

Significance
The G protein–coupled receptor Smoothened (SMO) interacts biochemically with Gi proteins and guides cellular differentiation in both embryogenesis and cancer. We report here the fully activated structure of SMO coupled to a Gi protein and bound to sterol ligands, provided by all-atom molecular dynamics simulations. We find that G protein binding to SMO is sufficient to break the molecular lock that regulates receptor activation, form anchors with Gi subunits, tilt the extracellular cysteine-rich domain, and cause a sterol ligand in the transmembrane region to migrate to an activated position. We expect that this structure will provide the basis for designing selective small molecules to regulate SMO function.
Keywords: Smoothened receptor, G protein–coupled receptor, molecular dynamics, sterols, activation
Abstract
Smoothened (SMO) is an oncoprotein and signal transducer in the Hedgehog signaling pathway that regulates cellular differentiation and embryogenesis. As a member of the Frizzled (Class F) family of G protein–coupled receptors (GPCRs), SMO biochemically and functionally interacts with Gi family proteins. However, key molecular features of fully activated, G protein–coupled SMO remain elusive. We present the atomistic structure of activated human SMO complexed with the heterotrimeric Gi protein and two sterol ligands, equilibrated at 310 K in a full lipid bilayer at physiological salt concentration and pH. In contrast to previous experimental structures, our equilibrated SMO complex exhibits complete breaking of the pi-cation interaction between R4516.32 and W5357.55, a hallmark of Class F receptor activation. The Gi protein couples to SMO at seven strong anchor points similar to those in Class A GPCRs: intracellular loop 1, intracellular loop 2, transmembrane helix 6, and helix 8. On the path to full activation, we find that the extracellular cysteine-rich domain (CRD) undergoes a dramatic tilt, following a trajectory suggested by positions of the CRD in active and inactive experimental SMO structures. Strikingly, a sterol ligand bound to a shallow transmembrane domain (TMD) site in the initial structure migrates to a deep TMD pocket found exclusively in activator-bound SMO complexes. Thus, our results indicate that SMO interacts with Gi prior to full activation to break the molecular lock, form anchors with Gi subunits, tilt the CRD, and facilitate migration of a sterol ligand in the TMD to an activated position.
The Hedgehog (Hh) signaling pathway is an essential regulator of cellular differentiation and renewal in both embryonic and adult processes (1). Faulty Hh signaling has a variety of consequences, including birth defects and cancers such as basal cell carcinoma and medulloblastoma (2). As a primary signal transducer of the Hedgehog pathway (3), Smoothened (SMO) is a logical drug target for treatment of such cancers. Although several SMO inhibitors, including sonidegib and vismodegib, are well established in the clinic, there is considerable interest in developing additional small molecule antagonists that avoid known resistance mechanisms and target noncanonical SMO activities (4). A thorough understanding of ligand binding and SMO activation is necessary to develop such agents through structure-guided drug design.
A high-resolution structure of active SMO bound to its putative sterol ligands and its coupled heterotrimeric Gi protein would provide key details about responses to endogenous and synthetic molecules. Indeed, numerous cryogenic electron microscopy (cryo-EM) and X-ray crystal structures of SMO are available, with resolutions between 2.45 and 3.96 Å (5–15). However, these structures often do not resolve important salt bridge and hydrogen bonding interactions, and several are missing key components. Most SMO constructs used for structure determination contain the transmembrane domain (TMD) and extracellular linker domain (LD), but only a subset contains the cysteine-rich domain (CRD), N-linked glycans, or a coupled Gi protein. Moreover, experimental structures of activated SMO complexed with Gi proteins are of modest resolution, ranging from 3.14 to 3.96 Å (7, 9). Currently, the only structure of unmutated SMO bound to a heterotrimeric Gi protein that resolves part of the CRD has a resolution of 3.96 Å (7). Since the positions of only two CRD helices were visible in this structure, a full map was derived by docking the CRD from an inactive X-ray structure (12) onto the cryo-EM density.
To perform an optimized analysis of the SMO–Gi protein complex with sterol ligands, we started with the 3.84 Å cryo-EM structure of the human SMO TMD bound to a heterotrimeric Gαi protein, which contained the ligand 24(S),25-epoxycholesterol (CO1) bound to a shallow TMD pocket (PDB 6OT0) (9). We removed the Fab-G50 antibody present in the experiment and added a palmitoyl group to the N terminus of Gαi and a myristoyl group to the C-terminus of Gγ. We then embedded the complex in a phosphatidylcholine (POPC) bilayer. We combined this structure with the CRD from the 3.20 Å X-ray structure of an inactive V329F SMO mutant containing a cholesterol ligand bound to the CRD (PDB 5L7D) (12). After optimizing the side chains using the side-chain rotamer excitation analysis method (SCREAM) (16), we conducted long (up to 370 ns) molecular dynamics (MD) simulations to predict the fully active structure of SMO. Because the CRD is known to be mobile the protein is dynamic, making the time-dependent nature of MD particularly appropriate for these studies.
Our predicted structure provides a full atomistic map of active SMO with a bound Gi protein. We find that intracellular loop 1 (ICL1), intracellular loop 2 (ICL2), transmembrane helix 6 (TM6), and helix 8 (H8) of SMO make strong contacts to the coupled Gi protein at seven anchor points, positioning the Gαi5 helix for extensive interactions with the receptor. Significantly, we show that Gi interacts with SMO prior to full activation, rotating TM6 of SMO outward, breaking the pi-cation lock, and causing a sterol bound at a shallow TMD site to migrate downward to a fully activated location. We also identify numerous salt bridge and hydrogen bonding interactions that link the motions of the TMD, LD, and CRD. These findings reveal key similarities and differences between SMO and other GPCRs, offering detailed insight into SMO activation.
Results
Strong SMO–Gi Protein Contacts Accompany Receptor Activation.
In our SMO–Gαi–ligand structure (Fig. 1A) equilibrated with 370 ns of MD at 310 K, SMO is coupled to the Gi protein at seven strong anchor points, none of which were resolved experimentally. Prior to allowing changes in the protein backbone, only two of these salt bridges could be identified via SCREAM: namely, between K4406.21 and D341Gαi5 (Fig. 1B) and R2612.38 and D350Gαi (Fig. 1C) (superscripts refer to Class F Wang numbering) (13). After allowing full equilibration of the system, we find that most interactions between SMO and the Gi protein involve the Gαi subunit, specifically the Gαi5 helix (SI Appendix, Fig. S1). In particular, K4406.21 near the cytoplasmic end of TM6 forms a stable system of salt bridges with both D341Gαi5 and E318Gαi-β5/β6 loop in the RAS-like domain (Fig. 1B). This strongly couples TM6 to the Gαi subunit, consistent with the role of TM6 in receptor activation (17). In addition, the Gαi terminal carboxylate, F354Gαi5, forms salt bridges to K4446.25 in TM6 and K5398.48 in H8 (Fig. 1B), while D350Gαi5 forms a stable salt bridge to R2612.38 near ICL1 (Fig. 1C). These five salt bridges shift the Gαi5 helix both toward TM6 and deeper into the receptor, facilitating interactions with SMO, which include stable hydrogen bonds between the backbone of A2642.41 and D350Gαi5 and between W3393.50 and the backbone of C351Gαi5 (Fig. 1C). These contacts are reinforced by variable water-mediated hydrogen bonds between N347Gαi5 and Y350ICL2 and between N347Gαi5 and the backbone of S3423.53 (Fig. 1C).
Fig. 1.
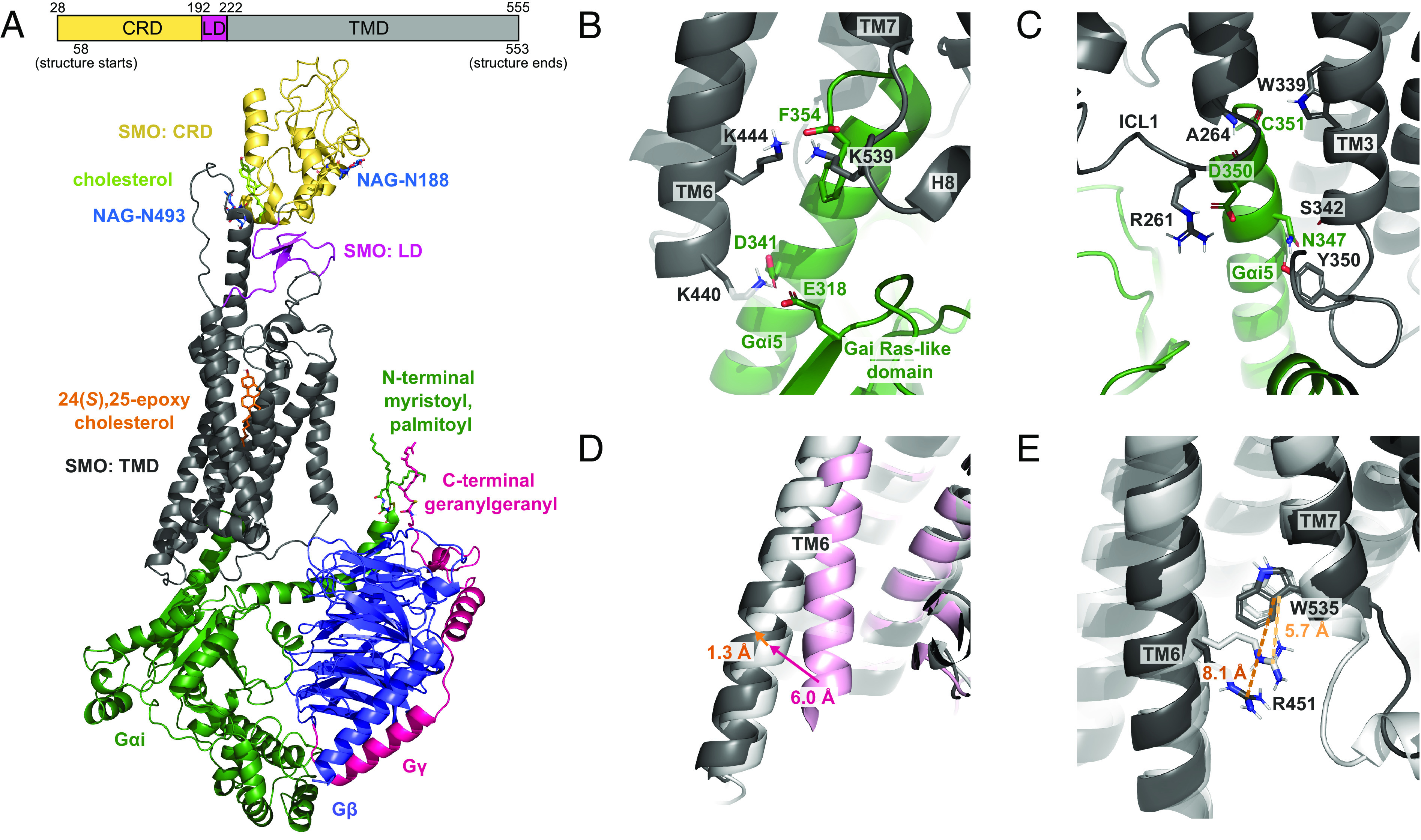
Formation of seven strong anchor points in the equilibrated MD structure of the human SMO-24(S),25-epoxycholesterol (CO1)-cholesterol-Gi complex. (A) Schematic of the SMO protein and side view of the complex. (B) Interaction between the Gαi5 helix and residues in TM6 and H8. (C) Interaction between the Gαi5 helix and residues in ICL1 and TM3. (D) Outward movement of the cytoplasmic end of TM6 between the inactive (PDB 5L7D, pink), initial (0 ns, white), and equilibrated structures (369 ns, dark gray). (E) Breaking of the R4516.32-W5357.55 pi-cation lock between the initial (0 ns, white) and equilibrated structure (369 ns, dark gray).
Notably, these anchors cause the Gαi5 helix to insert 3.4 Å further into SMO (Fig. 2A), suggesting that our predicted SMO–Gi protein complex is more fully activated than the starting cryo-EM structure. Furthermore, the cytoplasmic end of TM6 moves an additional 1.3 Å outward from the starting cryo-EM structure to the final equilibrated structure (Fig. 1D). The overall outward displacement of TM6 relative to the inactive structure (12) is 6.0 Å, following a pattern of receptor activation that is also seen in Class A GPCRs (18). Finally, the pi-cation interaction between R4516.32 and W5357.55, which acts as a molecular switch for the activation of Class F GPCRs (19), is present in the starting structure but breaks during equilibration (Fig. 1E), indicating that our final structure is fully activated.
Fig. 2.
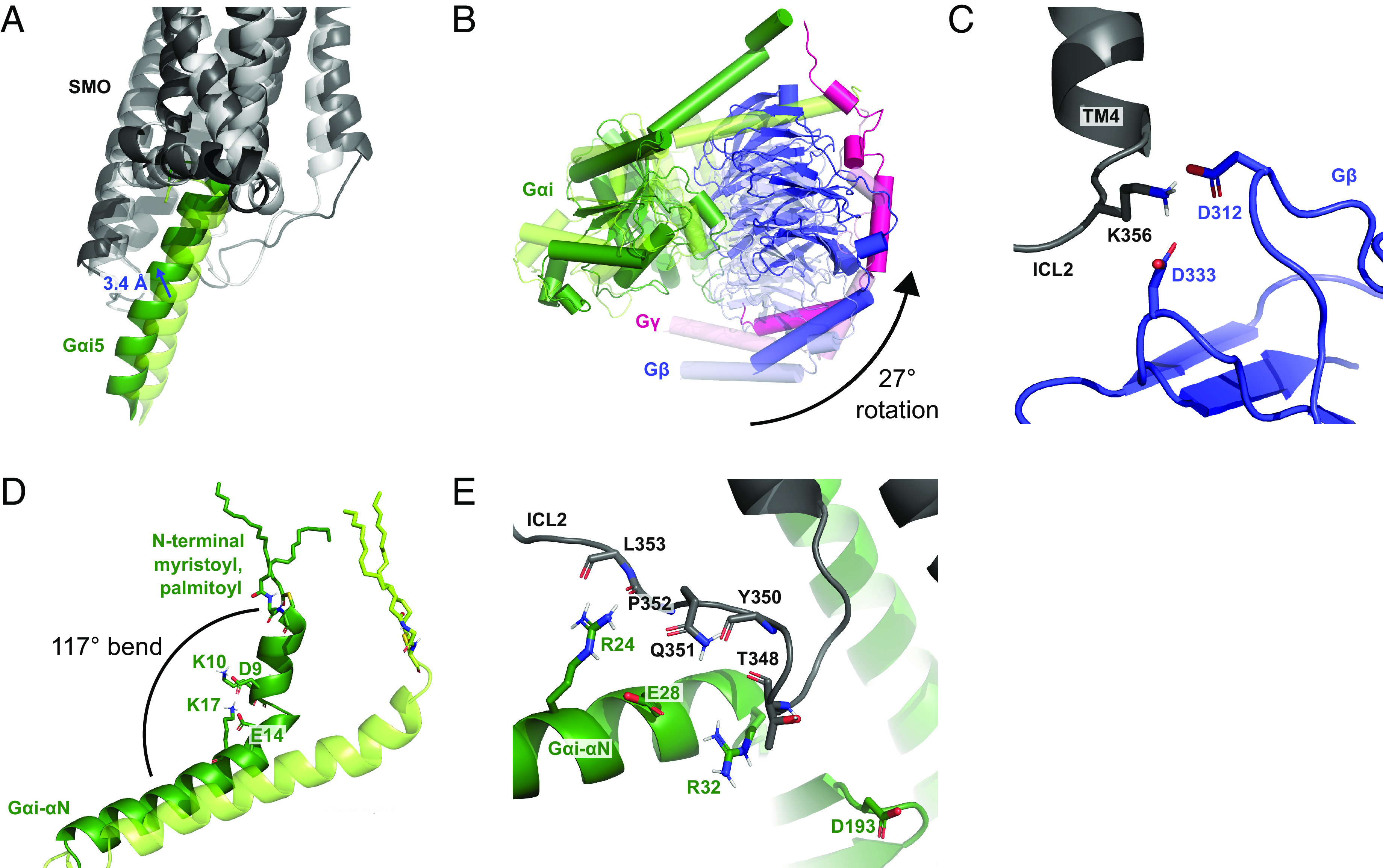
Reorientation of the heterotrimeric Gi relative to SMO over a 370-ns MD simulation. (A) Comparison of the Gαi5 binding position in the initial (0 ns, light tint) and final structure (370 ns, dark tint). (B) Comparison of the Gi protein subunits between the initial (0 ns, light tint) and final structure (370 ns, dark tint). (C) Interaction between ICL2 and Gβ at 370 ns. (D) Movement of the Gαi-αN helix toward the membrane between the initial (0 ns, light tint) and final structure (370 ns, dark tint). (E) Interaction between ICL2 and Gαi at 370 ns.
Over the course of our 370 ns of MD simulations, several components of the heterotrimeric G protein reorient relative to the SMO receptor (Fig. 2 A and B and SI Appendix, Figs. S2 and S3). Interestingly, an interaction that was resolved in the original cryo-EM structure between D312Gβ of the Gβ subunit and R25712.48 of SMO (9) was lost, despite computational attempts to preserve it by temporarily imposing harmonic restraints. Instead, we found that D312Gβ and D333Gβ of Gβ form two new, highly stable salt bridges with K3564.41 in ICL2 of the SMO receptor (Fig. 2C). These two salt bridges pull the Gi protein toward ICL2, rotating it by 27° from the starting structure. This corresponds to a shift in Gβ toward ICL2 by 3.48 Å, measured from the center of mass (Fig. 2B). In addition, the N-terminal end of the Gαi-αN helix bends toward the lipid bilayer, making a 117° angle with the C-terminal end of the helix. This bending is promoted by a network of salt bridges among D9Gαi-αN, K10Gαi-αN, E14Gαi-αN, and K17Gαi-αN, and may be facilitated by the lipid anchors on Gαi (Fig. 2D). These movements create new interactions between ICL2 of SMO and the Gαi-αN helix, which often fluctuate and become water-mediated (Fig. 2E). Specifically, R24Gαi-αN of Gαi-αN forms hydrogen bonds to both Q351ICL2 and the backbone atoms of P352ICL2 and L353ICL2 of SMO. Hydrogen bonds also form between E28Gαi-αN and Q351ICL2, between R32Gαi-αN and the backbone of Y350ICL2, and between D193Gαi-β2/β3 loop and both the backbone and side chain of T348ICL2. Strong SMO–G protein interactions coincide with full outward rotation of TM6 in replicate simulations. Significantly, control simulations without a G protein show only partial movement of TM6, suggesting that this aspect of SMO activation may be facilitated by G protein coupling (SI Appendix, Fig. S4).
Several SMO–G protein interactions are similar to those revealed in previous studies of Class A GPCRs, in particular the human κ-opioid receptor (20). For example, both receptors share a close interaction between TM6 and Gαi, which in SMO encompasses salt bridges between K4406.21 and both D341Gαi5 and E318Gαi-β5/β6 loop (Fig. 1B). This interaction parallels a salt bridge between K2656.26 and E318Gαi-β5/β6 loop in the κ-opioid receptor (20). Furthermore, both receptors form salt bridges at the Gαi5 terminal carboxylate with TM6, which involve K4446.25 in SMO (Fig. 1B) and R2716.32 in the κ-opioid receptor (20). A significant difference is that, while Class A GPCRs form salt bridges between D350Gαi5 of Gαi and ICL2 (21), SMO does not, instead forming a salt bridge between D350Gαi5 and R2612.38 near ICL1 (Fig. 1C) and hydrogen bonds between Gαi-αN and ICL2 (Fig. 2E). Indeed, the position of the Gαi5 helix in SMO is unusually close to both TM6 and TM7 compared to other GPCRs, and shifts even further in this direction as SMO becomes increasingly activated (SI Appendix, Fig. S5) (22). Despite the fact that Class A and Class F GPCRs share few structural motifs, the similarities in receptor-Gi contacts suggest that important features of Gi protein coupling are conserved between diverse GPCR families.
G Protein Coupling Exposes a Buried TMD Sterol Pocket.
While the positions of the TM helices change considerably at the beginning of the simulation (Fig. 3A), they remain quite stable once the structure has equilibrated (SI Appendix, Fig. S6A). At the extracellular portion of the helical bundle, TM1 and TM7 move inward by 3.0 Å and 1.1 Å (Fig. 3A, pink arrows) while TM2, TM5, and TM6 move outward by 2.0 Å, 2.1 Å, and 1.7 Å (Fig. 3A, orange arrows). At the intracellular portion, TM4 and TM6 move outward by 3.0 Å and 1.3 Å while TM5 moves inward by 1.3 Å (Fig. 3A, green arrows). TM3 bulges out near the middle and H8 undergoes a lateral movement of 2.6 Å toward TM1 (Fig. 3A, blue arrows). The movement of TM6, which is involved in activation of the receptor, also affects salt bridges between K4406.21 and both the Gαi5 helix and the Gαi Ras-like domain (Fig. 1B). As discussed below, the unusually long TM6 of SMO links the intracellular and extracellular domains, coupling the movement of TM6 to the position of the CRD.
Fig. 3.
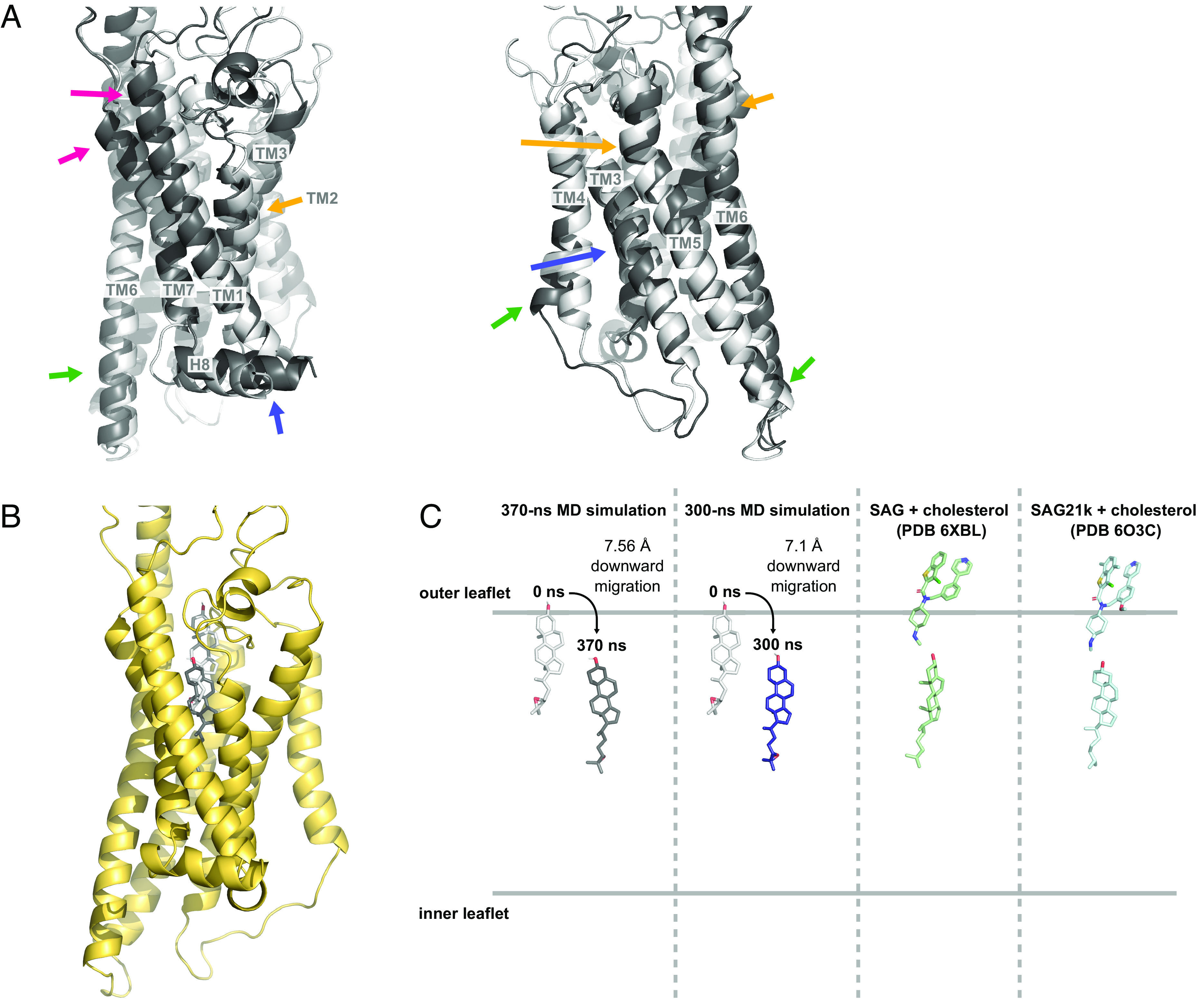
Repositioning of the TMD and downward migration of a sterol ligand. (A) Comparison of the TMD between the initial (0 ns, white) and final structure of the SMO–ligand–Gi complex (370 ns, dark gray). Colored arrows correspond to movements described in the text. (B) Position of 24(S),25-epoxycholesterol (CO1) in the initial (0 ns, white) and final structure (370 ns, dark gray). (C) Locations of CO1 in the initial and final computed structures in two independent MD simulations (0 ns, light gray; 370 ns, dark gray; 300 ns, navy) compared to the positions of SAG/SAG21k and cholesterol in cryo-EM structure (6XBL, green) and crystal structure (6O3C, blue).
Changes in the TM region of SMO profoundly influence its sterol-binding site. Remarkably, over the course of our 370 ns simulation, the CO1 ligand starting in a shallow TMD pocket in the initial cryo-EM structure migrates approximately 7.56 Å downward in the receptor (Fig. 3 B and C). The final position of the ligand coincides with the location of a deeply bound cholesterol observed in a 2.8 Å SAG21k-bound SMO stabilized by an activating nanobody (8) and a putative sterol in a 3.96 Å structure of a SAG-bound SMO–Gαi-antibody complex (7) (Fig. 3C). Our attempts to locate hydrogen bonds to the C3-hydroxyl group or the C24-C25 epoxide oxygen of CO1 at various positions in this trajectory failed to identify significant contacts, indicating that ligand interactions during migration are dominated by hydrophobic and steric factors (SI Appendix, Fig. S6B). A second 300-ns simulation with SMO–CO1–cholesterol–Gi shows the same the downward movement of the CO1 ligand by 7.1 Å, measured from the center of mass (Fig. 3C).
Notably, the observed downward movement of the sterol during equilibration provides specific information regarding SMO activation. While previous models have proposed a trajectory of sterol flipping and lateral access to an SMO tunnel between the lower and upper TMD sites (7), our results raise the possibility of alternative dynamics. Indeed, our observations align with proposals involving initial sterol entry at the upper TMD site via the outer leaflet, followed by descent within the TMD during activation (23–25). However, the order of events in G protein binding and TMD sterol repositioning is unknown for SMO (6, 26). Our finding that Gαi coupling and structural hallmarks of SMO activation coincide with opening of an activated sterol binding site suggests that G protein coupling may be a key parameter in SMO-sterol interactions.
The Position of the CRD Is Flexible and Is Coupled to the TMD.
Because it is locked in place by five disulfide bridges, the overall shape of the CRD is largely static (27, 28). However, the flexibility of the CRD relative to the TMD makes it difficult to resolve experimentally. While it has been shown that SMO can be activated by binding an oxysterol at the CRD (29, 30), a mutant SMO missing its CRD exhibits higher levels of basal signaling (31), highlighting the need for dynamic models of CRD-TMD interactions.
Most experimental structures, whether activated or inactive, show the CRD positioned somewhat similarly relative to the TMD, with a slight tilt toward parallel in more activated structures (8) (SI Appendix, Fig. S7). Previous MD simulations to investigate the dynamics of this movement suggest that the CRD of inactive apo-hSMO also fluctuates but becomes constrained upon ligand binding to the CRD (11, 12). Our studies that include the Gi protein show that CRD flexibility is restored in fully active sterol-bound SMO. Over the course of our 370 ns MD calculations, the CRD undergoes a wide range of motion both during equilibration and in the fully active state, where its center of mass swings by 8.5 Å between the 340 ns and 360 ns frames (Fig. 4A). Although the motion of the CRD fluctuates in a hinge-like manner, the overall trend is that activation leads to a change from bent to linear with respect to the TMD (Fig. 4B). Thus, by 369 ns, the CRD has moved 10.7 Å from the starting structure, as measured from the center of mass. A second simulation of SMO–ligand–Gαi complex similarly showed a 19.7 Å movement toward a linear position by the 300 ns mark (SI Appendix, Fig. S7B). In contrast, in two control simulations without the Gi protein the CRD tilts in a bent direction, moving by 15.9 Å in one case and 9.2 Å in the other (SI Appendix, Fig. S7A). This is consistent with previous MD simulations of SMO in the absence of a G protein, which also identified TMD/CRD interactions that stabilized a bent conformation (12). Together, these results imply that the CRD is flexible and that G protein coupling may favor an equilibrium toward the upright orientation.
Fig. 4.
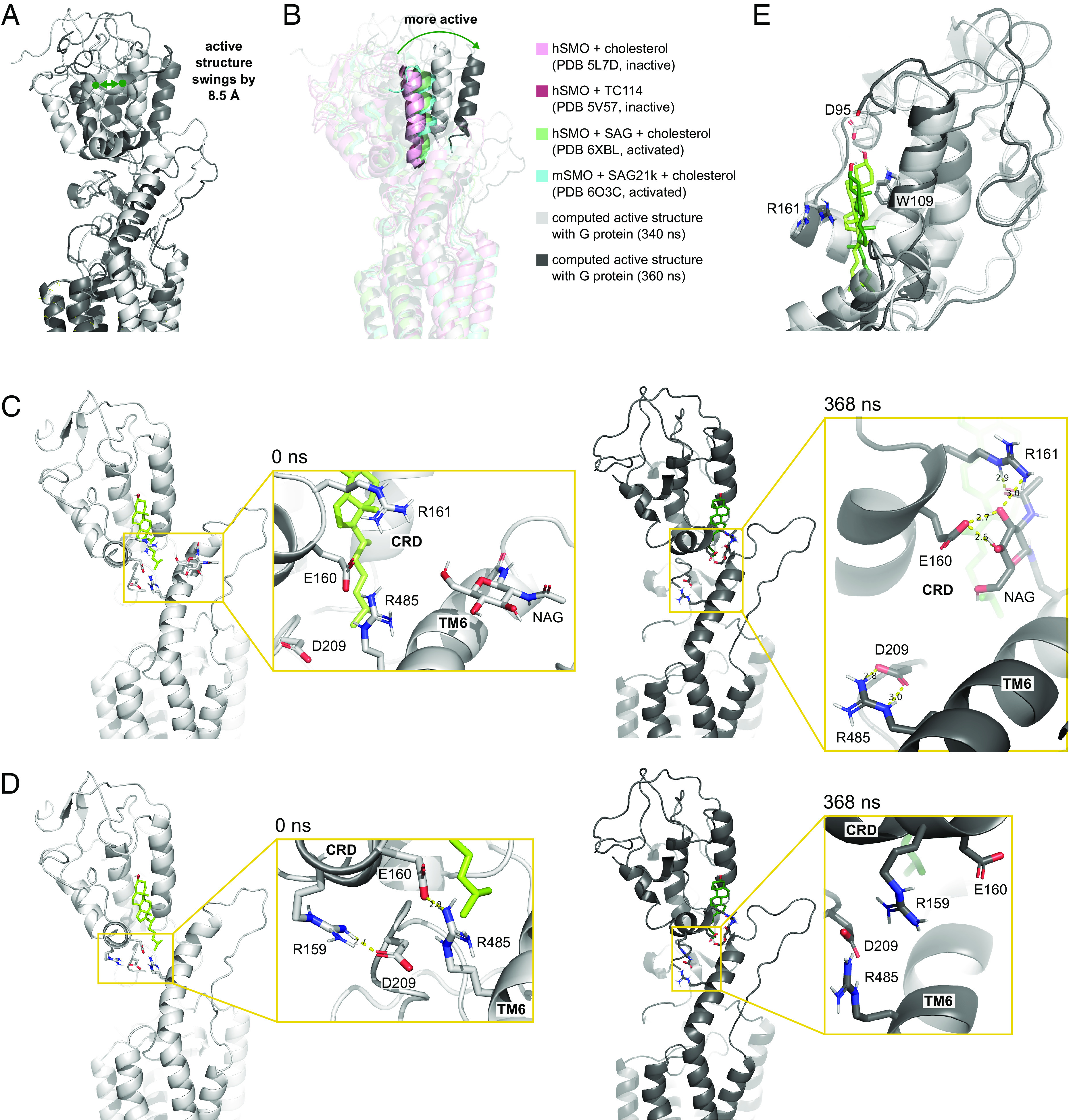
CRD movements and interactions with the LD and TMD. (A) Comparison between the computed SMO structure at 340 (white) and 360 ns (dark gray). (B) A representative CRD helix showing the relative positions of the computed structures (340 ns, light gray; 360 ns, dark gray), inactive (PDB 5L7D, light pink; PDB 5V57, dark pink), and active (PDB 6XBL, green; PDB 6O3C, blue) experimental structures. (C) Exchange of salt bridges between the CRD and residues in the TMD and LD during CRD motion. (D) Weakening of an interaction between D209LD and R159CRD and breaking of the interaction at E160CRD and R4856.66 between the initial (0 ns, light gray) and final structure (370 ns, dark gray). (E) Comparison of CRD-cholesterol interactions between the initial (0 ns, light tint) and final structure (370 ns, dark tint).
The orientation of the CRD is intimately connected to the TMD via the LD, which consists of a number of flexible loops that make hydrogen bonds to both the CRD and the TMD. We find that the CRD is also linked to TM6 through a network of salt bridges between R159CRD, E160CRD, R161CRD, D209LD, E2261.28, R4856.66, D4866.67, E508ECL3, and K510ECL3, as well as through hydrogen bonding with the N-linked glycan (NAG) at N4936.74. Interactions between NAG, E160CRD, D209LD, and R4856.66 can also be seen in the initial structure (12).
Several interactions within this large network of salt bridges may be critical to the conformational changes that we observe in the CRD. Two strong interactions emerge between TM6 and the LD and CRD, including a salt bridge between D209LD and R4856.66 and a hydrogen bond between NAG at N4936.74, E160CRD, and R161CRD (Fig. 4C). In order for these interactions to form, contacts present at the start of the simulation weaken or break; namely, a salt bridge between D209LD and R159CRD weakens (Fig. 4D) and a salt bridge between E160CRD and R4856.66 breaks (Fig. 4C).
Notably, the glycan at N4936.74 forms the side of the CRD binding pocket at the interface between the TMD and CRD. Hydrogen bonds to NAG at this position shield the CRD binding pocket from the solvent and weakly anchor the CRD to TM6. Without this anchor, it is possible that the CRD would oscillate even more wildly (10). It has been shown in mice that although N-linked glycosylation of SMO is not necessary for canonical Hh signaling, it is required for noncanonical Hh signaling through Gαi (32). These observations indicate that the N493 glycan occupies a key position linking the movements of the TMD and CRD and may act to coordinate multiple signaling outputs.
The CRD also contains cholesterol at a binding site near the TM6 extension (Fig. 4E). Over the course of our simulations, the cholesterol rotates and moves slightly (1.4 Å) downward relative to the CRD. This movement breaks the stacking interactions with Y109CRD found in the inactive experimental structure (12). The rotation also results in loss of a hydrogen bond between D95CRD and the C3-hydroxyl group of the cholesterol, despite computational attempts to preserve their interaction. By contrast, a pi-cation interaction between cholesterol and R161CRD that is present in the initial structure is maintained throughout our calculations. Overall, these results highlight the highly hydrophobic nature of the CRD sterol binding pocket and emphasize the importance of nonpolar interactions in stabilizing the bound cholesterol ligand.
Discussion
The seven anchor points between SMO and a bound Gαi protein found in our MD simulations fall into two main categories: 1) interactions at Gαi which pull the Gαi5 helix toward ICL1 and TM6, and 2) interactions at Gβ which pull Gβ and Gαi-αN toward ICL2. Both types of anchors cause the Gi protein to insert more fully into the receptor, implying that SMO becomes more fully activated over the course of our MD simulations. This conclusion is also supported by the extensive interactions between the Gi protein and TM6, which rotate TM6 outward and break the pi-cation activation lock between R4516.32 and W5357.55.
The progression of the structures in the MD simulation over time suggests a possible trajectory for SMO activation. Anchors first begin to form between SMO and the Gi protein at the Gαi5 helix. As the Gi protein moves toward SMO, interactions with Gβ shift from ICL1 to ICL2. Progressive formation of each Gαi5 interaction inserts Gαi5 more deeply into the receptor, allowing additional anchors to form. As the contacts between the Gi protein and SMO increase, the TM helices move to expand the lower region of the TMD, enabling the sterol ligand to move downward by 7.6 Å in the binding pocket.
Our fully equilibrated SMO–ligand–Gi complex shares several key interactions with Class A GPCRs. Both types of receptors interact with the G protein through many of the same Gαi residues, including multiple interactions with TM6. However, while Class A receptors anchor to Gαi via salt bridges to ICL2, SMO does not. These differences coincide with the atypical position of the Gαi5 helix relative to SMO as compared to other classes of GPCR. Thus, although SMO engages many of the contacts identified in Class A GPCRs, SMO features unique Gi protein interactions that characterize its distinct activation mechanism (22).
Previous models of SMO activation have been ligand-centric, suggesting that activation is caused by binding of an agonist or dynamic occupancy of endogenous ligands at specific positions (6, 7). Our results shed light on this analysis, demonstrating that G protein coupling may be sufficient to activate sterol-bound SMO. Indeed, MD equilibration of the partially active SMO bound to a heterotrimeric Gi protein fully breaks the molecular lock, tilts the CRD, and reshapes the sterol binding site in the TMD. These enhanced G protein interactions favor sterol binding at a location occupied by cholesterol exclusively when SMO is bound to the synthetic agonists SAG21k/SAG and stabilized in an activated state (8, 9). This sterol repositioning occurs despite the fact that neither SAG nor a second TMD sterol is present in our structure. Significantly, our simulations without the G protein show attenuated TM6 movement associated with activation and a bias toward a “bent” CRD conformation. Our proposed activation model thus suggests that the TMD ligand is mobile and that the nature of the TMD binding site may be coupled to SMO activation. In this scenario, the position of the sterol in the TMD binding pocket may influence the activation energy of the receptor in a manner similar to how partial agonists affect the function of Class A GPCRs (33–35). Ultimately, our simulations demonstrate that the equilibria involved in SMO activation depend on more than just ligand identity and concentration, showing that G protein coupling may play a key role in these dynamics.
Methods
Assembly of the Human SMO–CO1–cholesterol–Gi Protein Complex.
To build the initial complex of the full SMO receptor combined with the Gαi protein, we obtained the following:
The CRD from the crystal structure of the human SMO receptor in complex with cholesterol (PDB 5L7D, residues 58–189) (12) and
The TMD from the cryo-EM structure of the human SMO receptor in complex with CO1 and a Gi protein (PDB 6OT0, residues 190–553) (9). This structure contains a dominant-negative human Gαi1 mutant (S47N, G204A, E246A, and A327S) that mimics an active, GDP-bound Gαi in the absence of GDP/GTP (36). We did not mutate Gαi back to the wild-type sequence in our complex.
We superimposed these structures using PyMOL (37) and reconstructed the peptide bond using Maestro (38). Then, we fixed the protein backbone while allowing residues 184–195 to relax and conducted 1 cycle of simulated annealing. This simulated annealing was performed over a span of 10 ps, during which time the residues in the loops were cyclically heated and cooled from 50 to 600 K. The resulting structure was then minimized for 300 steps.
Refining the Activated Human SMO in Complex with the Gi Protein.
Using the Maestro protein preparation function on the complex assembled above, we removed all hydrogens and N-linked glycans. We also reconstructed disulfide bridges between C217/C295 and C314/C390, choosing rotamers to create perpendicular CβS-S-Cβ dihedrals. Next, the missing hydrogens were replaced using LinGraf (39).
We then optimized the side chain conformations using the side chain rotamer excitation analysis method (SCREAM) (16) and minimized the resulting structure for 400 steps. To identify hydrogen bonding interactions with the ligand and to resolve unfavorable interactions, we again used SCREAM to generate 10 possible rotamers for each of the following residues: N521, Q477, R400, M525, L325, F391, D95, K105, W163, L112, I156, and V210. We selected the optimal configuration for each residue based on the lowest potential energy and maximal hydrogen bonding, and then, we minimized the entire complex. At the end of this process, we fixed the protein backbone and performed an additional energy minimization to relax the ligand.
Finally, to replace the previously removed N-linked glycans, we superimposed the glycans from the original crystal structure (PDB 5L7D) (12) onto our refined structure in PyMOL. Using Maestro, we selected the original rotamers for N188 and N493 and then reconstructed the covalent bonds.
Gi Protein and System Environment Preparation.
We added residues 63–68 to the Gγ subunit of the Gi protein missing from the original cryo-EM structure (PDB 6OT0) (9). Next, we added lipid anchors to residues G2 and C3 of the Gαi subunit and C68 of the Gγ subunit.
Using the CHARMM-GUI input generator, we placed the refined SMO–ligand–Gi protein complex with lipid anchors into a 100 × 100 Å3 palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer with a 70-Å layer of water on both the intracellular and extracellular sides of the membrane. While previous studies have demonstrated that SMO and other GPCRs can interact with cholesterol and anionic lipids in the membrane (23, 40, 41), we used POPC as a representative bilayer for simplicity. Sodium and chloride ions were added to a physiological concentration of 0.15 M to balance the charge. The lipid anchors and N-linked glycans were manually rotated to the optimal orientation using Maestro. Notably, our initial sterol-like ligands were identical to cholesterol and CO1 except that the C5-C6 double bonds were saturated.
Initial Equilibration.
All equilibration was done using the CHARMM36 force field (42) and the GROMACS MD package (43) at a temperature of 310 K. For pre-equilibration, we conducted 10,000 steps of steepest descent energy minimization and then performed short NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) equilibrations for 250 ps and 325 ps, respectively. During this process, we temporarily imposed various positional restraints on heavy atoms and distance restraints between hydrogen bonding interactions and then gradually relaxed these restraints. In total, we equilibrated the system for ~240 ns, with no constraints for the final 150 ns.
Replacement of the Sterol Ligands.
After this initial 240 ns equilibration, we replaced our C5-C6-saturated sterol-like ligands in the CRD and the TMD with cholesterol and CO1, respectively. To accomplish this, we took the ligand coordinates from the last frame (240 ns) and then added the C=C bonds with Maestro. Using PyMOL, we next aligned the new ligands to their appropriate positions at 240 ns in the entire protein-solvent-ion-membrane system. We generated new input files for cholesterol and CO1 using the CHARMM-GUI input generator.
Final Equilibration.
We continued equilibration at a temperature of 310 K using the CHARMM36 force field and the GROMACS MD package. First, two steepest descent energy minimizations were performed on the new system: 1) minimization with positional restraints on all heavy atoms of the protein (force constant 10,000 kJ mol−1 nm−2) except those within 4 Å of the ligands and 2) minimization without positional restraints. These minimizations converged to maximum force under 1,000 kJ mol−1 nm−1 in 729 and 7 steps, respectively.
Finally, we equilibrated the system without restraints for an additional 130 ns.
Duplicate and Control Simulations.
To ensure reproducibility of results, we conducted an additional MD simulation of the SMO–CO1–cholesterol–Gi protein complex for 300 ns. We started from the system prepared in “Gi Protein and System Environment Preparation” of Methods but used native CO1 and cholesterol structures as ligands throughout. We performed a steepest descent energy minimization with positional restraints on all heavy atoms of the protein (force constant 10,000 kJ mol−1 nm−2) except those within 4 Å of the ligands. The energy minimization converged in 1,038 steps. We equilibrated the system for ~20 ns as described in steps 2−4 of SI Appendix, then removed constraints and equilibrated for a further 70 ns. For (h) and (i), we subsequently equilibrated for an additional 20 ns with restraints and 190 ns without restraints.
As a control, we conducted two MD simulations of the SMO–CO1–cholesterol complex without the Gi protein for 120 ns each. We used the protein complex described in “Refining the Activated Human SMO in Complex with the Gi Protein” of the Methods with the Gi protein removed. We used the CHARMM-GUI input generator as described to place the complex in solvent and membrane (box size 80 × 80 × 150 Å3) and then manually rotated the N-linked glycans to the correct positions. We performed a steepest descent energy minimization with positional restraints on all heavy atoms of the protein (force constant 10,000 kJ mol−1 nm−2) except those within 4 Å of the ligands. The energy minimization converged in 3,370 steps for each independent simulation. We equilibrated the system as described in steps 2−4 of SI Appendix, then removed constraints and equilibrated for a further 100 ns.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
W.A.G. thanks NIH (R01HL155532 and R35HL150807) for support.
Author contributions
A.E.O. and W.A.G. designed research; A.-D.P.V. and S.-K.K. performed research; A.-D.P.V., S.-K.K., and M.Y.Y. analyzed data; and A.-D.P.V., S.-K.K., A.E.O., and W.A.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: M.S.S., University of Oxford; and C.Z., University of Pittsburgh School of Medicine.
Contributor Information
Alison E. Ondrus, Email: aondrus@uic.edu.
William A. Goddard, III, Email: wag@caltech.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Wu F., Zhang Y., Sun B., McMahon A. P., Wang Y., Hedgehog signaling: From basic biology to cancer therapy. Cell Chem. Biol. 24, 252–280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pak E., Segal R. A., Hedgehog signal transduction: Key players, oncogenic drivers, and cancer therapy. Dev. Cell 38, 333–344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong J. H., Siebold C., Rohatgi R., Biochemical mechanisms of vertebrate hedgehog signaling. Development 146, dev166892 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arensdorf A. M., Marada S., Ogden S. K., Smoothened regulation: A tale of two signals. Trends Pharmacol. Sci. 37, 62–72 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang K., Wu H., Hoppe N., Manglik A., Cheng Y., Fusion protein strategies for cryo-EM study of G protein-coupled receptors. Nat. Commun. 13, 4366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kinnebrew M., et al. , Patched 1 regulates Smoothened by controlling sterol binding to its extracellular cysteine-rich domain. Sci. Adv. 8, 37 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi X., Friedberg L., De Bose-Boyd R., Long T., Li X., Sterols in an intramolecular channel of Smoothened mediate Hedgehog signaling. Nat. Chem. Biol 16, 1368–1375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshpande I., et al. , Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 571, 284–288(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qi X., et al. , Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 571, 279–283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang P., et al. , Structural basis of Smoothened activation in Hedgehog signaling. Cell 174, 312–324.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., et al. , Crystal structure of a multi-domain human Smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 8, 15383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrne E. F. X., et al. , Structural basis of Smoothened regulation by its extracellular domains. Nature 535, 517–522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C., et al. , Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 5, 4355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weierstall U., et al. , Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 5, 3309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C., et al. , Structure of the human Smoothened receptor bound to an antitumour agent. Nature 497, 338–343 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tak Kam V. W., Goddard W. A. III, Flat-bottom strategy for improved accuracy in protein side-chain placements. J. Chem. Theory Comput. 4, 2160–2169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weis W. I., Kobilka B. K., Structural insights into G-protein-coupled receptor activation. Curr. Opin. Struct. Biol. 18, 734–740 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauser A. S., et al. , GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 28, 879–888(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright S. C., et al. , A conserved molecular switch in Class F receptors regulates receptor activation and pathway selection. Nat. Commun. 10, 667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mafi A., Kim S.-K., Goddard W. A. III, The atomistic level structure for the activated human κ-opioid receptor bound to the full Gi protein and the MP1104 agonist. Proc. Natl. Acad. Sci. U.S.A. 117, 5836–5843 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong S. S., Goddard W. A. III, Abrol R., Conformational and thermodynamic landscape of GPCR activation from theory and computation. Biophys. J. 110, 2618–2629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen C., et al. , Structural basis of GABAB receptor-Gi protein coupling. Nature 594, 594–598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedger G., et al. , Cholesterol interaction sites on the transmembrane domain of the Hedgehog signal transducer and Class F G protein-coupled receptor Smoothened. Structure 27, 549–559.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bansal P. D., Dutta S., Shukla D., Activation mechanism of the human Smoothened receptor. Biophys. J. 22, 1400–1413 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumari S., Mitra A., Bulusu G., Structural dynamics of Smoothened (SMO) in the ciliary membrane and its interaction with membrane lipids. Biochim. Biophys. Acta Biomembr. 1864, 183946 (2022). [DOI] [PubMed] [Google Scholar]
- 26.Myers B. R., Neahring L., Zhang Y., Roberts K. J., Beachy P. A., Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. U.S.A. 114, E11141–E11150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nachtergaele S., et al. , Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. ELife 2, e01340 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang P., et al. , Cellular cholesterol directly activates Smoothened in Hedgehog signaling. Cell 166, 1176–1187.e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nachtergaele S., et al. , Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 8, 211–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nedelcu D., Liu J., Xu Y., Jao C., Salic A., Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat. Chem. Biol. 9, 557–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers B. R., et al. , Hedgehog pathway modulation by multiple lipid binding sites on the Smoothened effector of signal response. Dev. Cell 26, 346–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marada S., et al. , Functional divergence in the role of N-linked glycosylation in Smoothened signaling. PLoS Genet. 11, e1005473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mafi A., Kim S.-K., Goddard W. A. III, The mechanism for ligand activation of the GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A. 119, e2110085119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Audet M., Bouvier M., Restructuring G-protein-coupled receptor activation. Cell 151, 14–23 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Mafi A., Kim S.-K., Goddard W. A. III, The dynamics of agonist-β2-adrenergic receptor activation induced by binding of GDP-bound Gs protein. Nat. Chem. 15, 1127–1137 (2023). [DOI] [PubMed] [Google Scholar]
- 36.Liang Y. L., et al. , Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 1, 12–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeLano W. L., The PyMOL Molecular Graphics System (Version 2.4.1, Schrödinger, LLC; ). https://pymol.org/2/. Accessed 13 November 2023. [Google Scholar]
- 38.Maestro (Schrödinger LLC, New York, NY, 2021). [Google Scholar]
- 39.Olafson B. D., Goddard W. A., Lingraf release 3.23 (Materials and Process Simulation Center, Caltech, 2007). [Google Scholar]
- 40.Arensdorf A. M., et al. , Sonic Hedgehog activates phospholipase A2 to enhance Smoothened ciliary translocation. Cell Rep. 19, 2074–2087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thakur N., et al. , Anionic phospholipids control mechanisms of GPCR-G protein recognition. Nat. Commun. 14, 794 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang J., et al. , CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abraham M. J., et al. , GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.