity (<5% ee).
Scheme 32.

Oxidative coupling approaches for synthesis of carpanone.
However, the Kozlowski group recently developed a vanadium(V)-catalyzed system that induces modest levels of enantioselectivity in the constructions of benzoxanthenone backbones, a feat that was unachievable with prior methods.141 Due to the sensitive nature of the substrate, identification of a terminal oxidant that would not directly react with the substrates proved challenging. Ultimately, AgCl was identified as an oxidant that allows the vanadium-catalyzed oxidation to occur selectively, but it was difficult to scale the reaction with this heterogenous oxidant. In this work, the authors were able to access unnatural exo carpanone (39i) in a 64% yield and 36% ee with CuCl2/(−)-sparteine (Scheme 32b). The natural carpanone (39) (endo) was also obtained in 20% yield, but only 6% ee. It was concluded that the vanadium catalyst provided moderate control of facial selectivity in the initial C–C bond forming event but remained coordinated for the subsequent Diels-Alder reaction directing toward an exo cyclization mode. A strong correlation (R2=0.88) was found between reaction solvent dielectric and the observed enantioselectivity, where a lower solvent dielectric enhances the enantioselectivity. Studies on other derivatives led to 44–50% yields and 36–49% ee. A recent publication from the Kozlowski group detailed a photochemical oxidative coupling of phenols and alkenyl phenols using a heterogenous TiO2 catalyst for the formation of the aforementioned natural products licarin A (37) and carpanone (39).142
8. Catalytic and electrochemical oxidative coupling of phenolic-type compounds
Phenolic-type compounds can also engage in oxidative homo- and cross-coupling reactions. Such methods are often more robust than conventional cross-coupling approaches across broader areas of chemical space. Once oxidized, carbon-centered phenoxyl radicals can be captured by a variety of electrophilic species to generate complex products. Other redox active moieties, like indoles, can also oxidatively couple to form C–C bonds. This ensuing section will focus on advances in oxidative coupling of hydroxycarbazoles, amines, indoles and vinylic species including alkynes and carbonyls.
8.1. Carbazole couplings
Carbazoles are nitrogen-containing tricyclic aromatic structures that are often found in natural products and agrochemicals. Similar to naphthols, these moieties can form axially chiral biaryl products when coupled. Derivatives of carbazoles, more specifically, hydroxycarbazoles contain phenolic backbones and can participate in similar redox chemistry to phenols. Biaryl hydroxycarbazoles are typically furnished via traditional cross-coupling techniques involving pre-functionalization with a halide or other electrophiles prior to coupling. Recently, oxidative techniques have been leveraged to enable the oxidative coupling of hydroxycarbazoles.
In 2015, Kozlowski and co-workers disclosed the first vanadium-catalyzed regioselective oxidative couplings of hydroxycarbazoles using molecular oxygen as the terminal oxidant to furnish various dimeric 2-hydroxycarbazoles.143 This study fueled their 2017 report on the asymmetric oxidative coupling of 2-hydroxycarbazoles as well as phenols.28 With N-benzyl protecting groups, the authors oxidatively coupled 40a derivatives to produce dimeric 2-hydroxycarbazoles (40) in up to 91% yields and 96% ee with monomeric vanadium(V) complexes (Scheme 33a). Studies into the ligand/catalyst design revealed that the addition of a Brønsted/Lewis acid increased both reactivity and enantioselectivity. Monomeric catalysts with these acid additives performed comparably to dimeric complexes. A follow-up experimental and computational study indicated that the additives help the catalysts aggregate to form “pseudo-dimeric” structures which aid in the enantioselectivity of the reaction.144
Scheme 33.

Oxidative naphthol coupling to afford carbazole dimers.
Takizawa and co-workers utilized a different monomeric chiral vanadium(V) catalysts to access a bis-2-hydroxycarbazole natural product, (+)-bi-2-hydroxy-3-methylcarbazole (41).145 Starting from cyclohexanone and an aniline derivative, the authors achieved 41a in two facile steps (Scheme 33b). The authors were able to oxidatively couple 41a to form (+)-bi-2-hydroxy-3-methylcarbazole (41) in 44–70% yields and 16–21% ee. The poor enantioselectivity was attributed to the ease of rotation about the biaryl bond when the nitrogens were not protected as erosion in the enantiomeric excess was seen at room temperature.
Hydroxycarbazole natural products sorazolon E (42a) and (+)-sorazolon E2 (42), extracted from Sorangium cellulosum myxobacteria, have been found to possess antibacterial and cytotoxic properties against mouse fibroblasts.146 In 2017, Sasai and Takizawa reported the first enantioselective coupling of 3-hydroxycarbazoles resulting in the total synthesis of (+)-sorazolon E2 (42).147 The use of a chiral vanadium(V) catalyst V3 enabled the homocoupling of sorazolon E (42a) in 71% yield and 60% ee (90% ee after recrystallization) to furnish (+)-sorazolon E2 (42) (Scheme 33c). Ultimately, the authors assembled sorazolon E (42a) in an efficient three step process (condensation/aromatization/C–C coupling) whereas prior art typically required up to eight steps with the use of stoichiometric toxic oxidants. It is important to note that Sasai and co-workers were able to complete this coupling in the presence of a basic nitrogen (free carbazole). These authors also reported a related enantioselective cross-coupling of 3-hydroxycarbazoles and naphthols.148
These authors were also able to oxidatively couple 4-hydroxycarbazoles with a dinuclear vanadium(V) complex in high yield and enantioselectivity (Scheme 33d).149 Specifically, the V3 catalyst affords 4-hydroxycarbazole dimers (43) under mild conditions with up to quantitative yields and 90% ee. Substrates containing hydrogen or methyl groups on the nitrogen (R3 on 43a) were compatible with these conditions and led to the highest enantioselectivity, but utilization of aryl, benzyl, or allyl substituents eroded selectivity. Of note, mononuclear complexes did not impart significant enantiocontrol in these reactions.
8.2. Amine couplings
Lumb and co-workers disclosed the synthesis of benzoxazinone and benzoxazole backbones utilizing a biomimetic copper-catalyzed oxidative coupling of phenols and amines.150 Similar to prior work from the Lumb group, the reactivity of an ortho-quinone intermediate was harnessed to forge new C–O and C–N bonds via an amine condensation in one-pot. A sterically demanding catalyst enabled regioselective ortho-oxygenation of symmetrical and unsymmetrical phenols to form ortho-aminophenols. Using [Cu(MeCN)4]PF6/DBED and molecular oxygen, the authors condensed phenol 44a and indole amine 44b to form 44c which underwent lactonization to form 44d in a 72% yield (Scheme 34). The benzoxazine natural product cephalandole A (44) was formed after subsequent de-tert-butylation in a 71% yield. This method proceeded with mild conditions using an earth-abundant metal (Cu) catalyst and H2O as the only stoichiometric byproduct.
Scheme 34.

Oxidative coupling of amines and phenols in the synthesis of cephalandole A.
8.3. Indole couplings
Indoles are N-heterocyclic aromatic structures with a 5-membered pyrrole fused to a benzene ring. These moieties are prevalent in biologically active natural products as well as pharmaceuticals and have remained as one of the most common scaffolds in medicinal chemistry. Indole-containing compounds have profound activity at the 5-HT serotonin receptors, including psychedelic natural products like psilocybin and ergot alkaloids which have recently garnered interest for potential treatment of depression and PTSD.151–153
Electrochemical methods have been pioneered to tackle difficult oxidative couplings between two unsymmetrical partners. Both phenols and indoles have sufficiently low oxidation potentials to facilitate a union in an oxidative coupling. The Harran lab utilized electrolytic conditions to accomplish an intramolecular oxidative coupling of a phenols with an indole en route to cancer therapeutic, DZ-2384 (45).154 DZ-2384 (45) has garnered interest from the synthetic community as it showed a significant improvement in anti-cancer properties from the original natural product scaffold of (−)-diazonamide A (not shown). Conventional chemical methods involving hypervalent iodine failed to achieve the macrocyclic core of DZ-2384 (45) as the authors uncovered a competing oxidation of the phenolic portion (shown in red) in 45a to a spirocyclohexadienone (not shown). Anodic conditions were devised that avoided over-oxidation of the phenol and facilitated union of the two fragments. As shown in Scheme 35, oxidation of 45a at a potential of +1.6 V with portion-wise addition of (NH4)2CO3 gave DZ-2384 (45) in a 35% yield and 2.3:1 dr after 56 h. Overall, the authors were able to synthesize DZ-2384 (45) in 13 total steps and 6% overall yield with a key oxidative coupling as the final step to achieve the macrocyclic core.
Scheme 35.

Electrochemical oxidative coupling of indole and phenol to synthesize cancer therapeutic drug DZ-2384.
In a recent disclosure by Xu and co-workers, the spiroindimcin natural product family was accessed using a chemoenzymatic approach involving a biomimetic intramolecular oxidative coupling of two indole units.155 Spiroindimcins, isolated from deep-sea Streptomyces strains, are biosynthetically assembled via oxidative dimerization of tryptophan and have anti-cancer and anti-parasitic biological activities. Previous synthetic endeavors of this natural product family have relied on transition metal-catalyzed cross-coupling reactions to forge the spirocyclic skeleton.156, 157 In a different approach, Xu et al. synthesized the spiroindimcin D core (46i) in a 35% yield using catalytic trichloroisocyanuric acid (TCCA) and the spiroindimcin A core (46ii) with stoichiometric NIS/AgSbF6 conditions in a 98% yield (Scheme 36). With these regioselective 3′-2″ and 3′-4′ oxidative couplings, the authors were able to synthesize (±)-spiroindimcin A, D, G, and H (not shown) after one or two additional synthetic procedures. The common intermediate 46a was produced via an enzymatic dimerization of L-tryptophan.
Scheme 36.

Total synthesis of spiroindimcin cores via indole oxidative coupling.
8.4. Alkyne and alkene couplings
The use of low-energy visible light has emerged as a powerful synthetic technique to furnish new chemical bonds with mild, selective transformations. The Hwang lab reported the oxidative coupling of phenols and terminal alkynes using photocatalytic conditions and an earth-abundant copper catalyst.158 In the presence of O2, C≡C triple bonds are cleaved to assemble alkyl and aryl ketones, which are valuable functional groups in natural products as well as useful synthetic intermediates. In this reaction, the phenol is first oxidized to the para-quinone with CuCl/O2 and then undergoes a Paterno-Buchi [2+2] cycloaddition with a Cu(II)-acetylide which undergoes subsequent ring opening and addition with O2 to form the product after loss of CO2 (Scheme 37a). As suggested by the mechanism, the scope is limited to phenols with open para positions. This method allowed the authors to synthesize two pharmaceuticals in a high yielding two-step process. Alkyne 47a and phenol 47b were coupled using the photocatalytic conditions in a 82% yield and then subsequently engaged in an SN2 reaction to afford pitofenone (47) in a 72% overall yield (Scheme 37b). A similar approach was taken to form fenofibrate (48), a cholesterol drug, using 48a and 47b in a 76% overall yield (Scheme 37c).
Scheme 37.

Photocatalytic oxidative coupling of phenols and terminal alkynes.
In 2013, Pappo and co-workers developed an iron-catalyzed oxidative cross-coupling of phenols and alkenes for the construction of 2,3-dibenzofurans (49).159 These moieties are present in a plethora of natural products and it is proposed that nature constructs these backbones in a similar oxidative fashion.160 Previous methods have been established for the synthesis of 2,3-dibenzofurans (49), but they often rely upon stoichiometric metal oxidants with poor selectivity outcomes. Using a FeCl3/t-BuOOt-Bu/DCE system, the authors were able to couple a variety of phenols and naphthols (49a) with styrene and stilbene derivatives (49b) to furnish 2,3-dibenzofurans (49) in 27–94% yields (Scheme 38). For further examples of the use of oxidative couplings in the assembly of dihydrobenzofuran natural products, see review by Zhao et al.161
Scheme 38.

Photocatalytic oxidative coupling of phenols and terminal alkynes.
8.5. 1,3-Dicarbonyl couplings
A subsequent study from Pappo et al. in 2015 examined catalytic oxidative coupling of phenols and β-dicarbonyls to furnish benzofurans.162 The authors observed a dramatic enhancement in cross-coupling selectivity with fluoroalcohol solvents (HFIP/TFE) which is hypothesized to arise from their ability to stabilize radical cation intermediates. Partners 50a and 50b were oxidatively coupled using a Fe(ClO4)3 hydrate catalyst in TFE/DCE in 40% yield (Scheme 39). Three additional synthetic procedures resulted in the first total synthesis of natural product 2″-dehydroxycalodenin B (50) in a 17% overall yield. While a stoichiometric loading of catalyst was used in the total synthesis, the substrate scope disclosed by Pappo and co-workers used only a 10 mol% loading.
Scheme 39.
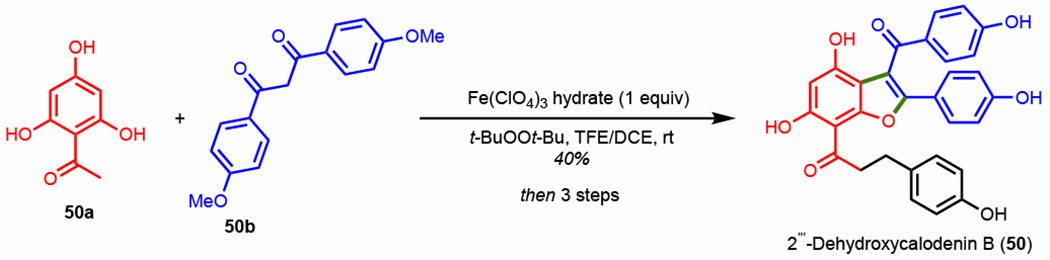
Oxidative coupling of phenols and β-dicarbonyl compounds in the synthesis of 2‴-dehydroxycalodenin B.
Expanding upon their work in iron-catalyzed oxidative couplings of phenols and β-dicarbonyls, Pappo et al. were able to access coumestrol analogues (51) for a structural activity relationship (SAR) study against breast cancer cell lines.163 In their synthesis of a library of coumestrol derivatives (51), the Pappo group leveraged a two-step approach involving the assembly of a 3-ester benzofuran intermediate followed by lactone formation via an intramolecular esterification. Scheme 39 illustrates their approach in which 51a and 51b are oxidatively coupled to form 51c derivatives in 51–77% yields using FeCl3 and t-BuOOt-Bu. Subsequent BBr3 deprotection of the methoxy groups and lactonization led to coumestrol analogues (51) in 67–97% yields. For the oxidative coupling, the use of molecular oxygen as the terminal oxidant led to inferior yields in comparison to t-BuOOt-Bu. Additionally, the authors were able to complete the gram scale total synthesis of natural product coumestrol (not shown) in a 59% overall yield.
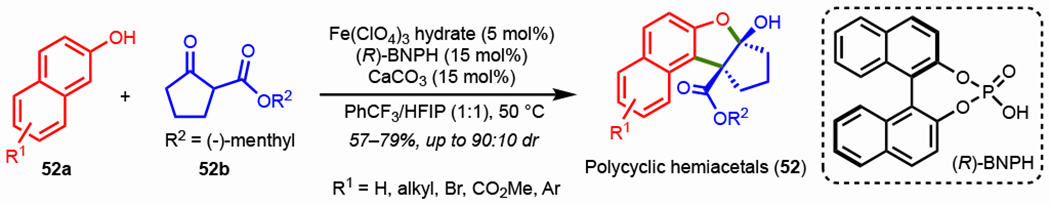
The Pappo group also reported a key advance with the stereoselective oxidative coupling of 2-naphthols with chiral β-ketoesters using a chiral iron phosphate complex to synthesize polycyclic hemiacetal analogues.164 Benzofuran-based polycyclic acetals are key backbones in many natural products with promising biological properties.165 The authors screened a diverse set of phosphoric acid ligands with an Fe(ClO4)3 hydrate catalyst to achieve both high yield and diastereoselectivity. Mechanistic studies revealed that the active catalyst was an iron monophosphate complex. Scheme 40 highlights the optimized conditions using an (R)-BNPH ligand in PhCF3/HFIP to couple derivatives of 52a and 52b to form polycyclic hemiacetals (52) in 57–79% yields and up to 90:10 dr. Motivated by this work, Sasai and Takizawa disclosed a catalytic enantioselective oxidative coupling between 3-hydroxycarbazoles and achiral β-ketoesters with their vanadium(V) catalysts.148
Scheme 40.

Synthesis of coumestrol derivatives via oxidative coupling of β-ketoesters with phenols.
The Pappo lab also developed an iron-catalyzed oxidative coupling of naphthols/phenols with β-ketoesters to favor the formation of spirolactones over polycyclic hemiacetals.166 The authors discovered that elevated temperatures (above 70 °C) facilitate the trans-esterification step required to form spirolactones and avoid formation of polycyclic hemiacetals using traditional FeCl3/t-BuOOt-Bu/DCE conditions. The utility of this method was highlighted in the biomimetic synthesis of the lachnanthospirone core (53), a pigment isolated from Lachnanthes tinctoria. The original isolation paper hypothesized that lachnanthospirone (not shown) could be biosynthetically assembled via an oxidative coupling of a phenol and β-ketoester.167 To probe the synthetic feasibility, Pappo and co-workers coupled 53a and 53b using their optimized oxidative conditions to afford the lachnanthospirone core (53) in a 43% yield (Scheme 41) with sequential C–C and C–O bond formation.
Scheme 41.
Asymmetric oxidative coupling of 2-naphthols and β-ketoesters with iron phosphate catalyst.
9. Concluding remarks
In conclusion, oxidative phenol couplings have significantly aided synthetic chemists in their quest to synthesize elusive natural products. Tremendous strides have been made within the last decade using innovate chemistries to push the boundaries and reach new chemical space. These powerful bond-forming reactions, requiring no prefunctionalization, have found wide applications with coupling diverse partners. Chemical approaches utilizing stoichiometric oxidants have fared well in the assembly of complex natural products, but their ultimate utility is limited by the presence of sensitive functional groups. Enzymatic oxidative couplings have been recently leveraged to conduct bench-top reactions with biological precision. Catalytic oxidative methods have combined atom economy, reaction efficiency, and functional group tolerance to achieve selective transformations in furnishing natural products. Of the transition metal catalysts, those derived from iron, vanadium, copper, and chromium have emerged as the most successful catalyst types in the field. Electrochemistry has gained prominence in oxidative coupling by succeeding under mild conditions and with high selectivity. Additionally, oxidative photoredox chemistry has recently proven advantageous in oxidative phenol couplings by using low-energy visible light.
While the regioselectivity of phenol homocoupling has been carefully analyzed in several studies, regioselective cross-coupling approaches remain a challenge and often rely upon the use of blocking/activating groups. The installation and subsequent removal of such moieties increases synthetic step counts. Discovering approaches that reduce these inefficiencies and allow direct regioselective cross-couplings will lead to shorter and even more efficient syntheses. Expanding phenol oxidative couplings to new heteroarenes is also of interest to the community. These bonds are often forged by stoichiometric Lewis acids and transition metals, which can create roadblocks in drug development on industrial scale. Overall, the oxidative coupling of phenols and phenolic-type compounds have enabled synthetic chemists to mimic biological pathways in the synthesis of complex natural products. Further advances will enhance efficiency and allow for use with non-natural substrates to access greater portions of chemical space.
Scheme 42.

Iron-catalyzed coupling of β-ketoesters with naphthols in synthesis of lachnanthospirone core.
11. Acknowledgements
We are grateful to the NSF (CHE2102626) and the NIH (R35 GM131902) for financial support of this research.
Footnotes
10. Conflicts of interest
There are no conflicts to declare.
12. References
- 1.Pummerer R and Frankfurter F, Ber. Dtsch. Chem. Ges, 1914, 47, 1472–1493. [Google Scholar]
- 2.Scott AI, Q. Rev. Chem. Soc, 1965, 19, 1. [Google Scholar]
- 3.Roche SP and Porco JA, Angew. Chem. Int. Ed, 2011, 50, 4068–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan R, Ding Q and Ye Y, Synthesis, 2012, 45, 1–16. [Google Scholar]
- 5.Huck CJ, Boyko YD and Sarlah D, Nat. Prod. Rep, 2022, 39, 2231–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassall CH and Lewis JR, J. Chem. Soc, 1961, 2312–2315. [Google Scholar]
- 7.O’Donovan DG and Horan H, J. Chem. Soc. C, 1969, 1737–1739. [DOI] [PubMed] [Google Scholar]
- 8.Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K and Robinson JA, J. Am. Chem. Soc, 2007, 129, 6887–6895. [DOI] [PubMed] [Google Scholar]
- 9.Tang S and Vincent G, Chem. Eur. J, 2021, 27, 2612–2622. [DOI] [PubMed] [Google Scholar]
- 10.Yeung CS and Dong VM, Chem. Rev, 2011, 111, 1215–1292. [DOI] [PubMed] [Google Scholar]
- 11.Kozlowski MC, Acc. Chem. Res, 2017, 50, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arkley V, Dean FM, Robertson A and Sidisunthorn P, J. Chem. Soc, 1956, 2322–2328. [Google Scholar]
- 13.Taguchi H, Sankawa U and Shibata S, Chem. Pharm. Bull, 1969, 17, 2054–2060. [DOI] [PubMed] [Google Scholar]
- 14.Barton DHR, Deflorin AM and Edwards OE, J. Chem. Soc, 1956, 530–534. [Google Scholar]
- 15.Pummerer R, Puttfarcken H and Schopflocher P, Ber. Dtsch. Chem. Ges, 1925, 58, 1808–1820. [Google Scholar]
- 16.Barton DHR, Deflorin AM and Edwards OE, Chem. Ind, 1955, 1039–1040. [Google Scholar]
- 17.Barton DHR and Cohen T, in Fetschrift ed. Stoll A, Basel, Birkhauser, 1957, pp. 117–143. [Google Scholar]
- 18.Wu J and Kozlowski MC, ACS Catal, 2022, 12, 6532–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlowski MC, Morgan BJ and Linton EC, Chem. Soc. Rev, 2009, 38, 3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libman A, Shalit H, Vainer Y, Narute S, Kozuch S and Pappo D, J. Am. Chem. Soc, 2015, 137, 11453–11460. [DOI] [PubMed] [Google Scholar]
- 21.Shalit H, Libman A and Pappo D, J. Am. Chem. Soc, 2017, 139, 13404–13413. [DOI] [PubMed] [Google Scholar]
- 22.Shalit H, Dyadyuk A and Pappo D, J. Org. Chem, 2019, 84, 1677–1686. [DOI] [PubMed] [Google Scholar]
- 23.Lee YE, Cao T, Torruellas C and Kozlowski MC, J. Am. Chem. Soc, 2014, 136, 6782–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nieves-Quinones Y, Paniak TJ, Lee YE, Kim SM, Tcyrulnikov S and Kozlowski MC, J. Am. Chem. Soc, 2019, 141, 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esguerra KVN, Fall Y, Petitjean L and Lumb J-P, J. Am. Chem. Soc, 2014, 136, 7662–7668. [DOI] [PubMed] [Google Scholar]
- 26.Schön F, Kaifer E and Himmel HJ, Chem. Eur. J, 2019, 25, 8279–8288. [DOI] [PubMed] [Google Scholar]
- 27.Takizawa S, Kodera J, Yoshida Y, Sako M, Breukers S, Enders D and Sasai H, Tetrahedron, 2014, 70, 1786–1793. [Google Scholar]
- 28.Kang H, Lee YE, Reddy PVG, Dey S, Allen SE, Niederer KA, Sung P, Hewitt K, Torruellas C, Herling MR and Kozlowski MC, Org. Lett, 2017, 19, 5505–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niederer KA, Gilmartin PH and Kozlowski MC, ACS Catal, 2020, 10, 14615–14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar A, Sasai H and Takizawa S, Acc. Chem. Res, 2022, 55, 2949–2965. [DOI] [PubMed] [Google Scholar]
- 31.Lumb J-P and Esguerra K, Synlett, 2015, 26, 2731–2738. [Google Scholar]
- 32.Kirste A, Schnakenburg G, Stecker F, Fischer A and Waldvogel SR, Angew. Chem. Int. Ed, 2010, 49, 971–975. [DOI] [PubMed] [Google Scholar]
- 33.Kirste A, Schnakenburg G and Waldvogel SR, Org. Lett, 2011, 13, 3126–3129. [DOI] [PubMed] [Google Scholar]
- 34.Kirste A, Elsler B, Schnakenburg G and Waldvogel SR, J. Am. Chem. Soc, 2012, 134, 3571–3576. [DOI] [PubMed] [Google Scholar]
- 35.Lipp A, Ferenc D, Gütz C, Geffe M, Vierengel N, Schollmeyer D, Schäfer HJ, Waldvogel SR and Opatz T, Angew. Chem. Int. Ed, 2018, 57, 11055–11059. [DOI] [PubMed] [Google Scholar]
- 36.Lipp A, Selt M, Ferenc D, Schollmeyer D, Waldvogel SR and Opatz T, Org. Lett, 2019, 21, 1828–1831. [DOI] [PubMed] [Google Scholar]
- 37.Geffe M and Opatz T, Org. Lett, 2014, 16, 5282–5285. [DOI] [PubMed] [Google Scholar]
- 38.Baker Dockrey SA, Lukowski AL, Becker MR and Narayan ARH, Nat. Chem, 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez Benítez A, Tweedy SE, Baker Dockrey SA, Lukowski AL, Wymore T, Khare D, Brooks CL, Palfey BA, Smith JL and Narayan ARH, ACS Catal, 2019, 9, 3633–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zetzsche LE and Narayan ARH, Nat. Rev. Chem, 2020, 4, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zetzsche LE, Yazarians JA, Chakrabarty S, Hinze ME, Murray LAM, Lukowski AL, Joyce LA and Narayan ARH, Nature, 2022, 603, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hüttel W and Müller M, ChemBioChem, 2007, 8, 521–529. [DOI] [PubMed] [Google Scholar]
- 43.Mazzaferro LS, Hüttel W, Fries A and Müller M, J. Am. Chem. Soc, 2015, 137, 12289–12295. [DOI] [PubMed] [Google Scholar]
- 44.Hüttel W and Müller M, Nat. Prod. Rep, 2021, 38, 1011–1043. [DOI] [PubMed] [Google Scholar]
- 45.Baker Dockrey SA and Narayan ARH, Org. Lett, 2020, 22, 3712–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Péault L, Planchat A, Nun P, Le Grognec E and Coeffard V, J. Org. Chem, 2021, 86, 18192–18203. [DOI] [PubMed] [Google Scholar]
- 47.Carson MC, Orzolek BJ and Kozlowski MC, Org. Lett, 2022, 24, 7250–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Afanasenko A, Kavun A, Thomas D and Li CJ, Chem. Eur. J, 2022, 28. [DOI] [PubMed] [Google Scholar]
- 49.Denizot N, Pouilhès A, Cucca M, Beaud R, Guillot R, Kouklovsky C and Vincent G, Org. Lett, 2014, 16, 5752–5755. [DOI] [PubMed] [Google Scholar]
- 50.Poupon E, Evanno L, Vincent G, Denizot N, Lachkar D and Kouklovsky C, Synthesis, 2018, 50, 4229–4242. [Google Scholar]
- 51.Morimoto K, Chem. Pharm. Bull, 2019, 67, 1259–1270. [DOI] [PubMed] [Google Scholar]
- 52.Tissot M, Phipps RJ, Lucas C, Leon RM, Pace RDM, Ngouansavanh T and Gaunt MJ, Angew. Chem. Int. Ed, 2014, 53, 13498–13501. [DOI] [PubMed] [Google Scholar]
- 53.Taber DF, Neubert TD and Rheingold AL, J. Am. Chem. Soc, 2002, 124, 12416–12417. [DOI] [PubMed] [Google Scholar]
- 54.Magnus P, Sane N, Fauber BP and Lynch V, J. Am. Chem. Soc, 2009, 131, 16045–16047. [DOI] [PubMed] [Google Scholar]
- 55.Richieu A, Peixoto PA, Pouységu L, Deffieux D and Quideau S, Angew. Chem. Int. Ed, 2017, 156, 13833–13837. [DOI] [PubMed] [Google Scholar]
- 56.Michihata N, Kaneko Y, Kasai Y, Tanigawa K, Hirokane T, Higasa S and Yamada H, J. Org. Chem, 2013, 78, 4319–4328. [DOI] [PubMed] [Google Scholar]
- 57.Wakamori S, Osada R, Matsumoto S, Kusuki R and Murakami K, Org. Lett, 2022, 24, 8130–8135. [DOI] [PubMed] [Google Scholar]
- 58.Peters DS, Romesberg FE and Baran PS, J. Am. Chem. Soc, 2018, 140, 2072–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Z-W, Salamonczyk GM, Han K, Machiya K and Sih CJ, J. Org. Chem, 1997, 62, 6700–6701. [Google Scholar]
- 60.Zhang X and Li S, Nat. Prod. Rep, 2017, 34, 1061–1089. [DOI] [PubMed] [Google Scholar]
- 61.Liu W-T, Kersten RD, Yang Y-L, Moore BS and Dorrestein PC, J. Am. Chem. Soc, 2011, 133, 18010–18013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Molinaro C, Kawasaki Y, Wanyoike G, Nishioka T, Yamamoto T, Snedecor B, Robinson SJ and Gosselin F, J. Am. Chem. Soc, 2022, 144, 14838–14845. [DOI] [PubMed] [Google Scholar]
- 63.Dumas VG, Defelipe LA, Petruk AA, Turjanski AG and Marti MA, Proteins: Struct. Funct. Genet, 2014, 82, 1004–1021. [DOI] [PubMed] [Google Scholar]
- 64.Cochrane JR, White JM, Wille U and Hutton CA, Org. Lett, 2012, 14, 2402–2405. [DOI] [PubMed] [Google Scholar]
- 65.Zhu X, Mcatee CC and Schindler CS, Org. Lett, 2018, 20, 2862–2866. [DOI] [PubMed] [Google Scholar]
- 66.Girol C. Gil, Fisch KM, Heinekamp T, Günther S, Hüttel W, Piel J, Brakhage AA and Müller M, Angew. Chem. Int. Ed, 2012, 51, 9788–9791. [DOI] [PubMed] [Google Scholar]
- 67.Zetzsche LE, Chakrabarty S and Narayan ARH, ACS Chem. Bio, 2022, 17, 2986–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Podlesny EE and Kozlowski MC, Org. Lett, 2012, 14, 1408–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Podlesny EE and Kozlowski MC, J. Org. Chem, 2013, 78, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai J, Liu Y, Zhou Y-D and Nagle DG, J. Nat. Prod, 2007, 70, 1824–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Podlesny EE and Kozlowski MC, J. Nat. Prod, 2012, 75, 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ben‐Lulu M, Gaster E, Libman A and Pappo D, Angew. Chem. Int. Ed, 2020, 59, 4835–4839. [DOI] [PubMed] [Google Scholar]
- 73.Phillips M, Science, 1931, 73, 568–570. [DOI] [PubMed] [Google Scholar]
- 74.Iwahara K, Honda Y, Watanabe T and Kuwahara M, Appl. Microbiol. Biotechnol, 2000, 54, 104–111. [DOI] [PubMed] [Google Scholar]
- 75.Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, González A, Cully D, Bramhill D and Singh S, J. Biol. Chem, 2003, 278, 44424–44428. [DOI] [PubMed] [Google Scholar]
- 76.Park YS, Grove CI, González-López M, Urgaonkar S, Fettinger JC and Shaw JT, Angew. Chem. Int. Ed, 2011, 50, 3730–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shaw J, Grove C and Fettinger J, Synthesis, 2012, 44, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng J, Yang XB, Lang S, Zhang J and Yu XQ, Tetrahedron Lett, 2013, 54, 355–357. [Google Scholar]
- 79.Wu X, Iwata T, Scharf A, Qin T, Reichl KD and Porco JA, J. Am. Chem. Soc, 2018, 140, 5969–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hollóczki O, Berkessel A, Mars J, Mezger M, Wiebe A, Waldvogel SR and Kirchner B, ACS Catal, 2017, 7, 1846–1852. [Google Scholar]
- 81.Vershinin V, Dyadyuk A and Pappo D, Tetrahedron, 2017, 73, 3660–3668. [Google Scholar]
- 82.Gilmartin PH and Kozlowski MC, Org. Lett, 2020, 22, 2914–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwartz MA, Rose BF, Holton RA, Scott SW and Vishnuvajjala B, J. Am. Chem. Soc, 1977, 99, 2571–2578. [Google Scholar]
- 84.Schwartz MA and Zoda MF, J. Org. Chem, 1981, 46, 4623–4625. [Google Scholar]
- 85.Schubert M, Wehming K, Kehl A, Nieger M, Schnakenburg G, Fröhlich R, Schollmeyer D and Waldvogel SR, Eur. J. Org. Chem, 2016, 2016, 60–63. [Google Scholar]
- 86.Hamamoto H, Anilkumar G, Tohma H and Kita Y, Eur. J. Chem, 2002, 8, 5377–5383. [DOI] [PubMed] [Google Scholar]
- 87.Blasko G and Cordell GA, Heterocycles, 1988, 27, 445–452. [Google Scholar]
- 88.Li M-M, Wu Y and Liu B, Org. Lett, 2019, 21, 575–578. [DOI] [PubMed] [Google Scholar]
- 89.Greenberg M, Dodds M and Tian M, J. Agric. Food Chem, 2008, 56, 11151–11156. [DOI] [PubMed] [Google Scholar]
- 90.Sakaue Y, Domon H, Oda M, Takenaka S, Kubo M, Fukuyama Y, Okiji T and Terao Y, Microbiol. Immunol, 2016, 60, 10–16. [DOI] [PubMed] [Google Scholar]
- 91.a) Ochoa CR, Roenfanz HF and Kozlowski MC, ChemMedChem, 2022, 17, e202100783. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roenfanz HF, Ochoa CR and Kozlowski MC, ChemMedChem, 2023, 18, e202200521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jaracz S, Kozlowski MC, Lee YE, Kim SM, WO/2017/070568, Improved Synthesis of Honokiol, 2017. Apr 27.
- 93.Reddy BVS, Rao RN, Reddy NSS, Somaiah R, Yadav JS and Subramanyam R, Tetrahedron Lett, 2014, 55, 1049–1051. [Google Scholar]
- 94.Srinivas J, Singh PP, Varma YK, Hyder I and Kumar HMS, Tetrahedron Lett, 2014, 55, 4295–4297. [Google Scholar]
- 95.Harada K, Arioka C, Miyakita A, Kubo M and Fukuyama Y, Tetrahedron Lett, 2014, 55, 6001–6003. [Google Scholar]
- 96.Solinski AE, Ochoa C, Lee YE, Paniak T, Kozlowski MC and Wuest WM, ACS Infect. Dis, 2018, 4, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ming Ge H, Yun Zhang W, Ding G, Saparpakorn P, Chun Song Y, Hannongbua S and Xiang Tan R, Chem. Commun, 2008, 45, 5978–5980. [DOI] [PubMed] [Google Scholar]
- 98.Kang H, Torruellas C, Liu J and Kozlowski MC, Org. Lett, 2018, 20, 5554–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart KL, Chem. Rev, 1971, 71, 47–72. [DOI] [PubMed] [Google Scholar]
- 100.Hamamoto H, Shiozaki Y, Nambu H, Hata K, Tohma H and Kita Y, Eur. J. Chem, 2004, 10, 4977–4982. [DOI] [PubMed] [Google Scholar]
- 101.Hara H, Komoriya S, Miyashita T and Hoshino O, Tetrahedron-Asymmetry, 1995, 6, 1683–1692. [Google Scholar]
- 102.Stadler R, Kutchan TM, Loeffler S, Nagakura N, Cassels B and Zenk MH, Tetrahedron Lett, 1987, 28, 1251–1254. [Google Scholar]
- 103.Bhakuni DS and Jain S, J. Chem. Soc., Perkin Trans 1, 1988, 1447–1449. [Google Scholar]
- 104.Schwartz MA and Kim PPT, J. Org. Chem, 1988, 53, 2318–2322. [Google Scholar]
- 105.Schwartz MA, Synth. Commun, 1973, 3, 33–35. [Google Scholar]
- 106.Vanderlaan DG and Schwartz MA, J. Org. Chem, 1985, 50, 743–747. [Google Scholar]
- 107.Qin Y, Zhu L and Luo S, Chem. Rev, 2017, 117, 9433–9520. [DOI] [PubMed] [Google Scholar]
- 108.Yoshimura A and Zhdankin VV, Chem. Rev, 2016, 116, 3328–3435. [DOI] [PubMed] [Google Scholar]
- 109.Dohi T and Kita Y, PATAI’S Chemistry of Functional Groups, 2018, DOI: 10.1002/9780470682531.pat0952, 1–84. [DOI] [Google Scholar]
- 110.Quideau S, Deffieux D and Pouységu L, Comprehensive Organic Synthesis II, 2014, DOI: 10.1016/B978-0-08-097742-3.00318-9, 656–740. [DOI] [Google Scholar]
- 111.Xiong Z, Weidlich F, Sanchez C and Wirth T, Org. Biomol. Chem, 2022, 20, 4123–4127. [DOI] [PubMed] [Google Scholar]
- 112.Bridi H, Meirelles GD and von Poser GL, Phytochemistry, 2018, 155, 203–232. [DOI] [PubMed] [Google Scholar]
- 113.Dohi T, Minamitsuji Y, Maruyama A, Hirose S and Kita Y, Org. Lett, 2008, 10, 3559–3562. [DOI] [PubMed] [Google Scholar]
- 114.Su B, Deng M and Wang Q, Org. Lett, 2013, 15, 1606–1609. [DOI] [PubMed] [Google Scholar]
- 115.Evano G, Wang J and Nitelet A, Org. Chem. Front, 2017, 4, 2480–2499. [Google Scholar]
- 116.Pitsinos EN, Vidali VP and Couladouros EA, Eur. J. Org. Chem, 2011, 2011, 1207–1222. [Google Scholar]
- 117.Esguerra KVN, Fall Y and Lumb J-P, Angew. Chem. Int. Ed, 2014, 53, 5877–5881. [DOI] [PubMed] [Google Scholar]
- 118.Huang Z, Askari MS, Esguerra KVN, Dai T-Y, Kwon O, Ottenwaelder X and Lumb J-P, Chem. Sci, 2016, 7, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang Z and Lumb J-P, Angew. Chem. Int. Ed, 2016, 55, 11543–11547. [DOI] [PubMed] [Google Scholar]
- 120.Huang Z, Ji X and Lumb J-P, Org. Lett, 2019, 21, 9194–9197. [DOI] [PubMed] [Google Scholar]
- 121.Blank N and Opatz T, J. Org. Chem, 2011, 76, 9777–9784. [DOI] [PubMed] [Google Scholar]
- 122.Xu W, Huang Z, Ji X and Lumb J-P, ACS Catal, 2019, 9, 3800–3810. [Google Scholar]
- 123.Kupchan SM, Liepa AJ, Kameswaran V and Sempuku K, J. Am. Chem. Soc, 1973, 95, 2995–3000. [DOI] [PubMed] [Google Scholar]
- 124.Chang J and Inui T, Chem. Pharm. Bull, 2005, 53, 1051–1053. [DOI] [PubMed] [Google Scholar]
- 125.Neuhaus WC and Kozlowski MC, Angew. Chem. Int. Ed, 2020, 59, 7842–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Feofilova EP and Mysyakina IS, Appl. Biochem. Microbiol, 2016, 52, 573–581. [Google Scholar]
- 127.Sun Z, Fridrich B, De Santi A, Elangovan S and Barta K, Chem. Rev, 2018, 118, 614–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Neuhaus WC, Jemison AL and Kozlowski MC, Org. Biomol. Chem, 2021, 19, 8205–8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nair V, Mathew J, Kanakamma PP, Panicker SB, Sheeba V, Zeena S and Eigendorf GK, Tetrahedron Lett, 1997, 38, 2191–2194. [Google Scholar]
- 130.Gavezzotti P, Navarra C, Caufin S, Danieli B, Magrone P, Monti D and Riva S, Adv. Synth. Catal, 2011, 353, 2421–2430. [Google Scholar]
- 131.Kishimoto T, Takahashi N, Hamada M and Nakajima N, J. Agric. Food Chem, 2015, 63, 2277–2283. [DOI] [PubMed] [Google Scholar]
- 132.Neuhaus WC, Jemison AL and Kozlowski MC, ACS Catal, 2019, 9, 11067–11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pan J-Y, Chen S-L, Yang M-H, Wu J, Sinkkonen J and Zou K, Nat. Prod. Rep, 2009, 26, 1251. [DOI] [PubMed] [Google Scholar]
- 134.Dong K, Zhao C-Y, Wang X-J, Wu L-Z and Liu Q, Org. Lett, 2021, 23, 2816–2820. [DOI] [PubMed] [Google Scholar]
- 135.Magoulas GE and Papaioannou D, Molecules, 2014, 19, 19769–19835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Poupon E and Nay B, Biomimetic Organic Synthesis, 2011, DOI: 10.1002/9783527634606. [DOI] [Google Scholar]
- 137.Chapman OL, Engel MR, Springer JP and Clardy JC, J. Am. Chem. Soc, 1971, 93, 6696–6698. [Google Scholar]
- 138.Matsumoto M and Kuroda K, Tetrahedron Lett, 1981, 22, 4437–4440. [Google Scholar]
- 139.Nihon K, Bull. Chem. Soc. Jpn, 1992, 65, 1662–1664. [Google Scholar]
- 140.Daniels RN, Fadeyi OO and Lindsley CW, Org. Lett, 2008, 10, 4097–4100. [DOI] [PubMed] [Google Scholar]
- 141.Neuhaus WC and Kozlowski MC, Chem. Asian J, 2020, 15, 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu J and Kozlowski MC, Org. Lett, 2023, 25, 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu L, Carroll PJ and Kozlowski MC, Org. Lett, 2015, 17, 508–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kang H, Herling MR, Niederer KA, Lee YE, Vasu Govardhana Reddy P, Dey S, Allen SE, Sung P, Hewitt K, Torruellas C, Kim GJ and Kozlowski MC, J. Org. Chem, 2018, 83, 14362–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sako M, Sugizaki A and Takizawa S, Bioorg. Med. Chem. Lett, 2018, 28, 2751–2753. [DOI] [PubMed] [Google Scholar]
- 146.Herrmann J, Fayad AA and Müller R, Nat. Prod. Rep, 2017, 34, 135–160. [DOI] [PubMed] [Google Scholar]
- 147.Sako M, Ichinose K, Takizawa S and Sasai H, Chem. Asian J, 2017, 12, 1305–1308. [DOI] [PubMed] [Google Scholar]
- 148.Sako M, Higashida K, Kamble GT, Kaut K, Kumar A, Hirose Y, Zhou D-Y, Suzuki T, Rueping M, Maegawa T, Takizawa S and Sasai H, Org. Chem. Front, 2021, 8, 4878–4885. [Google Scholar]
- 149.Kamble GT, Salem MSH, Abe T, Park H, Sako M, Takizawa S and Sasai H, Chem. Lett, 2021, 50, 1755–1757. [Google Scholar]
- 150.Esguerra KVN, Xu W and Lumb J-P, Chem, 2017, 2, 533–549. [Google Scholar]
- 151.Heinrich T, Böttcher H, Gericke R, Bartoszyk GD, Anzali S, Seyfried CA, Greiner HE and Van Amsterdam C, J. Med. Chem, 2004, 47, 4684–4692. [DOI] [PubMed] [Google Scholar]
- 152.López-Giménez JF and González-Maeso J, in Behavioral Neurobiology of Psychedelic Drugs, Springer Berlin Heidelberg, 2017, DOI: 10.1007/7854_2017_478, pp. 45–73. [DOI] [Google Scholar]
- 153.Singleton SP, Luppi AI, Carhart-Harris RL, Cruzat J, Roseman L, Nutt DJ, Deco G, Kringelbach ML, Stamatakis EA and Kuceyeski A, Nat. Commun, 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ding H, Deroy PL, Perreault C, Larivée A, Siddiqui A, Caldwell CG, Harran S and Harran PG, Angew. Chem. Int. Ed, 2015, 54, 4818–4822. [DOI] [PubMed] [Google Scholar]
- 155.Zheng X, Li Y, Guan M, Wang L, Wei S, Li YC, Chang CY and Xu Z, Angew. Chem. Int. Ed, 2022, 61. [DOI] [PubMed] [Google Scholar]
- 156.Blair LM and Sperry J, Chem. Commun, 2016, 52, 800–802. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Z, Ray S, Imlay L, Callaghan LT, Niederstrasser H, Mallipeddi PL, Posner BA, Wetzel DM, Phillips MA and Smith MW, Chem. Sci, 2021, 12, 10388–10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sagadevan A, Charpe VP, Ragupathi A and Hwang KC, J. Am. Chem. Soc, 2017, 139, 2896–2899. [DOI] [PubMed] [Google Scholar]
- 159.Kshirsagar UA, Regev C, Parnes R and Pappo D, Org. Lett, 2013, 15, 3174–3177. [DOI] [PubMed] [Google Scholar]
- 160.Quideau S, Deffieux D, Douat-Casassus C and Pouységu L, Angew. Chem. Int. Ed, 2011, 50, 586–621. [DOI] [PubMed] [Google Scholar]
- 161.Zhao J, Xiao J, Wang Y and Peng Y, Chinese J. Org. Chem, 2021, 41, 2933. [Google Scholar]
- 162.Gaster E, Vainer Y, Regev A, Narute S, Sudheendran K, Werbeloff A, Shalit H and Pappo D, Angew. Chem. Int. Ed, 2015, 54, 4198–4202. [DOI] [PubMed] [Google Scholar]
- 163.Kshirsagar UA, Parnes R, Goldshtein H, Ofir R, Zarivach R and Pappo D, Eur. J. Chem, 2013, 19, 13575–13583. [DOI] [PubMed] [Google Scholar]
- 164.Narute S and Pappo D, Org. Lett, 2017, 19, 2917–2920. [DOI] [PubMed] [Google Scholar]
- 165.Yang J, Qiu G, Jiang J, Hu Y, Chen S, Zhang S and Zhang Y, Adv. Synth. Catal, 2017, 359, 2184–2190. [Google Scholar]
- 166.Parnes R, Kshirsagar UA, Werbeloff A, Regev C and Pappo D, Org. Lett, 2012, 14, 3324–3327. [DOI] [PubMed] [Google Scholar]
- 167.Edwards JM, Mangion M, Anderson JB, Rapposch M and Hite G, Tetrahedron Lett, 1979, 4453–4456. [Google Scholar]