

Abstract
Polyolefins are the most important and largest volume plastics produced. Unfortunately, the enormous use of plastics and lack of effective disposal or recycling options have created a plastic waste catastrophe. In this work, we report an approach to create chemically recyclable polyolefin-like materials with diverse mechanical properties through the construction of multiblock polymers from hard and soft oligomeric building blocks synthesized with ruthenium-mediated ring-opening metathesis polymerization of cyclooctenes. The multiblock polymers exhibit broad mechanical properties, spanning elastomers to plastomers to thermoplastics, while integrating a high melting transition temperature (Tm) and low glass transition temperature (Tg), making them suitable for use across diverse applications (Tm as high as 128°C and Tg as low as −60°C). After use, the different plastics can be combined and efficiently deconstructed back to the fundamental hard and soft building blocks for separation and repolymerization to realize a closed-loop recycling process.
Polyolefins make up over half of all plastics produced with diverse applications that impact nearly every aspect of modern life (1, 2). However, the inert characteristics that provide plastics their useful materials properties make them resilient to degradation in the environment, with an estimated lifetime of hundreds of years and unknown environmental consequences (3, 4). Mechanical recycling of plastics is largely unsuccessful for preserving their value or as a waste remediation strategy; only around 9% of all plastics have been recycled (5). A substantial challenge in mechanical recycling is the need for physical separation because mixing different types of plastics leads to macrophase-segregated materials with compromised materials properties (6). Even if separation is achieved, mechanical recycling initiates cross-linking and chain-scission reactions that abate materials properties. Valorization of plastics is an alternative approach to recapture resources but yields new chemicals rather than original monomer feedstocks (7, 8). For example, poly-ethylene (PE) can be converted to low–molecular weight hydrocarbons for new applications including liquid fuels or waxes (9–11). An inspirational approach for a closed-loop life cycle of sustainable plastics is to design systems that are chemically depolymerizable back to the original monomers for purification and repolymerization (12). These technologies are not yet economically or technologically viable replacements for polyolefins because the complete depolymerization back to monomer maximizes the number of chemical bonds that must be broken and reformed during the recycling process, necessitating extreme reaction efficiency and increased energy inputs (13, 14). A more efficient strategy would de-construct plastics back to oligomeric units to allow for purification, yet minimize the number of bond transformations that must occur during chemical recycling.
Despite strong motivations, depolymerizable polyolefin-like materials have not yet been established as a broad replacement for the rich spectrum of polyolefin applications (15). The deconstruction of polyolefins is thermodynamically challenging, requiring energy-intensive conditions to break strong carbon-carbon bonds, leading to substantial side reactivity. As such, the incorporation of more chemically reactive functional groups into the polyolefin main chain to facilitate degradation has gained increasing attention (16, 17). Insertion of ketones into PE through the copolymerization of ethylene with carbon monoxide results in photo-degradable PE derivatives that can break down to smaller fragments but falls short of creating a circular plastics life cycle (18). The step-growth condensation polymerization of diester-functionalized alkanes with diols results in PE-like materials possessing ester functionality that enables depolymerization back to the building blocks for repolymerization (19). However, this approach produces a limited scope of materials and yields polymers that possess high ester content and lower thermal and materials properties relative to PE. The ester functionality does not drastically impact the unit-cell identity relative to PE but does lead to decreased lamellar thickness, destabilization of the crystal lattice, and lowering of the melting transition temperature (Tm) (20). Although condensation polymerization of dicarboxyl aliphatic oligomeric building blocks with small-molecule diols affords plastics with lower ester content for improved thermal and materials properties more comparable to PE (21), traditional step-growth polymerizations require precise stoichiometry matching of complimentary polymerizable groups (i.e., esters and alcohols) of the monomers to be coupled to achieve high molecular weight and technologically useful polymers. Even slight imbalances in stoichiometry result in drastically reduced polymer molecular weights and associated properties (22). Therefore, because of this constraint it is difficult to access polymers that possess disparate properties through a step-growth polymerization without completely changing the chemical identity of the building blocks.
We recognized that a major challenge to realizing diverse and recyclable polyolefin-like materials through a step-growth polymerization would require a polymerization methodology able to overcome the constraints of precise stoichiometry matching of chain-end groups and allow for deviations in monomer feed ratios. Notably, a step-growth polymerization of monomers or oligomers possessing identical chain-end groups would overcome this challenge and allow for modulation of the monomer feed composition to produce diverse high–molecular weight polymers with functionality designed for recyclability (Fig. 1A). The polymerization of oligomers presents the opportunity to readily produce multiblock polymers that can integrate multiple properties into a single macromolecule. For example, multiblock polyolefins with sequences of crystalline high-density PE and amorphous low-density polyolefin have both a high Tm and a low glass transition temperature (Tg), which provide strength and elasticity across a large window of operating temperatures and conditions (23). By contrast, the Tm of ethylene and α-olefin statistical copolymers decreases with increasing a-olefin incorporation (23). With this objective in mind, we were inspired by the ruthenium-catalyzed dehydrogenation polymerization of alcohols to produce polyesters (24). Furthermore, in the presence of hydrogen, the ruthenium complex can catalyze the depolymerization of polyesters to diol building blocks (25, 26), which suggests that a system for closed-loop chemical recycling could be developed using this methodology. This proposed circular plastics life cycle would be driven by the closed-loop removal and incorporation of hydrogen, the simplest and smallest molecule possible for a condensation polymerization and a sought-after foundation for green energy (27). Here, we report an approach for synthesizing multiblock polyolefin-like materials constructed from hard and soft aliphatic oligomeric building blocks with highly tunable materials properties dictated by the ratio of the two types of blocks. Notably, the different multiblock polymers can be mixed and efficiently depolymerized back to their building blocks for purification and repolymerization, demonstrating the recycling of these different plastics.
Fig. 1. Overview of chemically recyclable polyolefin-like multiblock materials with tunable properties from polymerization of hard and soft oligomers.
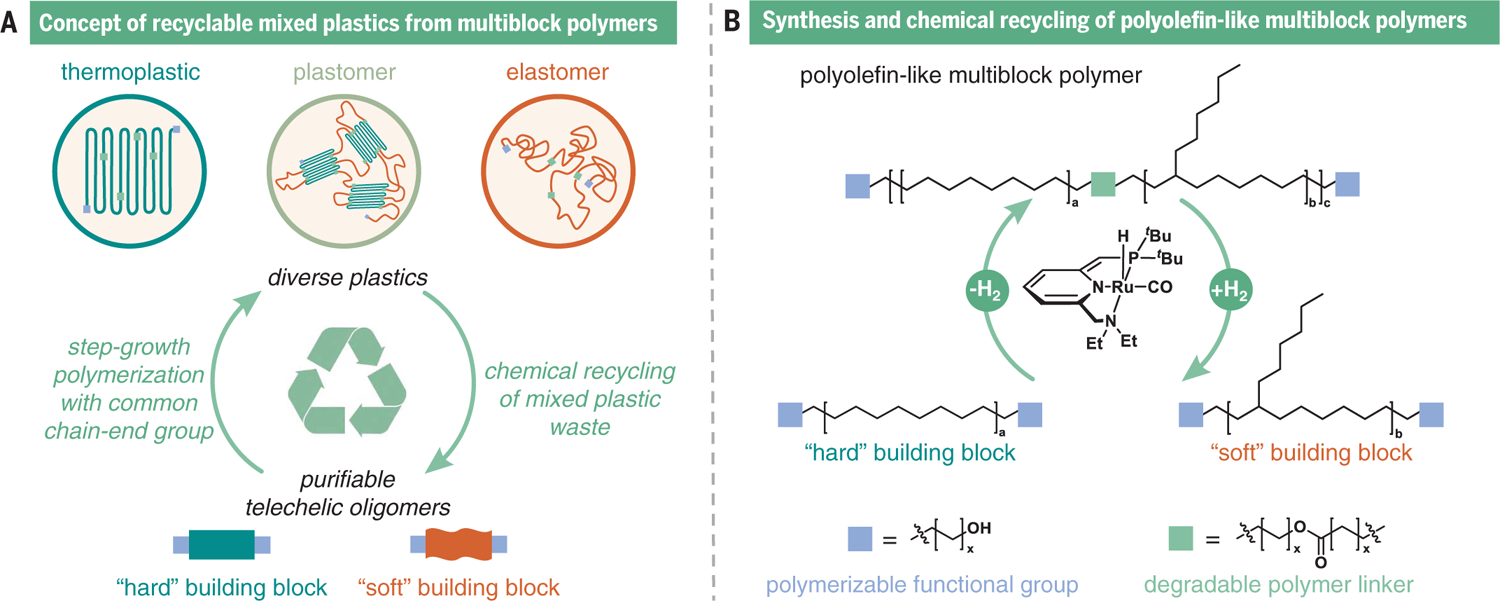
This strategy (A) successfully builds on an approach for the synthesis of ester-linked polyolefin-like multiblock polymers to create a platform for closed-loop recycling of mixed plastics catalyzed by a ruthenium complex (B).
Oligomer synthesis and copolymerization
To produce the required telechelic diol-functionalized hard [HO-HB-OH; HB, hydrogenated poly(cyclooctene)] and soft [HO-SB-OH; SB, hydrogenated poly(3-hexylcyclooctene)] oligomeric blocks, we used ruthenium-mediated ring-opening metathesis polymerization (28) of cyclooctene or 3-hexylcyclooctene, respectively, in the presence of cis-hexadec-6-ene-1,16-diol as a chain-transfer agent (fig. S1). These oligomers were subsequently hydrogenated (40 bar H2, 100°C for 36 hours), catalyzed by residual ruthenium in the unpurified product, to yield the hard HO-HB-OH [weight-average molecular weight (Mw) = 3.3 kDa] and soft HO-SB-OH (Mw = 3.0 kDa) building blocks (figs. S2 to S4). The crystallizable hard block was designed to imbue high Tm and modulus into the polymer, whereas the hexyl-functionalized soft block was designed to introduce controlled short-chain C6 branching to create noncrystallizable, elastomeric soft domains. Branching has substantial influences on polyolefin thermal and mechanical properties, primarily by reducing the allowable degree of crystallinity and increasing free volume (29). The oligomers were polymerized in high yields (>90%) with varying ratios of hard and soft blocks to modulate the hard content from 0 to 100% (figs. S5 to S10) by using ruthenium-catalyzed dehydrogenation polymerization to high–molecular weight polymers (Mw = 62.7 to 90.4 kDa) (figs. S11 to S14 and table S1) with high dispersity (Ð > 2.2), as expected for polymers produced through a step-growth polymerization (Fig. 1B and table S2). The branching content of the polymers was determined by proton nuclear magnetic resonance spectroscopy (1H NMR) and was consistent with the block feed ratios (Fig. 2A). Although the catalyst proved difficult to remove and colored the polymers (tables S3 and S4), advantageously, depolymerization could be successfully performed with this residual catalyst (table S9, entry S22). From sustainability and practical perspectives, precious metal catalysts for commodity plastic production are undesirable, and the residual ruthenium contamination could impede use in biomedical or food packaging applications, especially those in which transparency is desired. Toward addressing this concern, lowering the catalyst concentration or using a different ruthenium complex produced considerably less colored materials (table S5 and figs. S15 and S16).
Fig. 2. Properties of multiblock polymers can be modulated over diverse regimes.
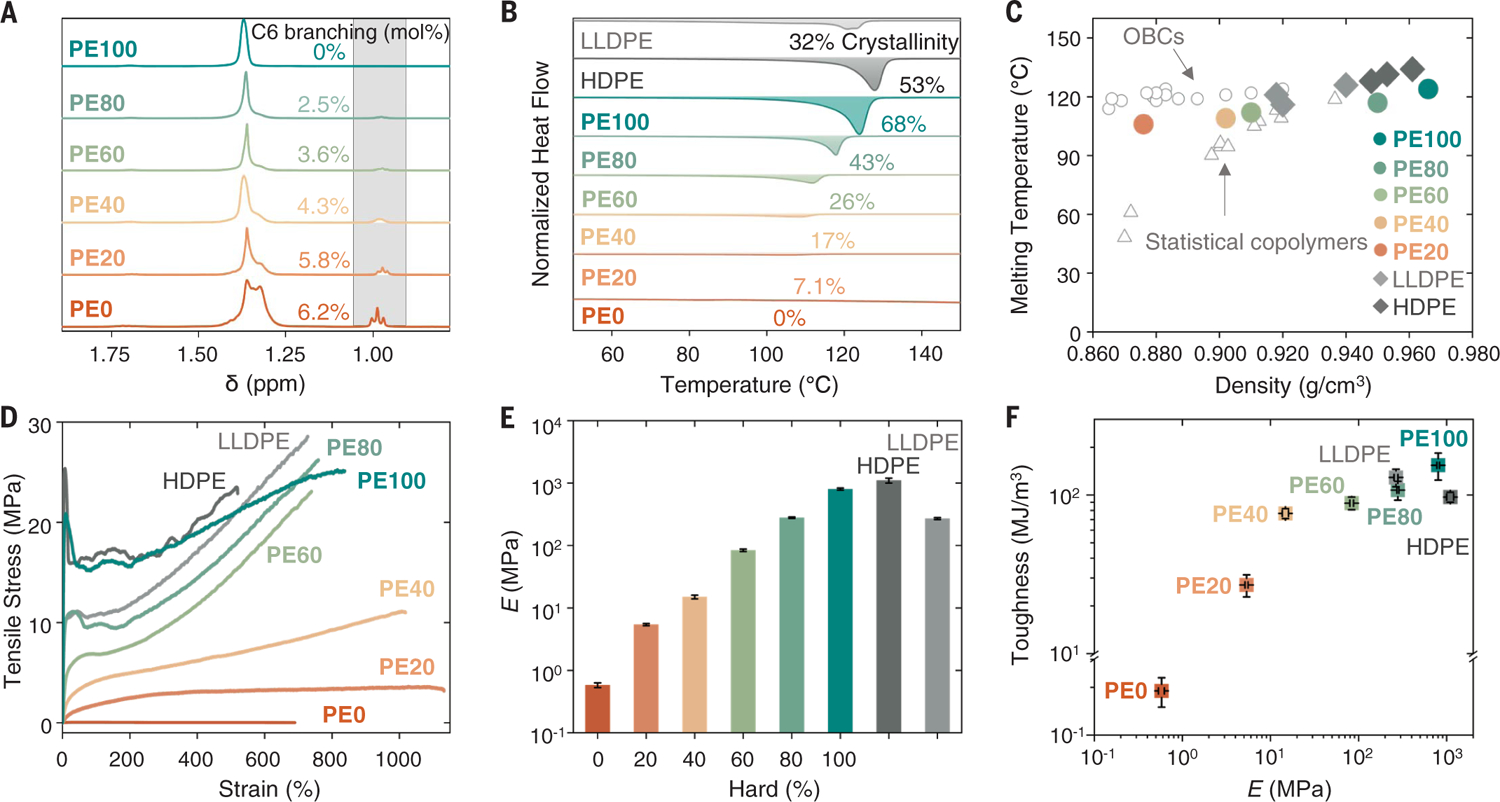
(A) Stacked 1H NMR spectra, (B) normalized differential scanning calorimetry traces, (C) melting temperature of multiblock polymers, commercial olefin block copolymers (OBCs), HDPE, LLDPE, and statistical olefin copolymers as a function of density, (D) representative stress-strain curves, (E) modulus, and (F) property comparison of toughness and modulus between multiblock polymers, HDPE, and LLDPE. Error bars represent standard deviations from the mean value of four to six samples.
Polymer properties
The resulting polymers were thermally stable, with high decomposition temperatures at 5% weight loss (Td5 = 406° to 421°C; fig. S17), consistent with commercially available high-density PE (HDPE; Td5 = 430°C) or linear low-density PE (LLDPE; Td5 = 434°C) samples (Table 1). With increasing hard content, the crystallinity of the polymers increased from 0 to 68% (Fig. 2B). Polymers with hard content <80% exhibited low glass transition temperatures (Tg = −46.5° to −60.0°C; figs. S18 and S19 and table S6), whereas a high melting transition temperature (Tm = 106° to 124°C) was observed for all multiblock polymers containing hard blocks, similarly to olefin-block copolymers (OBCs). (Fig. 2C and table S7). Further examination of the crystallinity by wide-angle x-ray scattering (WAXS) showed peaks corresponding to the orthorhombic unit cell of PE (fig. S20), although polarized light optical microscopy revealed that the crystallites were smaller in size than LLDPE or HDPE samples because of the presence of the ester group (fig. S21) (20). Thus, the multiblock polymers possessed both a high Tm and low Tg to allow for thermoplastic and elastic properties across a wide operating temperature.
Table 1. Properties of multiblock polymers tuned by controlling the hard-block incorporation.
See supplementary materials for details. For PEx samples, x represents the feed ratio of hard block in copolymers. Weight average molecular weight, Mw; dispersity, Ð; degree of crystallinity, Xc; glass transition temperature, Tg; melting transition temperature, Tm; decomposition temperature at 5% weight loss, Td5; Young’s modulus, E; tensile strength, σUTS; elongation at break, εb; tensile toughness, UT; zero-shear viscosity, η0. Average values for E, σUTS, εb, and UT are provided with standard deviations from the mean value of four to six samples.
| PE0 | 71.7 | 2.4 | 62 | 0 | −59.1 | - | 409 | 0.58 ± 0.05 | 0.041 ± 0.006 | 700 ± 100 | 0.19 ± 0.04 | 3.45 |
| PE20 | 90.4 | 2.5 | 58 | 7.1 | −60.0 | 106 | 406 | 5.4 ± 0.2 | 3.3 ± 0.3 | 1000 ± 100 | 27 ± 4 | 8.88 |
| PE40 | 82.4 | 2.4 | 43 | 17 | −50.6 | 108 | 413 | 15 ± 1 | 12.0 ± 0.7 | 1030 ± 30 | 77 ± 6 | 21.1 |
| PE60 | 62.7 | 2.7 | 36 | 26 | −46.5 | 112 | 421 | 84 ± 4 | 23 ± 1 | 710 ± 40 | 89 ± 8 | 68.7 |
| PE80 | 81.0 | 2.5 | 25 | 43 | - | 117 | 416 | 279 ± 8 | 24 ± 2 | 720 ± 60 | 110 ± 10 | 21.4 |
| PE100 | 80.7 | 2.2 | 0 | 68 | - | 124 | 415 | 800 ± 30 | 25 ± 2 | 800 ± 100 | 150 ± 30 | 447 |
| HDPE | 92.2 | 5.4 | 1.9 | 53 | - | 128 | 430 | 1100 ± 100 | 24 ± 1 | 530 ± 40 | 97 ± 7 | 2.47 |
| LLDPE | 90.4 | 2.8 | 30 | 32 | −27.4 | 121 | 434 | 270 ± 10 | 28 ± 2 | 750 ± 60 | 130 ± 20 | 1.30 |
Rheology was performed to investigate the creep compliance and zero-shear viscosity (η0) of the multiblock polymers. Generally, η0 increased with increasing hard content from PE0 to PE100 (η0 = 3.45 to 447 ×105 Pa·s), indicating that the introduction of short-chain branching reduced entanglement (fig. S22 and table S8). The molecular weight variations, combined with different hard and soft compositions and statistical block sequence formations, led to imprecise correlations with these properties (30). In comparison with polyolefins of similar molecular weights, these multiblock polymers exhibit increased η0, which hints at different interactions that may prove advantageous for use at temperatures approaching the Tm with reduced concern for material deformation (table S8). We postulate that this result may be due to structural effects imparted by functional group interactions from the addition of esters or from the block-polymer organization and effects due to the disparity in block sequences across varying compositions.
The mechanical properties of the multiblock polymers were investigated by uniaxial tensile testing (Fig. 2D and figs. S23 and S24). Polymer properties spanned drastic regimes, from elastomers to plastomers to thermoplastics, by increasing the hard content. Both Young’s modulus (E) and tensile strength (σUTS) increased by over three orders of magnitude (E = 580 kPa for PE0 to 800 MPa for PE100; σUTS = 40.5 kPa for PE0 to 24.7 MPa for PE100) (Fig. 2E and fig. S25). Furthermore, all samples spanning these regimes demonstrated excellent extensibility, with the average strain at break (εb) for all multiblock polymers exceeding 700%, with PE20 and PE40 achieving >1000% strain at break. These materials also exhibited tunable tensile toughness (UT) ranging from 0.19 to 150 MJ•m−3 (Fig. 2F). PE60, PE80, and PE100 all exhibited thermoplastic behavior with yield points and strain hardening, whereas a yield point was not observed for elastomeric polymers containing lower PE content. The overall thermal and materials properties of these polymers are akin to commercial OBCS. They also exhibit greater strain at break relative to commercially available HDPE and LLDPE samples tested in this work, and the toughness of PE80 and PE100 are comparable to or exceed that of very tough engineering plastics (>100 MJ•m−3) (table S7).
Recycling studies
To complete the closed-loop chemical recycling process, the depolymerization of PE0 was optimized to convert the polymer back to the fundamental oligomeric building blocks (fig. S26 and table S9). Although the residual ruthenium catalyst in the polymer with 40 bar H2 at 160°C in toluene was found sufficient to catalyze the depolymerization after 24 hours (entry S22), additional catalyst was added to ensure sufficient active species in recycling studies (table S3). The closed-loop chemical recycling process of mixed-plastic waste was demonstrated; in doing so, the multiblock polymers of all compositions following tensile testing were combined for simultaneous depolymerization (>99% conversion) and the hard and soft oligomeric building blocks were separated and purified in 91.7% isolated yield with no observable signs of oligomer decomposition by 1H NMR (Fig. 3A and figs. S27 and S28). The hard and soft blocks were readily separated through industrially viable selective-solvent isolation (31) by precipitation of the hard block and purified to remove catalyst residues to low parts-per-million levels such that ruthenium levels did not increase during subsequent polymerizations. These recovered oligomers were successfully repolymerized to PE80, which was subsequently depolymerized and repolymerized back into PE80 for two additional cycles with all steps proceeding in high yields (tables S10 and S11). The molecular weights of the recycled PE80 multiblock polymers remained consistent (Mw = 73.8 to 96.6 kDa) (figs. S29 and S30), demonstrating the robustness of this closed-loop recycling process. Notably, tensile testing of all recycled PE80 samples revealed that the modulus, tensile strength, elongation at break, and toughness were comparable to the virgin PE80 sample (Fig. 3, B and C, and fig. S31). We demonstrated that these multiblock polymers can be separated from technologically important isotactic polypropylene (PP) (Fig. 3C and figs. S32 and S33). Mixtures of PE and PP are difficult to separate, do not homogeneously blend, and result in materials with compromised properties, emphasizing the importance of being able to separate mixed plastics or devise strategies for compatibilization (32). In the presence of commercially available PP, the depolymerization of PE60 was efficient, and the oligomers were readily separated from the PP in high yield (92%) (fig. S32).
Fig. 3. Chemical recycling of multiblock polymers.
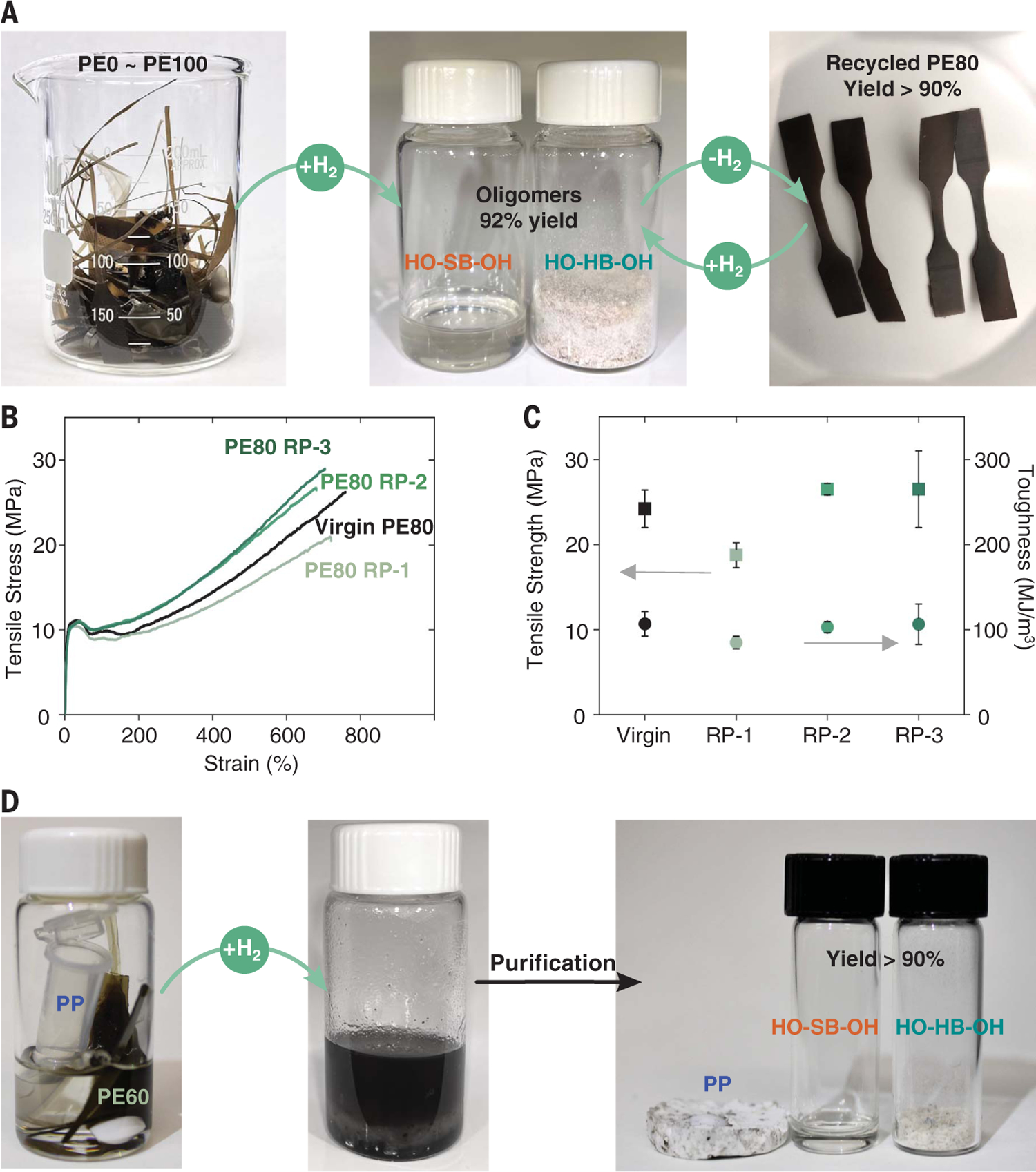
(A) Photographs of the chemical recycling of mixed multiblock polymers. (B) Representative stress-strain curves of virgin PE80 and repolymerized PE80 RP-1, PE80 RP-2, and PE80 RP-3. (C) Tensile strength and toughness of virgin PE80 and repolymerized PE80 RP-1, PE80 RP-2, and PE80 RP-3. Error bars represent standard deviations from the mean value of four to five samples. (D) A mixture of PP and PE60 as an example of a potential polyolefin waste stream. Photographs of mixed plastics in toluene (left), reaction after 72 hours (middle), and the separated PP, HO-HB-PH, and HO-SB-OH (right). See supplementary materials for details.
Despite vast synthetic strategies available to produce polymers, the ability to synthesize multiblock polymers remains a challenge most commonly met through tedious sequential monomer additions (33). Synthesizing multiblock polymers from hard and soft aliphatic oligomers possessing identical chain-end groups creates a platform to produce highly tunable polyolefin-like materials and their closed-loop chemical recycling process, including in the presence of other commercially important plastics. The modularity of this approach coupled with improvements focused on enhancing sustainability will further allow realization of other chemically recyclable plastics enabled by the multiblock polymer architecture for the union of otherwise unobtainable polymer compositions (34). Although this approach provides a promising resolution to pressing challenges of plastic recycling, we foresee that forthcoming endeavors will uncover nonprecious-metal catalyst systems that exhibit enhanced performance to enable commercial impact.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Analytical Resources Core (RRID: SCR_021758) at Colorado State University for instrument access and training; C. Josephitis and M. Nicki for their assistance in early hydrogenation experiments; B. Newell and M. Mailhot for their generous assistance with instrument configuration and training; and E. Quinn, M. Price, and X. Liu for insightful discussions throughout the experimental design.
Funding:
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award no. R35GM144356. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge support from RePLACE (Redesigning Polymers to Leverage a Circular Economy) funded by the Office of Science of the US Department of Energy through award no. DE-SC0022290. Funding for N.A.R. and J.M. was provided in part by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Manufacturing Office (AMO), and Bioenergy Technologies Office (BETO). This work was performed as part of the BOTTLE™ Consortium and was supported by AMO and BETO under contract no. DE-AC36-08GO28308 with the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC.
Footnotes
Competing interests: The Colorado State University Research Foundation with Y.Z., E.M.R., K.L.H., and G.M.M. have filed a provisional patent application on this technology.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Geyer R, Jambeck JR, Law KL, Sci. Adv 3, e1700782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli P, Vecellio G, J. Polym. Sci. A Polym. Chem 42, 396–415 (2004). [Google Scholar]
- 3.Chamas A et al. , ACS Sustain. Chem. & Eng 8, 3494–3511 (2020). [Google Scholar]
- 4.MacLeod M, Arp HPH, Tekman MB, Jahnke A, Science 373, 61–65 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Vogt BD, Stokes KK, Kumar SK, ACS Appl. Polym. Mater 3, 4325–4346 (2021). [Google Scholar]
- 6.Li H et al. , Green Chem. 24, 8899–9002 (2022). [Google Scholar]
- 7.Conk RJ et al. , Science 377, 1561–1566 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F et al. , Science 370, 437–441 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Kots PA, Vance BC, Danielson A, Vlachos DG, Sci. Adv 7, eabf8283 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia X, Qin C, Friedberger T, Guan Z, Huang Z, Sci. Adv 2, e1501591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason AH et al. , Nat. Commun 13, 7187 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J-B, Watson EM, Tang J, Chen EY-X, Science 360, 398–403 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Coates GW, Getzler YDYL, Nat. Rev. Mater 5, 501–516 (2020). [Google Scholar]
- 14.Shi C et al. , Chem 7, 2896–2912 (2021). [Google Scholar]
- 15.Kakadellis S, Rosetto G, Science 373, 49–50 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Stempfle F, Ortmann P, Mecking S, Chem. Rev 116, 4597–4641 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Shiono T, Naga N, Soga K, Makromol. Chem., Rapid. Commun 12, 387–392 (1991). [Google Scholar]
- 18.Baur M, Lin F, Morgen TO, Odenwald L, Mecking S, Science 374, 604–607 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Haeussler M, Eck M, Rothauer D, Mecking S, Nature 590, 423–427 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Pepels MPF, Hansen MR, Goossens H, Duchateau R, Macromolecules 46, 7668–7677 (2013). [Google Scholar]
- 21.Arrington AS, Brown JR, Win MS, Winey KI, Long TE, Polym. Chem 13, 3116–3125 (2022). [Google Scholar]
- 22.Stille JK, J. Chem. Educ 58, 862–866 (1981). [Google Scholar]
- 23.Arriola DJ, Carnahan EM, Hustad PD, Kuhlman RL, Wenzel TT, Science 312, 714–719 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Hunsicker DM, Dauphinais BC, Mc Ilrath SP, Robertson NJ, Macromol. Rapid Commun 33, 232–236 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Krall EM et al. , Chem. Commun 50, 4884–4887 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Westhues S, Idel J, Klankermayer J, Sci. Adv 4, eaat9669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crabtree GW, Dresselhaus MS, Buchanan MV, Phys. Today 57, 39–44 (2004). [Google Scholar]
- 28.Hilf S, Kilbinger AFM, Nat. Chem 1, 537–546 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Gahleitner M, Prog. Polym. Sci 26, 895–944 (2001). [Google Scholar]
- 30.Stadler FJ, Karimkhani V, Macromolecules 44, 5401–5413 (2011). [Google Scholar]
- 31.Walker TW et al. , Sci. Adv 6, eaba7599 (2020).33219017 [Google Scholar]
- 32.Eagan JM et al. , Science 355, 814–816 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Bates FS et al. , Science 336, 434–440 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Hawker CJ, Wooley KL, Science 309, 1200–1205 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.