

Abstract
Wax apple (Syzygium samarangense) is an economically important fruit crop with great potential value to human health because of its richness in antioxidant substances. Here, we present a haplotype-resolved autotetraploid genome assembly of the wax apple with a size of 1.59 Gb. Comparative genomic analysis revealed three rounds of whole-genome duplication (WGD) events, including two independent WGDs after WGT-γ. Resequencing analysis of 35 accessions partitioned these individuals into two distinct groups, including 28 landraces and seven cultivated species, and several genes subject to selective sweeps possibly contributed to fruit growth, including the KRP1-like, IAA17-like, GME-like, and FLACCA-like genes. Transcriptome analysis of three different varieties during flower and fruit development identified key genes related to fruit size, sugar content, and male sterility. We found that AP2 also affected fruit size by regulating sepal development in wax apples. The expression of sugar transport-related genes (SWEETs and SUTs) was high in ‘ZY’, likely contributing to its high sugar content. Male sterility in ‘Tub’ was associated with tapetal abnormalities due to the decreased expression of DYT1, TDF1, and AMS, which affected early tapetum development. The chromosome-scale genome and large-scale transcriptome data presented in this study offer new valuable resources for biological research on S. samarangense and shed new light on fruit size control, sugar metabolism, and male sterility regulatory metabolism in wax apple.
Introduction
Wax apple (Syzygium samarangense (Blume) Merr. and Perry), also known as Java apple and wax jambu, is a nonclimacteric tropical fruit tree from the Myrtaceae family and is native to the Malay Archipelago [1]. The Myrtaceae family is composed of approximately 80 genera and 3000 or more species [2]. According to a few studies of Myrtaceae genomes [3, 4], the phylogenetic position of this species remains uncertain. The Myrtaceae family has traditionally been divided into two main groups: fleshy-fruited and dry-fruited [2]. As one of the largest genera of fleshy fruits in Myrtaceae, Syzygium species exhibit complex genetic diversity [5]. The species of Syzygium include Syzygium aqueum (water apple, 2n = 44), Syzygium cumini (java plum, 2n = 66), and S. samarangense (wax apple, 2n = 33, 42, 44, 66, and 88) [2]. The available phylogenetic topology information based on chloroplast genomes is inconsistent with geographical and morphological classification to some degree [6]. Few Syzygium species genomes are available to provide clear genetic relationships. Accordingly, it is necessary to study the genome information of wax apple to construct a more reliable Syzygium species phylogenetic tree. The acquisition of long contigs from autopolyploid or highly heterozygous plants is the major obstacle to obtaining accurate genome information, which therefore remains a major challenge [7, 8].
Wax apple fruit, which is usually eaten fresh, is bell-shaped and narrow at the base, with four fleshy calyx lobes at the apex. Because of its strong flowering ability, wax apple can fruit in any given season under proper cultivation measures. The fruit has the characteristics of apple-like crispness, an aroma of roses, low-acidity flavor and richness in antioxidant compounds that are beneficial to human health, and it has therefore become a popular exotic fruit [9, 10]. According to statistics from relevant Chinese authorities, the total production of wax apple fruit in Taiwan and Hainan provinces was 89 800 tons in 2019, with great benefit for local farmers and the country’s economy (data from http://www.stats.gov.cn/). To meet the needs of consumers and enrich the diet with high-quality wax apples with a composition that guarantees high nutritional value, it is important to maintain a suitable sugar content and good size. Among annual crops, the Fruit Weight 2.2 (FW2.2) and Physalis Organ Size (POS) genes have been shown to modulate fruit size by regulating cell division or expansion in tomato [11, 12], and FRUITFULL (FUL)-like MADS-box gene CsFUL1 was shown to modulate cucumber fruit size elongation through auxin transportation [13]. However, genetic information related to fruit size regulation in perennial fruit trees is still unclear. In addition, there are low sugar and sour component contents in the fruit of most wax apple varieties. The regulatory mechanism of sugar and acid metabolism in wax apple are also unknown. Therefore, genome assembly and whole-genome re-sequencing are needed to further clarify the regulatory mechanisms related to fruit quality in wax apple.
Seedlessness is an important target trait in fruit breeding [14–16]. Consumers prefer wax apple with the seedless trait, which are largely selected from bud mutation of wild-types. It is a great challenge for breeders to obtain new seedless wax apple cultivars by cross-breeding, and no new cultivars have been bred for decades. There is still a lack of research on the genetic regulatory mechanisms of wax apple. The seedless trait caused by male sterility has been developed in grape, tomato, and citrus. In plants, male sterility refers to the inability to produce dehiscent anthers, viable male gametes, and functional pollen. Previous studies have confirmed that there are two major categories of male sterility. Male sterility resulting from the genes in both mitochondria and nuclei is identified as cytoplasmic male sterility (CMS); male sterility resulting from nuclear genes alone is known as genetic male sterility (GMS) [17]. For years, wax apple breeding efforts were hampered due to the complex genetic diversity of this species and the lack of genome information. Therefore, an accurate reference genome of wax apple is essential for understanding the mechanisms regulating fertility and for accelerating genomic selection breeding efforts.
In previous work, a superior clone, ‘Tub Ting Jiang’ (‘Tub’), with large and seedless fruit, high sweetness (total soluble solids, ‘TSS’, content of approximately 11.81%) and beautiful colour, was selected [18]. We also collected two special wax apple varieties, ‘DongKeng 3’ (‘DK’) and ‘ZiYu’ (‘ZY’). ‘DK’ is a rootstock variety with rich seeds in all of its fruits. The fruit of ‘ZY’ is bright red and small but highly sweet (TSS of approximately 14.56%) with 0 to 2 seeds inside. These varieties will be good materials for studying the genome information of wax apple. Through the study of the wax apple genome, we hope to accelerate the breeding process and produce more new cultivars that are larger, sweeter and more colourful.
In this study, we aim to sequence and assemble ‘Tub’, which is an autotetraploid wax apple variety, to fill existing genomic information gaps in wax apple. This genome was used to conduct a comparative genomic analysis to further insight into the functional and structural features of the S. samarangense genome. Furthermore, we identified key genes associated with fruit size, sugar content, and male sterility, which are important breeding traits of wax apple. This genome will provide a valuable resource for further molecular functional analyses and benefit the acceleration of wax apple breeding.
Results
Genome assembly and annotation
To investigate the features of the S. samarangense genome, we performed genome survey analysis, K-mer analysis, and 5S rDNA FISH experiment. The results verified that it was an autotetraploid genome with 44 chromosomes (2n = 4x = 44), and monoploid genome size was 420 Mb ((Figs SS1 andSS2, see online supplementary material). To generate a haplotype-resolved genome assembly, we sequenced a total of 92.0 Gb of PacBio subreads (~220 x of the estimated monoploid genome size), 90.0 Gb of Illumina short reads, and 92.40 Gb of high-throughput chromatin conformation capture (Hi-C) reads (Table SS1, see online supplementary material). A 1.49-Gb assembly with a contig N50 of 304.5 kb was assembled by using the CANU assembler [19] (Table SS2, see online supplementary material). All contigs were further anchored onto 44 pseudochromosomes with 11 homologous groups based on ALLHiC phasing. Finally, a total of 1.59 Gb of phased assembly sequences were obtained after gap filling, representing an allele-ware, chromosome-scale genome assembly with 98.9% completeness as evaluated by BUSCO (Fig. 1a; Fig. SS3, Table SS3, see online supplementary material) [20]. In addition, approximately 95.6% of the Illumina clean data could be aligned onto the genome assembly, covering 97.9% of the genomic regions (Table SS4, see online supplementary material), suggesting that high-quality genome sequences were acquired.
Figure 1.

Alignment of the Syzygium samarangense monoploid genome with the S. samarangense genome and summary of the genome assembly. (a) A set of four homologous chromosomes aligned to a single monoploid chromosome. (b) From the outermost to innermost layer, the rings indicate the haplotype genomes in Mbp (i), GC content (ii), gene density (iii), SNP density (iv), SV density (v), expression (iv) and synteny blocks, respectively.
To obtain high-fidelity gene annotations, we applied two rounds of the MAKER pipeline to produce a set of 74 888 high-quality protein-coding gene models (Table 1). BUSCO analysis showed a completeness of 90.7%, with 69.3% duplication (Table SS5, see online supplementary material), indicating that the annotation included a mixture of genes and alleles. We adopted the pipeline developed in our previous project [21] to separate genes and alleles, resulting in a total of 24 016 genes with defined alleles. In addition, we annotated 952 tandemly duplicated genes and 11 161 dispersed duplicated paralogues (Table SS6, see online supplementary material).
Table 1.
Statics of genome assembly in wax apple
| Assembly feature | Syzygium samarangense |
|---|---|
| Total length of contigs (Gb) | 1.49 |
| Number of contigs | 25 179 |
| Contig N50 (Kbp) | 304.5 |
| Sequence anchored to chromosomes (Gb) | 1.46 |
| Anchor rate (%) | 98 |
| Monoploid genome assembly size (Gb) | 0.41 |
| Genes with defined alleles | 24 016 |
The wax apple genome contained about 38.10% (593.25 Mb) of repetitive sequences. Long terminal retrotransposons (LTRs) were the predominant type of transposable elements (TEs) and accounted for 24.74% of the genome (Table SS7, see online supplementary material). The high proportion of LTRs was likely due to a recent large-scale burst that occurred ~0.1 million years ago (Mya) (Fig. SS4, see online supplementary material).
Evolutionary history and whole-genome duplication
We identified 344 single-copy genes from 12 sequenced genomes by using OrthoFinder and subsequently employed them to construct a phylogenetic tree. The results clearly showed that S. samarangense, Eucalyptus grandis, Psidium guajava, Rhodomyrtus tomentosa, and Punica granatum belonged to the same branch of Myrtales. A significantly closer genetic relationship was observed between S. samarangense and E. grandis, which both belong to the Myrtaceae family. We further estimated divergence times and found that Myrtales arose 79.4 million years ago (Mya). Within the Myrtaceae family, S. samarangense and E. grandis diverged from each other 26 million years ago (Mya). According to a CAFE analysis, we characterized 1328 expanded gene families and 5363 contracted gene families (Fig. 2a). Gene Ontology (GO) enrichment analysis showed that the 1328 expanded gene families were primarily enriched in DNA polymerase activity, retrotransposon nucleocapsid, and mitochondrial fission. In contrast, the 5363 contracted gene families were primarily enriched in protein serine/threonine kinase activity, floral organ senescence, and secondary metabolite biosynthetic process (Figs SS5–S6, see online supplementary material). In comparison with other species, 537 unique gene families were identified (Fig. 2b) within the S. samarangense genome. These gene families were mainly enriched in a series of functional items, including catalytic activity, acting on DNA, retrotransfer, nucleocapsid, transfer, and RNA mediated (Fig. SS7, see online supplementary material).
Figure 2.

Phylogenetic and comparative analysis of Syzygium samarangense. (a) Phylogenetic tree of S. samarangense, Eucalyptus grandis, Punica granatum, Arabidopsis thaliana, Vitis vinifera, Oryza sativa, Nyctophila colorata, Solanum lycopersicum, Psidium guajava, Prunus persica, Rhodomyrtus tomentosa, and Amborella trichopoda. Gene family expansion/contraction analysis of the S. samarangense genome. The divergence times of S. samarangense and the other species are labelled at the bottom. (b) Orthologous and species-specific gene families in S. samarangense and the other species. (c) Distribution of synonymous substitution rates (Ks) among S. samarangense paralogues and orthologues with other species. (d) Alignment of the S. samarangense genome with the V. vinifera genome.
Comparisons among the four haplotypes revealed 4.53 million SNPs, 0.49 million short indels, and 10 925 structural variations (SVs), and these genetic variations were evenly distributed along the 44 chromosomes (Fig. 1b; Table SS8, see online supplementary material). The clustering of chromosome-specific 13-mers partitioned each set of four haplotypes together (Fig. SS8, see online supplementary material), which was inconsistent with the allotetraploid Miscanthus genome and showed the separated distribution of subgenomes. The smudge plot analysis identified that the AAAB pattern was the dominant component, accounting for 56% of the examined K-mers (Fig. SS9, see online supplementary material). These results collectively indicated that S. samarangense has an autotetraploid genome with a high level of heterozygosity.
The distribution of synonymous substitutions per synonymous site (Ks) of the homologous gene pairs clearly illustrated that the genome of S. samarangense experienced three rounds (WGT-γ, WGD-1, and WGD-2) of whole-genome duplication events (Fig. 2c). In addition to WGT-γ which is commonly found in the evolutionary history of grape and E. grandis, we discovered that S. samarangense and E. grandis had also undergone an independent whole-genome duplication (WGD-1). Compared with the situation in E. grandis, the specific WGD-1 event that appeared in the genome of S. samarangense was more complex. Moreover, the synteny relationship between S. samarangense and Vitis vinifera was further analysed to verify that WGD-1 and WGD-2 occurred after WGT-γ. As shown in Fig. 2d, the collinear relationship between S. samarangense and V. vinifera was 8:1, indicating that the occurrence of the two lineage-specific WGDs in S. samarangense.
Genetic variations and population structure
We resequenced 35 accessions of S. samarangense at the whole-genome level and identified 2 891 846 variants, including 2 630 417 SNPs and 261 429 indels (Table SS9, see online supplementary material). A total of 67 430 synonymous and 78 424 nonsynonymous mutations were identified (Table SS10, see online supplementary material). Phylogenetic analysis demonstrated that these S. samarangense were partitioned into two distinct groups. The commercially cultivated accessions were clustered together as the first group, and the remaining accessions, forming the second group, were landraces with limited or not artificial selection (Fig. 3a; Table SS11, see online supplementary material). The results of both principal component analysis (PCA) and genome structure analysis were consistent with phylogenetic analysis (Fig. 3b and c).
Figure 3.

Phylogenetic splits and population genetic structure of 35 Syzygium samarangense accessions. (a) Maximum-likelihood tree of 35 resequenced S. samarangense individuals constructed based on 2 630 417 SNPs. (b) PCA plots of S. samarangense accessions showed two subgroups (Group_2, cultivars; Group_1, landraces). PC, principal component. (c) ADMIXTRUE analysis among the accessions revealed the distribution of K = 2 genetic clusters with the lowest cross-validation error. (d) Comparison of fruit weight between landraces and cultivars.
To identify the candidate genes that might have undergone natural or artificial selection during the evolutionary history of wax apple, we conducted a selective sweep-based SweeD analysis [22] in the 35 resequenced individuals. A total of 22.0 Mb of genomic sequences, covering 1299 and 1109 protein-coding genes, showed evidence of selective sweeps in the landraces and cultivars, respectively. These selectively swept regions were distributed along the 11 representative chromosomes that were selected from each set of homologous chromosomes, with some chromosomes having a higher density (Figs SS10–S11, see online supplementary material). GO enrichment analysis revealed that the swept genes in landraces were significantly enriched in the second-messenger-mediated signalling and calcium-mediated signalling pathways. However, the swept genes in cultivars were enriched in metabolic process and zygote asymmetric cell division (Figs SS12–S13, see online supplementary material).
Phenotypic analysis showed that the cultivated wax apples exhibited increased fruit weight compared with the landraces, leading to the hypothesis that fruit growth-related genes are likely under artificial selection (Fig. 3d). To verify this, we collected 30 homologous genes related to fruit growth in wax apple (Table SS12, see online supplementary material) based on the published genes in tomato [23]. We observed that the landraces contained three genes in the selectively swept genomic regions, namely KRP1-like, IAA17-like, and GME-like, which have been demonstrated to be involved in cell expansion, including endocycle control, auxin signalling, and ascorbate biosynthesis. In addition, the FLACCA-like gene, which is involved in ABA biosynthesis, was indicated to be under selection in cultivars [23] (Figs SS14–SS15, see online supplementary material).
Genes contributing to fruit size and sugar content
We conducted fruit quality analysis on the fruits of all 35 S. samarangense materials. Based on the data of fruit size and TSS content we obtained (Table SS13, see online supplementary material), three representative accessions (‘Tub’, great size and medium sweetness; ‘ZY’, medium size and high sweetness; ‘DK’, little size and low sweetness) were selected for further studies (Fig. SS16, see online supplementary material). The ‘Tub’ variety exhibited the greatest fruit weight, with an average of 124.60 g per single fruit. This was almost two times than that of ‘ZY’ (68.47 g on average) and four times that of ‘DK’ (35.41 g on average), indicating that the fruit sizes of the three accessions were significantly different. A previous study indicated that the sepal development gene APETALA (AP) controls the fruit size in apples [24], which the fruit structure was similar to wax apples. Based on comparative RNA-Seq data, we found that the expression of AP1 and AP2 genes was highest in the ‘Tub’ accession, which had the greatest fruit weight, followed by the ‘ZY’ and ‘DK’ accessions, which had much reduced fruit sizes (Fig. 4; Figs SS17–S19 and Table SS14, see online supplementary material). The AP1 gene was highly expressed in ‘Tub’Fr_T1 and ‘Tub’Fr_T3 samples, suggesting that AP1 may play a role in promoting fruit growth at the early stage of fruit development.
Figure 4.

. Genes related to fruit growth and sugar content. (a) The expression of sepal development homologues (AP1 and AP2) in ‘DK’, ‘ZY’, and ‘Tub’ during fruit development. (b) Comparison of fruit weight among ‘DK’, ‘ZY’, and ‘Tub’. ****P-value <0.0001, t-test, n = 10. (c) Comparison of sugar content between ‘Tub’ and ‘ZY’ fruit at maturity. ***P-value <0.001, t-test. (d) Expression of the candidate genes related to sugar transport (SWEETs, ERDLs, and TST) in the pink module in ‘DK’ and ‘Tub’ during fruit development. ‘DK’: ‘Dongkeng’; ‘Tub’: ‘Tub Ting Jiang’. FrT1, FrT2, FrT3, FrT4, and FrT5 represent 10 to 50 DAFB (days after full bloom) at approximately 10-day intervals.
In fruits, sugar content is usually defined as the TSS content that determines sweetness and is an important index in determining fruit quality. We observed that the fruits of ‘ZY’ presented a significantly higher soluble solid content than those of ‘Tub’ and ‘DK’ (14.56% vs. 11.81% vs. 4.67; Fig. 4c;Fig. SS16, see online supplementary material). We further queried the meaningful genes contributing to the elevated sugar content through a comparative RNA-seq analysis of fruit samples between ‘ZY’ and ‘Tub’. WGCNA identified 14 co-expressed modules (Fig. SS20, see online supplementary material), and a total of 400 genes were co-expressed in the ‘ZY’Fr_T5 sample, which is the most mature stage of ‘ZY’ fruit and presumably shows the highest sugar content (Fig. SS21, see online supplementary material). We observed that a list of important sugar transporter genes exhibited significantly high levels of expression in ‘ZY’Fr_T5 (Fig. 4d).
Genes associated with male sterility
Seedless fruits are highly desirable due to their commercial value. This trait likely results from abnormal ovule and pollen development [25]. The results showed that the anther contained abundant pollen and exhibits normal dehiscence in ‘DK’, but the anther contained only a small amount of pollen and exhibited abnormal dehiscence in ‘Tub’ (Fig. 5a and b). Subsequently, we determined the pollen germination rate in ‘DK’, ‘ZY’, and ‘Tub’. The results showed that the pollen germination rates were 11.73% and 45.06% for ‘ZY’ and ‘DK’, respectively, but the pollen of ‘Tub’ was not collected because of abnormal anther dehiscence (Fig. 5c).
Figure 5.
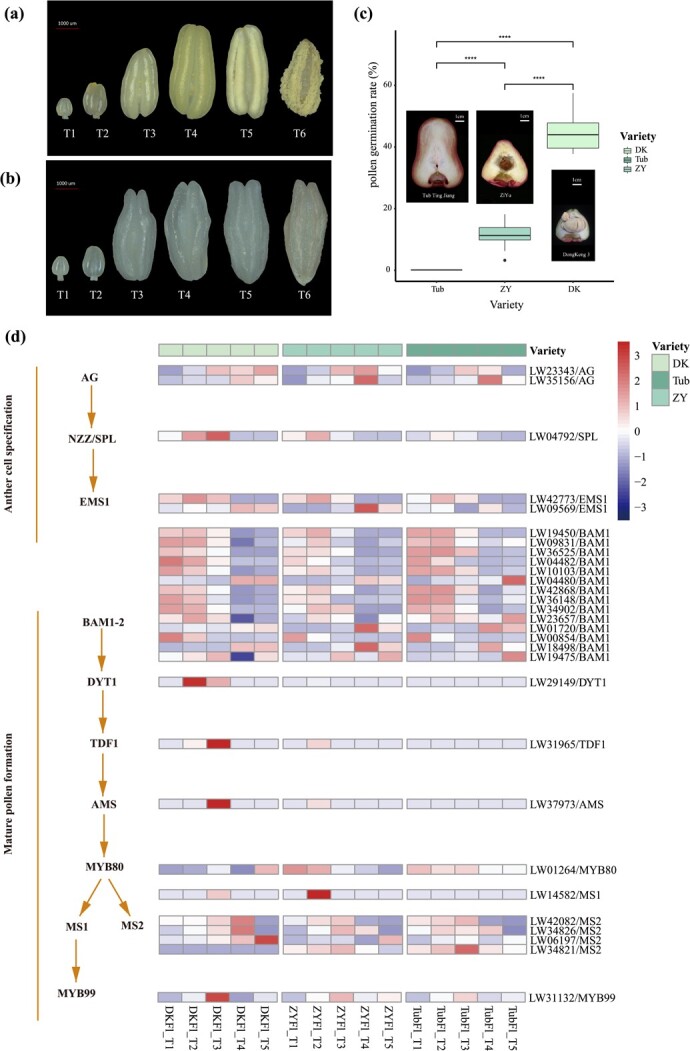
Anther development, pollen germination rate, and expression of anther and pollen development-related genes in ‘DK’, ‘ZY’, and ‘Tub’. (a) Anther development and dehiscence in ‘DK’. T1-T5 are consistent with FlT1-FlT5, and T6 represents 12 hours after blooming. (b) Anther development in ‘Tub’. T1-T5 are consistent with FlT1-FlT5, and T6 represents 12 hours after blooming. (c) Pollen germination rate of ‘Tub’, ‘ZY’, and ‘DK’. ****P-value <0.0001, t-test, n = 10. (d) Expression (FPKM) of anther and pollen development-related genes in ‘DK’, ‘ZY’, and ‘Tub’ from flowers at different stages, including FlT1, FlT2, FlT3, FlT4, and FlT5.
In our study, samples from different flowering stages were used for further RNA-seq analysis to identify key genes involved in the development of pollen and anthers. WGCNA was performed to explore the potential genes related to male sterility in ‘Tub’. The coexpression network was constructed based on the correlation of gene expression in all samples. Finally, 16 different modules, defined as highly interconnected gene clusters, were identified and marked with different colours (Fig. SS22, see online supplementary material). Among these modules, three potential pollen and anther development-associated module eigengenes were characterized (Table SS15, see online supplementary material). In ‘DK’, the turquoise, tan, and darkgreen modules were correlated with the development of pollen and anthers (Figs SS23–S25, see online supplementary material). Interestingly, the turquoise module contained highly connected hub genes, including LBD10, RPG1, RBOHE, CALS5, SK32, and MYB33, which are known genes involved in pollen development (Table SS16 and Fig. SS26, see online supplementary material).
Furthermore, we identified a total of 29 homologous genes that played an important role in male sterility in Arabidopsis. These genes were mainly involved in anther cell specification and mature pollen formation pathways, and many of them showed differential expression at five different flower developmental stages (FlT1 to FlT5) among the three examined varieties (Fig. 5; Fig. SS27, see online supplementary material). The anther cell specification related gene nozzle/sporocyteless (NZZ/SPL) was found to be more highly expressed in ‘DK’ than in ‘Tub’. We also observed that dysfunctional tapetum 1 (DYT1), tapetum development and function 1 (TDF1), and abortive microspore (AMS) genes were specifically expressed in the FlT2 and FlT3 stages in ‘DK’, and barely expressed in ‘Tub’. In addition, the expression of three male sterile 2 (MS2) homologous genes in ‘DK’ was much higher than that in ‘Tub’ at the FlT4 and FlT5 stages. The expression pattern of these pollen development-related genes was consistent with the results of the pollen germination rate (Fig. 5d).
Discussion
Wax apple is an economically important fruit crop that is widely cultivated throughout the Southeast Asian countries. Here, we generated a high-quality fully phased autotetraploid genome assembly and 35 resequencing accessions. These data represent comprehensive genomic resources for this species, facilitating the investigation of meaningful genetic variations and evolutionary history. Comparative genomics and transcriptomic analysis also revealed key genes underlying fruit growth, fruit size, and sugar content as well as factors related to male sterility caused by aborted pollen.
The assembly of the wax apple genome has been severely hindered by the high level of repetitive sequences and polyploidy. To date, only a few autotetraploid genomes have been assembled at the chromosome level, including those of sugarcane (Saccharum spontaneum) [21], cultivated alfalfa (Medicago sativa L.) [26], a potato cultivar (Solanum tuberosum, ‘Otava’) [27], and Rehmannia glutinosa [28]. Among these, only the sugarcane and cultivated alfalfa genomes were assembled by combining the developed sequencing technologies and chromosome phasing algorithm, whereas the developed sequencing technologies and pollen genome were used in the potato cultivar assembly. Here, we generated a haplotype-resolved chromosome-level genome of S. samarangense consisting of 44 allelic chromosomes by combining sequencing technologies and a chromosome phasing algorithm. The high percentage of assembled genome size relative to the monoploid estimate and anchor rate indicated a high-quality, allele-ware, and chromosome-scale genome assembly, benefiting downstream analysis and molecular breeding.
Fruit size and sugar content affect consumer preference. Emerging evidence shows that floral organ development-related genes participate in fruit development and play different roles among species, mainly depending on the type of floral organ that develops into fruit tissues [29]. Previous studies have shown that AP2 governs seed yield [30] and floral development (especially sepal development [31, 32]) in Arabidopsis and can affect fruit growth in apples [24]. Intriguingly, AP2 inhibits fruit size in Arabidopsis but promotes fruit size in apple [24]. In apple, miR172 inhibits the expression of AP2, and the overexpression of miR172 reduces fruit size, which indicates that miR172 plays a vital role in fruit size via AP2 [24]. The highest expression of sepal development genes (AP1 and AP2) according to our results was found in the ‘Tub’ group, which showed the greatest fruit weight, suggesting that AP1/2 may play an important role in the regulation of wax apple fruit size. Considering that wax apple is recognized as false fruit that develops from the ovary and sepals, genes regulating sepal development are likely related to fruit size. APETALA2 (AP2) governs sepal development, and APETALA2 (AP1) acts downstream of AP2 [33]. The main reason for this phenomenon is that unlike the fruits of apple and S. samarangense, which grow from hypanthium (and sepals, receptacle), the siliques of Arabidopsis develop from ovary tissues [34]. In apple, the overexpression of MdERDL6–1 improves glucose (Glc), fructose (Fru), and sucrose (Suc) concentrations in transgenic apple fruit and increases the expression of TST1/TST2, indicating that the sugar content in vacuoles is mediated by the coordinated action of MdERDL6–1 and MdTST1/2 [35]. In our study, ERDL6–1 and TST2 were mainly expressed in the ‘ZY’ variety, which has a higher sugar content, indicating that sugar accumulation in the ‘ZY’ variety could be attributed to the higher expression of ERDL6–1 and TST2. Through a comparative RNA-seq analysis of fruit samples for meaningful genes contributing to the elevated sugar content observed in ‘ZY’ and ‘Tub’, we identified 14 co-expressed modules, and a total of 400 genes were found to be co-expressed in the ‘ZY’Fr_T5 sample, representing the most mature stage of ‘ZY’ fruit, presumably containing the highest sugar content; these genes were significantly enriched in a series of molecular functions, particular in sugar transporter activity items. In addition, high expression levels were observed in ‘ZY’Fr_T5 for a list of important sugar transporter genes, including those encoding sucrose transporters (SUTs), monosaccharide transporters (MSTs), and sugars will eventually be exported transporters (SWEETs) and TMT2. Our results collectively indicated that these sugar transporter-related genes contributed to the elevated sugar content in the fruit of wax apple.
Seedless fruit occupies an important position in domestic and international markets. In Arabidopsis, the LBD10 orthologue can interact with LBD27 to form a heterodimer and plays an essential role in pollen development [36], strongly suggesting its potential role in the regulation of male sterility in wax apple, and many species have been developed, such as grape and citrus [37, 38]. Male sterility caused by aborted pollen is the main pathway targeted to cultivate seedless fruit. Based on this evidence, we speculate that the male sterility of ‘Tub’ is possibly attributed to functional defects in a few key genes, especially DYT1, TDF1, and AMS, affecting early tapetum development. In Arabidopsis, previous investigations showed that DYT1, TDF1, and AMS mutants all display a fully male sterile phenotype [39–41]. The DYT1-TDF1-AMS-MS188 genetic network was suppressed upon the mutation of Fatty Acid Export 1, causing defective pollen formation [42]. Trace concentrations of imazethapyr (IM) significantly decrease the gene expression of DYT1, TDF1, and AMS, which affects anther and pollen biosynthesis in Arabidopsis [43]. Here, we revealed that DYT1, TDF1, and AMS were highly expressed in the male-fertile variety ‘DK’ but were expressed at lower levels in ‘Tub’ and especially the male sterile variety ‘ZY’. Therefore, these genes may play a potential role in the regulation of fertility in wax apple. Together, the results indicated that male sterility produces seedless fruit and may be caused by the decreased expression of DYT1, TDF1, and AMS. The results suggest that these genes could play important roles in seedless phenotype formation, and the relative expression levels of LBD10, RPG1, RBOHE, CALS5, SK32, and MYB33 versus those of DYT1, TDF1, and AMS seem to be key factors in this process in wax apple.
Conclusion
Here, a haplotype-resolved autotetraploid genome assembly of wax apple was generated, and comparative genomic analysis revealed that S. samarangense has experienced three different rounds of WGD events, including two independent WGDs after WGT-γ. Transcriptome analysis was used to identify genes related to fruit size, sugar content, and male sterility. Combined with fruit weight, fruit development characteristics, and transcriptome data analysis, the AP1 and AP2 genes may regulate fruit size by regulating sepal development. Sugar transport-related genes (SWEETs and SUTs) were found to be more highly expressed in varieties with a higher sugar content in ‘ZY’. The low expression of DYT1, TDF1, and AMS in ‘Tub’ may be the main reason for its sterility. Our results provide a foundation for further study of the regulatory mechanisms of fruit quality and male sterility and can be used in the molecular-assisted breeding of wax apple, especially for seedless traits.
Materials and methods
Illumina short-read sequencing and genome survey
We chose the ‘Tub’ accession for de novo genome sequencing and assembly. The plant materials were maintained by the Fujian Academy of Agricultural Sciences, and young leaves were collected from an individual tree planted in the wax apple GenBank Field for of the Fujian Academy of Agricultural Sciences, Fujian Province, China (Coordinates: 26°7′53″N; 119°20′6″E) under voucher number GPLWFJGSS0058. Genomic DNA was isolated from young leaves using the Qiagen Plant Genomic DNA Kit according to the manufacturer’s instructions. Then, the qualified DNA samples were randomly fragmented with a Covaris S-series Instrument, and Illumina PCR-free libraries with an insert size of 350 bp were constructed using the TruSeq Nano DNA HT Sample preparation Kit (Illumina, San Diego, California, USA). Finally, the constructed libraries were sequenced with 150-bp paired-end sequencing using Illumina HiSeq PE. Using Illumina short reads, the genome size, repeat contents, and heterozygosity rate of S. samarangense were estimated using jellyfish2.2.7 software [44].
Genome sequencing
A combination of single-molecule real-time sequencing (SMRT), Illumina sequencing, and Hi-C sequencing with error correction was applied to assemble the complete genome sequence of S. samarangense. For SMRT, genomic DNA was disrupted randomly with 6 kb–20 kb fragments by using g-TUBE (Covaris, Woburn, Massachusetts, USA) and sequenced on the PacBio Sequel platform, generating 110 coverage. For Illumina sequencing, six libraries (300 bp) were constructed using the Illumina TruSeq Nano DNA Library Prep kit, and the libraries were sequenced on the Illumina Hi-Seq 2000 platform. For Hi-C sequencing, two Hi-C libraries were constructed using a standard procedure and sequenced using the Illumina HiSeq X Ten sequencer.
Genome assembly
The contig-level assembly of the wax apple genome incorporated Illumina short reads and PacBio CLR subreads. The PacBio subreads were subjected to the whole pipeline of Canu assembler v1.9 [19], followed by polishing using the Pilon program [45] to increase assembly accuracy. To construct the haplotype-resolved genome assembly, we first mapped the Hi-C reads to the polished contig assembly using BWA MEM (-5SPM) and extracted the uniquely mapped paired reads. The resulting BAM files were applied to haplotype phasing and scaffolding using the ALLHiC pipeline [46]. In addition, the chimeric scaffolds were manually corrected based on the Hi-C signals in the juicebox. To fill the gaps, first, TGS GapCloser [47] software was used to fill the gaps in the wax apple genome with 30X ultralong ONT data. After filling the genome, the number of gaps was significantly reduced. Then, we used Merqury [48] software to check the gap-filled genome and found that some errors were introduced compared with the previous filling. To correct these errors, we extracted all gap sequences filled by TGS GapCloser and checked the QV quality value of each gap and the error rate of the corresponding sequence in the genome using Mercury software. Finally, we filled the correct GAP in the initial chromosomal level genome. The quality of chromosome-scale assembly was assessed using a Hi-C heatmap.
Genome annotation
To annotate protein-coding genes, we followed the method described in a previous study [49]. Briefly, we integrated evidence from RNA-seq, orthologous proteins, and ab initio gene prediction by carrying out two rounds of the MAKER pipeline. In the first round of MAKER, Trinity was used for de novo assembly by using the RNA-seq data [50], and RSEM was applied to calculate transcript abundance [51]. After filtering the valid transcripts, the rest were imported into the PASA program, and the candidate proteins were trained by ab initio gene prediction [52]. In the second round of MAKER, the candidate proteins were retrained ab initio. Hisat2 and StringTie were used for reassembly [53, 54]. Finally, we selected the better annotation of the two rounds of annotation. BUSCO (v5) software was applied to calculate the degree of annotation complement. We used the same method reported for in an autopolyploid sugarcane genome to construct a monoploid genome, identify alleles, and analyse allelic variations [21].
RNA library construction and sequencing
To improve the prediction of gene annotations, we performed RNA-seq using different tissues of S. samarangense including flesh, flower, leaf, ovary, root, and stem. All tissues were collected and subsequently frozen in liquid nitrogen. Total RNA was extracted with the RNAprep Pure Plant Plus Kit (Tiangen) following the manufacturer’s procedure. Transcriptome libraries were constructed using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB, UK) according to the manufacturer’s instructions and sequenced with 150-bp paired-end sequencing using the Illumina NovaSeq 6000 (Illumina, USA) platform.
Phylogenetic analysis and estimation of divergence times
To construct the phylogenetic tree, single-copy orthologous genes were defined by OrthoFinder v2.3.1 [55] from protein sequences of seven species (E. grandis, P. granatum, Arabidopsis thaliana, V. vinifera, Oryza sativa, Nymphaea colorata, and Amborella trichopoda). Thereafter, protein sequences aligned by MUSCLE [56] and GBLOCKS [57] were used to trim ambiguous parts of the alignment. A phylogenetic tree was constructed using RAxML [58] utilizing the JTT + I + G + F model and 1000 bootstrap analyses. The divergence times among these species were estimated by r8s [59]. Whether the gene families had undergone the expansion or contraction events in the eight sequenced species was evaluated using CAFE2.2 [60].
Synteny and whole-genome duplication analysis
To investigate the whole-genome duplication (WGD) events in S. samarangense, synteny analysis of the S. samarangense and V. vinifera genomes was performed. The V. vinifera genome and annotation were downloaded from Phytozome (https://phytozome-next.jgi.doe.gov/). We applied the MCScan (python version) pipeline [61] following the suggested best workflow. The syntenic regions in the S. samarangense and V. vinifera genomes indicated that S. samarangense experienced two WGD events after WGT-γ.
To test the reliability of this result, the synonymous nucleotide substitutions at synonymous site (Ks) values were estimated for S. samarangense, V. vinifera, and E. grandis genomes by using the YN00 program in the WGDi package with the Nei-Gojobori approach [62]. Because the base substitution rate differs in the three species, the method applied by Jinpeng Wang [63] was used to correct the evolutionary rates of duplicated genes. After fit and merge operations, the Ks peaks caused by the same WGD event could be found in the same place.
Resequencing and population analysis
A total of 35 accessions were resequenced, including 28 landraces and seven cultivars. All accessions were collected from the wax apple GenBank Field of the Fujian Academy of Agricultural Sciences, Fujian Province, China. Young leaves were collected from each accession and flash frozen in liquid nitrogen for DNA isolation. Genomic DNA from each sample was isolated, and paired-end reads were sequenced on the Illumina NovaSeq platform. The adaptors and low-quality reads were trimmed using Trimmomatic [64], and clean reads were aligned to the reference genome of S. samarangense using BWA with the default parameters [65]. We identified variants following the GATK [66] best practices pipeline. HaplotypeCaller and GenotypeCaller were used to call variants from all samples. Maximum-likelihood trees were constructed using VCF2Dis (https://github.com/BGI-shenzhen/VCF2Dis).
To infer the subgroups among the resequenced S. samarangense accessions, admixture [67] was used with different k values (from 1 to 3), the optimal value determined in this study was k = 2. PLINK1.9 and VCFtools [68] were used to perform PCA. Finally, we used SweeD [22] to identify complete selective sweeps in the S. samarangense genome with the default settings.
Transcriptome sequencing and identification of coexpression modules
The fruits from three wax apple accessions, ‘ZY’, ‘Tub’, and ‘DK’, were sampled from 10 to 50 DAFB (days after full bloom) at approximately 10-day intervals, representing five developmental stages, namely T1 to T5. Total RNA was extracted from flowers and fruits using the RNAprep Pure Plant Plus Kit (Tiangen), and cDNA libraries were constructed and sequenced by the Illumina NovaSeq 6000 (Illumina, USA) platform. Subsequently, we evaluated read quality using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), and removed sequencing adapters and low-quality bases using Trimmomatic [64]. The clean data were aligned to the S. samarangense genome using HISAT2 (v2.0.5) [69], and the fragments per kilobase per million mapped fragments (FPKM) value was calculated using StringTie (v.1.2.3) [70]. The R package weighted gene coexpression network analysis (WGCNA) was used to cluster genes with similar expression based on the FPKM data [71]. Genes with |MM| > 0.8 and |GS| > 0.2 were selected for further analysis, and the network was represented and displayed using Cytoscape (v3.6.0) [72]. Male sterility- and flower development-related genes were retrieved from Arabidopsis (https://www.arabidopsis.org/), and the homologues of S. samarangense were identified by a BLASTP search of these sequences against all S. samarangense protein sequences.
Fruit quality analysis and pollen viability determination
For fruit weight analysis, the fruits of all 35 S. samarangense materials (including 28 landraces and seven cultivars) were collected. Ten fruits were randomly selected from three trees for each S. samarangense material. Fruit weight was measured with a QUINTIX213-1CN electronic balance (Sartorius, Germany). To determine the total soluble solids (TSS) content, 1 cm3 of tissue was taken from the upper, middle, and lower parts of each fruit sample. Then, the sample were mixed and homogenized thoroughly with a mortar and pestle. The supernatant of the homogenate was used for soluble solids content determinations with a hand-held Brix metre PAL-1 (ATAGO, Japan). To analyse pollen viability, the pollen tube germination rate was measured. At 35°C, pollen was cultured for 12 hours in medium with a sucrose concentration of 15%, boric acid concentration of 50 mg/L, and agar concentration of 1%. Then, optical microscopy was used to observe pollen tube germination. Three fields of vision were randomly selected, and the total number of pollen grains and the number of germinated pollen grains were counted at the same time. The germination rate was calculated. The standard for defining budding pollen was that the length of the pollen tube exceeded the diameter of the pollen.
For each experiment, the significance of between-group differences was analysed using t-test. All statistical analyses were performed using IBM SPSS software. P-values <0.001 were considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (2020 J011361), and the High-quality Development beyond the ‘5511’ Collaborative Innovation Project in Fujian Province (XTCXGC2021016-4). We thank Ping Zhou (Fujian Academy of Agricultural Sciences) for the data analysis program.
Contributor Information
Xiuqing Wei, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China; Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China.
Min Chen, Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China.
Xijuan Zhang, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China.
Yinghao Wang, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China.
Liang Li, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China.
Ling Xu, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China.
Huanhuan Wang, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China.
Mengwei Jiang, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China.
Caihui Wang, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China.
Lihui Zeng, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China.
Jiahui Xu, Fruit Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China.
Author contributions
J.X. and L.Z. designed the experiments; X.W. performed the most of the experiments; M.C. performed the genome assembly, annotation and the transcriptome data analysis; X.Z. and L.X. performed phenotype analysis; L.L. collected the materials for sequencing; H.W. and C.W. analysed the resequenced data; M.J. conducted comparative genomic analysis. Y.W. filled the gaps in the wax apple genome.
Data availability
The raw sequencing reads used for de novo whole-genome assembly and the final genome and annotation files have been deposited in the Genome Warehouse in National Genomics Data Center, under accession number GWHDUEN00000000 and GWHDVFZ00000000 which are publicly accessible at https://ngdc.cncb.ac.cn/gwh. Raw resequencing data were uploaded to the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/), submission ID: WGS034963; BioProject access number: PRJCA011822; BioSample access number: SAMC1129200; GSA access number: CRA010157.
Conflict of interest statement
The authors declare no competing interests.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
References
- 1. Morton JF. Java apple in fruit of warm climates. In: Morton JF, ed. Creative Resources Systems. Miami, USA, 1987,381–2 [Google Scholar]
- 2. Paull RE, Duarte O. Tropical Fruits. Vol. II, 2nd edn. UK: CABI, 2010 [Google Scholar]
- 3. Feng C, Feng C, Lin X. et al. A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol J. 2021;19:717–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo X, Li H, Wu Z. et al. The pomegranate (Punica granatum L.) draft genome dissects genetic divergence between soft- and hard-seeded cultivars. Plant Biotechnol J. 2020;18:955–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vasconcelos TNC, Proença CEB, Ahmad B. et al. Myrteae phylogeny, calibration, biogeography and diversification patterns: increased understanding in the most species rich tribe of Myrtaceae. Mol Phylogenet Evol. 2017;109:113–37 [DOI] [PubMed] [Google Scholar]
- 6. Wei X, Li L, Xu L. et al. Complete chloroplast genome sequence of Syzygium samarangense (Myrtaceae) and phylogenetic analysis. Mitochondrial DNA B Resour. 2022;7:977–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edger PP, Poorten TJ, VanBuren R. et al. Origin and evolution of the octoploid strawberry genome. Nat Genet. 2019;51:541–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen C, du H, Chen Z. et al. The chromosome-level genome sequence of the autotetraploid alfalfa and resequencing of core germplasms provide genomic resources for alfalfa research. Mol Plant. 2020;13:1250–61 [DOI] [PubMed] [Google Scholar]
- 9. Supapvanich S, Pimsaga J, Srisujan P. Physicochemical changes in fresh-cut wax apple (Syzygium samarangenese [Blume] Merrill & L.M. Perry) during storage. Food Chem. 2011;127:912–7 [DOI] [PubMed] [Google Scholar]
- 10. FAO . Growing pains for tropical fruit market. Vol. 2022 (ed. FAO) (Food and Agricultural Organization of the United Nations, 2005). Italy [Google Scholar]
- 11. Wang L, He L, Li J. et al. Regulatory change at Physalis organ size 1 correlates to natural variation in tomatillo reproductive organ size. Nat Commun. 2014;5:4271. [DOI] [PubMed] [Google Scholar]
- 12. Frary A, Nesbitt TC, Frary A. et al. fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science. 2000;289:85–8 [DOI] [PubMed] [Google Scholar]
- 13. Zhao J, Jiang L, Che G. et al. A functional allele of CsFUL1 regulates fruit length through repressing CsSUP and inhibiting auxin transport in cucumber. Plant Cell. 2019;31:1289–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H, Zhang H, Liang F. et al. PbEIL1 acts upstream of PbCysp1 to regulate ovule senescence in seedless pear. Hort Res. 2021;8:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amato A, Cardone MF, Ocarez N. et al. VviAGL11 self-regulates and targets hormone- and secondary metabolism-related genes during seed development. Horticulture Research. 2022;9. uhac133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharif R, Su L, Chen X. et al. Hormonal interactions underlying parthenocarpic fruit formation in horticultural crops. Horticulture Research. 2022;9:uhab024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen L, Liu YG. Male sterility and fertility restoration in crops. Annu Rev Plant Biol. 2014;65:579–606 [DOI] [PubMed] [Google Scholar]
- 18. Xu J, Wei X, Xu L. et al. Introduction and supporting cultivation techniques of 'Zihong' wax apple in Fujian. China Fruits. 2016;90–3 [Google Scholar]
- 19. Koren S, Walenz BP, Berlin K. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manni M, Berkeley MR, Seppey M. et al. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J, Zhang X, Tang H. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet. 2018;50:1565–73 [DOI] [PubMed] [Google Scholar]
- 22. Pavlidis P, Živkovic D, Stamatakis A. et al. SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Mol Biol Evol. 2013;30:2224–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Azzi L, Deluche C, Gévaudant F. et al. Fruit growth-related genes in tomato. J Exp Bot. 2015;66:1075–86 [DOI] [PubMed] [Google Scholar]
- 24. Yao JL, Xu J, Cornille A. et al. A microRNA allele that emerged prior to apple domestication may underlie fruit size evolution. Plant J. 2015;84:417–27 [DOI] [PubMed] [Google Scholar]
- 25. Lora J, Hormaza JI, Herrero M. et al. Seedless fruits and the disruption of a conserved genetic pathway in angiosperm ovule development. Proc Natl Acad Sci U S A. 2011;108:5461–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen H, Zeng Y, Yang Y. et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat Commun. 2020;11:2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun H, Jiao WB, Krause K. et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat Genet. 2022;54:342–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma L, Dong C, Song C. et al. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput Struct Biotechnol J. 2021;19:3954–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yao JL, Kang C, Gu C. et al. The roles of floral organ genes in regulating Rosaceae fruit development. Front Plant Sci. 2022;12:644424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jofuku KD, Omidyar PK, Gee Z. et al. Control of seed mass and seed yield by the floral homeotic gene APETALA2. Proc Natl Acad Sci U S A. 2005;102:3117–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yant L, Mathieu J, Dinh TT. et al. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell. 2010;22:2156–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomson B, Wellmer F. Molecular regulation of flower development. Curr Top Dev Biol. 2019;131:185–210 [DOI] [PubMed] [Google Scholar]
- 33. Weigel D, Meyerowitz EM. The ABCs of floral homeotic genes. Cell. 1994;78:203–9 [DOI] [PubMed] [Google Scholar]
- 34. José Ripoll J, Bailey LJ, Mai QA. et al. microRNA regulation of fruit growth. Nat Plants. 2015;1:15036. [DOI] [PubMed] [Google Scholar]
- 35. Zhu L, Li B, Wu L. et al. MdERDL6-mediated glucose efflux to the cytosol promotes sugar accumulation in the vacuole through up-regulating TSTs in apple and tomato. Proc Natl Acad Sci U S A. 2020;118:e2022788118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim MJ, Kim M, Lee MR. et al. LATERAL ORGAN BOUNDARIES DOMAIN (LBD)10 interacts with SIDECAR POLLEN/LBD27 to control pollen development in Arabidopsis. Plant J. 2015;81:794–809 [DOI] [PubMed] [Google Scholar]
- 37. Ye W, Qin Y, Ye Z. et al. Seedless mechanism of a new mandarin cultivar ‘Wuzishatangju’ (Citrus reticulata Blanco). Plant Sci. 2009;177:19–27 [Google Scholar]
- 38. Wang D, Cai J, Zhu BQ. et al. Study of free and glycosidically bound volatile compounds in air-dried raisins from three seedless grape varieties using HS-SPME with GC-MS. Food Chem. 2015;177:346–53 [DOI] [PubMed] [Google Scholar]
- 39. Sorensen AM, Kröber S, Unte US. et al. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003;33:413–23 [DOI] [PubMed] [Google Scholar]
- 40. Zhang W, Sun Y, Timofejeva L. et al. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development. 2006;133:3085–95 [DOI] [PubMed] [Google Scholar]
- 41. Zhu J, Chen H, Li H. et al. Defective in Tapetal development and function 1 is essential for anther development and tapetal function for microspore maturation in Arabidopsis. Plant J. 2008;55:266–77 [DOI] [PubMed] [Google Scholar]
- 42. Zhu L, He S, Liu Y. et al. Arabidopsis FAX1 mediated fatty acid export is required for the transcriptional regulation of anther development and pollen wall formation. Plant Mol Biol. 2020;104:187–201 [DOI] [PubMed] [Google Scholar]
- 43. Qian H, Li Y, Sun C. et al. Trace concentrations of imazethapyr (IM) affect floral organs development and reproduction in Arabidopsis thaliana: IM-induced inhibition of key genes regulating anther and pollen biosynthesis. Ecotoxicology. 2015;24:163–71 [DOI] [PubMed] [Google Scholar]
- 44. Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walker BJ, Abeel T, Shea T. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, Zhang S, Zhao Q. et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on hi-C data. Nat Plants. 2019;5:833–45 [DOI] [PubMed] [Google Scholar]
- 47. Xu M, Guo L, Gu S. et al. TGS-GapCloser: fast and accurately passing through the Bermuda in large genome using error-prone third-generation long reads. GigaScience 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rhie A, Walenz BP, Koren S. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang X, Chen S, Shi L. et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat Genet. 2021;53:1250–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haas BJ, Papanicolaou A, Yassour M. et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haas BJ, Delcher AL, Mount SM. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pertea M, Kim D, Pertea GM. et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77 [DOI] [PubMed] [Google Scholar]
- 58. Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90 [DOI] [PubMed] [Google Scholar]
- 59. Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–2 [DOI] [PubMed] [Google Scholar]
- 60. De Bie T, Cristianini N, Demuth JP. et al. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–71 [DOI] [PubMed] [Google Scholar]
- 61. Tang H, Bowers JE, Wang X. et al. Synteny and collinearity in plant genomes. Science. 2008;320:486–8 [DOI] [PubMed] [Google Scholar]
- 62. Sun P, Jiao B, Yang Y. et al. WGDI: a user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Molecular Plant. 2022;15:1841–51 [DOI] [PubMed] [Google Scholar]
- 63. Wang J, Yuan J, Yu J. et al. Recursive Paleohexaploidization shaped the durian genome. Plant Physiol. 2019;179:209–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McKenna A, Hanna M, Banks E. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Patterson N, Moorjani P, Luo Y. et al. Ancient admixture in human history. Genetics. 2012;192:1065–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Danecek P, Auton A, Abecasis G. et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim D, Paggi JM, Park C. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Garber M, Grabherr MG, Guttman M. et al. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–77 [DOI] [PubMed] [Google Scholar]
- 71. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shannon P, Markiel A, Ozier O. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing reads used for de novo whole-genome assembly and the final genome and annotation files have been deposited in the Genome Warehouse in National Genomics Data Center, under accession number GWHDUEN00000000 and GWHDVFZ00000000 which are publicly accessible at https://ngdc.cncb.ac.cn/gwh. Raw resequencing data were uploaded to the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn/), submission ID: WGS034963; BioProject access number: PRJCA011822; BioSample access number: SAMC1129200; GSA access number: CRA010157.