

Significance
Hidradenitis suppurativa (HS) is a chronic inflammatory skin disease characterized by a complex heterogeneous histologic phenotype, that includes epidermal psoriasiform hyperplasia, follicular occlusion and hyperkeratosis, immune cell infiltration, dermal fibrosis, and the development of epithelialized sinus tract. Here, we delineated the reprogrammed transcriptional profiles in HS basal stem/progenitor cells and identified two keratinocyte populations (POSTN/ASS1 and S100A clusters) that amplify immune responses in HS. We present a database uncovering distinct chromatin accessibility patterns in epidermal progenitors in healthy and HS skin. The primed/active enhancers significantly affect the expression of inflammatory genes in HS. This study identifies a paradigm for targeted intervention in HS by combining biological therapies with pharmacological agents that block the chromatin landscape of inflammatory genes.
Keywords: skin epithelial cell, epigenetic regulation, S100A family genes, IRF3, hidradenitis suppurativa
Abstract
Hidradenitis suppurativa (HS) is a complex inflammatory skin disease with undefined mechanistic underpinnings. Here, we investigated HS epithelial cells and demonstrated that HS basal progenitors modulate their lineage restriction and give rise to pathogenic keratinocyte clones, resulting in epidermal hyperproliferation and dysregulated inflammation in HS. When comparing to healthy epithelial stem/progenitor cells, in HS, we identified changes in gene signatures that revolve around the mitotic cell cycle, DNA damage response and repair, as well as cell–cell adhesion and chromatin remodeling. By reconstructing cell differentiation trajectory and CellChat modeling, we identified a keratinocyte population specific to HS. This population is marked by S100A7/8/9 and KRT6 family members, triggering IL1, IL10, and complement inflammatory cascades. These signals, along with HS-specific proinflammatory cytokines and chemokines, contribute to the recruitment of certain immune cells during the disease progression. Furthermore, we revealed a previously uncharacterized role of S100A8 in regulating the local chromatin environment of target loci in HS keratinocytes. Through the integration of genomic and epigenomic datasets, we identified genome-wide chromatin rewiring alongside the switch of transcription factors (TFs), which mediated HS transcriptional profiles. Importantly, we identified numerous clinically relevant inflammatory enhancers and their coordinated TFs in HS basal CD49fhigh cells. The disruption of the S100A enhancer using the CRISPR/Cas9-mediated approach or the pharmacological inhibition of the interferon regulatory transcription factor 3 (IRF3) efficiently reduced the production of HS-associated inflammatory regulators. Our study not only uncovers the plasticity of epidermal progenitor cells in HS but also elucidates the epigenetic mechanisms underlying HS pathogenesis.
Hidradenitis suppurativa (HS) is a chronic, inflammatory skin disease characterized by recurrent abscesses, painful nodules, and scaring in high-density hair follicles and apocrine-gland-rich regions of the body (1, 2). Disease severity is an outcome of multiple interdependent factors including genetic mutations, hormonal status, lifestyle factors, and immune dysregulation (3–7). HS presents a heterogeneous histologic phenotype, which includes epidermal hyperplasia, follicular occlusion and hyperkeratosis, immune cell infiltration, dermal fibrosis, and development of epithelialized sinus tract (1, 8, 9). Overall, the disease activity and progression are likely propagated by dysregulated adaptive and innate immune responses, altered keratinocyte functions, along with changes in the microbiome of lesional skin hair follicles, sweat glands, and other areas of the skin (2, 10–13). Adalimumab (ADA), a fully humanized monoclonal antibody targeting soluble and transmembrane TNF-α, is currently the US Food and Drug Administration (FDA) approved biologic drug for the treatment of moderate-to-severe HS (14, 15). Surgical resection of affected tissue remains the ultimate option for patients who do not respond to available treatments.
The currently proposed models of plasticity of keratinocytes are 1) self-renewal and stochastic differentiation of interfollicular epidermal stem cells (IFESCs) in epidermal homeostasis (16–18); 2) generation of differentiated lineages by the differential rate of cell division of heterogeneous IFESC populations under homeostatic conditions; and 3) the balance between proliferation and differentiation co-contributed by stem cells and their committed progenitors (19–21). More recently, single-cell RNA sequencing (sc-RNA seq) revealed remarkable heterogeneity in human IFESCs/progenitors that differ in capabilities for stratified programming under physiological conditions (22, 23). Apart from studies describing homeostasis and tissue repair, a detailed understanding of epithelial cell reprogramming in the context of chronic inflammatory skin diseases is underdeveloped.
Lineage-specific chromatin structure is established during somatic stem cell commitment and is further remodeled in terminal differentiation (24). The topology of cis-regulatory elements and their coordinated TFs are the key factors in orchestrating normal epithelial stem/progenitor cell fate, immune cell plasticity, and neurodevelopment (25–30). However, the contribution of similar epigenetic mechanisms underpinning chronic skin inflammation remains undefined.
In this study, we defined altered transcriptional states in HS lesional epidermis at single-cell resolution and determined that the fidelity of basal stem/progenitor cells was reprogrammed contributing to epithelial cell hyperproliferation and inflammation. Pseudotime analysis revealed that the plasticity of HS basal progenitors gives rise to a distinct subcluster, serving as a key inflammatory response center. Moreover, our data indicated that the HS-associated transcription profiles are mediated by the coordinated effects of genome-wide chromatin rewiring, altered epigenetic modifications, and switched regulatory TFs. Lastly, we experimentally validated the epigenetic regulation as a critical molecular mechanism involved in HS pathogenesis.
Results
sc-RNA Seq Analysis Revealed Heterogeneous Populations in HS Lesional Epidermis.
To define transcriptional states in the HS epidermis at single-cell resolution and assess the remodeling of cis-regulatory landscape in basal stem/progenitor cells under the disease condition, we performed sc-RNA seq to characterize cell heterogeneity and dual-omics profiling with a limited number of CD49fhigh cells for chromatin accessibility (ATAC-sequence) and gene expression (31) as well as CUT&RUN sequencing for multiple chromatin modification states (Fig. 1A).
Fig. 1.
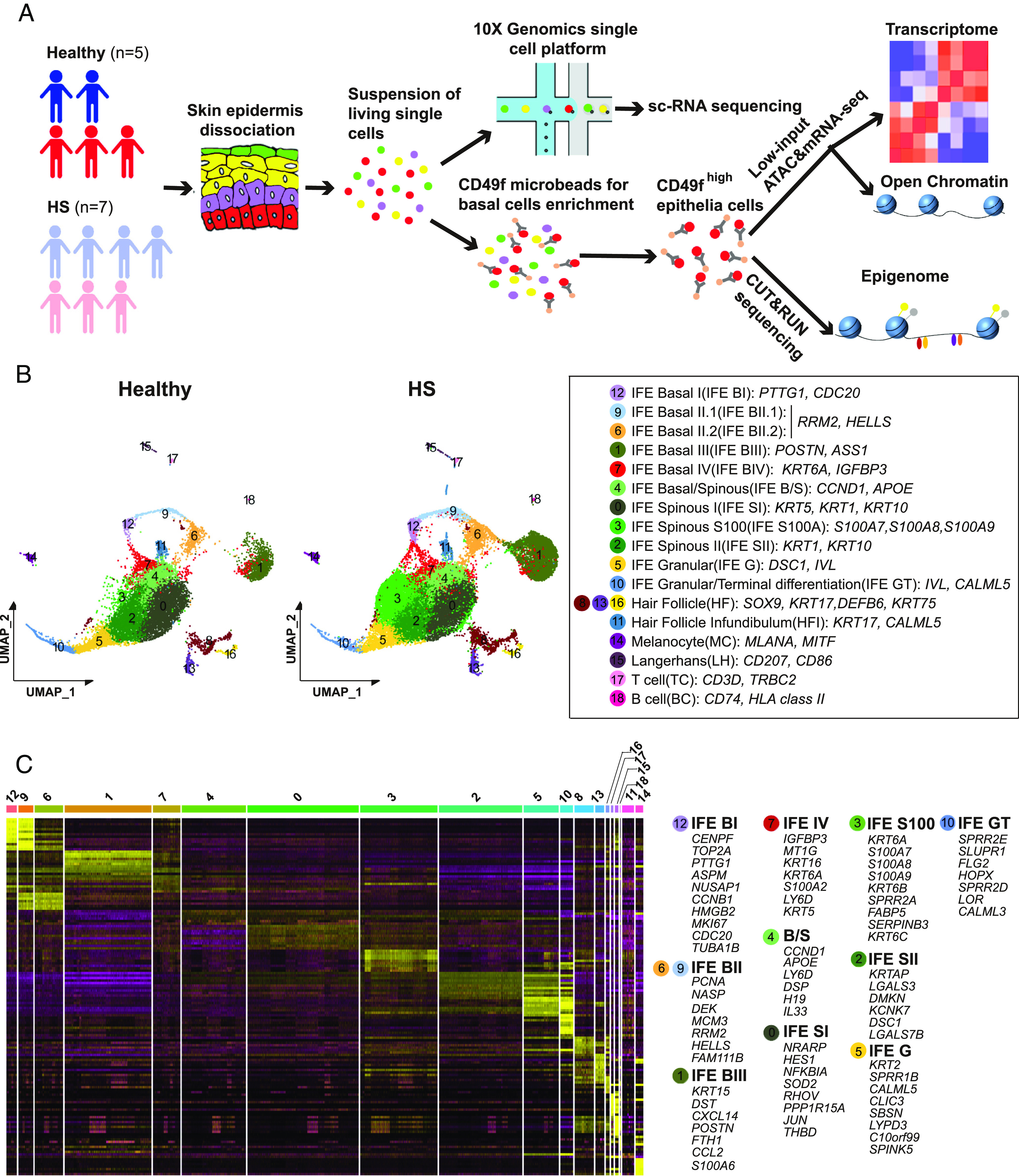
Defining the heterogeneity of epidermal cell population in healthy and HS lesional skin. (A) Experimental strategy for multimodal molecular analysis of HS epidermal cells. (B) Merged epidermal cell transcriptomes at single-cell resolution for healthy (n = 5 samples) and HS lesional skin (n = 7 samples). Data were visualized with uniform manifold approximation and projection (UMAP) and colored according to unsupervised clustering. Identity of cell types (Left in box) defined using established marker genes (Right in box). (C) Row-normalized heat map of top marker genes from integrated healthy and HS scRNA-seq datasets for indicated cell types. Representative genes for keratinocyte clusters are displayed on the right.
Following quality control filtering, sequencing generated single-cell transcriptomes of 12,942 cells from healthy libraries and 24,867 cells from HS ones (SI Appendix, Fig. S1 A and B). We identified 19 highly distinct clusters of epidermal and immune cells using uniform manifold approximation and projection (UMAP). Except for cluster 3, all other clusters were annotated based on their expression of known markers (Fig. 1B and SI Appendix, Fig. S1 C–G). Further analysis resolved 10 clusters within the IFE population, including the previously described IFE basal I-IV cells (basal I cell—PTTG1/CDC20, basal II cell—RRM2/HELLS, basal III cell—POSTN/ASS1, and basal IV cell—IGFBP3/KRT6A), IFE basal/spinous transition cells (CCND1/APOE), two stages of IFE spinous cells marked by different levels of KRT1/KRT10, and IFE granular and terminal differentiation cells (DSC1 & IVL and CALML5, respectively) (22, 23, 32). Remarkably, when we overlaid HS and healthy clusters, UMAP revealed a marked disease-associated cluster within IFE cells, characterized by S100A inflammatory genes (S100A7/A8/A9, hereafter referred to as IFE S100A). We determined the IFE S100A cluster was related to the basal/spinous transitional and early-stage spinous clusters (SI Appendix, Fig. S1H). The distribution of cell populations/clusters was strikingly consistent across two cohorts (SI Appendix, Fig. S1I).
We subsequently identified cluster-specific gene expression signatures (Fig. 1C). IFE basal I cells were enriched for genes related to chromosome separation and mitotic spindle organization (CENPF, TOP2A, PTTG1, and NUSAP1), IFE basal II cells were featured with chromatin-remodeling related genes (NASP, MCM3, and HELLS), and IFE basal III cells highly expressed genes with regulatory roles in adhesion junctions and tissue regeneration (DST and POSTN). IFE basal IV cells were enriched with the growth inhibition gene IGFBP3. In contrast, basal/spinous transitional cells expressed markers such as APOE (basal cell marker), CCND1 (G1/S phase regulator), and keratinocyte differentiation genes (LY6D and DSP). Spinous I and II cells were enriched with keratinocyte differentiation determinants (HES1 and JUN) and keratin-associated gene (KRTAP), respectively. In addition to S100A genes, high expression of KRT6 family genes (KRT6A/6B/6C) and SERPINB3 was associated with the S100A cluster. Lastly, the granular and terminal clusters expressed genes consistent with differentiation and cornification of keratinocytes (CLIC3, SPRRIB, and FLG2). Altogether, our data revealed a meaningful gene expression landscape for basal cell compartments and epidermal heterogeneity in healthy and HS skin, providing a useful reference for studying human skin inflammatory diseases.
The Fidelity of Basal Stem/Progenitor Cells Was Reshaped in HS Skin.
Having identified the different cellular populations in healthy and HS epidermis, we sought to examine to what extent the gene signature within each basal stem/progenitor population reflects disease pathogenesis (33). Interestingly, the proportion of basal cells (BI to BIV) in HS was approximately twofold greater than that in healthy individuals (SI Appendix, Fig. S2A). Clusters representing T cells and B cells, but not melanocytes and Langerhans cells, were also increased in HS (SI Appendix, Fig. S2A). Using flow cytometry as an independent approach, we determined that CD49fbri/CD71dim cells, a characteristic feature of keratinocyte stem cells (34, 35), were significantly higher in HS skin compared to healthy one (SI Appendix, Fig. S2 B–D).
We next performed immunofluorescent (IF) assays to quantify the stem/progenitor cells within the HS epithelium. Histopathological analysis showed that HS lesion contained thicker epidermal layers than perilesional and healthy skin, with increased irregularity of rete ridges (SI Appendix, Fig. S2E). K14-expressing cells (basal keratinocytes) localized in the basal layer and K10-positive cells (suprabasal keratinocytes) were in the suprabasal layers within the normal epidermis (SI Appendix, Fig. S2F). However, HS K14+ cells were present in basal and spinous layers in the perilesional skin and throughout the epithelial layers in the lesional skin. The expansion of K14+ cells was accompanied by a marked decrease in K10+ cells (SI Appendix, Fig. S2F). Compared to healthy skin, the perilesional and lesional area in HS had a significantly expanded number of PTTG1+ and HELLS+ cells (Fig. 2 A and B). Notably, these cells could be readily identified scattered throughout the irregular rete ridges and nonbasal areas (Fig. 2 A and B). Moreover, COL17A1+ cells, expressing COL17A1 evenly across BI to BIII clusters (SI Appendix, Fig. S2G), showed distinct behavioral traits. These cells visibly proliferated from the rete ridges and interridges, some of which continuously migrated upward and produced new clonal zones (Fig. 2C). Together, these results reveal two critical aspects in the context of hyperproliferative keratinocytes: 1) the remodeling of the lesional epidermis is spatially fueled by basal stem-like populations, and 2) the dispersion of COL17A+ clones is suggestive of the niche-like compartments favoring the recruitment of HS-associated immune cells (36).
Fig. 2.

Identification of basal stem/progenitor cells in HS lesional skin. (A–C) High-resolution images of healthy and HS skin biopsies immunostained for PTTG1 (A), HELL (B), and COL17A1(C), respectively, along with DAPI. Right Upper, representative images with high magnification of HS lesional tissue sections are depicted. The experiment was performed with six skin tissue each from healthy and HS individuals, respectively, with similar results. Right Bottom, percentage of the indicated number of positive cells per 104 square micrometers (A and B) or the ratio of the positive cell area to the total area (C) determined from histological sections of respective skin (n = 8 areas per sample). The dashed lines demarcate epidermal–dermal boundaries. The experiment was repeated two independent times with similar results (n = 3 biological replicates per group). (D) GO enrichment analysis showing top-ranked molecular functions of DEGs in HS vs. healthy condition within indicated basal clusters. (E) Dot plot showing representative GO genes differentially expressing across basal clusters in healthy and HS samples. Data are presented as mean ± SD, and significance was calculated using the unpaired Student t test (A–C). Magnification, 20× (A–C). [Scale bars, 50 μm (A–C).]
We further performed Gene Ontology (GO) analysis based on the differentially expressed genes (DEGs) within our sc-RNA datasets (Fig. 2 D and E). The biological processes of the mitotic cell cycle and chromosome segregation were specifically enriched in the HS BI cluster. Examples of genes include TOP2A, CDC20, PTTG1, and BRCA1. Genes associated with the DNA damage response (PARP1, WDR76, and CHEK1) were mainly enriched in HS BII cells and down-regulated in the microtubule-based process (TUBB2A, TUBB, and DCTN3). Interestingly, integrin molecules (ITGA3/6 and CCN1) were highly expressed in HS BIII; and the expression of genes associated with tight junction (RAB10 and TJP1) was decreased in HS BI and BII. In addition, HS BIV cells showed enrichment of chromatin remodeling genes (HDAC1/2, JARID2, and SMARCA5).
Pseudotime Analysis Revealed Keratinocyte Fate Plasticity in HS Skin.
Having defined the transcriptional program of IFE subtypes in healthy and HS skin, we reconstructed their cell-fate trajectory, respectively. The trajectory of the basal I cluster (the initial node) in the healthy epidermis had two bifurcating branches (Fig. 3 A, Left). Branch 1 showed BI cells differentiated to BII, BIV, B/S, S, G, and eventually to terminally differentiated cells. In branch 2, BI primed the prodifferentiation program into BII and BIII at the branch end. In contrast, five different trajectories over HS keratinocyte lineage commitment were identified (Fig. 3 A, Right). Except for branches 1 and 2 which were similar to healthy skin, in HS, the BI cluster gave rise to S100A and transit amplifying (TA) populations as shown by branches 3 and 4, respectively. The disruption of the progenitor state was also predicted in the HS BIII cluster, as pseudotime values in this population correspond to intermediate and terminal states over differentiation order (branch 2). Branch 5 was derived from HS spinous cells toward HF populations. Of note, these HF cells expressed elevated levels of spinous cell markers (KRT1/10), and decreased expression of HF stem cell markers (SOX9 and KRT17) (SI Appendix, Fig. S3A). These data support that in HS, HF stem cells lose their identity and differentiated into keratinocytes to favor the formation of a keratin-filled epidermal cyst (12).
Fig. 3.
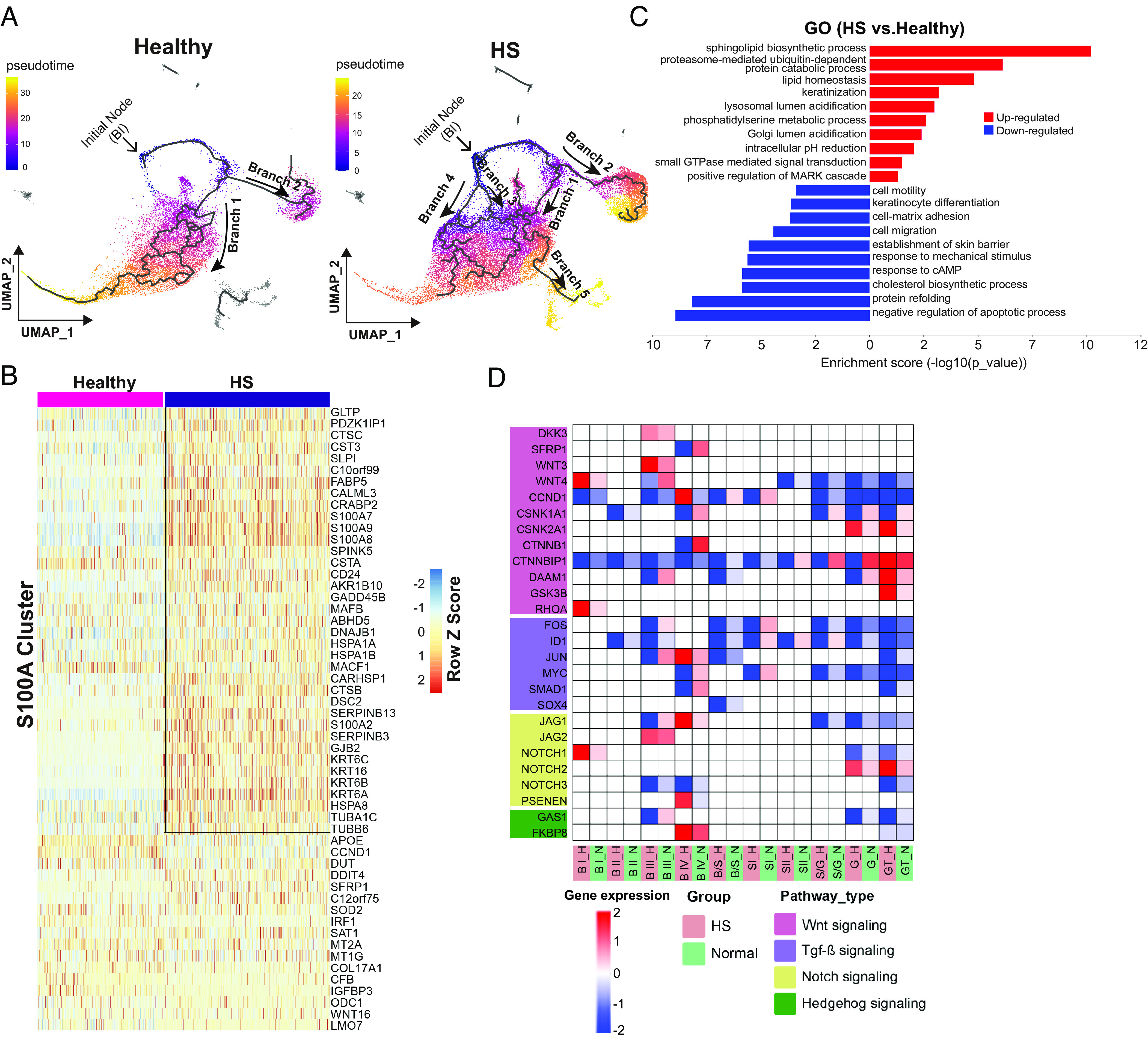
Transcriptomic trajectory analysis of HS lesional keratinocytes in pseudotime. (A) Pseudotime transcriptional trajectories of clustered epithelial cells. Branch trajectories are depicted with arrows. (B) The heatmap displaying a scaled expression of the top DEGs from individual cells in HS S100A cluster compared to the healthy counterpart. Box, top up-regulated genes in HS S100A cluster. (C) GO enrichment analysis showing top-ranked molecular functions within suprabasal clusters in HS vs. healthy condition. (D) Expression of genes linked to indicated signaling pathways for each keratinocyte population.
Next, we sought to identify distinct signatures within the HS-specific S100A cells (Fig. 3B). Relative to healthy skin, HS S100A cells were characterized by the higher expression of proinflammatory response genes including CTSC, C10orf99, and S100A7/8/9. This population also expressed high levels of barrier alarmin molecules (KRT6A/B/C and KRT16) and genes associated with barrier development (CARHSP1, CALML3, and GJB2). Among the down-regulated genes in HS S100A cells was the cholesterol carrier gene, APOE. We further confirmed the expression of S100A7 and S100A8 by IF assays. Despite these proteins were not detectable in healthy epidermis, their expression showed a gradual increase in hyperproliferative keratinocytes from HS perilesions to lesions (SI Appendix, Fig. S3 B and C).
We performed GO analysis and evaluation of DEGs in suprabasal keratinocytes (Fig. 3C and SI Appendix, Fig. S3D). Keratinization pathway genes, including KRT 77/78/80, EVPL, and SPRR2D, had higher expression in HS granular cells than in healthy counterparts. These results favor the recently proposed disease pathogenesis concept where HS was proposed to be a dysregulated keratinization disease (37). Several genes involved in lipid homeostasis (CEBPA, IRS2, and PNPLA1/2/3) were up-regulated in terminally differentiated keratinocytes in HS, confirming previously described lipid abnormalities in HS (38). However, the expression of molecules for cell mobility (DST and ACTB), migration (EFNA1, ITGB1/4, and LAMB4), and response to mechanical stimuli (FOSL1, JUND, and PPL) were down-regulated across all suprabasal cells in HS. While focusing on epidermal differentiation pathways, we observed heterogeneity in the expression of Wnt signaling components in HS skin compared to healthy skin, demonstrating Wnt pathway was activated in HS basal layer and inactivated in suprabasal cells (Fig. 3D). All clusters in HS manifested significant diminution in TGF-β, Notch, and Hedgehog signaling pathways, representing severe dysregulation in HF-regulated signaling (Fig. 3D).
S100A8/9 proteins have been reported to be as transcriptional coactivators during breast cellular transformation (39). As we found that S100A8 expression was predominantly in the nuclei of HS keratinocytes (SI Appendix, Fig. S3C), we next characterized its nuclear function in HS. Compared with healthy controls, we detected 3,738 peaks from S100A8 CUT&RUN sequencing in HS epithelial cells, with 435 specific genes (absolute log2FoldChange > 1.5, FDR-adjusted P < 0.25) (Dataset S1). The molecular occupancy map of S100A8 was highly enriched up and downstream 2.5kb around the translation start site (TSS) regions and dropdown at the TSS center in HS versus healthy condition (SI Appendix, Fig. S3E), suggesting its potential role in mediating chromatin priming for target gene expression. Indeed, we found that ~15.4% of S100A8 targets (67 out of 435) increased their expression in HS (Fig. 4A). The GO analysis of genes bound by S100A8 revealed categories mainly relevant to metabolic processes for organic substances, nucleobase-containing compounds, and nitrogen compounds (Fig. 4B). For instance, the locus containing SWAPS1 (encoding a DNA-stimulated ATPase) and SF1 (encoding splicing factor 1, involved in spliceosome assembly), respectively, showed S100A8 CUT&RUN peaks enriched in HS samples (Fig. 4C). The increased accessibility of the binding sites was consistent with the high expression of these two genes in HS (SI Appendix, Fig. S3 F and G).
Fig. 4.

S100A8 molecular function in HS keratinocytes. (A) Venn diagram showing the number of S100A8-bounded genes up-regulated in HS. (B) Top-scoring GO analyses for genes bound by S100A8 in HS vs. healthy condition. (C) Genome browser representative views of SWAPS1 and SF1 gene loci characterized by the enrichment of S100A8 CUT&RUN peaks (green box) in healthy and HS epithelial cells.
Proinflammatory Networks Defined Intercellular Communications in HS Lesional Keratinocytes.
To investigate potential intercellular communications among keratinocyte subtypes, we employed the CellChat program to visualize the expression levels of the inflammatory factors differentially expressed in HS lesions versus healthy skin (40). In HS lesions, the IL1 family (IL1A, IL1B, and IL33) and IL10 family (IL19 and IL20) cytokines were enhanced mainly within basal populations (Fig. 5 A and B). HS-basal cells were characterized by elevated expression of the macrophage-derived chemokines (CCL2, CCL4, and CCL22), the antigen-engaged B cell chemoattractant (CCL19), and the neutrophil chemo-attractants (CXCL3 and CXCL10) (41) (Fig. 5 A and B). In HS suprabasal cells, the expression of a subset of inflammatory genes were enhanced, including CCL20,CXCL1/8/14/16/17 for intraepidermal neutrophilic micro abscess and recruitment of active nature killer (NK) cells (41–43). Interestingly, IL1R2, the IL1 signaling negative regulator, was reduced across HS epidermal populations indicating an impaired monocyte and Langerhans cell trafficking in HS (44) (Fig. 5C).
Fig. 5.
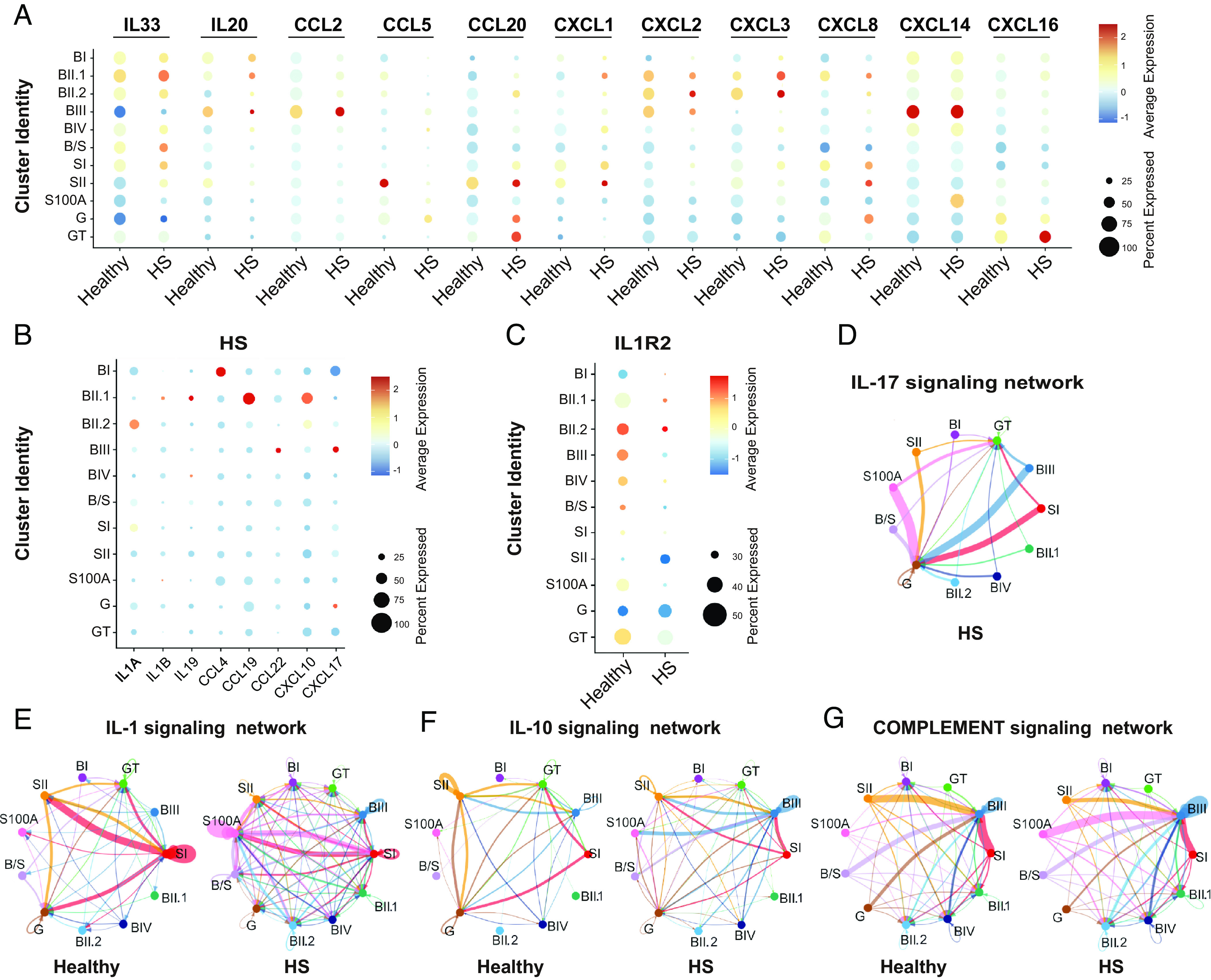
The inflammatory program reshaping in HS epidermal cells. (A and B) Dot plot showing the log2-transformed expression of inflammatory ligand genes characteristic of each keratinocyte subset. Percentage expression per spot (dot size) confidently assigned to each epithelial population (color intensity) in healthy and HS conditions or HS alone. (C) Dot plot showing log2-transformed expression of inflammatory receptor gene IL1R2 in healthy and HS epithelial clusters. (D–G) Circle plots displaying the inferred networks of HS-associated inflammation signaling pathways. The total number of interactions among different keratinocyte populations in HS (D) or between healthy and HS (E–G).
We next calculated the differential outgoing and incoming interaction strengths of the defined signaling pathways using the CellChatDB database (40). IL17 signaling network was exclusively expressed in the HS clusters (Fig. 5D and SI Appendix, Fig. S4A), addressing its possible druggability previously implicated in psoriasis (43). Compared to healthy counterparts, all HS keratinocyte subpopulations showed elevated IL1 signaling, indicating enhanced priming of innate immunity. In the dataset from healthy skin, SI cells sent abundant IL1 signaling onto differentiated subsets (SII and G clusters) (Fig. 5E and SI Appendix, Fig. S4B). In contrast, this communicating pattern shifted in HS, showing increased outgoing and incoming interaction strength between SI and S100A cells (Fig. 5E and SI Appendix, Fig. S4B). Interestingly, an anti-inflammatory cytokine network IL10 was also present in healthy and HS groups across BIII cells and SI/SII/G cells (Fig. 5F and SI Appendix, Fig. S4C). In HS, SII and S100A cells were transformed into new signaling centers for receiving intensive IL10 signals from SI and BIII cells, respectively (Fig. 5F). Altered complement signaling interactions were inferred within HS populations, where the S100A cluster sent significantly increased outgoing signals to BIII cells compared to healthy control (Fig. 5G and SI Appendix, Fig. S4D). Overall, HS BIII and S100A clusters gained more outgoing interaction weights and strength and SII and G/GT clusters lost cell–cell communication signals (SI Appendix, Fig. S4 E and F).
To assess the inflammatory signatures of the basal keratinocytes (BKs), we first cultured primary BKs from healthy and HS skin and characterized them with the expression of KRT14 (SI Appendix, Fig. S4G). We validated the relative transcriptional abundances of several inflammatory factors in HS BKs, including IL1A, IL33, CCL2, CXCL10, and S100A7/8/9 (SI Appendix, Fig. S4 H and I). However, we did not observe the differential expression of CXCL14/16 between healthy and HS BKs (SI Appendix, Fig. S4J), indicating the specific immune response in HS.
HS Environment Impacted the Chromatin Landscape of Basal Keratinocytes.
Recent findings have shown that epithelial stem cells may contribute to the persistence of human disease by serving as repositories for inflammatory memories (45). To assess whether the inflammatory environment in HS affects the regulatory landscape of BKs, we enriched CD49fhigh basal cells and performed low-input ATAC&RNA-seq (SI Appendix, Fig. S5A). Principal component analysis (PCA) indicated a strong correlation within the intragroups (SI Appendix, Fig. S5B). Compared with healthy control, HS gained and lost 1,434 and 3,722 high-confidence peaks, respectively, (absolute log2FoldChange > 0.5 and FDR-adjusted P < 0.1), highlighting substantial chromatin remodeling in HS genomic landscape (Dataset S2). Heatmap displayed top differential accessibility based on ATAC-seq signal intensity comparing HS data to healthy cohort (Fig. 6A). GO analysis revealed that open chromatin-related genes in HS were highly enriched for the wound healing process, stress response, and inflammatory activity (Fig. 6B), as exemplified at the KRT6B locus (encoding Keratin 6B, involved in wound healing) (Fig. 6C). However, genes for cell adhesion and keratinocyte differentiation, such as the GJD4 locus (encoding Gap Junction Protein Delta 4), had restricted accessibility in HS (Fig. 6D). In parallel, 1,287 genes were differentially expressed (absolute log2FoldChange > 1, P < 0.05) between healthy and HS groups, which predominantly enriched in inflammatory signaling, innate immune response, and impaired epithelium development (SI Appendix, Fig. S5C and Dataset S3). The gene expression patterns for the 80 top-ranked DEGs are shown in a heatmap (SI Appendix, Fig. S5D). Importantly, our data showed that ATAC-seq and RNA-seq datasets were significantly correlated (P<0.0001, Chi-squared test) (Fig. 5E).
Fig. 6.
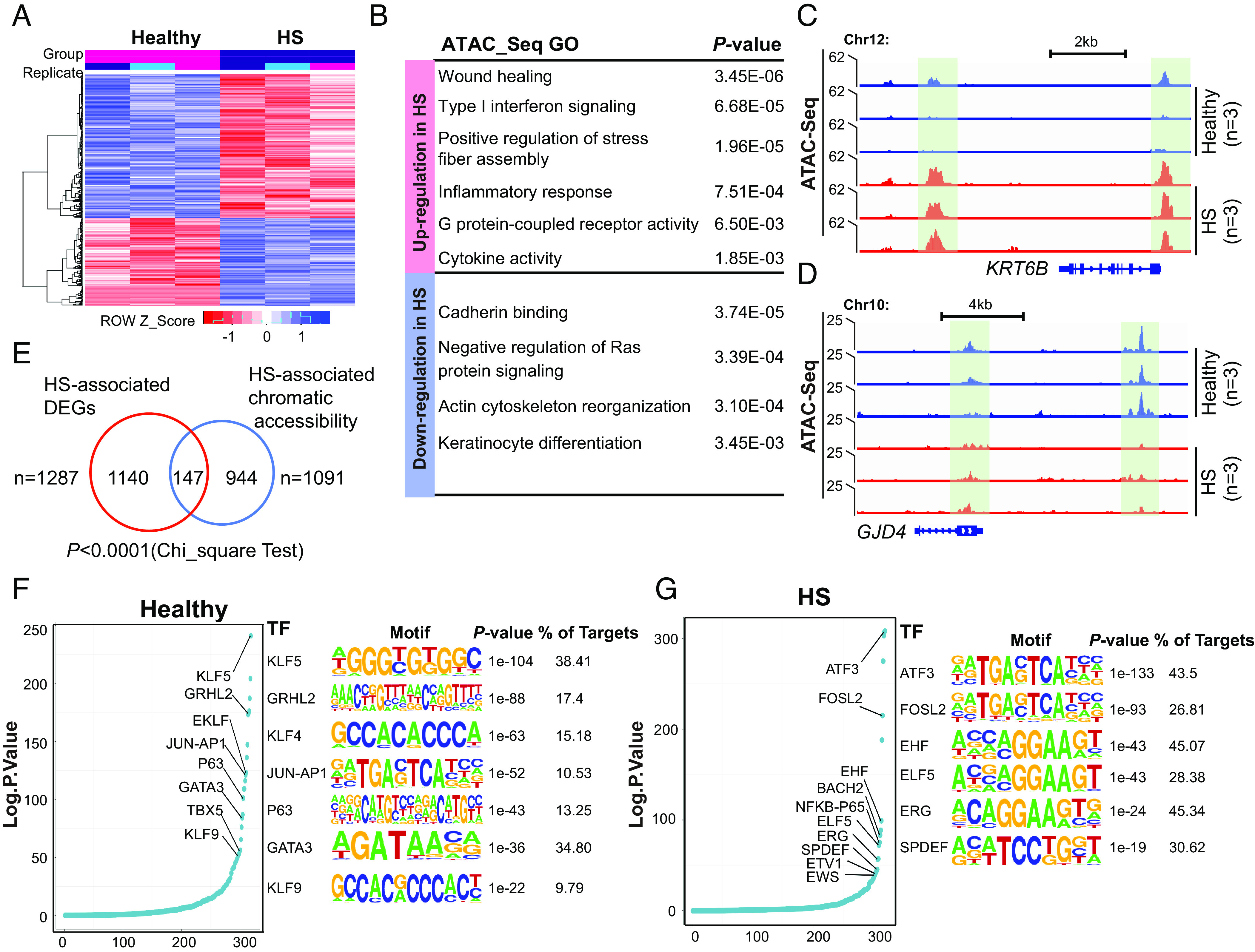
The remodeling of the chromatin landscape in HS CD49fhigh basal cells. (A) Heatmap representation of top 200 ATAC-seq peaks gained or lost in HS vs. healthy conditions (n = 3 each for heathy and HS). (B) Top-scoring GO analyses showing genes associated with ATAC-seq peaks for all peaks present in healthy and HS samples, respectively. (C and D) Representative views of KRT6B (C) and GJD4 (D), illustrating their upregulation and downregulation characterized by gain and loss in chromatin accessibility (green boxes) in HS. (E) Venn diagram showing significant overlap numbers between HS-associated DEGs and HS-specific chromatin states. (F and G) Left, the ranked curve for enriched TFs in healthy (F) and HS (G) CD49fhigh basal cells, respectively. Right, logo visualization of the top HOMER motif outputs generated from healthy (F) and HS (G) merged ATAC seq datasets, respectively. The fraction of target regions and respective P value with each motif are displayed.
By performing TF motif analysis, we identified top-ranking TFs enriching at accessible regions. Healthy CD49fhigh cells were enriched in TF motifs, including KLF family members (KLF4/5/9, JUN-AP1, p63, and GATA3) known to maintain an undifferentiated state (46) (Fig. 6F). Interestingly, we found expression of this subset of TFs was not limited to basal cells, but also, found in suprabasal cells, suggestive of their multifaceted functions in epidermal homeostatic regulation (SI Appendix, Fig. S5E). In HS, open chromatin regions were enriched for the motifs of immune response TFs (ATF3 and FOSL2) (47, 48) and ETS family members for epithelial cell differentiation and malignant progression (49) (Fig. 6G). Except for ERG, these TF genes did not express in a cell state–specific manner, implying that their DNA binding activity was dispensable for the induction of transcription in HS pathogenesis (SI Appendix, Fig. S5F).
Proinflammatory Enhancers Were Activated in HS Basal Keratinocytes.
To investigate whether the transcription of HS-related genes was amplified by an epigenetic mechanism, we assessed the activity of the cis-regulatory elements using CUT&RUN sequencing. Enriched CD49fhigh keratinocytes were incubated with three histones posttranslational modification (PTM) antibodies: anti-H3K4me1, -H3K4me3, and -H3K27ac. We identified highly reproducible peak density within replicates of H3K4me1, H3K4me3, and H3K27ac, which were well correlated in healthy and HS groups (SI Appendix, Fig. S6A). There was no significant genome-wide bias enrichment of PTM between healthy and HS conditions (SI Appendix, Fig. S6B). Notably, HS-associated H3K4me1 and H3K27ac densities were more profoundly enriched up or downstream 1.5kb around TSS regions as compared to healthy counterparts (Fig. 7A). Conversely, a sharp reduction of H3K4me3 occupancy immediately downstream of the TSS was shown in the disease groups (Fig. 7A), indicating the decreased transcriptional activity. In fact, our bulk RNA-seq indeed revealed more down-regulated DEGs (993 out of 1,827, 54.3%) than up-regulated ones (834 out of 1,827, 45.7%) (Dataset S3).
Fig. 7.
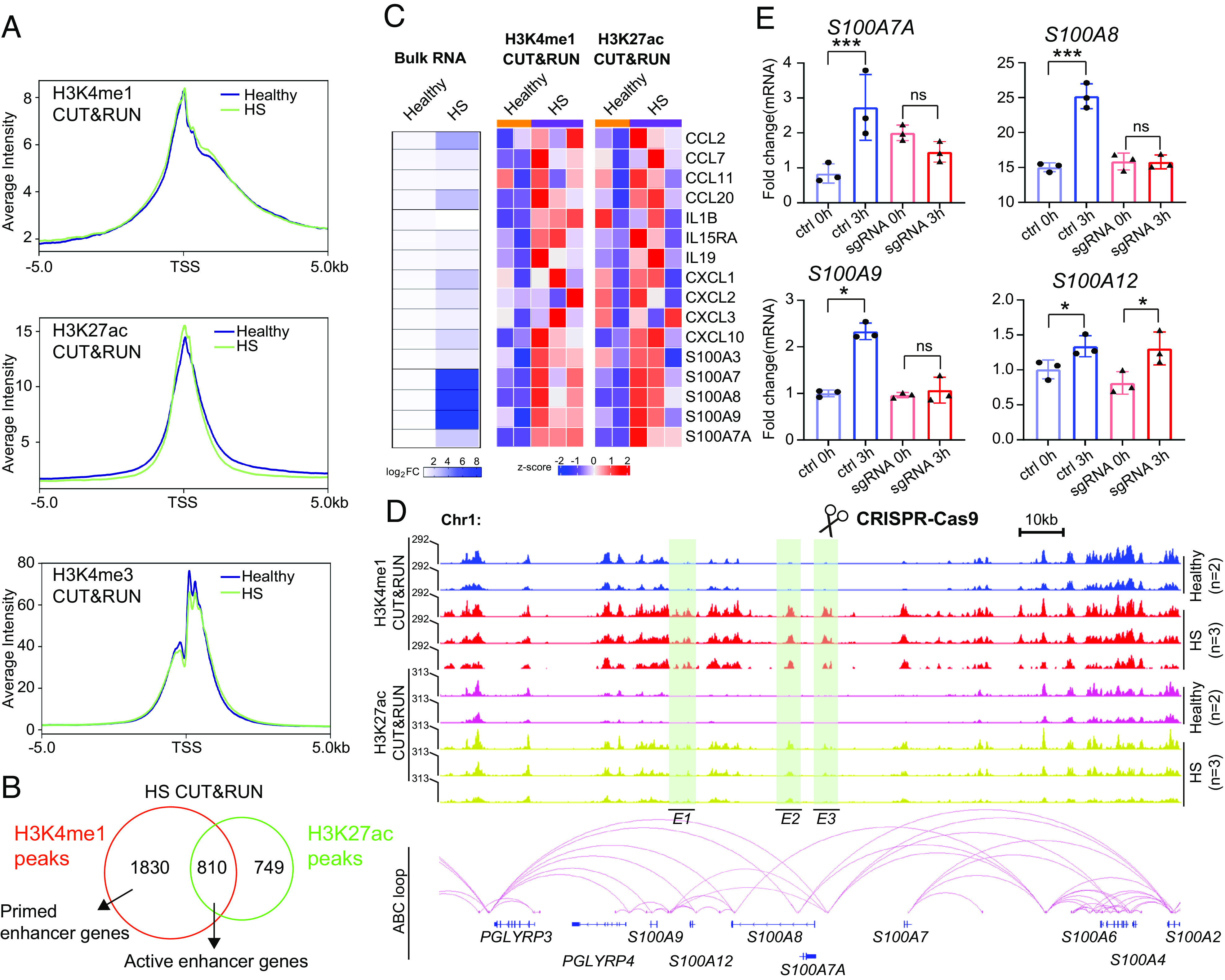
Identification of inflammation enhancers in HS. (A) Distribution of indicated histone marks on binding genes (from −5 kb of TSS to +5kb) in healthy and HS CD49fhigh basal cells. (B) Venn diagram showing HS-associated active enhancer genes common in H3K4me1 and H3K27ac CUT&RUN peaks. (C) Left, heatmaps summarizing the mRNA expression for indicated inflammatory genes in healthy and HS CD49fhigh basal cells, respectively. Middle and Right, heatmaps displaying normalized H3K4me1 and H3K27ac CUR&RUN signals at the active enhancer and promoter regions of certain inflammatory genes between healthy and HS CD49fhigh basal cells (n = 2 for healthy, n = 3 for HS). (D) Upper, genome browser representative view of PGLYRP and S100A gene clusters characterized by enrichment in histone markers (green boxes) in healthy vs. HS CD49fhigh basal cells. Bottom, chromatin loops and putative enhancers (E1-3) predicted by the ABC program are shown. (E) qRT-PCR quantifying the relative mRNA levels of indicated genes between control and sgRNA keratinocytes. P values based on unpaired Student t test. Error bars indicate the mean ± SD from n = 3 replicates. The experiment was repeated three times independently showing similar results. *P < 0.05 and ***P < 0.001. ns, nonsignificant.
Gene enhancer elements are thought to exist in either active (presence of both H3K4me1 and H3K27ac) or primed (presence of H3K4me1 and absence of H3k27ac) states (50). We overlaid HS-enriched H3K4me1 and H3K27ac data based on their closest genes and identified 810 active-enhancer (AE) genes and 1830 primed-enhancer (PE) genes (absolute log2FoldChange > 0.75, FDR-adjusted P < 0.2) (Fig. 7B and Dataset S4). GO analysis on the AE genes revealed that their biological processes were predominantly involved in lymphocyte migration, G protein-coupled receptor pathway, and cytokine responses (SI Appendix, Fig. S6C). Also, we profiled normalized read counts for H3K4me1 and H3K27ac at a subset of cytokine and chemokine genes (Fig. 7C). We found the transcriptional activation of these genes coincided with their active enhancers in the disease progression, indicating that HS-specific enhancers may orchestrate chromatin accessibility and drive transcription of proinflammatory genes. We indeed identified such changes in IL19/20, CCL2, and CCL20 loci (SI Appendix, Fig. S6D), whose mRNA expression was abundant in HS CD49fhigh basal cells (Fig. 7C).
To determine whether these HS-specific enhancers are truly associated with the inflammatory state, we employed a genetic perturbation approach to interrupt target genomic proximity. We focused on the linear region (153,300 to 153,500 kb) of human chromosome 1, where S100A and PGLYRP gene clusters are located. We used Activity-by-Contact (ABC) tool to construct genome-wide maps of enhancer-promoter connections based on integrating the profiles from chromatin accessibility and H3K27ac CUT&RUN sequences (51). We were able to distinguish three putative enhancers (E1, E2, and E3) in the above-defined regions (Fig. 7D). Next, we examined the role of E3, which is specifically activated in HS and enriched in proximal promoters of S100A7A and S100A8 along with a distal connection close to the S100A9 locus (Fig. 7D). CRISPR– Cas9 mediated genetic excision of E3 was achieved using dual guide RNAs (gRNAs) in neonatal human epidermal keratinocytes (NHEK) (SI Appendix, Fig. S6 E–G). Lipopolysaccharide (LPS) (40 μg/mL) stimulation of sgRNA-control NHEK successfully induced the production of S100A7A/8/9 mRNA after 3 h of the treatment. However, under the same condition, the disruption of E3 significantly blocked the expression of S100A7A/8/9 but not its nontarget gene, S100A12 (Fig. 7E).
Next, we focused on identifying TFs on HS-associated inflammation cis-regulatory elements. We utilized our H3K4me1 CUT&RUN profile as a reference dataset and created a comprehensive database for annotating 255 typical enhancers associated with 81 proinflammatory genes in HS CD49fhigh basal cells (Dataset S5). TF motif similarity was predicted with the TOMTOM tool, resulting in four significantly enriched TF models (Fig. 8A), including TFs regulating interferon-dependent immune response (IRF3), immune cell differentiation (IRF4), and vascular inflammation (KLF15 and VEZF1) (52–54). Notably, IRF3 displayed high gene expression across heterogeneous populations of the HS epidermis (SI Appendix, Fig. S7A). Subsequently, we investigated whether the inactivity of IRF3 could repress HS-associated inflammatory products. We evaluated the effects of BAY-985, an inhibitor known to mediate the inactivation of IRF3 and NF- κB signaling (55), on NHEKs. BAY-985 demonstrated antiproliferative activity in NHEKs, with approximately 50% inhibition observed at 850 nM through MTT cytotoxicity assay (SI Appendix, Fig. S7B). Treatment with 50 nM of BAY-985 resulted in a ~45% reduction in IL6 mRNA levels in LPS-induced NHEKs (SI Appendix, Fig. S7C). We thus selected this concentration to treat healthy and HS keratinocytes for 24 h, followed by a multiplex Luminex assay. When compared to healthy controls, HS groups exhibited an activated proinflammatory pattern, characterized by higher production of cytokines (IL10, IL15, IL1RA, and IFNgamma), chemokines (CCL2 and CXCL9), and inflammation mediators (VEGFa, PDGF-AB/BB, and GCSF) (56–58). Remarkably, treatment of HS keratinocytes with BAY-985 significantly decreased the expression of these proteins (Fig. 8 B and C). Notably, three genes encoding IL10, IL15, and CCL2 were predicted as targets of IRF3 through sequence-specific sites (Fig. 8A).
Fig. 8.

The role of IRF3 in regulating the production of HS inflammatory genes. (A) Upper, de novo motifs significantly enriched at inflammatory enhancer groups. TOMTOM outputs motifs ranked by P value. Bottom, representative genes with inflammation enhancers potentially targeted by TFs from upper panel. (B and C) Dot plots showing the overall comparison of indicated cytokines/chemokines/growth factors protein between BAY-985 treated healthy and HS keratinocytes (n = 3 for healthy, n = 4 for HS). P values based on the unpaired Student t test. Error bars indicate the mean ± SD from n = 3 or 4 biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001. ns, nonsignificant.
Discussion
Recently, Navrazhina et al. demonstrated that the epithelialized tunnels in HS recapitulate the psoriasiform epidermal hyperplastic morphology of the overlying epidermis, including the expression of S100A7 (Psoriasin) (59). These tunnels also exhibit features typically found in epidermal skin, such as the expression of LORICRIN, FILAGGARIN, LIPOCALIN, and others (59). Thus, our CD49fhigh epithelial cells represent populations of interfollicular, follicular, and tunnel-associated cells. Pseudotime analysis showed that there is no single-cell trajectory reconstruction against the classic keratinocyte lineage commitment in HS, hence ruling out the possibility of induced dedifferentiation of IFE- lineage progenies in HS pathogenesis (Fig. 3A). Additionally, HS basal I and II clusters were featured by enhanced activation of Wnt signals along with low expression of TGF-beta genes (Fig. 3D), a requirement for gaining a hyperproliferative phenotype. It was demonstrated that Wnt signaling activation in stem cells regulates core pluripotency factors (60). Cell–cell adhesion genes were preferentially down-regulated in the basal II subtype (Fig. 2E). Thus, these genotypic changes may partially explain the phenotype of high-proliferation and differential localization observed in HS basal I, II, and III, clusters (Fig. 2 A–C).
Although the IL1 pathway has been reported to be hyperactive in HS and represents a potential therapeutic target, the producer cell type was undefined (61). Our cell–cell communication analysis suggested that HS-S100A and -BIII cells contribute to this pathway, as evidenced by their intensive outgoing IL1 signals to other keratinocyte subpopulations (Fig. 4E). S100A8/9 heterodimers and S100A7 are members of the S100A family of Ca2+-binding proteins which are expressed at basal levels in multiple cell types under physiological conditions (62). The increased expression of S100A family proteins in HS is known (63, 64); however, the physiological functions of these S100 proteins and their underlying mechanism in HS pathogenesis are largely unknown. Here, we not only observed elevated levels of S100A7/A8 proteins in HS lesional skin (SI Appendix, Fig. S3 B and C) but also found that HS-S100A cells marked by S100A7/8/9 are the new signaling center for exacerbating the production of inflammatory cytokines (Fig. 4).
Here, we identified numerous poised enhancers within HS gene loci uncoupled with their transcriptional regulation, such as CCL11, IL1B, and IL15RA (Fig. 7C). We reasoned that these epigenetic changes occurred due to one or more of the following: 1) poised chromatin elements in HS BKs increase their susceptibility and are transcribed rapidly upon new stress challenges in disease progression; 2) these chromatin signatures are induced by early inflammatory signals, which drive transcriptional activation of HS-associated genes during recurrent inflammatory insults; and 3) since these open chromatins are marked by H3K4me1, we cannot rule out the possibility that some of these putative enhancers may represent a distinct set of regulatory elements to control their distal target via a topological loop (65).
We determined that the upstream cues driving the distinct gene expression profile in HS disease are modulated by a diverse range of TFs and cis-regulatory elements (Fig. 6A). Additionally, our genome-wide profiling of epigenetic histone marks revealed a subset of high-confidence binding sites for TFs on HS inflammatory enhancers (Fig. 8A). Consistently, pharmacological inhibition of IRF3 significantly reduced the production of its target genes (Fig. 8 B and C).
HS is a complex and heterogenous disease; the mechanisms underlying the pathogenic processes are limited. Follicular occlusion caused by infundibular keratosis and hyperplasia of the follicular epithelium is thought to be an early event in the onset of HS, which eventually ruptures leading to dermal inflammatory infiltration of myeloid cells and T helper (Th) cells (2, 7, 13). Current HS therapy targets several molecules associated with exaggerated inflammatory response, including TNF‐α, IL-10, IL-17, and IL‐1β as well as IL12/23 (15, 66). However, there is a lack of effective biological interventions for moderate-to-severe HS patients (66). In this study, we characterized the cellular and molecular signatures of HS epithelial cells and elucidated the epigenetic mechanisms underlying the pathogenic inflammatory plasticity of basal progenitors in HS lesional skin (Fig. 9). Targeting inflammatory enhancers and key TFs could complement current biological drug-based therapies and pave the way for precision medicine in HS.
Fig. 9.

Schema representation of the proposed model for the heterogeneity of basal keratinocyte populations in HS epidermis and the current targeted immunomodulation treatment for HS. Left panel, The remodeling of HS lesional epidermis is fueled by heterogeneous basal stem/progenitor populations. Mechanistically, HS cues reshape the chromatin accessibility of basal undifferentiated cells and activate inflammatory enhancers, resulting in cell-specific transcriptome and phenotypic traits in HS lesional epithelium. Right panel, the current clinical biology-associated therapy for HS. Abbreviations: BI, basal I subtype; BII, basal II subtype; BIII, basal III subtype; BIV, basal IV subtype; S, spinous subtype; G, granular subtype; TG, granular/terminal subtype; S100A, HS-specific S100A subtype; Ab, antibody.
Materials and Methods
Human Subjects.
The Institutional Review Board of the University of Alabama at Birmingham approved the protocol (IRB-300005214) for obtaining surgically discarded skin tissues from healthy (n = 8) and HS (under 40, Hurley late II/III stage) (n = 10) subjects. The patient information and the usage of each sample are included in Supplementary material (SI Appendix, Table S1).
Please see SI Appendix, Materials and Methods, for specific details of experimental procedures used in this study, including descriptions of isolation of epidermal cells for single-cell sequencing, primary basal keratinocytes culture, enrichment of CD49fhigh keratinocytes, flow cytometry, immunofluorescence staining, quantitative RT-PCR, CRISPR/Cas9-mediated enhancer editing, low-input ATAC&mRNA library preparation, CUT&RUN library preparation, MTT cell viability assay, multiplex Luminex assay, and data analysis for next-generation sequencing.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Acknowledgments
We thank the Sequencing Core Facility at the La Jolla Institute for their scientific and technical assistance. We are grateful to Leslie M. Roop for the final editing of the manuscript. This work is supported by NIH/NIEHS grant R01 ES026219 and NIH/NCI grant 5P01CA210946 to M.A. and by intramural UAB funds to L.J. and M.A.
Author contributions
L.J. and M.A. designed research; L.J., Y.C., S.M., C.L., C.A.M.-A., J.K., M.P.K., S.L., and R.S. performed research; C.L. and T.M.T. contributed new reagents/analytic tools; L.J., Y.C., S.M., C.A.M.-A., J.S.D., B.E.E., C.A.E., D.K.C., C.R., and M.A. analyzed data; M.A. supervised the project; and L.J., D.K.C., C.R., and M.A. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Lin Jin, Email: ljin@uabmc.edu.
Mohammad Athar, Email: mohammadathar@uabmc.edu.
Data, Materials, and Software Availability
The processed data of this study have been deposited in the GEO database under accession code GSE226428 (67). All other data are included in the manuscript and/or supporting information.
Supporting Information
References
- 1.Prens E., Deckers I., Pathophysiology of hidradenitis suppurativa: An update. J. Am. Acad. Dermatol. 73, 8–11 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Goldburg S. R., Strober B. E., Payette M. J., Hidradenitis suppurativa: Epidemiology, clinical presentation, and pathogenesis. J. Am. Acad. Dermatol. 82, 1045–1058 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Wang B., et al. , Gamma-secretase gene mutations in familial acne inversa. Science 330, 1065 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Riis P. T., Ring H. C., Themstrup L., Jemec G. B., The role of androgens and estrogens hidradenitis suppurativa - a systematic review. Acta Dermatovenerol. Croat 24, 239–249 (2016). [PubMed] [Google Scholar]
- 5.Kaleta K. P., et al. , Metabolic disorders/obesity is a primary risk factor in hidradenitis suppurativa: An immunohistochemical real-world approach. Dermatology 238, 251–259 (2022). [DOI] [PubMed] [Google Scholar]
- 6.Jafari S. S., Knüsel E., Cazzaniga S., Hunger R. E., A retrospective cohort study on patients with hidradenitis suppurativa. Dermatology 234, 71–78 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kelly G., Sweeney C. M., Tobin A. M., Kirby B., Hidradenitis suppurativa: The role of immune dysregulation. Int. J. Dermatol. 53, 1186–1196 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Gill L., Williams M., Hamzavi I., Update on hidradenitis suppurativa: Connecting the tracts. F1000Prime Rep. 6, 112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frew J. W., Navrazhina K., Marohn M., Lu P. J. C., Krueger J. G., Contribution of fibroblasts to tunnel formation and inflammation in hidradenitis suppurativa/acne inversa. Exp. Dermatol. 28, 886–891 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giamarellos-Bourboulis E. J., et al. , Altered innate and adaptive immune responses in patients with hidradenitis suppurativa. Br. J. Dermatol. 156, 51–56 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Schell S. L., et al. , Keratinocytes and immune cells in the epidermis are key drivers of inflammation in hidradenitis suppurativa providing a rationale for novel topical therapies. Br. J. Dermatol. 188, 407–419 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Hotz C., et al. , Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. J. Invest. Dermatol. 136, 1768–1780 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ring H. C., et al. , The follicular skin microbiome in patients with hidradenitis suppurativa and healthy controls. JAMA Dermatol. 153, 897–905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narla S., et al. , Identifying key components and therapeutic targets of the immune system in hidradenitis suppurativa with an emphasis on neutrophils. Br. J. Dermatol. 184, 1004–1013 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Goldburg S. R., Strober B. E., Payette M. J., Hidradenitis suppurativa: Current and emerging treatments. J. Am. Acad. Dermatol. 82, 1061–1082 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Clayton E., et al. , A single type of progenitor cell maintains normal epidermis. Nature 446, 185–189 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Lim X., et al. , Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Science 342, 1226–1230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rompolas P., et al. , Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mascré G., et al. , Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489, 257–262 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Cockburn K., et al. , Gradual differentiation uncoupled from cell cycle exit generates heterogeneity in the epidermal stem cell layer. Nat. Cell Biol. 24, 1692–1700 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Centonze A., et al. , Heterotypic cell–cell communication regulates glandular stem cell multipotency. Nature 584, 608–613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S., et al. , Single cell transcriptomics of human epidermis identifies basal stem cell transition states. Nat Commun. 11, 4239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng J. B., et al. , Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep. 25, 871–883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubin A. J., et al. , Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet 49, 1522–1528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin A. J., et al. , Coupled single-cell cRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saini A., Ghoneim H. E., Lio C. W. J., Collins P. L., Oltz E. M., Gene regulatory circuits in innate and adaptive immune cells. Annu. Rev. Immunol. 40, 387–411 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarropoulos I., et al. , Developmental and evolutionary dynamics of cis-regulatory elements in mouse cerebellar cells. Science 373, 4696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wierda R. J., Geutskens S. B., Jukema J. W., Quax P. H. A., Elsen P. J., Epigenetics in atherosclerosis and inflammation. J. Cell Mol. Med. 14, 1225–1240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasquali L., et al. , Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 46, 136–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li P., et al. , Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 10, 2246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li R., Grimm S. A., Wade P. A., Low-input ATAC&mRNA-seq protocol for simultaneous profiling of chromatin accessibility and gene expression. STAR Protoc. 2, 100764 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou Z., et al. , A single-cell transcriptomic atlas of human skin aging. Dev. Cell 56, 383–397 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Naik S., et al. , Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds G., et al. , Developmental cell programs are co-opted in inflammatory skin disease. Science 371, 6500 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abreu C. M., Pirraco R. P., Reis R. L., Cerqueira M. T., Marques A. P., Interfollicular epidermal stem-like cells for the recreation of the hair follicle epithelial compartment. Stem Cell Res Ther. 12, 62 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laffert M. V., et al. , Hidradenitis suppurativa (acne inversa): Early inflammatory events at terminal follicles and at interfollicular epidermis. Exp. Dermatol. 19, 533–537 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Nomura T., Hidradenitis suppurativa as a potential subtype of autoinflammatory keratinization disease. Front Immunol. 11, 847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penno C. A., et al. , Lipidomics profiling of hidradenitis suppurativa skin lesions reveals lipoxygenase pathway dysregulation and accumulation of proinflammatory leukotriene B4. J. Invest. Dermatol. 140, 2421–2432 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Song R., Struhl K., S100A8/S100A9 cytokine acts as a transcriptional coactivator during breast cellular transformation. Sci Adv. 7, 5357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin S., et al. , Inference and analysis of cell-cell communication using Cell Chat. Nat. Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montaldo E., et al. , Human NK cells at early stages of differentiation produce CXCL8 and express CD161 molecule that functions as an activating receptor. Blood 119, 3987–3996 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Metzemaekers M., Gouwy M., Proost P., Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol. Immunol. 17, 433–450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furue M., Furue K., Tsuji G., Nakahara T., Interleukin-17A and Keratinocytes in Psoriasis. Int. J. Mole. Sci. 21, 1275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pilkington S. M., et al. , Lower levels of interleukin-1β gene expression are associated with impaired Langerhans’ cell migration in aged human skin. Immunology 153, 60–70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montanes J. O., et al. , Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavazza A., et al. , Dynamic transcriptional and epigenetic regulation of human epidermal keratinocyte differentiation. Stem Cell Rep. 6, 618–632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jadhav K., Zhang Y., Activating transcription factor 3 in immune response and metabolic regulation. Liver Res. 1, 96–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renoux F., et al. , The AP1 transcription factor Fosl2 promotes systemic autoimmunity and inflammation by repressing Treg development. Cell Rep. 31, 107826 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Luk I. Y., Reehors C. M., Mariadason J. M., ELF3, ELF5, EHF and SPDEF transcription factors in tissue homeostasis and cancer. Molecules 23, 2191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calo E., Wysocka J., Modification of enhancer chromatin: What, how and why? Mol. Cell. 49, 825–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fulco C. P., et al. , Activity-by-Contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yanai H., et al. , Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 5253–5258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nam S., Lim J. S., Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch Pharm Res. 39, 1548–1555 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Lu Y., et al. , Kruppel-like factor 15 is critical for vascular inflammation. J. Clin. Invest. 123, 4232–4241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lefranc J., et al. , Discovery of BAY-985, a highly selective TBK1/IKKε inhibitor. J. Med. Chem. 63, 601–612 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Schonthaler H., Huggenberger R., Wculek S. K., Detmar M., Wagner E. F., Systemic anti-VEGF treatment strongly reduces skin inflammation in a mouse model of psoriasis. Proc. Natl. Acad. Sci. U.S.A. 106, 21264–21269 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He C., et al. , PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 6, 7770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lotfi N., et al. , Roles of GM-CSF in the pathogenesis of autoimmune diseases: An update. Front Immunol. 10, 1265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navrazhina K., et al. , Epithelialized tunnels are a source of inflammation in hidradenitis suppurativa. J. Allergy Clin. Immunol. 147, 2213–2224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lien W., Fuchs E., Wnt some lose some: Transcriptional governance of stem cells by Wnt/ β-catenin signaling. Genes Dev. 28, 1517–1532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Händel E. W., et al. , The IL-1 pathway is hyperactive in hidradenitis suppurativa and contributes to skin infiltration and destruction. J. Invest. Dermatol. 139, 1294–1305 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Singh P., Ali S. A., Multifunctional role of S100 protein family in the immune system: An update. Cells 11, 2274 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Batycka-Baran A., Matusiak L., Nowicka-Suszko D., Szepietowski J. C., Baran W., Increased serum levels of S100A4 and S100A15 in individuals suffering from hidradenitis suppurativa. J. Clin. Med. 10, 5320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Batycka-Baran A., et al. , Serum concentration and skin expression of S100A7 (Psoiasin) in patients suffering from hidradenitis suppurativa. Dermatology 237, 733–739 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Crispatzu G., et al. , The chromatin, topological and regulatory properties of pluripotency-associated poised enhancers are conserved in vivo. Nat. Commun. 12, 4344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alikhan A., et al. , North American clinical management guidelines for hidradenitis suppurativa: A publication from the United States and Canadian Hidradenitis Suppurativa Foundations. J. Am. Acad. Dermatol. 81, 91–101 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin L., et al. , Epigenetic switch reshapes epithelial stem/progenitor cells signatures and contributes to inflammatory programming in hidradenitis suppurativa. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse226428. Deposited 1 March 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Data Availability Statement
The processed data of this study have been deposited in the GEO database under accession code GSE226428 (67). All other data are included in the manuscript and/or supporting information.