

Abstract
Myelin protein zero (MPZ/P0) is a major structural protein of peripheral nerve myelin. Disease‐associated variants in the MPZ gene cause a wide phenotypic spectrum of inherited peripheral neuropathies. Previous nerve biopsy studies showed evidence for subtype‐specific morphological features. Here, we aimed at enhancing the understanding of these subtype‐specific features and pathophysiological aspects of MPZ neuropathies. We examined archival material from two Central European centers and systematically determined genetic, clinical, and neuropathological features of 21 patients with MPZ mutations compared to 16 controls. Cases were grouped based on nerve conduction data into congenital hypomyelinating neuropathy (CHN; n = 2), demyelinating Charcot–Marie‐Tooth (CMT type 1; n = 11), intermediate (CMTi; n = 3), and axonal CMT (type 2; n = 5). Six cases had combined muscle and nerve biopsies and one underwent autopsy. We detected four MPZ gene variants not previously described in patients with neuropathy. Light and electron microscopy of nerve biopsies confirmed fewer myelinated fibers, more onion bulbs and reduced regeneration in demyelinating CMT1 compared to CMT2/CMTi. In addition, we observed significantly more denervated Schwann cells, more collagen pockets, fewer unmyelinated axons per Schwann cell unit and a higher density of Schwann cell nuclei in CMT1 compared to CMT2/CMTi. CHN was characterized by basal lamina onion bulb formation, a further increase in Schwann cell density and hypomyelination. Most late onset axonal neuropathy patients showed microangiopathy. In the autopsy case, we observed prominent neuromatous hyperinnervation of the spinal meninges. In four of the six muscle biopsies, we found marked structural mitochondrial abnormalities. These results show that MPZ alterations not only affect myelinated nerve fibers, leading to either primarily demyelinating or axonal changes, but also affect non‐myelinated nerve fibers. The autopsy case offers insight into spinal nerve root pathology in MPZ neuropathy. Finally, our data suggest a peculiar association of MPZ mutations with mitochondrial alterations in muscle.
Keywords: Charcot–Marie‐tooth, congenital hypomyelinating neuropathy, inherited neuropathy, MPZ, myelin protein zero
To enhance the understanding of subtype‐specific features and pathophysiological aspects of hereditary neuropathies due to mutations in the myelin protein zero (MPZ), we performed neuropathological analyses of archival nerve and muscle biopsies as well as autopsy material and present them along with clinical and genetic data. We confirmed fewer myelinated fibers, more onion bulbs and reduced regeneration in demyelinating Charcot‐Marie‐Tooth (CMT) 1 compared to CMT2/CMTi and observed significantly more denervated Schwann cells, more collagen pockets, fewer unmyelinated axons per Schwann cell unit and a higher density of Schwann cell nuclei in CMT1 compared to CMT2/CMTi. Furthermore, we found prominent neuromatous hyperinnervation of the spinal meninges in the autopsy case and marked structural mitochondrial abnormalities in skeletal muscle, the latter suggesting a peculiar association of MPZ mutations with mitochondrial alterations.
1. INTRODUCTION
Myelin protein zero (MPZ/P0) is the major protein component of myelin in peripheral nerves [1, 2]. The gene consists of six exons and is mainly expressed by Schwann cells. The MPZ protein is an integral membrane glycoprotein of 28 kDa that contains a single membrane‐spanning domain, a large, glycosylated immunoglobulin‐like extracellular domain, and a smaller basic intracellular domain; homotypic interactions between the extracellular domains of adjacent MPZ molecules on opposing myelin lamellae are required for myelin sheath compaction as demonstrated by several studies on MPZ knockout mice [2, 3, 4, 5, 6]. Hence, an essential role of MPZ in myelinating Schwann cells and myelination in the peripheral nervous system (PNS) has unequivocally been demonstrated.
MPZ mutations can cause a spectrum of hereditary neuropathies, including severe congenital hypomyelination neuropathy (CHN) as well as different forms of hereditary motor and sensory neuropathies (HMSN), called Charcot–Marie‐Tooth (CMT) diseases. The CMT classification has been based on a study by Dyck and Lambert who defined one group (HMSN‐I) with slowed nerve conduction velocity (NCV) and nerve hypertrophy (now called demyelinating CMT, CMT1) and one group (HMSN‐II) without these features but primarily axonal/neuronal degeneration (now called axonal CMT, CMT2). These groups were further subdivided by more specific clinical features and mode of inheritance [7, 8]. With the advance of molecular genetic analysis, more and more subtypes were identified. The most frequently identified CMT causing mutation is a duplication on chromosome 17p12, encompassing the PMP22 gene, accounting for around 40% of all CMTs, and termed CMT1A [9, 10, 11, 12, 13]. Demyelinating neuropathies due to MPZ mutations were called CMT1B and are less frequently observed (single‐digit percentage range of CMT patients) [12, 13]. However, for MPZ mutations, a bimodal distribution with early‐onset of symptoms and slowed NCV (CMT1B) as well as late‐onset axonal damage (CMT2) became apparent [3].
Applying the traditional CMT classification can be complex since CMT subtypes can be caused by numerous gene mutations and one gene can be involved in several subtypes. Novel classification systems have been suggested, which can overcome these obstacles [14]. Nevertheless, because no such system has as yet gained general acceptance, we here group our cases based on the traditional CMT classification systems into: infantile and juvenile neuropathy with mostly demyelinating phenotype (CMT1) with nerve conduction velocities (NCV) < 38 m/s, as well as late‐onset, adult patients with milder, predominantly axonal neuropathy (CMT2) with NCV ≥38 m/s [3, 15]. An intermediate type (CMTi) has been suggested in patients with NCV between 30 and 40 m/s [16] and more recently been used to describe families in which different affected family members have motor conduction velocities in both the CMT type 1 and 2 ranges (i.e., above and below 38 m/s) [17]. Morphologically, compared to CMT2 patients CMT1 patients showed reduced density of myelinated fibers, demyelinating pathology, tomacula, and onion bulbs [3, 18, 19, 20]. Based on the known function of MPZ in myelin formation and compaction, the demyelinating phenotype can easily be explained. As expected, CMT2 patients showed more axonal changes [3, 18, 19, 20]. In contrast, involvement of unmyelinated fibers has ‐ to our knowledge ‐ thus far not been reported even though, expression of MPZ protein is not restricted to myelinating Schwann cells. Actually, it was also demonstrated in multipotent glial precursor cells derived from dorsal root ganglia (DRG) and neural crest, suggesting a role in embryonic development of other components of the PNS [21]. This raises the possibility that MPZ could be involved in regulating development and/or maintenance of non‐myelinating Schwann cells and unmyelinated axons.
Nowadays, patients with inherited peripheral neuropathies, including those with mutations in the MPZ gene are routinely diagnosed genetically, and nerve biopsies are obsolete in most cases [22]. Thus, in order to understand genotype–phenotype correlations, it is important to screen the available archival material. Here, we re‐examined nerve biopsies from two Central European centers. Along this line, we retrospectively systematically examined nerve biopsies of 21 patients with MPZ‐related neuropathies and compared them to nerve biopsies of 16 control patients. Furthermore, muscle biopsies were available of six of the patients with MPZ mutation and one case was autopsied.
2. METHODS
2.1. Ethics statement
The study was approved by the ethics committees of the participating centers (UAntwerpen; Ethical Commission of the Medical Faculty of the RWTH Aachen, EK 125/18 and EK126/18).
2.2. DNA isolation and sequencing
Genomic DNA was isolated from blood leucocytes or paraffin‐embedded nerve biopsy tissues using the Qiagen Kit (QIAamp® DNA Mini Kit 250, Hilden, Germany) according to the manufacturer's protocols. The six MPZ exons were analyzed by PCR and Sanger sequencing as described [20, 23, 24, 25].
2.3. Patients and comparison with CMT mutation database
This study was a retrospective study of archival material of 21 unrelated individuals with MPZ mutation. Previous detection of an MPZ mutation was an inclusion criterion for the group of cases that had been biopsied between 1978 and 2014. Controls were biopsied between 1988 and 2012. Clinical and genetic information is summarized in Table 1, Table 2 contains the results of electrophysiological measurements. In all patients, nerve biopsies were available for light and electron microscopic examination. In most patients, the sural nerve was biopsied, except for MPZ patient #3 (saphenous nerve) and patient #9 (superficial fibular nerve). Four of the MPZ variants found by us have ‐ to the best of our knowledge ‐ not been reported so far (see Human Gene Mutation Database (HGMD, Qiagen), Inherited Neuropathy Variant Browser: https://neuropathybrowser.zuchnerlab.net/#/ and [15]). Sixteen sural nerve biopsies that had been classified as neuropathologically normal served as matched controls (Table S1).
TABLE 1.
Clinical findings in patients with MPZ variants.
| # | MPZ mutation | CMT type | AAO (years) | Gender | Symptoms at onset | ALE | Walking | Ws dist. LL | Ws prox. LL | Atr. dist. LL | Pes cavus | DTR Ach/knee | Vib LL | Ws dist. UL | Ws prox. UL | DTR UL | Additional features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.567_568insGGCC (p.L190GfsX46) | CHNP | 0.1 | m | Floppy infant, feeding problems, areflexia | 10 | Walking frame unaided (5.7 years) | + | + | + | − | − | − | + | + | − | Dysphagia, scoliosis, ametropia |
| 2 | c.626_630delCGTCG (p.A209EfsX24) | CHNP | Cong | f | Hypotonia, breathing difficulties | 27 | Wheelchair | + | + | NA | NA | −/− | ↓ | + | + | − | de novo mutation |
| 3 | c.292C > T (p.R98C) | CMT1 | NA | m | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | − |
| 4 | c.292C > T (p.R98C) | CMT1 | 1 | m | Abasia | 49 | Wheelchair | + | + | + | − | − | ↓ | + | + | − | Bronchial asthma, scoliosis, claw hand, hypesthesia, thenaratrophy; died at age of 49 years |
| 5 | c.293G > A (p.R98H) | CMT1 | 55 | f | Hypesthesia left hand | 57 | Toe/heel walking not possible | + | − | + | − | −/− | NA | − | − | + | Claw foot |
| 6 | c.293G > A (p.R98H) | CMT1 | 63 | m | Atrophy and weakness of lower limbs | 67 | Steppage gait, toe/heel walking not possible | +(0–2) | − | + | + | −/− | ↓ | + (2, 3) | − | − | Pupillary miosis, hypertrophic lumbar radiculopathy, hypesthesia lower limbs |
| 7 | c.371C > T (p.T124M) | CMT1 | 40 | m | Weakness in the lower limbs | 72 | Wheelchair | + | + | + | − | −/− | − | + | − | − | Deafness, diabetes mellitus |
| 8 | c.402C > A (p.D134E) | CMT1 | <1 | m | Delayed motor milestones | 14 | Wheelchair | +(0) | + (2) | + | Equino‐varus | −/− | NA | + (1) | + (1–3) | − | − |
| 9 | c.402C > A (p.D134E) | CMT1 | 4 | m | Gait difficulties | 28 | Steppage gate | + (4) | − | + | + | −/− | ↓ | + (4) | − | − |
Nystagmus |
| 10 | c.400G > A (p.D134N) | CMT1 | NA | f | NA | 33 | Ataxic, steppage gate | + | − | NA | + | −/− | NA | + | − | − |
Anisocoria |
| 11 | c.487G > A (p.G163R) | CMT1 | Child‐hood | m | Foot deformities | 51 | Steppage gait, heel walking not possible | +(0–5) | − | + | + | −/− | ↓ | + | − | − | Anisocoria left<right, pupils not reacting to light, hypertrophy of cauda equina, scoliosis |
| 12 |
c.543C > G (p.Y181X) |
CMT1 | >20 | f | Gait difficulties | 34 | Walks unaided | + | − | + | + | ↓/↓ | + | − | − | − | − |
| 13 | c.678delC (p.S226RfsX26) | CMT1 | 2 | m | Abnormal gait, foot deformities | 30 | Steppage gait, toe/heel walking not possible | +(0) | +(4+) | + | + | −/− | − | +(1–3+) | − | − | Neuropathic pain, hypesthesia, scoliosis, de novo mutation |
| 14 | c.182A > G (p.D61G) | CMTi | ~40 | m | Unsteady gait, pain in the legs | 72 | Ataxic, steppage gate | + | + (4) | − | − | −/− | ↓ | + (4) | − | + | Hypacusis, hypesthesia |
| 15 | c.182A > G (p.D61G) | CMTi | 63 | m | Unsteady on the feet | 78 | Impaired gait | +(0) | + | + | NA | −/− | ↓ | + | + | NA | Pain in extremities at night |
| 16 | c.371C > T (p.T124M) | CMTi | 40 | m | Shooting pain, paresthesia lower limbs, unsteady on the feet in darkness | 48 | Limited walking distance | + | + | + | − | − | − | − | − | − | Anisocoria left>right, paresthesia, hearing difficulty, facial nerve palsy (right) |
| 17 | c.73_74 delinsA (p.S25TfsX22) | CMT2 | 29 | f | Gait difficulties, foot drop | 49 | Ataxic gait | + (3, 4) | − | − | + | −/− | ↓ | + (4, 5) | − | + | Hypesthesia lower limbs |
| 18 | c.356A > G (p.Y119C) | CMT2 | 49 | f | Unsteady on the feet | NA | NA | + | NA | NA | NA | −/↓ | − | NA | NA | NA | Hyp−/paresthesia lower limb |
| 19 | c.371C > T (p.T124M) | CMT2 | 36 | f | Distal weakness LL | 85 | Wheelchair | + (4) | + (4) | + | + | −/− | + | + (4) | + (4) | − | Deafness, pupillary abnormalities |
| 20 | c.616G > T (p.G206X) | CMT2 | 0.33 | m | Hypotonia | 8 | At the age of 5 | + | + | + | − | ↓/↓ | NA | + | + | ↓ | Dystrophy, scapula alata, nanism, microcephaly, carnitine deficiency, heart failure, respiratory insufficiency; died at age of 8 years |
| 21 |
c.709G > A (p.A237T) |
CMT2 | 58 | f | Unsteady on the feet | 65 | Sensory ataxia | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
Abbreviations: AAO, age at onset; Ach, Achilles tendon; ALE, age at last examination; Atr, atrophy; CHN, congenital hypomyelinating neuropathy; CMT1, Charcot–Marie‐Tooth disease type 1; CMTi, Charcot–Marie‐Tooth disease intermediate type; CMT2, Charcot–Marie‐Tooth disease type 2; dist., distal; DTR, deep tendon reflexes; f, female; LL, lower limbs; m, male; MRC‐scale, Medical Research Council (indicating range of muscle weakness between brackets if available); NA, information not available; prox., proximal; UL, upper limbs; Vib, vibration; Ws, weakness; +, present; −, absent; ↓, reduced.
TABLE 2.
Electrophysiological data of patients with MPZ mutations. Results of neurography of different nerves as indicated on the R (right) and L (left) side; normal values are listed in the first row.
| # | CMT type | Age (years) | R/L | Median motor | Ulnar motor | Peroneal motor | Tibial motor | Median sensory | Ulnar sensory | Sural sensory | Comments | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amp | NCV | Amp | NCV | Amp | NCV | Amp | NCV | Amp | NCV | Amp | NCV | Amp | NCV | |||||
| 4.0 | 49.0 | 4.0 | 49.0 | 3.0 | 41.0 | 3.0 | 41.0 | 17.0 | 46.0 | 17.0 | 46.0 | 12.0 | 44.0 | |||||
| 1 | CHNP | 0.8/10 | R | A | A | A | A | A | A | A | A | A | A | A | A | A | A | |
| L | A | A | A | A | A | A | A | A | A | A | A | A | A | A | ||||
| 2 | CHNP | 20 | R | 1.4 | 2.8 | A | A | A | A | A | A | A | A | nom | nom | A | A | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 3 | CMT1 | – | R | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 4 | CMT1 | 41 | R | nom | nom | nom | 7.5 | nom | nom | A | A | A | A | A | A | nom | nom | |
| L | nom | nom | nom | 6.5 | nom | nom | A | A | A | A | A | A | nom | nom | ||||
| 5 | CMT1 | 57 | R | 5.0 | 25 | nom | nom | 0.2 | 15 | 1.0 | 16 | 4.0 | 18 | 2.0 | 20 | A | A | |
| L | 4.0 | 21 | 3.0 | 18 | 0.6 | 12 | 1.0 | 13 | A | A | A | A | A | A | ||||
| 6 | CMT1 | 65/73 | R | nom | nom | nom | nom | nom | nom | 1.0 | 11 | nom | nom | nom | nom | nom | nom | |
| L | 8.0/8.6 | 16/18 | 0.8/0.6 | 13/17 | nom | nom | 0.3 | 10 | 2.2 | 17 | nom | nom | nom | nom | ||||
| 7 | CMT1 | 72 | R | nom | nom | 0.7 | 30 | A | A | A | A | A | A | nom | nom | nom | nom | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 8 | CMT1 | 5 | R | 0.6 | 7.0 | 1.2 | 17 | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 9 | CMT1 | 21 | R | 2.0 | 13.0 | 2.5 | 13 | A | A | nom | nom | nom | nom | A | A | nom | nom | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 10 | CMT1 | 32 | R | NA | NA | NA | NA | A | NA | NA | NA | NA | NA | NA | NA | NA | Strongly reduced motor and sensory NCV | |
| L | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | |||||
| 11 | CMT1 | 51 | R | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | No muscle action potentials (legs, arms) |
| L | NA | 19 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | No nerve action potential (legs) | |||
| 12 | CMT1 | 33 | R | 6.7 | 30.0 | 8.4 | 36 | 4.0 | 26 | nom | nom | 13 | 31 | 11 | 32 | nom | nom | |
| L | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 13 | CMT1 | 23/27 | R | nom | nom | nom | nom | nom | nom | A | A | nom | nom | nom | nom | A | A | |
| L | A | A | 1.4 | 30 | nom | nom | A | A | 1.2 | 49 | A | A | A | A | ||||
| 14 | CMTi | 72 | R | 2.0 | 30 | 3.0 | 43 | A | A | A | A | A | A | nom | nom | nom | nom | |
| L | 3.0 | 30 | 3.0 | 46 | A | A | NA | 26 | A | A | nom | nom | nom | nom | ||||
| 15 | CMTi | 78 | R | NA | 21.9 | NA | 39.5 | NA | NA | NA | NA | NA | 37.2 | NA | NA | NA | NA | |
| L | NA | NA | NA | NA | NA | NA | NA | NA | NA | 6.2 | NA | NA | NA | NA | ||||
| 16 | CMTi | 64 | R | nom | nom | nom | nom | nom | 23.1 | nom | 18.6 | nom | nom | nom | nom | nom | 34.2 | |
| L | nom | nom | nom | nom | nom | 32.7 | 1.5 | 25.5 | nom | nom | nom | nom | nom | 28.5 | ||||
| 17 | CMT2 | 24/49 | R | nom | nom | nom | nom | 4.5 | 36.7 | 4.5 | 36.7 | nom | nom | nom | nom | nom | nom | |
| L | 4.2 | 41 | 12.9 | 42 | 6.1 | 34 | 6.1 | 34 | nom | nom | nom | nom | nom | nom | ||||
| 18 | CMT2 | 51 | R | 39 | 31 | 27 | NA | NA | NA | NA | A | |||||||
| L | NA | NA | NA | NA | ||||||||||||||
| 19 | CMT2 | 85 | R | 1.4 | 38.0 | 6.7 | 45 | nom | nom | nom | nom | A | A | A | A | nom | nom | |
| L | nom | nom | 0.4 | 26 | nom | nom | nom | nom | A | A | nom | nom | nom | nom | ||||
| 20 | CMT2 | 8 | R | A | A | nv | nv | nv | nv | nom | nom | nom | nom | nom | nom | nom | nom | |
| L | A | A | nv | nv | nv | nv | nom | nom | nom | nom | nom | nom | nom | nom | ||||
| 21 | CMT2 | – | R | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | At 1 year normal CV |
| L | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | CV almost normal | |||
Note: Patients with motor NCV < 38 m/s at the upper limb (ulnar or median nerve) were classified as CMT1 (10 patients). An eleventh patient was classified as CMT1 based on clinical information since precise electrophysiological data were not available. Those patients with normal motor NCV (>38 m/s) at the upper limb (ulnar or median nerve) were classified as CMT2 (five patients). Two patients with upper motor NCV < 38 m/s for the median nerve, but >38 m/s for the ulnar nerve were classified as CMTi. A third patient was classified as CMTi based on lower limb (peroneal and tibial) motor NCV between 19 and 33 m/s since lower limb motor NCVs are generally approximately 10 m/s slower than upper limb motor NCV and values for the upper limb were not available. For statistical assessment, we grouped the CMTi patients with the CMT2 patients, but still display them separately in tables and figures to consider their somewhat lower NCV compared to the “CMT2” group.
Abbreviations: A, absent response; Amp, amplitude ‐ in mV for motor nerves and in μV for sensory nerves; NA, no information available; NCV, nerve conduction velocity in m/s; nom, not measured; nv, normal value.
2.4. Tissue preparation and quantification
Nerve and muscle biopsies were processed as described previously [26, 27]. Briefly, sections from formalin‐fixed, paraffin‐embedded, and glutaraldehyde‐fixed, epoxy resin‐embedded nerve segments were examined by light microscopy using a Zeiss Axio Scope.A1. Ultrathin resin sections were examined by electron microscopy (EM) using a Philips 400 T or a Hitachi HT7700 transmission electron microscope (TEM). Likewise, autopsy tissue, including brain, spinal cord, dorsal root ganglia, and peripheral nerves was fixed in phosphate‐buffered 4% formaldehyde solution and processed for histology and immunohistochemistry as described previously [28].
Myelinated and unmyelinated axons were counted to determine fiber density and number of unmyelinated axons per nucleated Schwann cell unit. Nucleated Schwann cell unit was defined as Remak bundles with visible nucleus. To distinguish unmyelinated axons from Schwann cell processes by EM the following criteria were used: (i) higher density of microtubules, (ii) diameter of the cell process ranging from 1.0 to 1.6 μm [29], and (iii) less condensed cytoplasm of unmyelinated axons. Schwann cells were identified based on the presence of the basement membrane. Invaginations of collagen fibrils by Schwann cell cytoplasm were counted as collagen pockets [30]. Uncompacted myelin was defined as described [31, 32]. Based on previous reports [33], we defined classic onion bulb formations as concentrically arranged thin Schwann cell processes with occasionally interpositioned empty basement membranes that surround more than half of the axon; those which were mainly composed of paired basement membranes were considered basal lamina onion bulb formations (BLOB). Regenerative clusters were defined by the presence of three or more closely apposed myelinated axons—with or without a single basal lamina surrounding the group of axons [34]. Fiber and axon diameters were determined by measuring the fiber and axon area using ImageJ and calculating the diameter. G‐ratio as the fiber diameter divided by the axon diameter was determined as described [35]. Tomacula were specified as marked focal concentric myelin thickenings without discernible foldings as described [36], whereas focally folded myelin was define as more clear‐cut and less symmetric focal myelin folding. Microangiopathic changes of endoneurial blood vessels: pericyte loss, endothelial hyperplasia, reduplication/thickening of basal laminae, luminal occlusion were classified semi‐quantitatively as described [37, 38].
2.5. Statistics
Dot plots are presented for numerical data. Presence/absence of regenerative clusters, tomacula, focally folded myelin, BLOB, uncompacted myelin, and microangiopathy are displayed in stacked bar graphs (as percentage of patients). One‐way ANOVA tests with Tukey's multiple comparison correction were used for almost all statistical analyses by GraphPad Prism, except comparisons between presence and absence of uncompacted myelin and microangiopathy, Fisher's exact test was used (GraphPad online tool). p‐Values <0.05 were considered statistically significant.
2.6. Data availability
Data not provided in the article because of space limitations may be shared (anonymized) at the request of any qualified investigator for purposes of replicating procedures and results.
3. RESULTS
3.1. Classification, clinical, and genetic features of patients with MPZ‐neuropathies
Clinical and genetic data of the 21 patients included are summarized in Table 1. Results of electrophysiological recordings are shown in Table 2. Two patients (#1 and #2) were classified as CHN based on the congenital onset, severe disease and extremely slow or unobtainable nerve conduction velocity (NCV). The remaining patients were classified as CMT1 (11 cases), CMTi (CMT intermediate; three cases), and CMT2 (five cases) based on the results of electrophysiological recordings using established criteria (motor NCV of the median nerve) [39]. Details on the classification of your cohort are provided in the legend of Table 2.
CHN patient #1 presented as a floppy infant with areflexia and feeding problems. Additional features were dysphagia, scoliosis, and ametropia. Electrophysiological examination at 8 months and 10 years revealed absent motor and sensory action potentials at all extremities examined (Table 2). An insertion of four bases after codon 189 in exon 4 of the MPZ gene was found: c.567_568insGGCC (p.L190GfsX46). To our knowledge, this variant has not been previously reported. It may lead to nonsense‐mediated decay or truncation of the cytoplasmic domain based on a premature termination codon and is expected to be pathogenic (see discussion below). CHN patient #2 showed congenital hypotonia and breathing difficulties. The patient never learnt to walk and was wheelchair bound. Electrophysiological examination at the age of 20 years revealed a very slow NCV of 2.8 m/s of the median nerve (motor) and an absent response of the ulnar (motor), peroneal, tibial, median (sensory), and sural nerves. A frameshift mutation in the MPZ gene, affecting the amino acids from codon 209 has been detected (p.A209EfsX24) [24].
Eleven of the 21 patients (#3–13) were classified as CMT1 including five patients with childhood onset (one at age below one, one at 1 year, one at 2 years, one at 4 years, and one at an undefined age in childhood). Four patients had a reported onset in adulthood at the age of >20, 40, 55, and 63 years, respectively. In two patients, the age at onset was unknown. Symptoms and clinical features are summarized in Table 1. Distal weakness of lower limbs and gait disturbances as well as foot deformities were predominant features at onset. Three of the CMT1 patients showed pupillary abnormalities; one patient had nystagmus, three had scoliosis, and hearing was impaired in one patient (Table 2). Four patients had mutations of the amino acid 98 (two patients with p.R98C and two patients with p.R98H, the latter two with late onset disease at 55 and 63 years of age), one patient showed a p.T124M mutation, three patients with a mutation of amino acid 134 (two patients with p.D134E, one patient with p.D134N). The p.G163R mutation was detected in one patient. Another patient displayed a p.Y181X mutation and one presented with a novel c.678delC variant (p.S226RfsX26) that leads to a frame shift from amino acid 226 on, predicted to result in a longer a longer protein with a C‐terminal sequence unrelated to wild‐type MPZ. All the other observed mutations p.R98C/H, p.Y181X, p.D134, and p.G136R mutations have previously been associated with a CMT1 phenotype [40, 41, 42].
We classified three patients as CMTi based on NCV studies. Age at onset was between 40 and 63 years of age. Two of them showed p.D61G mutations, one a p.T124M mutation. Hearing was reduced in patient #14 and #16 and #16 also had anisocoria. Five patients were classified as CMT2 based on the NCV data. Age at onset ranged from 29 to 58 years with the exception of patient #20 with p.G206X mutation who had early onset hypotonia at 4 months of age and died at the age of 8 years with respiratory insufficiency and pulmonary heart disease, most likely due to concomitant muscle carnitine deficiency, lipid storage myopathy and mitochondrial myopathy (see below). Based on his normal NCV values at 1 year of age, he was classified as CMT2, although he might have developed slow NCV later in childhood similar to other patients with this mutation [23]. The p.D61G, p.Y119C, and p.T124M mutations have already been reported in CMT2 type MPZ‐related neuropathies [20, 43]. Patient #19 with p.T124M mutation in MPZ has been previously reported without morphological analysis of the biopsy [25]. She displayed pupillary abnormalities and hearing impairment. CMT2 patient #21 displayed a c.709G > A (p.A237T) variant. Starting from 58 years of age, she was unsteady on the feet and developed sensory ataxia. To our knowledge, neuropathy associated with this particular variant has not been reported to date. A mutation in the adjacent amino acid c.706A > G (p.K236E) was shown to cause CMT2 [44], but by prediction methods using the web service InMeRF (https://www.med.nagoya‐u.ac.jp/neurogenetics/InMeRF/main.php) [45], p.A237T is classified as “normal,” raising the possibility that additional factors determine manifestation of neuropathy in such patients as discussed below. We also detected a novel c.73_74 delinsA (p.S25TfsX22) variant that may truncate MPZ due to a premature stop or usage of an alternative start codon or lead to nonsense‐mediated decay in CMT2 patient #17. She developed gait difficulties and foot drop in the late third decade of life and had upper limb motor NCV above 40 m/s.
Overall, known genetic variants associated with specific CMT types were in line with previous reports. The predicted consequence of novel genetic variants on the MPZ protein matched the allocation to CHN (one case), CMT1 (one case), and CMT2 (two cases) in most patients and will be discussed further below.
3.2. Morphological characteristics of neuropathies associated with MPZ mutations
We performed histology and electron microscopy on nerve biopsies of control (Figure 1A) and of MPZ‐related neuropathy patients (Figure 1B, Figure 2A–F) and quantified the morphological alterations (Figure 1C–K, Figure 2G–M). We confirmed CMT‐type characteristic alterations previously established in smaller case series [18, 19, 20]. These included a reduced myelinated nerve fiber density in CMT1, but not in CMTi/CMT2 (Figure 1C) and no significant difference in the density of unmyelinated axons between controls and any CMT‐type (Figure 1D) [18]. Compared to previous studies, we extended the morphological investigations to particular features of Schwann cells and unmyelinated axons. The number of axons per nucleated Schwann cell unit (Remak bundles with visible nucleus) was significantly reduced in CMT1 compared to controls (Figure 1A,B,E). Nucleated Schwann cell profiles without axons corresponding to so‐called denervated Schwann cell bands were significantly increased in both the CMT1 and the CMTi/CMT2 group of patients, with the CMT1 patients showing the highest density of such denervated Schwann cell bands (Figure 1B,I). In contrast, this was not a feature in nerves of CHN patients (Figure 1I). Although we did not observe a significant difference in unmyelinated nerve fiber density indicative of a loss of unmyelinated axons, we detected a significantly elevated number of collagen pockets in CMT1 compared to the control group, but not in the other disease groups (Figure 1J). Another marked CMT1‐specific finding was a higher density of Schwann cells in CMT1 compared to CMTi/CMT2 (Figure 1F,H). In CHN, we observed a further increase in Schwann cell density (Figure 1F–H) and a very severe hypomyelination that was more pronounced than in CMT1 and CMT2 (Figure 1K).
FIGURE 1.
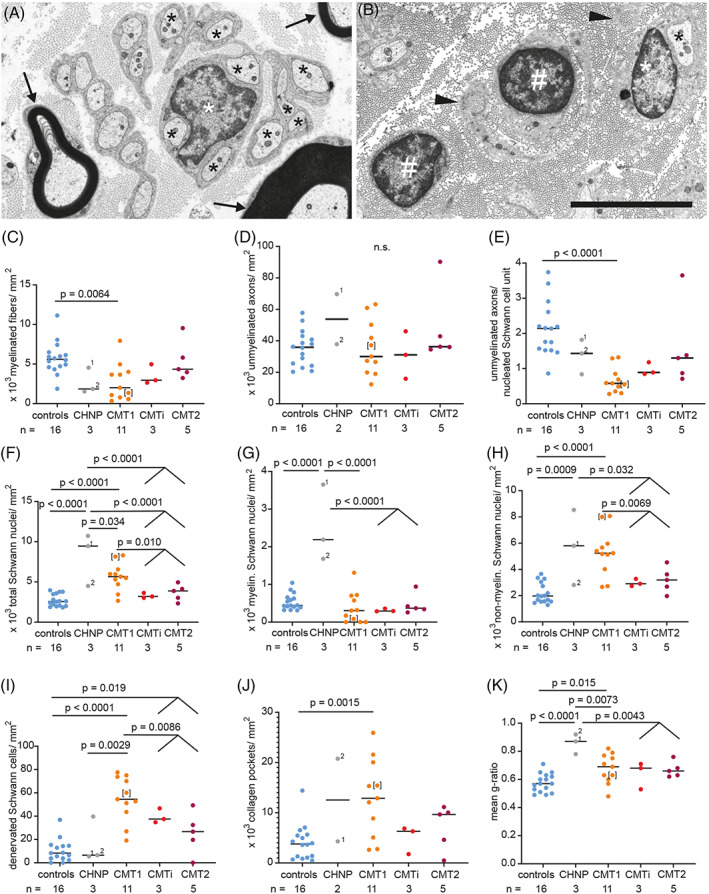
CMT‐type dependent differences in ultrastructural alterations of density of axons and Schwann cells as well as myelin thickness. (A, B) Electron microscopy images of cross sections through the sciatic nerve. Nerve of a control patient (A), showing normally myelinated axons (arrows) and several non‐myelinated axons (black asterisks) in one nucleated Schwann cells unit (white asterisk marking the nucleus). MPZ‐mutation‐induced neuropathy in CMT1‐patient #11 (B), showing two denervated nucleated Schwann cells (white pound key, #), one non‐myelinated axon (black asterisk) in a nucleated Schwann cell unit (white asterisk marking the nucleus) and collagen pockets (black arrowheads). (C–K) Graphs showing quantification of ultrastructural findings, sorted by CMT‐type: Densities of myelinated fibers (C) and unmyelinated axons (D), unmyelinated axons per nucleated Schwann cells units (E), density of all Schwann cell nuclei (F), myelinating Schwann cell nuclei (G), non‐myelinated Schwann cell nuclei (H), density of denervated Schwann cells (i.e., nucleated Schwann cell units without any axon; (I), density of collagen pockets (J), mean g‐ratio per case (K). One‐way ANOVA with Tukey's multiple comparison test was used as statistical test. Groups for analyses were: controls, CHN, CMT1, CMTi combined with CMT2. For CHN case #1, two nerve biopsies were available, taken 6 years apart from each other, at 7 months and 6 years of age, labeled with 1 and 2, respectively. Data points of clinically defined CMT1 case #3 are shown in brackets. Limited image quality did not allow to determine unmyelinated axon density and density of collagen pockets in CHN case #2. Numbers of analyzed biopsies are displayed below the graph.
FIGURE 2.
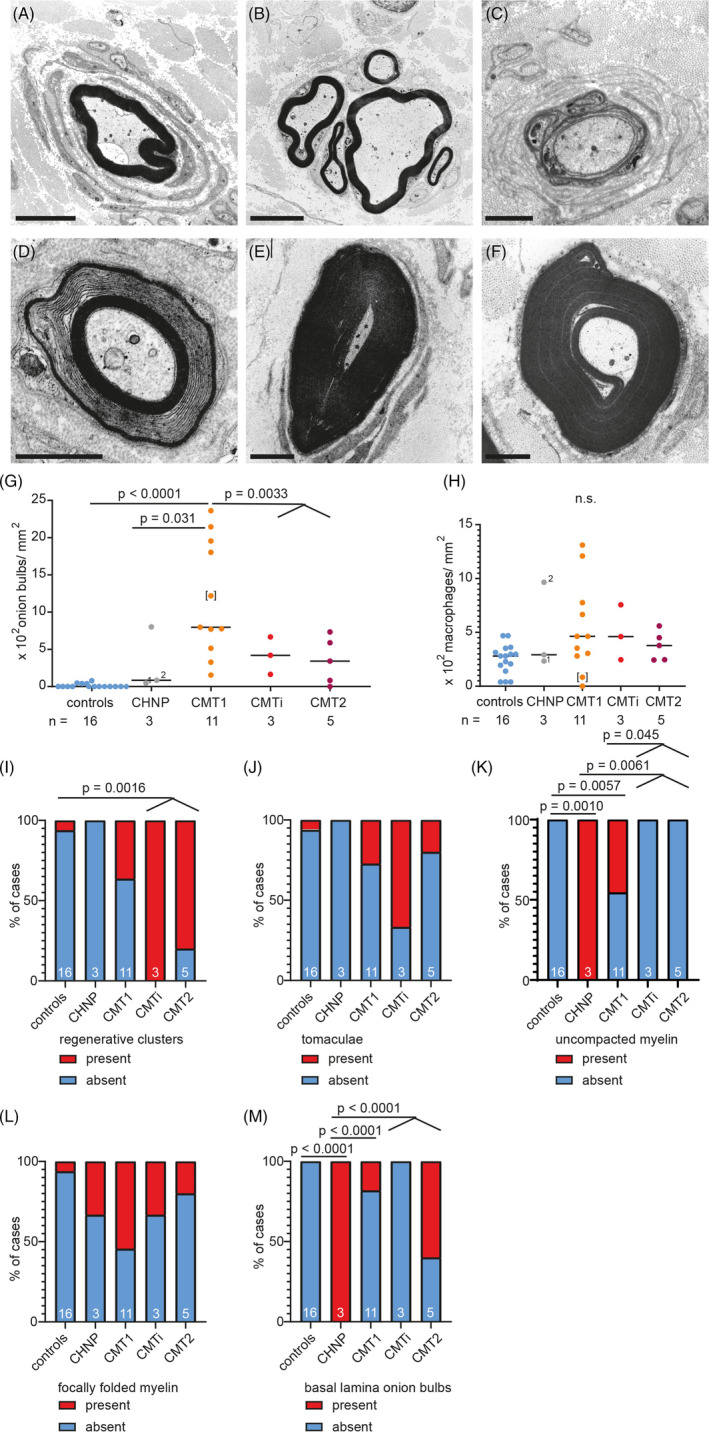
CMT‐type dependent differences in ultrastructural alterations of myelin structure, onion bulb formation and axon regeneration. (A–F) Electron microscopy images of cross sections through peripheral nerves of patients with MPZ‐neuropathy: Onion bulb formation around a large caliber axon in CMT1 patient #4 with lamellae containing Schwann cell cytoplasm in addition to basement membranes (A). Regenerative cluster in CMTi patient #16 (B). Basal lamina onion bulb (BLOB) formation around a severely hypomyelinated larger caliber axon in CHN case #1. In contrast to regular onion bulbs, lamellae of BLOB contain Schwann cell basal laminae without cytoplasm (C). Uncompacted myelin lamellae in CMT1 patient #3. Uncompacted myelin is caused by separation of myelin lamellae at the major dense line [32]. In the four MPZ patients reported here, the lack of compaction could affect either the central or peripheral parts or the entirety of the myelin sheath. While it was often the only anomaly observed at this specific location of a fiber, it sometimes occurred in the context of excessive myelin folding (D). Tomaculum (E) and focally folded myelin (F) in CMT1 patient #9. Scale bars in A and B are 5 μm, and 2 μm in C–F. (G–M) Graphs showing quantification of ultrastructural findings, sorted by CMT‐type: Density of onion bulbs (G) and macrophages (H). Stacked bar diagrams displaying presence or absence of regenerative clusters (I), tomacula (J), uncompacted myelin (K), focally folded myelin (L), and basal lamina onion bulbs (M). One‐way ANOVA with Tukey's multiple comparison test was used as statistical test, also for numerical data on the density of regenerative clusters, tomacula, focally folded myelin BLOB, shown in graph I, J and L, M. For K and M, Fisher exact test was used. Groups for analyses were: controls, CHN, CMT1, CMTi combined with CMT2. For CHN case #1, two nerve biopsies were available, taken 6 years apart from each other, at 1 and 6 years of age, labeled with 1 and 2, respectively. Data points of clinically defined CMT1 case #3 are shown in brackets. Numbers of analyzed biopsies are displayed below the graph or in the bars.
The higher frequency of onion bulbs in CMT1 compared to controls and CMTi/CMT2 patients (Figure 2A,G) was in line with previous observations [18]; we also confirmed that axonal regeneration is frequent in the combined group of CMTi and CMT2 (Figure 2B,I) [18, 19, 20]. Except for the higher density of denervated Schwann cells, axonal regeneration was in fact the only significant ultrastructural difference in CMTi/CMT2 patients compared to controls. In line with previous observations, we observed that basal lamina onion bulb formations are especially frequent in CHN (Figure 2C,M). In addition, classical onion bulb formations composed of redundant Schwann cell processes and their basal laminae were abundant in one of the two CHN patients (Figure 2G).
We frequently observed uncompacted myelin lamellae in both, CHN (2 of 2) and in 5 out of 11 CMT1 patients, but not in CMTi/CMT2 (Figure 2D,K). Uncompacted myelin lamellae have been described previously in nerve biopsies of patients harboring the p.R98C and p.R98H mutations [31, 46, 47]. We observed uncompacted myelin lamellae in two CMT1 patients with the p.R98C mutation, but not in those with p.R98H mutation. In addition, we observed myelin uncompaction in one of the CMT1 patients with a frame shifting mutation (#13), one with p.Y181X (#12), and in one of the two patients with the p.D134E mutation (#9). Focal thickenings of the myelin sheath (tomacula and focally folded myelin; Figure 2E,F) were significantly increased in the group of MPZ mutation patients as a whole (11 of 21 patients in MPZ neuropathy versus 2 of 16 in controls, p = 0.017), but in contrast to previous observations [18], there was no significant difference between CMT types. The low number of CMTi and CMT2 cases limits our study and may for example obscure the clear delineation of certain morphological alteration between groups, for example, density of denervated Schwann cells, for which there is a non‐significant trend to a lower value in CMT2 compared to CMTi.
We did not observe lymphocytic infiltrates or other overt signs of inflammation in any case. Endoneurial macrophages were infrequent in all patients. No amyloid deposits were found. Eight of 19 CMT patients (42%) showed moderate to marked microangiopathic changes of endoneurial blood vessels (as described in Section 2 and in [37, 38]) ‐ significantly more than in the controls (1 of 16; 6.3%, p = 0.022). None of the two patients with CHN showed signs of microangiopathy. In the CMT1 group, 4 of 11 patients (36.4%) and in the CMTi and CMT2 group 4 of 8 patients (50%) had moderate to severe microangiopathic changes. Hence, microangiopathy coincided with the manifestation of MPZ neuropathy, in particular in the late‐onset CMTi/CMT2 group.
3.3. Involvement of spinal nerve roots and meninges in a patient with MPZ mutation
Patient #4 (Figure 3) with early onset CMT1 died at the age of 49 years due to global respiratory insufficiency, infection‐induced exacerbation of bronchial asthma, and thoracic deformities due to severe scoliosis (Figure 3A). During his childhood, a combination of meningitis and poliomyelitis had been discussed, but at the age of 41 years, a severe sensorimotor neuropathy of unknown origin was diagnosed. Sural nerve biopsy revealed a severe demyelinating process with prominent onion bulb formation (Figure 3B). Postmortem molecular genetic investigation revealed the known pathogenic heterozygous mutation in exon 3 c.292C > T (p.R98C) of the MPZ gene. Neuropathological autopsy showed an advanced degree of demyelinating disease with numerous onion bulb formations in dorsal and ventral roots of the spinal cord and in peripheral nerves (Figure 3C). There was marked concentric thickening of perineurial cell layers of the nerve fascicles and Renaut‐body formation in the center of a median nerve fascicle (Figure 3D,E). The cauda equina was partially ossified. In addition, thickening of the meninges of the spinal cord associated with neuromatous sprouting of small nerve fascicles in the leptomeninges, resembling findings reported for multiple endocrine neoplasia, type 2b MEN2b [48] and surplus of perineurial septae in spinal nerve roots were observed (Figure 3F–J).
FIGURE 3.
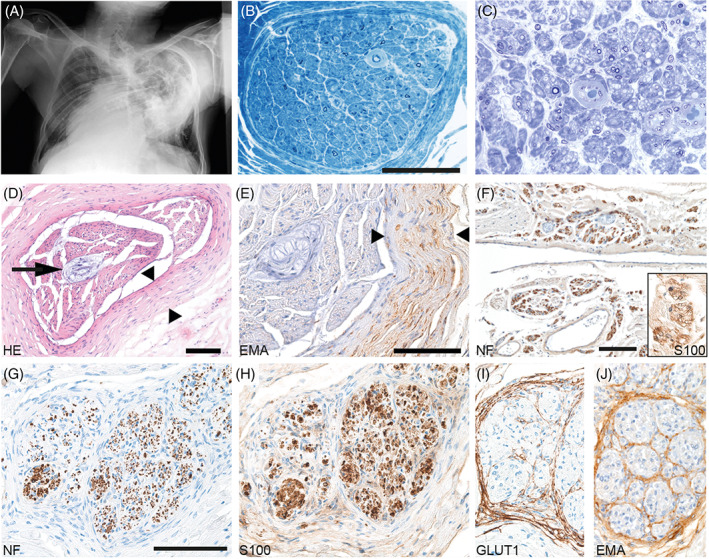
Biopsy and autopsy findings in a CMT1 patient with p.R98C mutation. (A) X‐ray (a.p.) of the chest: severe scoliosis of the thoracic vertebral column. Marked chest deformation. (B) Sural nerve biopsy in 1998 revealed severe demyelinating neuropathy and angiopathy of endoneurial capillaries; scale bar = 100 μm. (Semithin section: toluidine blue staining) (C) Sciatic nerve, taken in 2006 during autopsy, showing advanced stage of severe neuropathy with only few remaining thinly myelinated axons (semithin section: toluidine blue staining). (D, E) Post mortem investigation of median nerve: HE (D) and EMA immunohistochemistry (E) show marked concentric thickening of the perineurium of a nerve fascicle (between arrowheads). Renaut body in the center of the nerve fascicle (arrows). Scale bars = 100 μm. (F) Leptomeningeal neuromatous sprouting observed in autopsy material: Neurofilament‐positive axons (F) surrounded by S100‐positive Schwann cells (insert in F; 2.5‐fold zoomed) forming minifascicles. Nerve root showing surplus perineurial septae (G–J): neurofilament (G), S100 (H), GLUT1 (I), and EMA (J). Scale bars = 100 μm.
3.4. Involvement of skeletal muscle in patients with MPZ mutations
As a direct target organ, skeletal muscle is known to be affected in motor neuron diseases and peripheral neuropathies. Typically, skeletal muscle displays signs of grouped neurogenic atrophy with flattened and angular fibers and fiber type grouping. Further changes include altered autophagy and proteostasis, myofibrillar disintegration and focally diminished mitochondrial oxidation activities, resulting in target and targetoid fiber phenotypes [49, 50, 51, 52]. In six patients (#4, 5, 6, 13, 15, 20) muscle biopsies from combined nerve‐muscle biopsies were available (Figure 4). One patient showed typical signs of neurogenic atrophy (#4, Figure 4A) and there was mild, probably neurogenic Z‐band streaming in another case (#13, Figure 4B). CMTi patient #15 displayed massive accumulation of autophagic material (Figure 4C) beyond the levels normally encountered in in neurogenic muscular atrophy. This might be due to the denervation exaggerated by an additional genetic predisposition independent from the MPZ mutation. Interestingly, four of the patients who underwent muscle biopsy, two with CMT1 (#5 and 6, Figure 4D,E), one with CMTi (#15, Figure 4F) and one with CMT2 (#20, Figure 4G), showed marked structural mitochondrial alterations in muscle fibers, including paracrystalline inclusions and concentric layering of cristae membranes, both of which are typical signs of mitochondrial myopathies [53]. While patient #5 and #15 showed signs of neurogenic atrophy, patient #20 showed some not clear neurogenically atrophic fibers. Instead, patient #20 displayed some necrotic fibers, several ragged red fibers, carnitine deficiency, and massive accumulation of lipid droplets (Figure 4H). This led to the suspicion of a concomitant lipid storage myopathy in this case. However, respiratory chain activities were normal in this patient. In contrast to alterations in the skeletal muscle, none of these patients displayed mitochondrial alterations in the nerve fibers.
FIGURE 4.
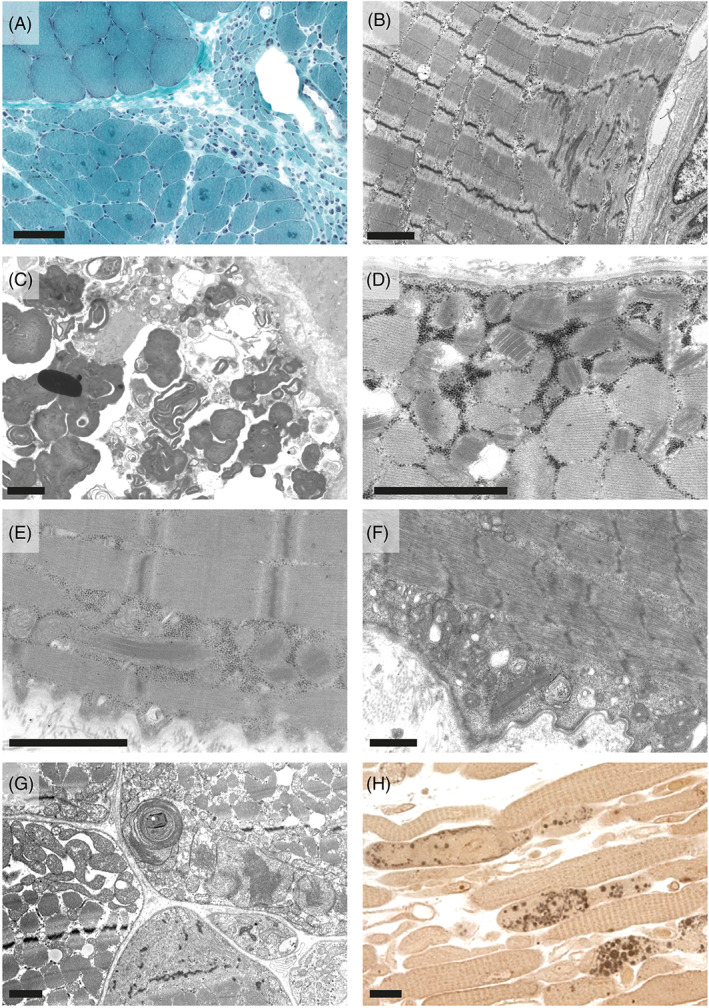
Skeletal muscle pathology in patients with MPZ neuropathies. Neurogenic atrophy with groups of atrophic fibers, angular atrophic fibers, and targetoid fibers in CMT1 patient #4 in trichrome stained cryo cross sections (A) and mild Z‐band streaming in CMT1 patient #13 (B). CMTi patient #15 showed massive accumulation of altered autophagic material. This material showed the predominantly membranous, myelin‐like morphology and was surrounded by partially membrane‐bound vacuolar structures together with degraded organelles including mitochondria, which is typical for autophagic vacuoles (C). Paracrystalline inclusions, so called “parking lots” in mitochondria of CMT1 patients #6 (D) and #5 (E) seen in electron microscopy (EM) images. CMTi patient #15 also showed paracrystalline inclusions in mitochondria (F). EM image of a cross section of skeletal muscle of CMT patient (#20) showing accumulation of structurally altered mitochondria with concentric christae formation (G). Paraphenylendiamine‐stained semithin longitudinal section of skeletal muscle of the same patient #20 showing accumulation of numerous large diameter lipid droplets within muscle fibers (H). Scale bars are: 100 μm in A, 2 μm in B–G, and 20 μm in H.
4. DISCUSSION
4.1. Schwann cell changes in MPZ neuropathies
To date, diagnosis of inherited peripheral neuropathies is usually achieved by molecular genetic methods without the need for nerve biopsy. Thus, only few mostly archival nerve biopsies from MPZ neuropathy patients are available. However, correlation of molecular genetic findings with the morphological phenotype provides useful information about the consequences of the respective mutations and about the normal and disturbed functions of MPZ. Therefore, we collected a series of 21 patients with MPZ mutation in which nerve biopsies were available at two central European centers for systematic analysis. To our knowledge, this is the largest systematic investigation of the morphological alterations in different forms of inherited peripheral neuropathies due to MPZ mutations. As expected, we confirmed previous observations of de‐ and re‐myelination and Schwann cell onion bulb formation combined with decreased myelinated nerve fiber density in CMT1, but not in CMT2. Axonal regeneration was significantly increased in the group of CMTi/CMT2 patients in line with previous reports [18, 19, 20]. Even though density of unmyelinated axons did not differ between CMT groups in line with previous reports [18], we observed a significant increase in collagen pockets and a decreased number of unmyelinated axons per nucleated Schwann cell unit specifically in CMT1 patients, suggesting that unmyelinated fibers are also affected in CMT1. This result is in line with the clinical finding that pupillary reaction, which is controlled by unmyelinated postganglionic autonomic nerve fibers [54], can be affected in patients with MPZ neuropathies. For the most part, such abnormalities have been reported in CMT2 patients with p.T124M mutation [25], except for one patient with demyelinating neuropathy due to p.I112T mutation [55]. In our cohort, five of 21 patients showed pupillary abnormalities: three patients with CMT1 due to p.D134N, p.R98H, and p.G163R mutation and two patients with p.T124M (one CMTi and one with CMT2). Hence, our data support the conclusion that pupillary abnormality is rather frequent in MPZ‐CMT1 and demonstrate a clinical involvement of unmyelinated fibers in these patients. We also observed significantly more denervated Schwann cells in all CMT groups compared to controls, especially in CMT1. Together, these data suggest a myelination‐independent function of MPZ in non‐myelinating Schwann cells. This is in accordance with the MPZ expression demonstrated in multipotent glial precursor cells in the developing PNS before the onset of myelination [21], the activation of the MPZ promotor in non‐myelinating Schwann cells [56] and the reported broad alteration in Schwann cell gene expression in mice lacking MPZ [4].
We observed an increased density in Schwann cell nuclei in CMT1 and CHN patients. In CMT1, this increase was mostly due to an increase of non‐myelinating Schwann cells, while in CHN patients, the density of myelinating Schwann cells was also increased. Since we did not observe an obvious shrinkage of the examined nerves, this was most likely due to an actual increase in the number of Schwann cells, possibly by Schwann cell proliferation. An increase in the number of Schwann cells along with a stable overall number of unmyelinated axons also explains the smaller number of unmyelinated axons per nucleated Schwann cell unit, the increase of denervated Schwann cells and possibly even the increase of collagen pockets as proliferated Schwann cells may start to enclose collagen bundles instead of axons. This combination of findings (higher density of Schwann cells, fewer unmyelinated axons per Schwann cell unit) is reminiscent to CMT1A due to PMP22 duplication, a condition in which Schwann cell proliferation is well established [57], suggesting that Schwann cells also proliferate in MPZ neuropathies. Schwann cells may be stimulated to proliferate in response to the degeneration of myelinated axons, which has been reported to produce signals that are mitogenic for non‐myelinating Schwann cells [58]. Non‐myelinating Schwann cells have not been implicated in the pathogenesis of MPZ‐related peripheral neuropathies so far, because mature non‐myelinating Schwann cells were not found to express MPZ. Nevertheless, non‐myelinating Schwann cells might be crucially involved as they might transdifferentiate to an immature state and hyperproliferate. Hyperproliferation could be compensatory, or alternatively, Schwann cells with MPZ mutation might fail to differentiate fully and remain in an immature state prone to hyperproliferation. Insufficient maturation and predisposition to hyperproliferation might contribute to the development of neuropathy. In fact, Schwann cell proliferation has been suggested as the cause of axonal degeneration and neuropathy in neurofibromatosis‐2 [59].
Excessive growth of peripheral nerve fibers/Schwann cells was also observed in the autopsy case (# 4), that is, we observed thickened spinal meninges containing neuromatous sprouting of small nerve fascicles resembling the spinal neuroma described in patients with multiple endocrine neoplasia, type 2b MEN2b [48]. Neuromatous sprouting has not been described in the few autopsies of patients with MPZ ‐neuropathies that have previously been reported [60, 61, 62, 63]. Although we have never observed such a phenomenon in any autopsy so far, we would like to emphasize that this is at this point solely an observation in a single case of MPZ neuropathy. It might be associated with the massive scoliosis in this patient. Scoliosis is often observed in CMT and particularly frequent in patients associated with MPZ mutation [64]. Scoliosis may have caused compression trauma of nerve roots [65], a possible cause of neuromatous nerve fiber outgrowth [66]. The question remains unsolved whether neuroma formation was directly linked to the MPZ mutation and associated hyperproliferation of Schwann cells or whether it was a consequence of nerve trauma due to the severe scoliosis; the two pathomechanisms might actually have combined to produce this peculiar phenotype.
4.2. Microangiopathy and mitochondrial myopathy in patients with MPZ mutation
Microangiopathy of endo‐ and epineurial blood vessels was a prominent feature of several patients, particularly in the CMTi/CMT2 group. The association with the predominantly late‐onset forms, CMTi and CMT2 could theoretically be explained by the overall higher incidence of microangiopathy in aged people [67, 68]. However, the incidence in CMT patients was higher than in our age‐matched controls, in line with previous findings of a study on vascular changes in diabetic neuropathy, in which nine non‐genetically characterized CMT1 patients served as disease controls and also displayed thickening of the pericapillary basal lamina and endothelial cell hyperplasia when compared to nerves of organ donors [69]. Hence CMT, including MPZ neuropathy may trigger manifestation of concomitant vascular disease, although repeated nerve fiber degeneration and regeneration alone does not induced basement membrane reduplication of endoneurial microvessels [70]. Alternatively, vascular disease may favor the manifestation of MPZ‐related neuropathy, for example, by exposing altered myelin to additional stress and/or because a common factor may favor manifestation of both diseases. For example, disturbed lipid metabolism is a pathogenic factor in microangiopathy and has also been suggested to determine clinical variability in CMT1A [71].
Surprisingly, considerable mitochondrial abnormalities were found in four of six MPZ neuropathy patients for which skeletal muscle biopsies were available. There was no CMT‐type‐specific association. At this point, this remains an observation of unknown causal relation to the MPZ neuropathy; mitochondrial abnormalities are to our knowledge not an established feature of MPZ neuropathy thus far. Nevertheless, mitochondrial alterations were clearly more frequent than generally observed in muscle biopsies and were in line with altered size, function, and distribution of axonal mitochondria in a mouse model of MPZ neuropathy [72]. Furthermore, denervation causes extensive muscle fiber changes including myofibrillar disintegration (target phenomena) and remodeling of the sarcoplasmic reticulum [49], which should also affect muscle fiber mitochondria. This assumption is supported by experimental evidence indicating that denervation can induce mitochondrial dysfunction and mitophagy [73]. In patient #20 carrying the p.G206X MPZ mutation, mitochondrial alterations most likely reflect a concomitant disease due to the additional muscle carnitine deficiency and excessive lipid storage. Thus, chronic denervation and reinnervation in the context of hereditary neuropathy might be a confounding factor in individuals predisposed to mitochondrial abnormalities. In fact, hereditary neuropathies can be accompanied by mitochondrial alterations, especially in the context of mitofusin 2 (MFN2) mutations (CMT2A/HMSNVI) [74, 75, 76].
4.3. Genotype–phenotype correlation
The CMT phenotype and with this the CMT subtype depends at least in part on location and type of the MPZ mutation. In general, mutations associated with CHN and CMT1 are regarded as (toxic) gain of function or dominant‐negative, while haploinsufficy/loss of function mutations are typical for CMT2 [77]. Experiments showed that certain CHN‐ and CMT1‐associated MPZ mutations either affect myelin compaction or lead to retention of mutated protein in the ER, inducing the unfolded protein response (UPR); both mechanisms contribute to the severe demyelination neuropathy in mice [41, 78, 79, 80, 81]. Other MPZ mutations associated with severe, early‐onset neuropathies cause aberrant MPZ trafficking to non‐myelin plasma membranes or even affect early developmental processes before myelination such as radial sorting of axons by Schwann cells [82] or hyperglycosylation and retention in the Golgi [83]. In most CMT1 patients in the present study, we identified MPZ mutations known to be associated with CMT1, including those in the extracellular domain, p.R98C/H and p.D134E/N; p.G163R in the transmembrane domain as well as p.Y181X in the cytoplasmic domain [40, 41, 42, 84]. Interestingly, although uncompacted myelin and focally folded myelin had been reported to be mutually exclusive nerve pathologies in MPZ neuropathies [85], patient #9 with p.D134E mutation displayed both pathologies simultaneously. One CMT1 patient (#13) displayed a novel c.678delC variant (p.S226RfsX26) that is predicted to cause a frame shift from amino acid 226 on and to result in a protein that is two amino acids longer than wild‐type MPZ. The patient presented with an early onset during early childhood, similar to a patient with the c.661_662dupGC mutation of established pathogenicity that is predicted to cause a similar frameshift, just a few amino acids closer to the N‐terminus (p.Ala221fs) and to lead to an extension of the protein by three amino acids [86]. The latter mutation was predicted to result in a completely different conformation of the intracellular part and to consequently interfere with the formation of the myelin major dense line [86], which likely occurs in c.678delC (p.S226RfsX26) as well. Frame shifting mutations in MPZ were also observed in our two CHN patients. Both lead to MPZ truncation; patient #1 displayed a novel four bases insertion after codon 189 in exon 4 of the MPZ gene (c.567_568insGGCC, p.L190GfsX46) and patient #2 had a five base pair deletion after codon 208 (c.626_630, p.A209EfsX24) [24]. The severe disease course induced by the c.567_568insGGCC, p.L190GfsX46 variant is in line with the early onset neuropathy and dominant negative effect described in a patient with a single nucleotide deletion (c.570delG) that is causing a frame shifting mutation starting at just one amino acid closer to the C‐terminus [87]. Basal lamina onion bulb (BLOB) formation and uncompacted myelin were frequent features in both CHN patients. However, in patient #1 with the p.L190GfsX46 variant virtually no normally myelinated axons were present, suggesting that myelin development was affected at a very early stage. Patient #2 with p.A209EfsX24 mutation displayed more mature onion bulbs. BLOB formation has been suggested to be caused by repeated loss and regeneration of non‐myelinating Schwann cells, while “classical” onion bulbs are thought to result from de‐ and remyelination [88]. These differences might be due to the presence of the protein kinase C (PKC) target motif between amino acids 193 and 206 of MPZ that is essential for its adhesive function [89] and preserved in patient #2, but not in patient #1.
While CMT1‐associated mutations strongly perturbed intercellular adhesions or were retained in the cytoplasm, CMT2‐associated mutations only moderately reduced intercellular adhesions in cultured cells [90] or mainly affect axo‐glial interactions [60, 72, 90, 91], including axo‐glial interactions to stabilize nodes and paranodes [72, 92]. In line with a loss of function mutation, CMT2 patient #17 with onset in the 3rd decade of life had a novel c.73_74 delinsA (p.S25TfsX22) variant. This could result in a shortened protein product due to truncation or usage of an alternative start codon. Alternatively, nonsense‐mediated decay could lead to very low amounts or absent protein. The p.A237T variant observed in CMT2 patient #21 with neuropathy onset at 40 years of age has not been associated with MPZ neuropathy before. In fact, this sequence variant has been classified as “apathogenic” by prediction tools (https://www.med.nagoya-u.ac.jp/neurogenetics/InMeRF/main.php) [45]. However, except for a moderate microangiopathy that was not sufficient to explain the extent of nerve fiber degeneration, no other potential cause of neuropathy was found in this patient. Therefore, this MPZ sequence variant might be considered to be mildly pathogenic.
The other CMTi/CMT2 mutations in our cohort are known from the literature to be associated with CMT2, including p.D61G, p.Y119C, and p.T124M [20, 25, 43]. These defects are located in the extracellular domain. This domain, in particular the region around D61, is important for homophilic adhesion [78]. MPZ p.T124M has been shown to be transported normally to the membrane; there, it leads to moderate impairment of intercellular adhesion, but does not delay myelination. Intercellular adhesion requires glycosylation of amino acid 122, which is unglycosylated in p.T124M [90]. In line with this, our three patients with p.T124M had a late disease onset (36, 40, and 40 years of age) and we did not observe myelin uncompaction in these patients. Nevertheless, based on NCV studies, of the three patients with p.T124M mutation, one was classified as CMT1, one as CMTi and one as CMT2 in line with previous observations that mutations at position 124 can cause both demyelinating and axonal neuropathy [20, 25]. Reduced hearing or deafness were observed in four patients, all with late onset—three with p.T124M mutation and one with p.D61G mutation—overall in four of 21 patients = 19%). This was independent of the CMT type (1 × CMT1, 2 × CMTi, and 1 × CMT2), but related to the specific mutations/late onset cases in line with previous observations of hearing loss in late‐onset MPZ neuropathies [18, 93, 94]. The frequency of hearing loss in our study was very similar to what has been observed in the USA (20.4%) [43] and much higher than in the East Asian population [95, 96], even when we directly compared the frequency of hearing loss in patients with p.T124M mutation (only one of four Asian patients [95] but in all three patients described here). This suggests that ethnical differences in genetics have an impact on these genotype–phenotype correlations.
4.4. Value of archival material and future molecular assessment
As hereditary neuropathies are nowadays usually diagnosed genetically, morphological analysis of MPZ‐mutation pathology in human patients is restricted to archival diagnostic material. Systematic assessment of such cases allowed us to uncover involvement of unmyelinated fibers, especially their Schwann cells, as well as concomitant mitochondrial abnormalities in skeletal muscle and neuromatous sprouting in spinal meninges. These findings highlight the relevance of such archival material. It should be stored indefinitely to enable future correlative pathoanatomical studies. Future molecular workup of archival biopsies may include whole exome sequencing or whole genome sequencing of hereditary neuropathies with yet unresolved genetic cause. Moreover, in hereditary neuropathies with a known genetic cause such as MPZ mutation these approaches would allow to detect additional genetic alterations that, for example, predispose to mitochondrial alterations, microangiopathy or determine age at onset/CMT type, for example, in families with MPZ pT124M mutation with variable onset and CMT type.
AUTHORS CONTRIBUTIONS
Juliane Bremer, Joachim Weis, Vincent Timmerman and Jonathan Baets designed the study. Juliane Bremer, Axel Meinhardt, Istvan Katona, Jan Senderek, Elke K. Kämmerer‐Gassler, Andreas Roos, Andreas Ferbert, J. Michael Schröder, Stefan Nikolin, Kay Nolte, Bernd Sellhaus, Klimentina Popzhelyazkova, Ulrike Schara‐Schmidt, Eva Neuen‐Jacob, Chantal Ceuterick de Groote, Peter de Jonghe, Vincent Timmerman, Jonathan Baets and Joachim Weis acquired the data. All the authors analyzed and interpreted at least part of the data. Juliane Bremer, Axel Meinhardt, Istvan Katona, Andreas Roos, Andreas Ferbert, Frank Tacke, Ulrike Schara‐Schmidt, Vincent Timmerman, Jonathan Baets and Joachim Weis wrote and/or revised the manuscript.
Supporting information
TABLE S1. Control nerves used in this study. Age at biopsy, gender (f = female, m = male) and clinical/pathological information if available.
ACKNOWLEDGEMENT
Open Access funding enabled and organized by Projekt DEAL.
Bremer J, Meinhardt A, Katona I, Senderek J, Kämmerer‐Gassler EK, Roos A, et al. Myelin protein zero mutation‐related hereditary neuropathies: Neuropathological insight from a new nerve biopsy cohort. Brain Pathology. 2024;34(1):e13200. 10.1111/bpa.13200
Contributor Information
Juliane Bremer, Email: jbremer@ukaachen.de.
Joachim Weis, Email: jweis@ukaachen.de.
DATA AVAILABILITY STATEMENT
All numerical data generated or analyzed during this study are included in this published article. Additional information is available from the corresponding author on reasonable request.
REFERENCES
- 1. Patzig J, Jahn O, Tenzer S, Wichert SP, De Monasterio‐Schrader P, Rosfa S, et al. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 2011;31(45):16369–16386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Urso D, Brophy PJ, Staugaitis SM, Stewart Gillespie C, Frey AB, Stempak JG, et al. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron. 1990;4(3):449–460. [DOI] [PubMed] [Google Scholar]
- 3. Shy ME, Jani A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain. 2004;127(Pt 2):371–384. [DOI] [PubMed] [Google Scholar]
- 4. Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71(4):565–576. [DOI] [PubMed] [Google Scholar]
- 5. Martini R, Mohajeri MH, Kasper S, Giese KP, Schachner M. Mice doubly deficient in the genes for P0 and myelin basic protein show that both proteins contribute to the formation of the major dense line in peripheral nerve myelin. J Neurosci. 1995;15(6):4488–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M. Protein zero (P0)‐deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat Genet. 1995;11(3):281–286. [DOI] [PubMed] [Google Scholar]
- 7. Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol. 1968;18(6):603–618. [DOI] [PubMed] [Google Scholar]
- 8. Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol. 1968;18(6):619–625. [DOI] [PubMed] [Google Scholar]
- 9. Lupski JR, de Oca‐Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot‐Marie‐tooth disease type 1A. Cell. 1991;66(2):219–232. [DOI] [PubMed] [Google Scholar]
- 10. Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogenduk JE, Baas F, et al. Duplication in chromosome 17p11.2 in Charcot‐Marie‐tooth neuropathy type 1a (CMT 1a). The HMSN collaborative research Group. Neuromuscul Disord. 1991;1(2):93–97. [DOI] [PubMed] [Google Scholar]
- 11. Patel PI, Roa BB, Welcher AA, Schoener‐Scott R, Trask BJ, Pentao L, et al. The gene for the peripheral myelin protein PMP‐22 is a candidate for Charcot‐Marie‐tooth disease type 1A. Nat Genet. 1992;1(3):159–165. [DOI] [PubMed] [Google Scholar]
- 12. Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, et al. Charcot‐Marie‐tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012;83(7):706–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot‐Marie‐tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69(1):22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: results of an international survey. Neurology. 2018;90(10):e870–e876. [DOI] [PubMed] [Google Scholar]
- 15. Callegari I, Gemelli C, Geroldi A, Veneri F, Mandich P, D'Antonio M, et al. Mutation update for myelin protein zero‐related neuropathies and the increasing role of variants causing a late‐onset phenotype. J Neurol. 2019;266(11):2629–2645. [DOI] [PubMed] [Google Scholar]
- 16. Bouche P, Gherardi R, Cathala HP, Lhermitte F, Castaigne P. Peroneal muscular atrophy. Part 1. Clinical and electrophysiological study. J Neurol Sci. 1983;61(3):389–399. [DOI] [PubMed] [Google Scholar]
- 17. Nicholson G, Myers S. Intermediate forms of Charcot‐Marie‐tooth neuropathy: a review. Neuromolecular Med. 2006;8(1–2):123–130. [DOI] [PubMed] [Google Scholar]
- 18. Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, et al. Demyelinating and axonal features of Charcot‐Marie‐tooth disease with mutations of myelin‐related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. 2003;126(Pt 1):134–151. [DOI] [PubMed] [Google Scholar]
- 19. Fabrizi GM, Tamburin S, Cavallaro T, Cabrini I, Ferrarini M, Taioli F, et al. The spectrum of Charcot‐Marie‐tooth disease due to myelin protein zero: an electrodiagnostic, nerve ultrasound and histological study. Clin Neurophysiol. 2018;129(1):21–32. [DOI] [PubMed] [Google Scholar]
- 20. Senderek J, Hermanns B, Lehmann U, Bergmann C, Marx G, Kabus C, et al. Charcot‐Marie‐tooth neuropathy type 2 and P0 point mutations: two novel amino acid substitutions (Asp61Gly; Tyr119Cys) and a possible "hotspot" on Thr124Met. Brain Pathol. 2000;10(2):235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagedorn L, Suter U, Sommer L. P0 and PMP22 mark a multipotent neural crest‐derived cell type that displays community effects in response to TGF‐beta family factors. Development. 1999;126(17):3781–3794. [DOI] [PubMed] [Google Scholar]
- 22. Lehmann HC, Wunderlich G, Fink GR, Sommer C. Diagnosis of peripheral neuropathy. Neurol Res Pract. 2020;2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Senderek J, Ramaekers VT, Zerres K, Rudnik‐Schoneborn S, Schroder JM, Bergmann C. Phenotypic variation of a novel nonsense mutation in the P0 intracellular domain. J Neurol Sci. 2001;192(1–2):49–51. [DOI] [PubMed] [Google Scholar]
- 24. Baets J, Deconinck T, De Vriendt E, et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain. 2011;134(Pt 9):2664–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Löfgren A, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot‐Marie‐tooth phenotype. Brain. 1999;122(Pt 2):281–290. [DOI] [PubMed] [Google Scholar]
- 26. Schroder JM, Kramer KG, Hopf HC. Granular nuclear inclusion body disease: fine structure of tibial muscle and sural nerve. Muscle Nerve. 1985;8(1):52–59. [DOI] [PubMed] [Google Scholar]
- 27. Nolte KW, Janecke AR, Vorgerd M, Weis J, Schroder JM. Congenital type IV glycogenosis: the spectrum of pleomorphic polyglucosan bodies in muscle, nerve, and spinal cord with two novel mutations in the GBE1 gene. Acta Neuropathol. 2008;116(5):491–506. [DOI] [PubMed] [Google Scholar]
- 28. Filezac de L'Etang A, Maharjan N, Cordeiro Brana M, Ruegsegger C, Rehmann R, Goswami A, et al. Marinesco‐Sjogren syndrome protein SIL1 regulates motor neuron subtype‐selective ER stress in ALS. Nat Neurosci. 2015;18(2):227–238. [DOI] [PubMed] [Google Scholar]
- 29. Ochoa J. Recognition of unmyelinated fiber disease: morphologic criteria. Muscle Nerve. 1978;1(5):375–387. [DOI] [PubMed] [Google Scholar]
- 30. Gamble HJ. Comparative electron‐microscopic observations on the connective tissues of a peripheral nerve and a spinal nerve root in the rat. J Anat. 1964;98:17–26. [PMC free article] [PubMed] [Google Scholar]
- 31. Vital C, Vital A, Bouillot S, Favereaux A, Lagueny A, Ferrer X, et al. Uncompacted myelin lamellae in peripheral nerve biopsy. Ultrastruct Pathol. 2003;27(1):1–5. [DOI] [PubMed] [Google Scholar]
- 32. Bilbao JM, Schmidt RE. Biopsy diagnosis of peripheral neuropathy. 2nd ed. Cham: Springer; 2015. [Google Scholar]
- 33. Gabreels‐Festen AA, Gabreels FJ, Jennekens FG, Janssen‐van Kempen TW. The status of HMSN type III. Neuromuscul Disord. 1994;4(1):63–69. [DOI] [PubMed] [Google Scholar]
- 34. Midroni G, Bilbao JM. Amyloid neuropathy. Biopsy diagnosis of peripheral neuropathy. Boston: Butterworth‐Heinemann; 1995. [Google Scholar]
- 35. Rushton WA. A theory of the effects of fibre size in medullated nerve. J Physiol. 1951;115(1):101–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Madrid R, Bradley WG. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): studies on the formation of the abnormal myelin sheath. J Neurol Sci. 1975;25(4):415–448. [Google Scholar]
- 37. Yasuda H, Dyck PJ. Abnormalities of endoneurial microvessels and sural nerve pathology in diabetic neuropathy. Neurology. 1987;37(1):20–28. [DOI] [PubMed] [Google Scholar]
- 38. Giannini C, Dyck PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol. 1995;37(4):498–504. [DOI] [PubMed] [Google Scholar]
- 39. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259–280. [DOI] [PubMed] [Google Scholar]
- 40. Nelis E, Timmerman V, De Jonghe P, Vandenberghe A, Pham‐Dinh D, Dautigny A, et al. Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene. Hum Genet. 1994;94(6):653–657. [DOI] [PubMed] [Google Scholar]
- 41. Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodny EH, Karpati G, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot‐Marie‐tooth type 1B, Dejerine‐Sottas, and congenital hypomyelination. Neuron. 1996;17(3):451–460. [DOI] [PubMed] [Google Scholar]
- 42. Nelis E, Van Broeckhoven C, De Jonghe P, Löfgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot‐Marie‐tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4(1):25–33. [DOI] [PubMed] [Google Scholar]
- 43. Sanmaneechai O, Feely S, Scherer SS, Herrmann DN, Burns J, Muntoni F, et al. Genotype‐phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain. 2015;138(Pt 11):3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi BO, Lee MS, Shin SH, Hwang JH, Choi KG, Kim WK, et al. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot‐Marie‐tooth neuropathy patients. Hum Mutat. 2004;24(2):185–186. [DOI] [PubMed] [Google Scholar]
- 45. Takeda JI, Nanatsue K, Yamagishi R, Ito M, Haga N, Hirata H, et al. InMeRF: prediction of pathogenicity of missense variants by individual modeling for each amino acid substitution. NAR Genom Bioinform. 2020;2(2):lqaa038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Komiyama A, Ohnishi A, Izawa K, Yamamori S, Ohashi H, Hasegawa O. De novo mutation (Arg98–>Cys) of the myelin P0 gene and uncompaction of the major dense line of the myelin sheath in a severe variant of Charcot‐Marie‐tooth disease type 1B. J Neurol Sci. 1997;149(1):103–109. [DOI] [PubMed] [Google Scholar]
- 47. Lagueny A, Latour P, Vital A, Rajabally Y, le Masson G, Ferrer X, et al. Peripheral myelin modification in CMT1B correlates with MPZ gene mutations. Neuromuscul Disord. 1999;9(6–7):361–367. [DOI] [PubMed] [Google Scholar]
- 48. Dyck PJ, Carney JA, Sizemore GW, Okazaki H, Brimijoin WS, Lambert EH. Multiple endocrine neoplasia, type 2b: phenotype recognition; neurological features and their pathological basis. Ann Neurol. 1979;6(4):302–314. [DOI] [PubMed] [Google Scholar]
- 49. Jesse CM, Bushuven E, Tripathi P, Chandrasekar A, Simon CM, Drepper C, et al. ALS‐associated endoplasmic reticulum proteins in denervated skeletal muscle: implications for motor neuron disease pathology. Brain Pathol. 2017;27(6):781–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krasnianski A, Deschauer M, Neudecker S, Gellerich FN, Müller T, Schoser BG, et al. Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain. 2005;128(Pt 8):1870–1876. [DOI] [PubMed] [Google Scholar]
- 51. Dubowitz V, Brooke MH, Neville HE. Muscle biopsy: a modern approach. London, Philadelphia: W. B. Saunders; 1973. [Google Scholar]
- 52. Boyd PJ, Gillingwater TH. Axonal and neuromuscular junction pathology in spinal muscular atrophy. In: Sumner CJ, Chien‐Ping Ko SP, editors. Spinal muscular atrophy. Academic Press; 2017. p. 133–151. [Google Scholar]
- 53. Vincent AE, Ng YS, White K, Davey T, Mannella C, Falkous G, et al. The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci Rep. 2016;6:30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crone C, Krarup C. Neurophysiological approach to disorders of peripheral nerve. Handb Clin Neurol. 2013;115:81–114. [DOI] [PubMed] [Google Scholar]
- 55. Murphy SM, Laurá M, Blake J, Polke J, Bremner F, Reilly MM. Conduction block and tonic pupils in Charcot‐Marie‐Tooth disease caused by a myelin protein zero p.Ile112Thr mutation. Neuromuscul Disord. 2011;21(3):223–226. [DOI] [PubMed] [Google Scholar]
- 56. Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0‐Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. [PubMed] [Google Scholar]
- 57. Koike H, Iijima M, Mori K, Yamamoto M, Hattori N, Katsuno M, et al. Nonmyelinating Schwann cell involvement with well‐preserved unmyelinated axons in Charcot‐Marie‐tooth disease type 1A. J Neuropathol Exp Neurol. 2007;66(11):1027–1036. [DOI] [PubMed] [Google Scholar]
- 58. Murinson BB, Archer DR, Li Y, Griffin JW. Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C‐fiber afferents. J Neurosci. 2005;25(5):1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Iwata A, Kunimoto M, Inoue K. Schwann cell proliferation as the cause of peripheral neuropathy in neurofibromatosis‐2. J Neurol Sci. 1998;156(2):201–204. [DOI] [PubMed] [Google Scholar]
- 60. Li J, Bai Y, Ianakova E, Grandis M, Uchwat F, Trostinskaia A, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol. 2006;498(2):252–265. [DOI] [PubMed] [Google Scholar]
- 61. Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, et al. Effect of an R69C mutation in the myelin protein zero gene on myelination and ion channel subtypes. Arch Neurol. 2006;63(12):1787–1794. [DOI] [PubMed] [Google Scholar]
- 62. Nadol JB Jr, Hedley‐Whyte ET, Amr SS, Oam JT, Kamakura T. Histopathology of the inner ear in Charcot‐Marie‐tooth syndrome caused by a missense variant (p.Thr65Ala) in the MPZ gene. Audiol Neurootol. 2018;23(6):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takase Y, Takahashi K, Takada K, Tatsumi H, Tabuchi Y. Hereditary motor and sensory neuropathy type 1 (HMSN1) associated with cranial neuropathy: an autopsy case report. Acta Neurol Scand. 1990;82(6):368–373. [DOI] [PubMed] [Google Scholar]
- 64. Horacek O, Mazanec R, Morris CE, Kobesova A. Spinal deformities in hereditary motor and sensory neuropathy: a retrospective qualitative, quantitative, genotypical, and familial analysis of 175 patients. Spine. 2007;32(22):2502–2508. [DOI] [PubMed] [Google Scholar]
- 65. Epstein JA, Epstein BS, Lavine LS. Surgical treatment of nerve root compression caused by scoliosis of the lumbar spine. J Neurosurg. 1974;41(4):449–454. [DOI] [PubMed] [Google Scholar]
- 66. Rajput K, Reddy S, Shankar H. Painful neuromas. Clin J Pain. 2012;28(7):639–645. [DOI] [PubMed] [Google Scholar]
- 67. Jacobs JM, Love S. Qualitative and quantitative morphology of human sural nerve at different ages. Brain. 1985;108(Pt 4):897–924. [DOI] [PubMed] [Google Scholar]
- 68. Hube L, Dohrn MF, Karsai G, Hirshman S, Van Damme P, Schulz JB, et al. Metabolic syndrome, neurotoxic 1‐deoxysphingolipids and nervous tissue inflammation in chronic idiopathic axonal polyneuropathy (CIAP). PLoS One. 2017;12(1):e0170583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bradley J, Thomas PK, King RH, Llewelyn JG, Muddle JR, Watkins PJ. Morphometry of endoneurial capillaries in diabetic sensory and autonomic neuropathy. Diabetologia. 1990;33(10):611–618. [DOI] [PubMed] [Google Scholar]
- 70. Baker MK, Bourque P, Dyck PJ. Microvessel basement membrane reduplication is not associated with repeated nerve fiber degeneration and regeneration. J Neurol Sci. 1996;136(1–2):31–36. [DOI] [PubMed] [Google Scholar]
- 71. Fledrich R, Schlotter‐Weigel B, Schnizer TJ, Wichert SP, Stassart RM, Meyer zu Hörste G, et al. A rat model of Charcot‐Marie‐tooth disease 1A recapitulates disease variability and supplies biomarkers of axonal loss in patients. Brain. 2012;135(Pt 1):72–87. [DOI] [PubMed] [Google Scholar]
- 72. Shackleford G, Marziali LN, Sasaki Y, Claessens A, Ferri C, Weinstock NI, et al. A new mouse model of Charcot‐Marie‐tooth 2J neuropathy replicates human axonopathy and suggest alteration in axo‐glia communication. PLoS Genet. 2022;18(11):e1010477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang X, Xue P, Chen H, Yuan M, Kang Y, Duscher D, et al. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR‐142a‐5p/MFN1 axis. Theranostics. 2020;10(3):1415–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schroder JM. Neuropathy associated with mitochondrial disorders. Brain Pathol. 1993;3(2):177–190. [DOI] [PubMed] [Google Scholar]
- 75. Zuchner S, Mersiyanova IV, Muglia M, Bissar‐Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. [DOI] [PubMed] [Google Scholar]
- 76. Verhoeven K, Claeys KG, Zuchner S, Schröder JM, Weis J, Ceuterick C, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot‐Marie‐tooth type 2. Brain. 2006;129(Pt 8):2093–2102. [DOI] [PubMed] [Google Scholar]
- 77. Howard P, Feely SME, Grider T, Bacha A, Scarlato M, Fazio R, et al. Loss of function MPZ mutation causes milder CMT1B neuropathy. J Peripher Nerv Syst. 2021;26(2):177–183. [DOI] [PubMed] [Google Scholar]
- 78. Shapiro L, Doyle JP, Hensley P, Colman DR, Hendrickson WA. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 1996;17(3):435–449. [DOI] [PubMed] [Google Scholar]
- 79. Wrabetz L, D'Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P, et al. Different intracellular pathomechanisms produce diverse myelin protein zero neuropathies in transgenic mice. J Neurosci. 2006;26(8):2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, et al. Ablation of the UPR‐mediator CHOP restores motor function and reduces demyelination in Charcot‐Marie‐tooth 1B mice. Neuron. 2008;57(3):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saporta MA, Shy BR, Patzko A, Bai Y, Pennuto M, Ferri C, et al. MpzR98C arrests Schwann cell development in a mouse model of early‐onset Charcot‐Marie‐tooth disease type 1B. Brain. 2012;135(Pt 7):2032–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fratta P, Ornaghi F, Dati G, Zambroni D, Saveri P, Belin S, et al. A nonsense mutation in myelin protein zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum Mol Genet. 2019;28(1):124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Prada V, Passalacqua M, Bono M, Luzzi P, Scazzola S, Nobbio LA, et al. Gain of glycosylation: a new pathomechanism of myelin protein zero mutations. Ann Neurol. 2012;71(3):427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, et al. CMT subtypes and disease burden in patients enrolled in the inherited neuropathies consortium natural history study: a cross‐sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86(8):873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gabreels‐Festen AA, Hoogendijk JE, Meijerink PH, Gabreëls FJM, Bolhuis PA, Van Beersum S, et al. Two divergent types of nerve pathology in patients with different P0 mutations in Charcot‐Marie‐tooth disease. Neurology. 1996;47(3):761–765. [DOI] [PubMed] [Google Scholar]
- 86. Rautenstrauss B, Nelis E, Grehl H, Pfeiffer RA, Van Broeckhoven C. Identification of a de novo insertional mutation in P0 in a patient with a Dejerine‐Sottas syndrome (DSS) phenotype. Hum Mol Genet. 1994;3(9):1701–1702. [DOI] [PubMed] [Google Scholar]
- 87. Kochanski A, Kabzinska D, Drac H, Ryniewicz B, Rowinska‐Marcinska K, Hausmanowa‐Petrusewicz I. Early onset Charcot‐Marie‐tooth type 1B disease caused by a novel Leu190fs mutation in the myelin protein zero gene. Eur J Paediatr Neurol. 2004;8(4):221–224. [DOI] [PubMed] [Google Scholar]
- 88. Bornemann A, Hansen FJ, Schmalbruch H. Nerve and muscle biopsy in a case of hereditary motor and sensory neuropathy type III with basal lamina onion bulbs. Neuropathol Appl Neurobiol. 1996;22(1):77–81. [PubMed] [Google Scholar]
- 89. Xu W, Shy M, Kamholz J, Elferink L, Xu G, Lilien J, et al. Mutations in the cytoplasmic domain of P0 reveal a role for PKC‐mediated phosphorylation in adhesion and myelination. J Cell Biol. 2001;155(3):439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Grandis M, Vigo T, Passalacqua M, Jain M, Scazzola S, La Padula V, et al. Different cellular and molecular mechanisms for early and late‐onset myelin protein zero mutations. Hum Mol Genet. 2008;17(13):1877–1889. [DOI] [PubMed] [Google Scholar]
- 91. Shy ME. Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci. 2006;242(1–2):55–66. [DOI] [PubMed] [Google Scholar]
- 92. Brugger V, Engler S, Pereira JA, Ruff S, Horn M, Welzl H, et al. HDAC1/2‐dependent P0 expression maintains paranodal and nodal integrity independently of myelin stability through interactions with neurofascins. PLoS Biol. 2015;13(9):e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kabzinska D, Korwin‐Piotrowska T, Drechsler H, Drac H, Hausmanowa‐Petrusewicz I, Kochanski A. Late‐onset Charcot‐Marie‐tooth type 2 disease with hearing impairment associated with a novel Pro105Thr mutation in the MPZ gene. Am J Med Genet A. 2007;143A(18):2196–2199. [DOI] [PubMed] [Google Scholar]
- 94. Kilfoyle DH, Dyck PJ, Wu Y, Litchy WJ, Klein DM, Dyck PJ, et al. Myelin protein zero mutation His39Pro: hereditary motor and sensory neuropathy with variable onset, hearing loss, restless legs and multiple sclerosis. J Neurol Neurosurg Psychiatry. 2006;77(8):963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lei L, Xiaobo L, Zhiqiang L, Yongzhi X, Shunxiang H, Huadong Z, et al. Genotype‐phenotype characteristics and baseline natural history of Chinese myelin protein zero gene related neuropathy patients. Eur J Neurol. 2023;30(4):1069–1079. [DOI] [PubMed] [Google Scholar]
- 96. Taniguchi T, Ando M, Okamoto Y, Yoshimura A, Higuchi Y, Hashiguchi A, et al. Genetic spectrum of Charcot‐Marie‐tooth disease associated with myelin protein zero gene variants in Japan. Clin Genet. 2021;99(3):359–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Control nerves used in this study. Age at biopsy, gender (f = female, m = male) and clinical/pathological information if available.
Data Availability Statement
Data not provided in the article because of space limitations may be shared (anonymized) at the request of any qualified investigator for purposes of replicating procedures and results.
All numerical data generated or analyzed during this study are included in this published article. Additional information is available from the corresponding author on reasonable request.