

Abstract
Background
Duloxetine is a balanced serotonin and noradrenaline reuptake inhibitor licensed for the treatment of major depressive disorders, urinary stress incontinence and the management of neuropathic pain associated with diabetic peripheral neuropathy. A number of trials have been conducted to investigate the use of duloxetine in neuropathic and nociceptive painful conditions. This is the first update of a review first published in 2010.
Objectives
To assess the benefits and harms of duloxetine for treating painful neuropathy and different types of chronic pain.
Search methods
On 19th November 2013, we searched The Cochrane Neuromuscular Group Specialized Register, CENTRAL, DARE, HTA, NHSEED, MEDLINE, and EMBASE. We searched ClinicalTrials.gov for ongoing trials in April 2013. We also searched the reference lists of identified publications for trials of duloxetine for the treatment of painful peripheral neuropathy or chronic pain.
Selection criteria
We selected all randomised or quasi‐randomised trials of any formulation of duloxetine, used for the treatment of painful peripheral neuropathy or chronic pain in adults.
Data collection and analysis
We used standard methodological procedures expected by The Cochrane Collaboration.
Main results
We identified 18 trials, which included 6407 participants. We found 12 of these studies in the literature search for this update. Eight studies included a total of 2728 participants with painful diabetic neuropathy and six studies involved 2249 participants with fibromyalgia. Three studies included participants with depression and painful physical symptoms and one included participants with central neuropathic pain. Studies were mostly at low risk of bias, although significant drop outs, imputation methods and almost every study being performed or sponsored by the drug manufacturer add to the risk of bias in some domains. Duloxetine at 60 mg daily is effective in treating painful diabetic peripheral neuropathy in the short term, with a risk ratio (RR) for ≥ 50% pain reduction at 12 weeks of 1.73 (95% CI 1.44 to 2.08). The related NNTB is 5 (95% CI 4 to 7). Duloxetine at 60 mg daily is also effective for fibromyalgia over 12 weeks (RR for ≥ 50% reduction in pain 1.57, 95% CI 1.20 to 2.06; NNTB 8, 95% CI 4 to 21) and over 28 weeks (RR 1.58, 95% CI 1.10 to 2.27) as well as for painful physical symptoms in depression (RR 1.37, 95% CI 1.19 to 1.59; NNTB 8, 95% CI 5 to 14). There was no effect on central neuropathic pain in a single, small, high quality trial. In all conditions, adverse events were common in both treatment and placebo arms but more common in the treatment arm, with a dose‐dependent effect. Most adverse effects were minor, but 12.6% of participants stopped the drug due to adverse effects. Serious adverse events were rare.
Authors' conclusions
There is adequate amounts of moderate quality evidence from eight studies performed by the manufacturers of duloxetine that doses of 60 mg and 120 mg daily are efficacious for treating pain in diabetic peripheral neuropathy but lower daily doses are not. Further trials are not required. In fibromyalgia, there is lower quality evidence that duloxetine is effective at similar doses to those used in diabetic peripheral neuropathy and with a similar magnitude of effect. The effect in fibromyalgia may be achieved through a greater improvement in mental symptoms than in somatic physical pain. There is low to moderate quality evidence that pain relief is also achieved in pain associated with depressive symptoms, but the NNTB of 8 in fibromyalgia and depression is not an indication of substantial efficacy. More trials (preferably independent investigator led studies) in these indications are required to reach an optimal information size to make convincing determinations of efficacy.
Minor side effects are common and more common with duloxetine 60 mg and particularly with 120 mg daily, than 20 mg daily, but serious side effects are rare.
Improved direct comparisons of duloxetine with other antidepressants and with other drugs, such as pregabalin, that have already been shown to be efficacious in neuropathic pain would be appropriate. Unbiased economic comparisons would further help decision making, but no high quality study includes economic data.
Plain language summary
Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia
Review question
Does duloxetine work to treat pain generated by nerves when they have been damaged in disease, or the pain caused by fibromyalgia?
Background
Duloxetine is a drug used to treat depression and urinary urge incontinence (leakage of urine) and it can be also be useful for certain types of pain. Pain can arise spontaneously when there is damage to nerves that carry pain information to the brain (neuropathic pain). When this damage is to nerves outside the spinal cord it is called a peripheral neuropathy. Another type of pain, nociceptive pain, occurs when the nerves sense damage to another tissue (for example, a pinprick in the skin). Some pain is of unclear origin and occurs without apparent nerve or tissue damage. This sort of pain happens, for example, in fibromyalgia. The objective of this review was to assess the benefits and harms of duloxetine for treating painful neuropathy and chronic pain of all sorts.
Study characteristics
We looked at all the published scientific literature and found 18 trials, involving a total of 6407 participants, that were of sufficient quality to include in this review. Eight trials tested the effect of duloxetine on painful diabetic neuropathy and six on the pain of fibromyalgia. Three trials treated painful physical symptoms associated with depression and one small study investigated duloxetine for the pain from strokes or diseases of the spinal cord (central pain).
Key results and quality of the evidence
The usual dose of duloxetine is 60 mg. At this dose, there was moderate quality evidence that duloxetine reduced pain in both painful diabetic peripheral neuropathy and fibromyalgia. In diabetic peripheral neuropathic pain, a 50% or better improvement with duloxetine 60 mg per day was just over one and a half times more likely than with placebo. Another way of saying this is that five people with painful diabetic peripheral neuropathy had to receive duloxetine to achieve a 50% or better response in one person. The effect on fibromyalgia was similar but the number needed to treat for one person to improve by 50% or more was eight. On the basis of a single study it is not possible to determine if a dose of 20 mg is effective, and 120 mg was no more effective than 60 mg.
We calculated that for diabetic neuropathy there have been enough trials to draw these conclusions and no more trials are needed. In fibromyalgia and the painful symptoms associated with depression, more trials are required to make convincing statements about the effectiveness of duloxetine.
Most people taking duloxetine will have at least one side effect. These are mostly minor and the most common are feeling sick, being too awake or too sleepy, headache, dry mouth, constipation or dizziness. About one in six people stop duloxetine because of side effects. Serious problems caused by duloxetine are very rare.
Although duloxetine is beneficial in the treatment of neuropathic pain and fibromyalgia there is little evidence from trials comparing duloxetine to other antidepressant drugs as to which is better.
We have concluded that duloxetine is useful for treating pain caused by diabetic neuropathy and probably fibromyalgia.
The information in this review is up to date to November 2013, the most recent search of the literature.
Summary of findings
Summary of findings for the main comparison. Duloxetine for the treatment of painful diabetic neuropathy.
| Duloxetine for painful diabetic neuropathy | ||||||
| Patient or population: patients with painful neuropathy or chronic pain from diabetic peripheral neuropathy Settings: primary and secondary care Intervention: duloxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Duloxetine | |||||
|
Number of patients with ≥ 50% improvement of pain at 12 weeks or less Duloxetine 60 mg daily 11‐point Likert score Follow‐up: 8 to 12 weeks |
257 per 1000 | 445 per 1000 (370 to 535) | RR 1.73 (1.44 to 2.08) | 908 (4 studies) | ⊕⊕⊕⊝ moderate1 | NNTB for ≥ 50% reduction in pain at 60 mg daily: 5 (95% CI 4 to 7) |
|
Mean improvement in pain at 12 weeks or less Duloxetine 60 mg daily 11‐point Likert score Scale from: 0 to 10 Follow‐up: 8 to 12 weeks |
The mean mean improvement in pain at 12 weeks or less ‐ duloxetine 60 mg daily in the control groups was ‐1.65 units | The mean mean improvement in pain at 12 weeks or less ‐ duloxetine 60 mg daily in the intervention groups was 0.96 lower (1.26 to 0.65 lower) | ‐ | 722 (4 studies) | ⊕⊕⊕⊝ moderate2 | |
|
Number of patients with ≥ 30% improvement in pain at 12 weeks or less Duloxetine 60 mg daily 11‐point Likert scale Follow‐up: 8 to 12 weeks |
411 per 1000 | 629 per 1000 (547 to 719) | RR 1.53 (1.33 to 1.75) | 799 (4 studies) | ⊕⊕⊕⊝ moderate1 | NNTB for ≥ 30% reduction in pain at 60 mg duloxetine daily: 5 (95% CI 3 to 7) |
|
Mean improvement in Patient Reported Global Impression of Change at 12 weeks or less Duloxetine 60 mg daily VAS Scale from: 0 to 10 Follow‐up: 8 to 12 weeks |
The mean mean improvement in patient reported global impression of improvement change at 12 weeks or less ‐ duloxetine 60 mg daily in the control groups was ‐3.06 units | The mean mean improvement in Patient Reported Global Impression of Improvement Change at 12 weeks or less ‐ duloxetine 60 mg daily in the intervention groups was 0.6 lower (0.77 to 0.44 lower) | ‐ | 1018 (5 studies) | ⊕⊕⊕⊝ moderate3 | |
|
Adverse event leading to cessation All neuropathic pain indications Duloxetine 60 mg daily |
56 per 1000 | 109 per 1000 (90 to 133) | RR 1.95 (1.6 to 2.37) | 4837 (14 studies) | ⊕⊕⊝⊝ low4 | NNTH for duloxetine 60 mg daily, all indications, and all adverse effects leading to cessation: 18 (95% CI 13 to 30) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Four trials, all company sponsored and performed but all trials pre‐registered on ClinicalTrials.gov have been published. No publication bias detected. 2 Two of four studies by company. Effect in Rowbotham nonsignificant, contributing some heterogeneity. 3 Five studies but wide CIs in the independent studies. 4 Variable quality of adverse event collection.
Summary of findings 2. Duloxetine for the treatment of the chronic pain of fibromyalgia.
| Duloxetine for the chronic pain of fibromyalgia | |||||||
| Patient or population: patients with the chronic pain of fibromyalgia Settings: Intervention: duloxetine | |||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | ||
| Assumed risk | Corresponding risk | ||||||
| Control | Duloxetine | ||||||
|
Number with ≥ 50% improvement of pain at 12 weeks or less Duloxetine 60 mg daily 11‐point Likert scale Follow‐up: 8 to 12 weeks |
233 per 1000 | 366 per 1000 (280 to 480) | RR 1.57 (1.2 to 2.06) | 528 (2 studies) | ⊕⊕⊝⊝ low1,2 | NNTB for ≥ 50% improvement of pain at duloxetine 60 mg daily: 8 (95% CI 4 to 21) | |
|
Number with ≥ 30% improvement of pain at 12 weeks or less Duloxetine 60 mg daily Follow‐up: 8 to 12 weeks |
347 per 1000 | 527 per 1000 (430 to 642) | RR 1.52 (1.24 to 1.85) | 528 (2 studies) | ⊕⊕⊝⊝ low1,2 | NNTB for ≥ 30% improvement of pain at duloxetine 60 mg daily: NNT 6 (95% CI 3 to 12) | |
|
Mean improvement in the Patient Reported Global Impression of Change at completion of trial Duloxetine 60 mg daily VAS Scale from: 0 to 10 Follow‐up: 12 weeks |
The mean mean improvement in the patient reported global impression of change at completion of trial ‐ duloxetine 60 mg daily in the control groups was 3.52 units | The mean mean improvement in the patient reported global impression of change at completion of trial ‐ duloxetine 60 mg daily in the intervention groups was 0.45 lower (0.73 to 0.18 lower) | ‐ | 519 (2 studies) | ⊕⊕⊝⊝ low1,2 | ||
|
Mean improvement in pain at 12 weeks or less Duloxetine 120 mg daily LikertScale from: 0 to 10 Follow‐up: 12 weeks |
The mean mean improvement in pain at 12 weeks or less ‐ duloxetine 120 mg daily in the control groups was ‐1.5 | The mean mean improvement in pain at 12 weeks or less ‐ duloxetine 120 mg daily in the intervention groups was 0.8 lower (1.35 to 0.25 lower) | ‐ | 507 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| Adverse events | See comment | See comment | See comment | ‐ | See comment | See pooled adverse events in 'Summary of findings' table 1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; NNTB: number needed to treat for an additional beneficial outcome | |||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||||
1 Substantial dropouts from all trials inform the outcomes. 2 Mostly female in some trials, all female in others.
Summary of findings 3. Duloxetine for the treatment of pain in major depressive disorder.
| Duloxetine for pain in major depressive disorder | ||||||
| Patient or population: patients with pain in major depressive disorder Settings: Intervention: duloxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Duloxetine | |||||
| Number with ≥ 50% pain relief at 12 weeks or less Follow‐up: 12 weeks | 360 per 1000 | 493 per 1000 (428 to 572) | RR 1.37 (1.19 to 1.59) | 1023 (2 studies) | ⊕⊕⊕⊝ moderate1 | NNTB for ≥ 50% pain relief at < 12 weeks 60 mg duloxetine daily: 8 (95% CI 5 to 14) |
| Number with ≥ 30% pain relief at 12 weeks or less | 467 per 1000 | 593 per 1000 (537 to 654) | RR 1.27 (1.15 to 1.4) | 1359 (3 studies) | ⊕⊕⊝⊝ low1,2 | NNTB for ≥ 30% pain relief at < 12 weeks 60 mg duloxetine: 8 (95% CI 4‐ to 14) |
| Mean improvement in pain at 12 weeks or less Visual analogue scale. Scale from: 0 to 10. Follow‐up: 12 weeks | The mean mean improvement in pain at 12 weeks or less in the control groups was 1.23 | The mean mean improvement in pain at 12 weeks or less in the intervention groups was 0.55 lower (0.75 to 0.35 lower) | 1359 (3 studies) | ⊕⊕⊝⊝ low1,2 | ||
| Mean improvement in Patient Reported Global Impression of Change at 12 weeks or less | See comment | See comment | Not estimable | ‐ | See comment | Outcome not measured |
| Adverse events | See comment | See comment | Not estimable | ‐ | See comment | See pooled adverse events in 'Summary of findings' table 1 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; NNTB: number needed to treat for an additional beneficial outcome | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Mixed causes for pain, not necessarily neuropathic. 2 Substantial dropouts partially accounted for by last observation carried forward and statistical manipulation.
Background
Description of the condition
Pain is common in peripheral nerve diseases such as the peripheral neuropathy associated with diabetes mellitus. Painful neuropathy is a particular example of neuropathic pain. Neuropathic pain is "pain arising as a direct consequence of a lesion or disease affecting the somatosensory system" (Treede 2008). Neuropathic pain is different from conventional or nociceptive pain. Nociceptive pain arises from the activation of primary pain receptors in response to injury or inflammation. Painful neuropathies are diseases of the peripheral nerves that cause neuropathic pain.
Chronic pain has been classified as pain exceeding three months' duration (Nagda 2004). Chronic pain is a major health problem affecting one in five people in Europe (Breivik 2006). A community based study in North West England estimated the prevalence of chronic painful peripheral neuropathy in people without diabetes as 4.9% (Daousi 2004).The prevalence in people with diabetes in the same community was 16.2%. In a large community study in the United Kingdom, the annual incidence of neuropathic pain was calculated as at least 84 per 100,000 by adding the incidence of the four commonest and most disabling causes (diabetic neuropathy, trigeminal neuralgia, postherpetic neuralgia and phantom limb pain) (Hall 2006).
The pain of painful peripheral neuropathy can be diverse and distressing. Descriptions include burning, cold, electric shocks, lancinating, tight or aching. Other spontaneous and evoked positive sensory symptoms include painful numbness, tingling or paraesthesiae. Stimuli that are not usually painful may be perceived as painful, a phenomenon called allodynia. Chronic pain can have serious complex adverse psychological and social effects.
Description of the intervention
Duloxetine is one of a newer type of antidepressant drug. It is a relatively balanced dual reuptake inhibitor of serotonin and noradrenaline (Schuessler 2006). Theoretically these actions should make it a good pain modulating agent (Bymaster 2001; Bymaster 2005). Serotonin modulates both pro‐nociceptive and anti‐nociceptive descending effects on central pain pathways from the brainstem. Noradrenaline has a predominantly anti‐nociceptive effect. Balance between facilitation and depression of pain pathways is important for normal function. Drugs that inhibit the reuptake of serotonin and noradrenaline potentiate monoamine neurotransmission in the descending inhibitory spinal pathways and so reduce nociceptive afferent transmission in the ascending spinal pain pathways. Potentiation of both serotonin and noradrenaline is required to produce effective analgesia. The action of drugs such as duloxetine is independent of their effects on depression (Perahia 2006). Onset of benefit occurs within days, earlier and at lower doses than in depression. Furthermore, they have similar effects on pain in depressed and non‐depressed people. Common side effects include nausea, headache, dry mouth, insomnia, constipation, dizziness, fatigue, somnolence, hyperhydrosis and diarrhoea (Gahimer 2007). These are mainly classified as mild to moderate and anecdotally appear less prevalent than the side effects with tricyclic antidepressants.
Why it is important to do this review
Duloxetine is licensed in the United States, European Union and United Kingdom for the treatment of major depressive disorder (Nose 2007), urinary stress incontinence (Mariappan 2009), and for the management of neuropathic pain associated with diabetic peripheral neuropathy. We do not know of a published systematic review of duloxetine for any pain condition and it was not included in a previous Cochrane review of antidepressants for neuropathic pain (Saarto 2007). This review aims to fill the gap. Painful neuropathy is the principal focus of this review because duloxetine has been chiefly tried for this indication. However, previous Cochrane reviews of interventions for pain have covered all forms of either neuropathic pain or acute and chronic pain. For conformity with these other reviews, we will include all forms of chronic pain which have a neuropathic component, chronic pain with no explanation and fibromyalgia, but not acute pain, for which duloxetine has not been proposed as a treatment, or pain from specific non‐neuropathic causes covered in other reviews (for example pain from osteoarthritis of the knee).
Objectives
To assess the benefits and harms of duloxetine for treating painful neuropathy and different types of chronic pain.
Methods
Criteria for considering studies for this review
Types of studies
For the detection of benefits, we included only double‐blind randomised trials of duloxetine for treating painful neuropathy or chronic neuropathic pain, chronic pain conditions without identified cause or fibromyalgia. Duloxetine was to have been administered for a minimum of eight weeks. We included eligible studies irrespective of publication status or language of publication.
Types of participants
We included participants with any form of painful peripheral neuropathy, chronic neuropathic pain, chronic pain conditions without identified cause, or fibromyalgia.
Types of interventions
We included all formulations and doses of duloxetine in comparison with placebo or other controls. We reported comparisons with placebo and with other controls separately.
Types of outcome measures
Primary outcomes
The primary outcome was short‐term (up to and including 12 weeks) improvement of pain compared with baseline using validated scales of pain intensity or pain relief. We accepted both visual analogue and categorical scales. Where reports expressed pain relief as none, minor, moderate, major or complete, we considered only moderate, major or complete as improvement. Where studies measured pain with a continuous scale, we took improvement to be an improvement of 50% or more from baseline on that scale. If studies reported results only as improvement on a continuous scale, we planned to try to obtain results from the authors to provide this dichotomous analysis.
Secondary outcomes
Long‐term (more than 12 weeks) improvement of pain compared with baseline, analysed as for the primary outcome.
Improvement in short‐term (up to and including 12 weeks) and long‐term (more than 12 weeks) pain of at least 30% compared with baseline using validated scales of pain intensity and or pain relief, analysed as for the primary outcome.
Improvement in any validated quality of life score of 30% or more compared to the baseline.
-
As the outcome measures for the assessment of pain were likely to be diverse and the majority of trials use standard subjective scales for pain intensity or pain relief or both, further results were to be analysed according to the third to sixth types in a hierarchy modified from Wiffen 2005. The full hierarchy of outcome measures is as follows.
Patient reported pain relief of 50% or greater.
Patient reported pain relief of 30% or greater.
Patient reported global impression of clinical change (PGIC).
Pain on movement.
Pain on rest.
Any other pain related measure.
Adverse events during treatment. We analysed categories of: all adverse events, severe or serious adverse events that led to hospitalisation or death, and adverse events leading to cessation of treatment.
We chose 30% and 50% as the percentage of pain improvement considered clinically important for dichotomous outcomes, in line with the recommendations made by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) (Dworkin 2008). Improvement in pain intensity of 30% or more is considered moderately important and 50% or more, substantial improvement.
Outcomes for inclusion in a 'Summary of findings' table
We created a 'Summary of findings' table for each included neuropathic pain condition for which meta‐analysis was possible, using the following outcomes.
Number of participants with ≥ 50% improvement of pain at 12 weeks or less.
Mean improvement in pain at 12 weeks or less.
Number of participants with ≥ 30% improvement in pain at 12 weeks or less.
Mean improvement in PGIC at 12 weeks or less.
Adverse event leading to cessation.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of a body of evidence (studies that contribute data for the prespecified outcomes). We used methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) using GRADEpro software (GRADEpro 2008). We justified decisions to down‐ or up‐grade the quality of studies using footnotes.
Search methods for identification of studies
Electronic searches
We searched the specialised registers of the Cochrane Neuromuscular Disease Group and the Cochrane Pain, Palliative and Supportive Care Group (PaPaS), CENTRAL (2013, Issue 11), MEDLINE (January 1966 to November 2013) and EMBASE (January 1980 to November 2013). We searched the National Institutes for Health clinical trials registry ClinicalTrials.gov (www.clinicaltrials.gov) up to April 2013 for current ongoing registered trials. We also searched DARE (Database of Abstracts of Reviews of Effects), HTA (Health Technology Assessment) and NHSEED (NHS Economic Evaluation Database) (2013, Issue 4 in The Cochrane Library), for papers for inclusion in the Discussion. The detailed search strategies are in the appendices: MEDLINE (Appendix 1), EMBASE (Appendix 2), CENTRAL (Appendix 3), Cochrane Neuromuscular Disease Group Specialized Register Appendix 4 and ClinicalTrials.gov Appendix 5.
Searching other resources
We also wrote to Eli Lilly who make duloxetine and to pain experts asking for information about other or ongoing trials. We searched the Lilly online trials database (http://www.lillytrials.com/) for other trials not identified in the above searches. We wrote to the authors of studies to clarify aspects of trial design that were unclear from the published papers.
Data collection and analysis
Selection of studies
Two review authors (MLand RACH) independently scrutinised all the titles and abstracts revealed by the searches and determined which trials fulfilled the selection criteria. They resolved disagreement by discussion without the need to involve the third review author (PW).
Data extraction and management
Two review authors (ML and RACH) extracted data independently onto a specially designed data extraction form. We would have resolved disagreements by discussion if necessary with the third review author (PW) but this was not necessary. One author (ML) entered data into the Cochrane software, Review Manager 5 (RevMan), and a second author (RACH) checked them.
Assessment of risk of bias in included studies
We used the methods of the Cochrane Collaboration to assess 'Risk of bias' as set out in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008, updated Higgins 2011) rather than those stipulated in the protocol for this review, which predated the new methods.
Measures of treatment effect
The effect measures of choice were the risk ratio (RR) for dichotomous data and the mean difference (MD) or standardised mean difference (SMD) for continuous data. We expressed uncertainty with 95% confidence intervals (CIs). We also expressed the most important results as numbers needed to treat for an additional beneficial outcome (NNTB) and numbers needed to treat for an additional harmful outcome (NNTH), where appropriate.
Unit of analysis issues
Cross‐over trials
We would have analysed cross‐over trials using the estimated differences in effects and their standard errors with the generic inverse variance (GIV) facility in RevMan if we had the necessary data. For results using dichotomous outcomes this would have been more difficult, but we would have used this approach if we could have converted the results to odds ratios (OR) on the log scale and calculated the standard errors. If necessary we would also have analysed the results following the methods of Elbourne 2002 with the assistance of a statistician.
Assessment of reporting biases
If there had been sufficient trials, we would have inspected funnel plots for asymmetry that might have been due to publication bias. We were aware that funnel plots and statistical tests based on them are not reliable indicators of publication bias and we would have treated any interpretations made from them with great caution.
Data synthesis
We undertook each meta‐analysis using a fixed‐effect model in the RevMan software. We used the I2 statistic for heterogeneity and if its value had been greater than 50% we would have inspected the trials, forest plots and L'Abbé plots for differences between trials that might have explained the heterogeneity. In the absence of any explanation, we would have repeated the analysis with a random‐effects model.
Subgroup analysis and investigation of heterogeneity
We reported results for painful diabetic neuropathy, fibromyalgia, chronic pain and non‐specific bodily pain associated with depression separately, and would have reported other specific causes of neuropathic pain, namely trigeminal neuralgia, postherpetic neuralgia and central ('thalamic') pain separately. In addition to reporting the results of all forms of painful neuropathy together, we would also have reported the results for the following different diagnostic subgroups separately: diabetic neuropathy, HIV neuropathy, and idiopathic painful neuropathy.
Sensitivity analysis
We intended to conduct the following sensitivity analyses:
trials that did and did not have perfect scores for 'Risk of bias';
trials with more than or less than 20% dropout or loss to follow‐up;
trials that were and were not led by the company producing the drug; and
trials with more than and fewer than 100 participants.
A sensitivity analysis was possible only in the trials of diabetic neuropathy and only in the context of studies with a less than a 20% dropout rate.
Trial sequential analysis
We performed trial sequential analysis (TSA) using software provided by the Copenhagen Trial Unit (Thorlund 2011). We used predefined limits to frame the statistical analysis, with a conventional analysis. Limits were alpha 0.05, beta 0.1, relative risk 0.66, and we defined the placebo rate according to that found in the extracted meta‐analysis data. We performed TSA on the primary outcome for each of the conditions included in the review (diabetic neuropathy, fibromyalgia, central pain, and painful physical symptoms in depression).
Adverse events
Randomised trials may not capture all important adverse events, but this systematic review now contains data from more than 6000 participants and the adverse effects reported were fairly consistent across all studies. It is noted that the drug manufacturer conducted all but one of the studies.
Economic issues
We considered costs in the Discussion.
We reported any changes from the published protocol of the review (Lunn 2008) in Differences between protocol and review.
Results
Description of studies
Results of the search
In 2009, for the original review, we identified 130 references to possible trials (MEDLINE 12, EMBASE 75, CENTRAL 19, Cochrane Neuromuscular Disease Group Specialized Register and PaPaS Group Register and Library 22, and handsearches 2). Following exclusion of duplicates and studies that were clearly irrelevant, two authors checked 37 titles and identified 14 RCTs or possible RCTs. From these we selected six trials for inclusion. In 2012 to 2013, we performed a database search extension to October 2012 and updated the search of www.clinicaltrials.gov to April 2013. The search for RCTsretrieved 298 new references (MEDLINE 120, EMBASE 156, CENTRAL 21, and the specialised registers of the Cochrane Neuromuscular Disease Group and PaPaS 1). We performed another search to November 2013 and identified a further 150 references including references to economic analyses and a number of other systematic reviews.
Two authors selected potentially eligible references from this list and after deduplication, 47 possible references remained. We found a further study from review of the reference lists of identified papers and another study from querying publications from studies ongoing in the first version of this review and now published. After discussion of titles and abstracts, we selected 27 new references for full‐text review, of which we included 12. There are therefore 18 trials in total in this update.
Included studies
The 18 studies in this Cochrane Systematic Review include a total of 6407 participants, and cover painful neuropathy, chronic neuropathic pain (in this review central pain from strokes or spinal cord disorders), fibromyalgia and painful physical symptoms (of unknown cause) in depressive disorders. We excluded trials of duloxetine in conditions where pain is from another disease where the pain is not neuropathic (for example, we excluded osteoarthritis and pelvic pain), but included neuropathic pain diagnoses associated with specific neural injury such as spinal cord injuries, multiple sclerosis, or stroke. The manufacturers of duloxetine, Eli Lilly, were the sponsors of all but one of the included studies (Vranken 2011).
We described the characteristics of the classified and included studies in Characteristics of included studies. Eight studies, including 2728 participants, looked at duloxetine in the treatment of painful diabetic peripheral neuropathy.
Six studies, involving 2249 participants, tested duloxetine for fibromyalgia. Four studies tested duloxetine for 12 weeks (Arnold 2004; Arnold 2005; Arnold 2010; Arnold 2012) and two for six months (Russell 2008; Chappell 2008).
Three studies (1382 participants) examined the effect of duloxetine in participants who had painful physical symptoms unexplained by any known alternative diagnosis in the context of major depressive disorder (Brecht 2007; Gaynor 2011a; Gaynor 2011b).
One study (Vranken 2011) examined the effect of duloxetine in 48 participants with central neuropathic pain.
Six of the eight studies in diabetic neuropathy compared duloxetine with placebo in parallel groups for two to three months (Goldstein 2005; Raskin 2005; Wernicke 2006; Gao 2010; Yasuda 2010; Rowbotham 2012). One study compared duloxetine to amitriptyline in a cross‐over design with only six weeks' treatment in each arm with a short two‐week washout (Kaur 2011), but because it is the only comparative trial of its type, we included some discussion of it but have not included it in meta‐analysis. Kaur 2011 and Gao 2010 had variable dosage schedules for duloxetine (Kaur 20 mg to 60 mg and Gao 60 mg to 120 mg). We carried out analyses for benefit as if all participants were on the higher dose and for harms as if all were on the lower dose. One study compared duloxetine to pregabalin in the randomised parallel group first arm of an enrichment trial design (Tesfaye 2013), but we used only the randomised parallel group study period II prior to enrichment in the meta‐analysis as it was unclear whether there was any rerandomisation in Study Period III.
Five of the included studies of diabetic peripheral neuropathy were broadly similar in design and were all conducted by the same drug company (Goldstein 2005; Raskin 2005; Wernicke 2006; Gao 2010; Yasuda 2010). Participants all had to be at least 18 years old, to have had a length‐dependent painful peripheral neuropathy caused by either type I or type II diabetes for at least six months, and had to have a diagnosis of diabetes on a validated published scale and a reasonable minimum average 24‐hour pain score (for example 4 on an 11‐point Likert scale or > 50% on a VAS of pain). The age, sex, pain severity and duration of pain at entry for the participants were similar in the treatment groups in each trial and between trials, except Yasuda 2010, in which three‐quarters of the participants were male. In these five trials participants were treated with duloxetine in oral capsule or tablet form. Doses varied between trials: in Yasuda 2010, participants were treated with doses of 40 mg or 60 mg; in Goldstein 2005, Raskin 2005 and Wernicke 2006, dosage was 60 mg once or twice per day or identical placebo, with the addition of a 20 mg once daily dose in the trial of Goldstein. Gao 2010 commenced with 60 mg duloxetine but this could increase to 120 mg after two weeks if the participant had an inadequate response. Treatment was for 12 weeks with a one week taper in four trials (Raskin 2005; Wernicke 2006; Gao 2010; Yasuda 2010). All five trials took place in healthcare and research centre settings. Tesfaye 2013 compared pregabalin or duloxetine at doses increasing to their maximum with a combination of the two drugs together. The seventh trial was of an α4ß2 neuronal nicotinic receptor agonist ABT‐894 and compared the efficacy of ABT‐894 to that of placebo and to duloxetine 60 mg over eight weeks (Rowbotham 2012).
The six studies of fibromyalgia included participants aged 18 or older who fulfilled American College of Rheumatology criteria for fibromyalgia. Five of the six studies stipulated minimum entry criteria: participants had significant pain at entry (≥ 4 on the pain intensity item of the Fibromyalgia Impact Questionnaire (Arnold 2004) or Brief Pain Inventory (Arnold 2005; Russell 2008; Arnold 2010; Arnold 2012)). One study (Chappell 2008) did not stipulate any criteria for pain at entry and participants could have, or not have, major depressive disorder. These trials were also conducted by the same drug company that performed the diabetic neuropathy studies. One study included only women (Arnold 2005) and the other five included over 90% women, despite being open to males and females, reflecting the epidemiology of this condition. In the five studies that gave ages (Arnold 2004; Chappell 2008; Russell 2008; Arnold 2010; Arnold 2012), the participants were approximately 10 years younger than in the diabetic peripheral neuropathy trials. Participants had similar levels of pain at entry in each trial even though there were no pain entry criteria for Chappell 2008. Only Arnold 2005 stated the duration of pain at entry (> 12 weeks). All six trials were blinded. Participants in Arnold 2004, Arnold 2005 and Russell 2008 received duloxetine in capsules or identical appearing placebos in identical dosage schedules in outpatient research facilities for 12 weeks (Arnold 2004; Arnold 2005), or for 28 weeks (Russell 2008). Arnold 2010 and Chappell 2008 used tablets and also a rather more complex dosing schedule, where the starting dose was 60 mg once daily with a 30 mg run‐in phase for one week. In Chappell 2008, there was then a randomised increase to 120 mg after 13 weeks if participants had not reached a reduction of > 50% in pain on the BPI average pain score. In Arnold 2010, after a one‐week 30 mg run‐in, all participants in the active arm received 60 mg; the treating physician then increased the dose after four weeks to 90 mg or 120 mg if there was less than 50% improvement on the BPI scale (participants were blinded to the increase in dose). The final doses at week 12 were: 60 mg, n = 137 (52.1%); 90 mg, n = 62 (23.6%); and 120 mg, n = 64 (24.3%). The Russell 2008 trial included a two‐week titration phase and a two‐week taper. Finally, Arnold 2012 used a low dose of 30 mg only, presented as capsules.
We included three studies of painful physical symptoms in major depressive disorder (MDD) (Brecht 2007; Gaynor 2011a; Gaynor 2011b). The trials included participants if they had a diagnosis of MDD and painful physical symptoms and were devoid of an alternative pain syndrome. Treatment in all three studies was eight weeks of duloxetine 60 mg or placebo and although there was a slightly higher proportion of men in the studies of Gaynor et al. than in Brecht 2007, the studies were otherwise well matched for pain, age, depression scores and other demographic characteristics. The studies provide no details of the quality or somatic distribution of the types of pain experienced.
One study (Vranken 2011) examined the effect of duloxetine compared versus placebo in participants with central neuropathic pain. Those eligible were over 18 years old with more than six months' severe neuropathic pain of spinal cord or cerebrovascular origin. Participants had a score of more than six on a 10‐point VAS. This trial was not company‐sponsored. The starting dose of duloxetine was initially 60 mg, which increased if participants did not improve by more than 1.8 points on VAS.
Excluded studies
We excluded 29 trials for a number of reasons (see Characteristics of excluded studies). Two excluded texts were summary reports of other studies that we have included (Raskin 2005a; Russell 2006). Of the trials that did not meet the pre‐defined criteria for inclusion in this review, seven were for conditions outside the remit of the review (NCT01451606; Chappell 2009; Skljarevski 2008; Skljarevski 2009; Skljarevski 2010; Skljarevski 2010b; Chappell 2011). Eight were open studies without blinding or a control group (NCT00125892; NCT00385671; NCT00552682; NCT00641719; Raskin 2006b; Skljarevski 2009a; Tanenberg 2011; Wu 2006). Canovas 2007 was neither randomised nor controlled. The Raskin 2006a trial (in diabetic peripheral neuropathic pain) was randomised but not blinded; it is mentioned in the Discussion of this review. The Wernicke 2006b study was a 52‐week extension of the Goldstein 2005 randomised trial with a similar design but without blinding. The NCT00266643, Brannan 2005, Vollmer 2011, Boyle 2012, Lavoie Smith 2012, Smith 2013 (abstract of Lavoie Smith 2012) and Harrison 2013 trials measured outcomes at durations of less than eight weeks, not the eight weeks stipulated in our protocol. Goldstein 2004 tested duloxetine for eight weeks for depression, but included pain scales as secondary outcome measures. However, it was not clear what sort of pain the participants had (for example musculoskeletal, neuropathic, or headache) and the levels of pain at baseline were low compared to the included trials. NCT00425230 was registered in ClinicalTrials.gov but was terminated before inclusion of participants.
Ongoing and completed but unpublished trials
A search of ClinicalTrials.gov revealed five ongoing studies in pain from multiple sclerosis (1 study), diabetic neuropathy (2 studies) and fibromyalgia (2 studies) (NCT00457730; NCT00619983; NCT01179672; NCT01237587; NCT01552057) (see Characteristics of ongoing studies). More importantly, we were unable to find results for five potentially eligible studies registered as 'completed' in ClinicalTrials.gov (NCT00125892, NCT00233025, NCT00489073, NCT00603265 and NCT01579279), as the results were not available in ClinicalTrials.gov, the trials were not identifiable as publications in the Lilly Trials register or a publication database, and we were unable to obtain the study reports through writing to the study investigators, where identifiable. Vollmer 2011 may be NCT00755807 on ClinicalTrials.gov, where results are presented, but although there is a published abstract there is no published full paper at the time of publication of this review.
Risk of bias in included studies
The 18 included studies were variable in their risk of bias (see Figure 1 and Characteristics of included studies). Eight of the studies were at a high risk of bias for at least one attribute. Seven of the studies had an unclear risk of bias in one or more domains, because of various problems. In only three studies was the risk of bias deemed to be low across all the attributes; one of the three being a study from the drug company. Interestingly, despite common authors across studies and development by the company of methodology and outcomes selection over time, the risk of bias in studies did not improve and some data are noticeably absent from later studies. Nearly all the studies had a dropout rate of more than 20% (only Raskin 2005, Gao 2010, Yasuda 2010 and Tesfaye 2013 had a dropout rate of less than 20%), which was deemed to change the overall risk of bias from low to unclear. Dropouts in Rowbotham 2012 (a relatively small study of the novel Abbott agent ABT‐894 versus duloxetine) and Arnold 2012 (a negative trial that used a subtherapeutic dose of duloxetine) were particularly low. The same company sponsored and performed all but one of the included studies (Vranken 2011).
1.
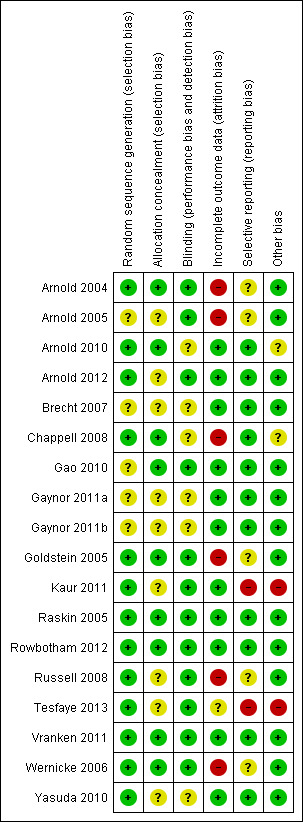
Methodological quality summary: review authors' judgements about each methodological quality item for each included study. Green = low risk of bias; yellow = unclear risk of bias; red = high risk of bias
The issue of incomplete outcome data in Arnold 2004 was unclear from the publication; 36% of the placebo group and 44% of the duloxetine group discontinued the study. From information provided by the authors, the analysis included all participants with at least one follow‐up measurement from baseline, with the last observation carried forward.
A number of trials used exploratory assessments of statistical processing including presentation of multiple statistical analyses, for example last observation carried forward (LOCF) and baseline observation carried forward (BOCF) data, leading to a suspicion of post hoc data mining using exploratory statistics.
Effects of interventions
See: Table 1; Table 2; Table 3
We analysed the effects of the interventions for painful peripheral neuropathy, fibromyalgia, central pain, and painful physical symptoms in major depressive disorder separately; we did not perform meta‐analysis combining all of the trials across the four 'conditions'. We identified no includable trials of duloxetine for other causes of neuropathic pain, although many low quality studies exist in other diseases such as post herpetic neuralgia.
All studies included adverse event data that had been sought prospectively and which the trial authors reported in detail. We analysed these across conditions.
None of the randomised trials included health economic data.
Painful peripheral neuropathy ‐ duloxetine versus placebo
Primary outcome: short‐term (up to and including 12 weeks) improvement of pain compared with baseline
Five trials in painful diabetic neuropathy reported data on the primary outcome measure of ≥ 50% improvement of pain compared with baseline at less than 12 weeks (Goldstein 2005; Raskin 2005; Wernicke 2006; Gao 2010; Yasuda 2010). Participants received duloxetine 20 mg, 40 mg, 60 mg or 120 mg per day. Combining data from all doses from the five trials together (1655 participants), the RR of ≥ 50% improvement with any dose was 1.53 (95% CI, 1.21 to 1.92) compared with placebo (see Figure 2, Analysis 1.1). The RR of improvement was significantly greater than placebo for the 40 mg, 60 mg and 120 mg daily doses but not the 20 mg daily dose, for which it was 1.43 (95% CI 0.98 to 2.09; the CIs for 20 mg were wide as only one study with few participants provided data). There was no significant difference nor a dose effect in the RR of improvement with increasing doses of duloxetine from 40 mg to 120 mg. Significant heterogeneity in the 'all doses' and the 120 mg dose analysis is explained by the inclusion of Gao 2010. As it was not clear how many participants completed that trial on doses of 60 mg or 120 mg daily, we assumed for the purposes of the analysis that the higher dose was reached by all. Removing Gao 2010 removed the heterogeneity and slightly increased the RR of benefit for 'all doses' (RR 1.68, 95% CI 1.41 to 2.02).
2.
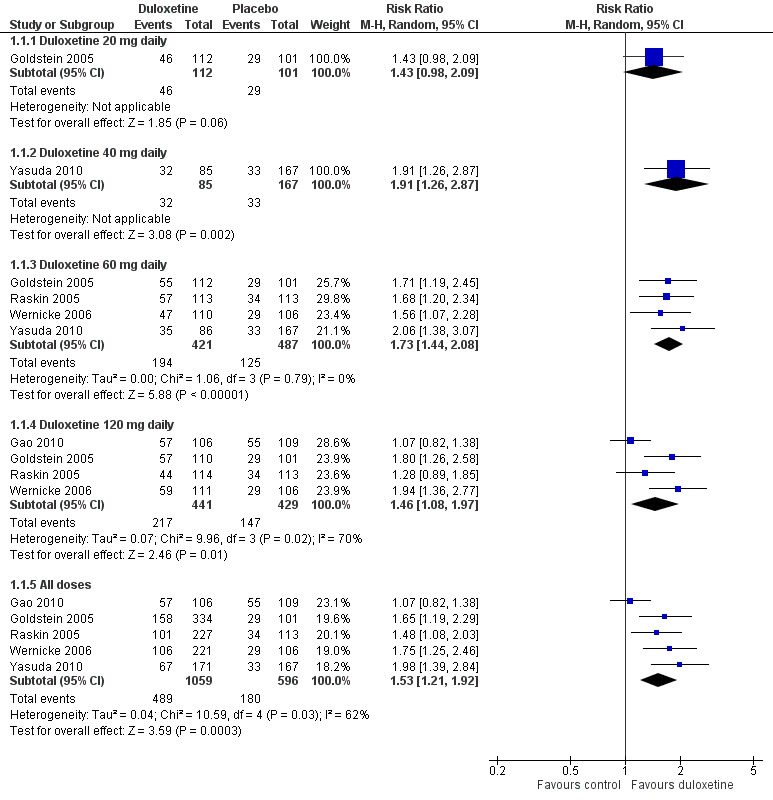
Duloxetine versus placebo in the treatment of painful neuropathy: Number of patients with >50% improvement of pain at <12 weeks.
1.1. Analysis.
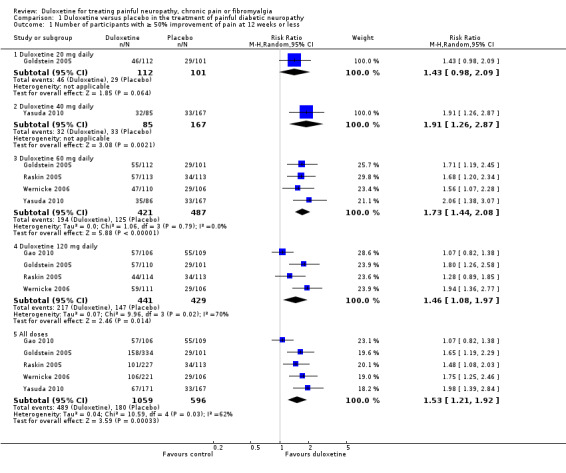
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 1 Number of participants with ≥ 50% improvement of pain at 12 weeks or less.
The mean improvement in pain at 12 weeks or less on an 11‐point Likert scale was significantly greater than placebo with the 60 mg dose of duloxetine (MD ‐0.96, 95% CI ‐1.26 to ‐0.65; 4 trials, 722 participants) and the 120 mg dose (MD ‐0.93, 95% CI ‐1.21 to ‐0.65; 4 trials, 828 participants), but not with the 20 mg dose (see Figure 3, Analysis 1.2, 1 trial, 179 participants, wide CIs from the single study). Removal of the Gao 2010 data removed the heterogeneity contributed by this study and the data then indicated a dose effect (MD ‐1.16, 95% CI ‐1.49 to ‐0.83; 3 trials, 612 participants) (see Analysis 1.2, Figure 3).
3.
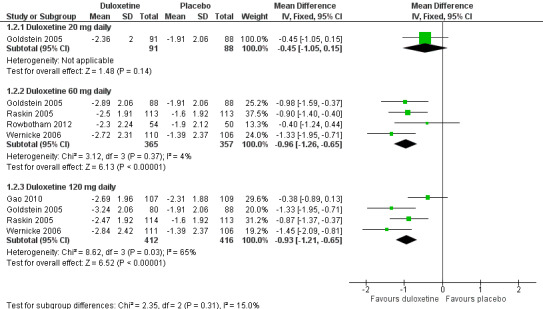
Duloxetine versus placebo in the treatment of pain: Mean improvement in pain at 12 weeks.
1.2. Analysis.
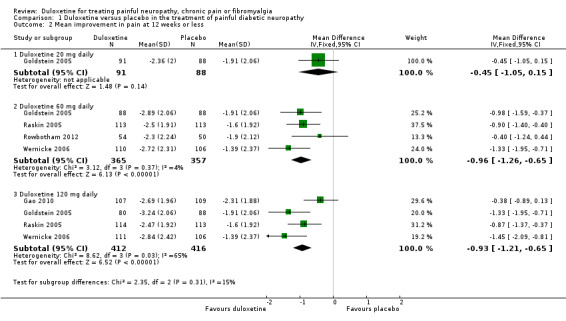
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 2 Mean improvement in pain at 12 weeks or less.
The quality of the evidence available for this outcome remains moderate, mainly as a result of relatively high dropout rates. Lilly sponsored and performed all of these studies but there is no significant suspicion of publication bias despite a number of trials remaining without an identified publication in ClinicalTrials.gov.
Secondary outcomes
None of the included trials of painful diabetic neuropathy reported outcomes at more than 12 weeks.
Five trials included data on ≥ 30% improvement of pain at 12 weeks or less (Raskin 2005; Wernicke 2006; Gao 2010; Yasuda 2010; Rowbotham 2012). The results were similar to those for at least 50% improvement, as was the heterogeneity introduced by Gao 2010. Relative rates of improvement were significantly greater than placebo with duloxetine for the 40 mg dose (RR 1.57, 95% CI 1.18 to 2.07; 1 trial, 252 participants), the 60 mg dose (RR 1.53, 95% CI 1.33 to 1.75; 4 trials, 799 participants), the 120 mg dose (RR 1.38, 95% CI 1.21 to 1.58; 3 trials, 659 participants) and for all three doses combined (RR 1.45, 95% CI 1.30 to 1.63; 4 trials, 1220 participants) (see Figure 4, Analysis 1.3). With Gao excluded for heterogeneity, the RR for 120 mg is 1.55, 95% CI 1.30 to 1.86 (444 participants), and for all doses, 1.57, 95% CI 1.37 to 1.80 (1005 participants). Data for this outcome for the 20 mg dose were not available.
4.
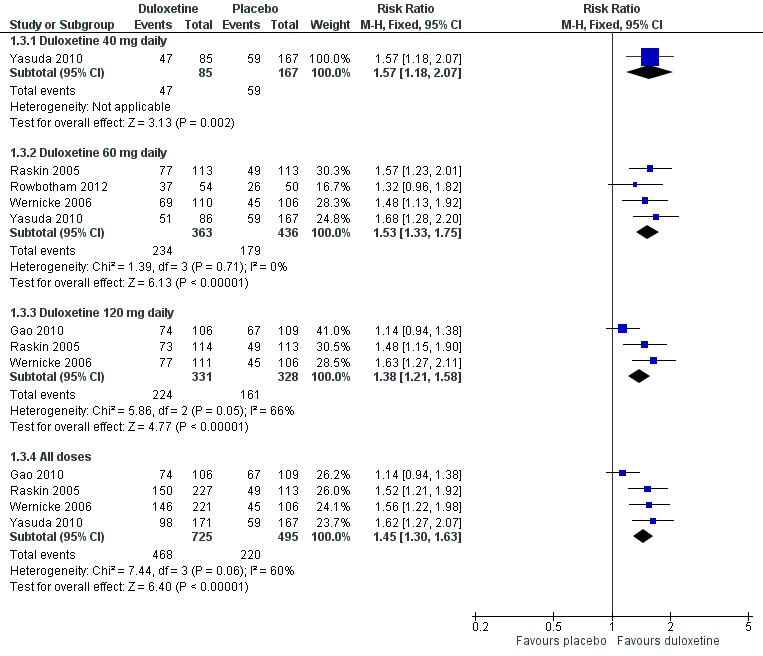
Duloxetine versus placebo in the treatment of pain: Number of patients with >30% improvement in pain at <12 weeks.
1.3. Analysis.
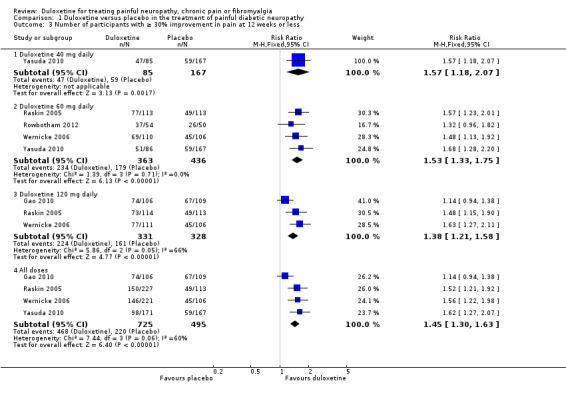
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 3 Number of participants with ≥ 30% improvement in pain at 12 weeks or less.
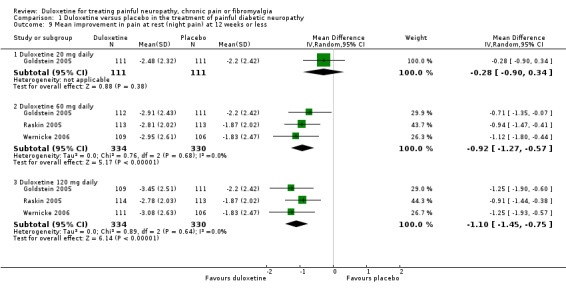
Trials that included quality of life information used the SF‐36. We included data on the relevant physical, mental and bodily pain subsections of the SF‐36. In painful diabetic neuropathy, the effect of 20 mg duloxetine was not significantly different from placebo on any of the selected SF‐36 subscores at 12 weeks or less (Raskin 2005; Wernicke 2006; Rowbotham 2012), or the mental subscore at 60 mg daily doses. The MD of improvement in the physical summary component was significantly greater than placebo with the 60 mg dose (2.65, 95% CI 1.38 to 3.92; 3 trials, 514 participants) and 120 mg dose (2.80, 95% CI 1.04 to 4.55; 2 trials, 409 participants) (see Analysis 1.4). The MD on the mental summary component was significantly greater than placebo only with the 120 mg dose (2.23, 95% CI 0.69 to 3.77; 2 trials, 409 participants) (see Analysis 1.5). The MD on the bodily pain subscale showed significantly more improvement than placebo with the 60 mg dose (5.58, 95% CI 1.74 to 9.42; 2 trials, 421 participants) and even more with the 120 mg dose (8.19, 95% CI 4.33 to 12.05; 2 trials, 420 participants) but not with the 20 mg dose (1 trial, 209 participants) (see Analysis 1.6).
1.4. Analysis.
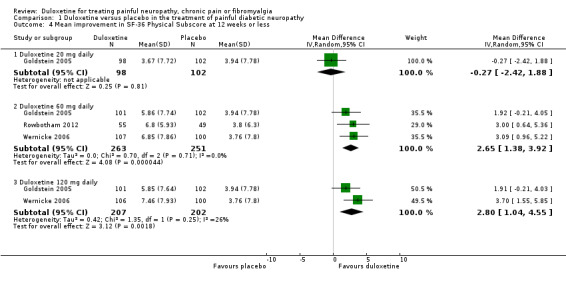
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 4 Mean improvement in SF‐36 Physical Subscore at 12 weeks or less.
1.5. Analysis.
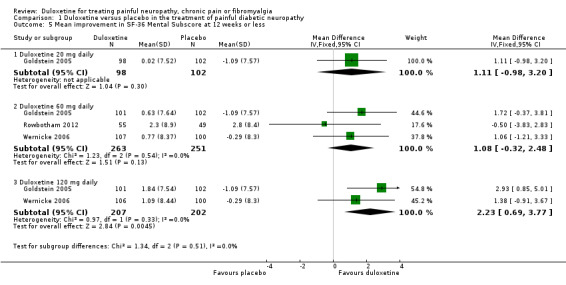
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 5 Mean improvement in SF‐36 Mental Subscore at 12 weeks or less.
1.6. Analysis.
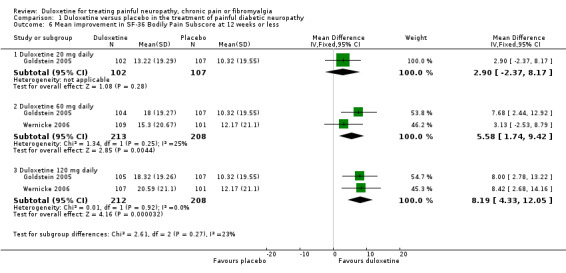
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 6 Mean improvement in SF‐36 Bodily Pain Subscore at 12 weeks or less.
Six studies reported the PGIC (Gao 2010; Goldstein 2005; Raskin 2005; Rowbotham 2012; Wernicke 2006; Yasuda 2010), three reported pain at rest (night pain) (Goldstein 2005; Raskin 2005; Wernicke 2006), and two reported the Brief Pain Inventory (BPI) (Raskin 2005; Wernicke 2006). Mean improvements only were reported. The MD versus placebo for each outcome was not significant for the 20 mg dose (1 study, 219 participants), but was significant and similar in magnitude for the 60 mg and 120 mg doses (see Figure 5, Analysis 1.7). However, a minimum clinically meaningful difference in the PGIC is suggested as one point (Dworkin 2008), and hence the change associated with 60 mg duloxetine (MD ‐0.60, 95% CI ‐0.77 to ‐0.44; 5 trials, 1018 participants) is unlikely to be clinically significant. The RR for the BPI with duloxetine 60 mg is statistically significantly reduced by ‐0.97 (95% CI ‐1.38 to ‐0.57; 2 trials, 433 participants), which borders on the change considered clinically significant (Dworkin 2008) (see Analysis 1.8; Figure 6). With duloxetine 120 mg, the MD reached the minimum clinically significant threshold (‐1.16, 95% CI ‐1.91 to ‐0.41; 2 trials, 428 participants). The mean difference of improvement in pain at rest at 12 weeks was significantly greater than placebo with duloxetine 60 mg and 120 mg daily (2 trials, 664 participants), but not with 20 mg daily (1 trial, 222 participants) (Analysis 1.9).
5.
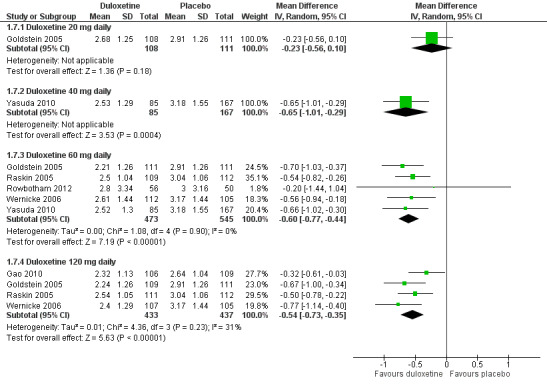
Duloxetine versus placebo in the treatment of pain: Patient reported global impression of change.
1.7. Analysis.
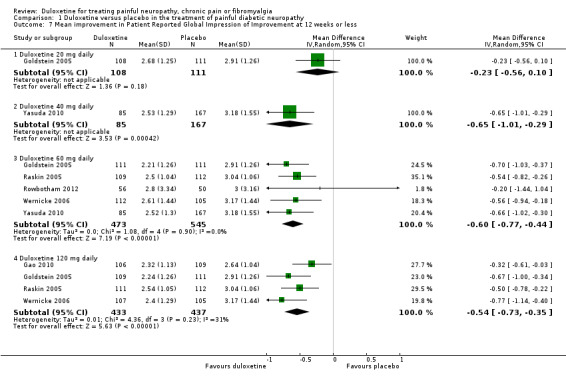
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 7 Mean improvement in Patient Reported Global Impression of Improvement at 12 weeks or less.
1.8. Analysis.
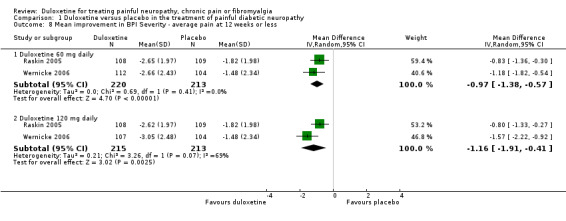
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 8 Mean improvement in BPI Severity ‐ average pain at 12 weeks or less.
6.
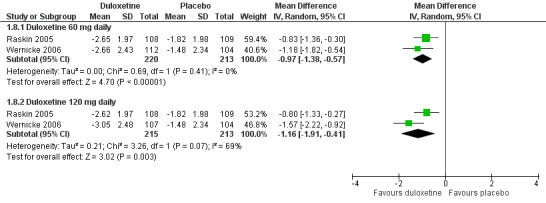
Duloxetine versus placebo in the treatment of pain: BPI severity ‐ average pain.
1.9. Analysis.
Comparison 1 Duloxetine versus placebo in the treatment of painful diabetic neuropathy, Outcome 9 Mean improvement in pain at rest (night pain) at 12 weeks or less.
Heterogeneity
Inclusion of data contributed by Gao 2010 caused heterogeneity in meta‐analyses. Where this was the case we repeated the analyses excluding the Gao 2010 data. The heterogeneity was probably the result of the estimated final doses of duloxetine used in the analysis figures.
Significant heterogeneity was not otherwise present except for the SF‐36 physical component summary, PGIC, bodily pain index and pain at rest. Heterogeneity was present in the subgroup analyses and also in the 'all doses' analysis, where doses were combined. The origin of this heterogeneity was not always clear and therefore we performed these analyses with a random‐effects model.
Sensitivity analysis
We attempted prespecified sensitivity analyses. All trials were carried out by the drug manufacturer and all had more than 100 participants. However, only three of the included studies had a dropout rate of less than 20%. When we included only these studies in the analysis, although duloxetine remained significantly effective (number of participants achieving ≥ 50% reduction in pain RR 1.83 (95% CI 1.41 to 2.36), the effect of duloxetine at 120 mg was lost and the effect of duloxetine at all doses was barely significant (RR 1.55, 95% CI 1.01 to 2.38).
Painful peripheral neuropathy ‐ duloxetine versus pregabalin
In the only comparison of duloxetine and pregabalin (Tesfaye 2013) (804 participants), the proportion of participants responding to duloxetine 60 mg by achieving 50% or more reduction in pain was significantly greater than those responding to pregabalin 300 mg daily (RR 1.46, 95% CI 1.19 to 1.80) (see Analysis 2.1). Both doses represent realistic therapeutic target doses for treatment. The magnitude of change was also greater for duloxetine than pregabalin (RR ‐0.62, 95% CI ‐0.92 to ‐0.32) (see Analysis 2.2). The number improved by 30% or more at 12 weeks was significantly greater with duloxetine than placebo (RR 1.42, 95% CI 1.20 to 1.68) (see Analysis 2.3).
2.1. Analysis.
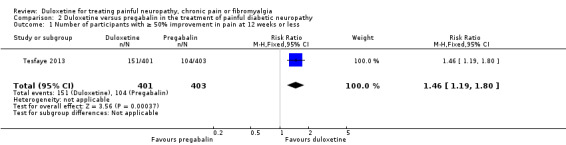
Comparison 2 Duloxetine versus pregabalin in the treatment of painful diabetic neuropathy, Outcome 1 Number of participants with ≥ 50% improvement in pain at 12 weeks or less.
2.2. Analysis.
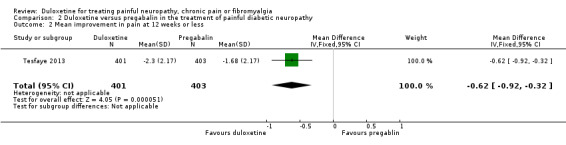
Comparison 2 Duloxetine versus pregabalin in the treatment of painful diabetic neuropathy, Outcome 2 Mean improvement in pain at 12 weeks or less.
2.3. Analysis.
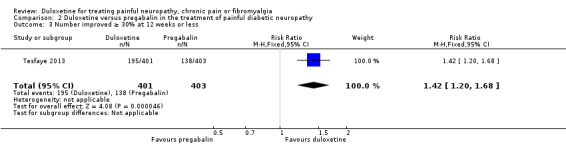
Comparison 2 Duloxetine versus pregabalin in the treatment of painful diabetic neuropathy, Outcome 3 Number improved ≥ 30% at 12 weeks or less.
The response rate for a ≥ 50% reduction in pain for duloxetine was 38%, whereas the 26% response rate to pregabalin was approximately the same as the placebo response rate in the other trials of duloxetine at 60 mg (compare Analysis 1.1 and Analysis 2.1). This raises questions about the similarity of the selected groups of participants or the efficacy of pregabalin, which is known, however, to be effective in other studies.
Painful peripheral neuropathy ‐ duloxetine versus amitriptyline
The only trial comparing duloxetine to amitriptyline (Kaur 2011) was a blinded cross‐over study with 62 participants comparing six weeks treatment with each active agent in escalating dose: duloxetine up to 60 mg and amitriptyline up to 50 mg. The trial did not meet our predefined inclusion criteria of eight weeks of study medication but is included here for completeness as it is the only comparative trial of its type. Significant carryover effects were evident (VAS pain scores only returned to 75% of baseline during washout (see Figure 2 of the Kaur 2011 paper)). In addition, a number of predefined outcome measures were not presented in the results, so there was an unclear risk of bias. Sixty‐five per cent of participants achieved 60 mg of duloxetine per day and 48% of participants 50 mg amitriptyline. The majority of participants (59% duloxetine and 55% amitriptyline) were reported to have achieved a 'good' (> 50% improvement) response to the interventions.
It is not possible to re‐analyse the data as no raw data are available. We contacted the authors by email to provide original data to enter into a GIV analysis. No reply was forthcoming.
Trial sequential analysis in painful peripheral neuropathy
We performed TSA for the primary outcome of a ≥ 50% reduction in pain at 12 weeks or less with at least 8 weeks of treatment with duloxetine, for the trials that compared duloxetine to placebo. There was not enough information to explore the pairwise trials. The TSA report demonstrated that although the optimal information size had not been reached, the Z‐score favoured duloxetine and diverged from futility (Figure 7).
7.

Trial sequential analysis of duloxetine versus placebo in the treatment of painful neuropathy ‐ 50% or more reduction in pain at 8‐12 weeks with at least 8 weeks of treatment
Fibromyalgia ‐ duloxetine versus placebo
Primary outcome
FIve trials reported data corresponding to the primary outcome for this review (Arnold 2004; Arnold 2005; Russell 2008; Arnold 2010; Arnold 2012) and the sixth reported data for the same outcome at more than 12 weeks (Chappell 2008). The studies used two scales; in Arnold 2004, the Fibromyalgia Impact Questionnaire pain score (Burckhardt 1991), and in the remainder, the BPI modified SF‐36 average pain severity score. The 20 mg dose of duloxetine in Russell 2008 (223 participants) did not show significant differences in any of the reported measures. The 30 mg dose, used only in Arnold 2012 (308 participants), was also negative on all measures of outcome except for a statistically significant benefit on the Patient Global Impression of Improvement (PGI‐I) (interchangeable with the Patient Global Impression of Change (PGI‐C)) and the mental component ot the SF‐36, neither of which were of a magnitude to be clinically significant. Both of these studies had wide CIs because of the small number of participants (Analysis 3.1). Five studies reported short‐term (up to and including 12 weeks) ≥ 50% improvement of pain compared with baseline. The RR of improvement was significantly greater with duloxetine 60 mg (1.57, 95% CI 1.20 to 2.06; 2 trials, 528 participants) and with 120 mg daily (1.69, 95% CI 1.40 to 2.03; 4 trials, 1234 participants) than with placebo (see Analysis 3.1). The RR of improvement compared with placebo for all doses in all five short‐term trials, which had a total of 1887 participants, was 1.50 (95% CI 1.29 to 1.75). Exclusion of the data for 111 participants in the Arnold 2012 study, which was responsible for the heterogeneity in the analysis, gave an RR of 1.68 (95% CI 1.41 to 2.01).
3.1. Analysis.
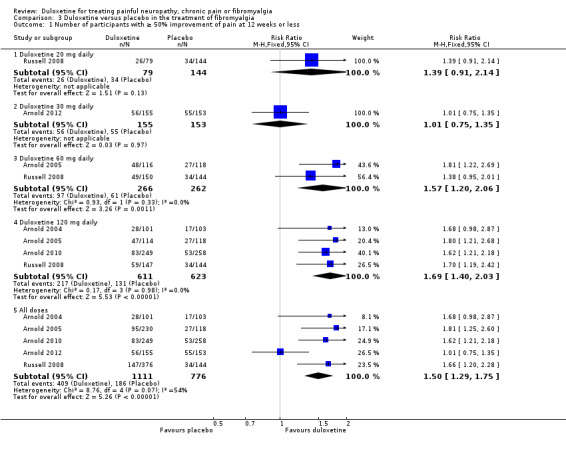
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 1 Number of participants with ≥ 50% improvement of pain at 12 weeks or less.
Secondary outcomes
Two studies looked at long‐term outcomes at more than 12 weeks (Chappell 2008; Russell 2008). These investigators documented outcomes at 28 weeks. Improvement of pain ≥ 50% compared with baseline at 27 to 28 weeks was similar between the 60 mg and 120 mg doses (no dose effect) and when all 989 participants were combined, despite the Chappell trial being negative, the RR for improvement was 1.40 (95% CI 1.09 to 1.79) (see Analysis 3.2).
3.2. Analysis.
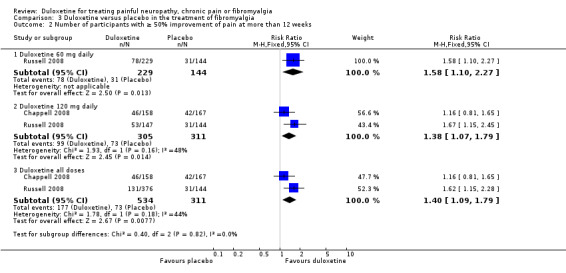
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 2 Number of participants with ≥ 50% improvement of pain at more than 12 weeks.
The RR of ≥ 30% improvement at 12 weeks or less was significantly greater than placebo with the duloxetine 60 mg dose (RR 1.52, 95% CI 1.24 to 1.85; 2 trials, 528 participants) and 120 mg dose (RR 1.46, 95% CI 1.26 to 1.69; 3 trials, 1020 participants) but not with the 20 mg or 30 mg doses (see Figure 8, Analysis 3.3). The RR for all doses combined was 1.38 (95% CI 1.22 to 1.56; 4 trials, 1673 participants). It is notable that no dose effect exists from 60 mg to 120 mg. There was no statistically significant improvement in pain at 30 mg duloxetine in the only trial presenting data for the mean improvement but at 120 mg there was a significant benefit in favour of duloxetine (MD ‐0.80, 95% CI ‐1.35 to ‐0.25; 1 trial, 507 participants) (see Analysis 3.4).
8.
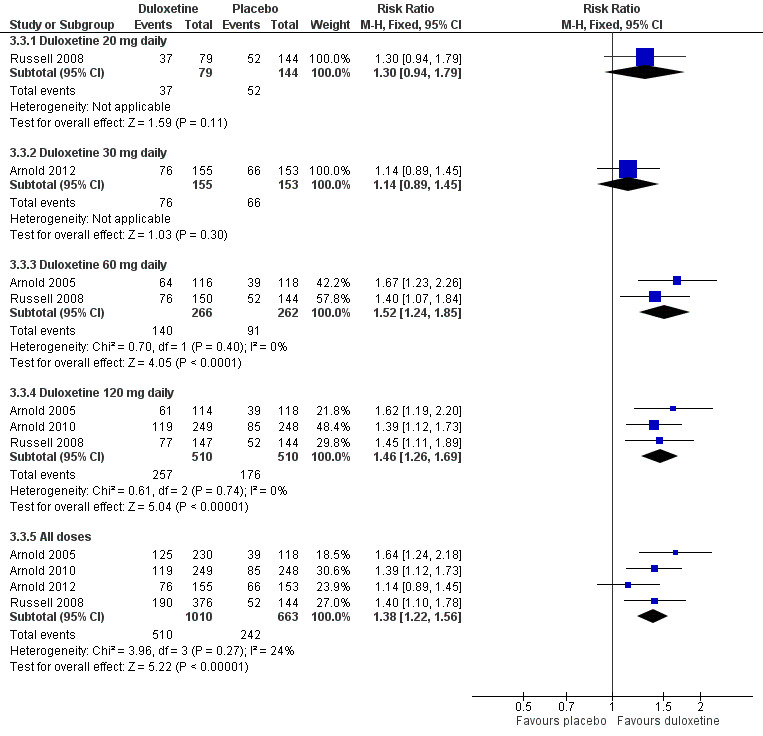
Duloxetine versus placebo in the treatment of fibromyalgia: >30% improvement <12 weeks.
3.3. Analysis.
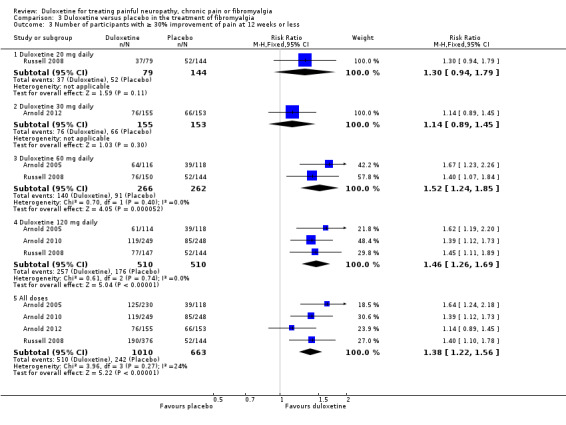
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 3 Number of participants with ≥ 30% improvement of pain at 12 weeks or less.
3.4. Analysis.
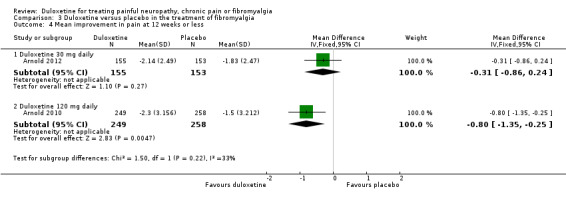
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 4 Mean improvement in pain at 12 weeks or less.
All six studies documented the physical component summary scores and bodily pain subscores of the SF‐36 and most reported the bodily pain subscore (Arnold 2004; Arnold 2005; Chappell 2008; Russell 2008; Arnold 2010; Arnold 2012). For the mental component summary score, the 30 mg, 60 mg and 120 mg doses had increasing effect compared to placebo (for the 120 mg dose, MD 4.22, 95% CI 2.43 to 6.02; 5 trials, 1531 participants) (see Analysis 3.5). Interestingly, the physical component summary score was only significant at the 120 mg dose of duloxetine (MD 2.13, 95% CI 0.95 to 3.30; 5 trials, 1531 participants) (see Analysis 3.6). For the bodily pain subscale, the RR of improvement from four studies (Arnold 2004; Arnold 2005; Chappell 2008; Arnold 2010) was significantly greater for duloxetine than placebo at both the 60 mg dose (MD 8.20, 95% CI 3.20 to 13.20; 1 trial, 221 participants) and the 120 mg dose (MD 5.96, 95% CI 3.76 to 8.16; 4 trials, 1243 participants) (Figure 9, Analysis 3.7). Again, it is notable that the 120 mg dose had less effect than the 60 mg dose.
3.5. Analysis.
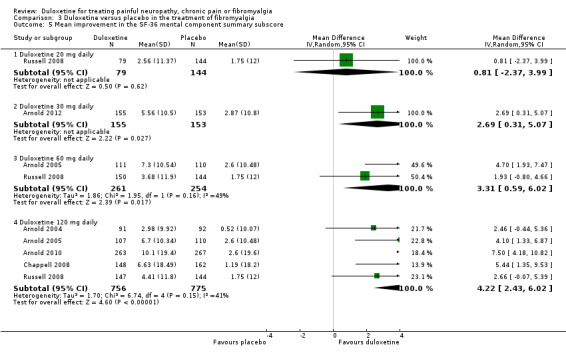
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 5 Mean improvement in the SF‐36 mental component summary subscore.
3.6. Analysis.
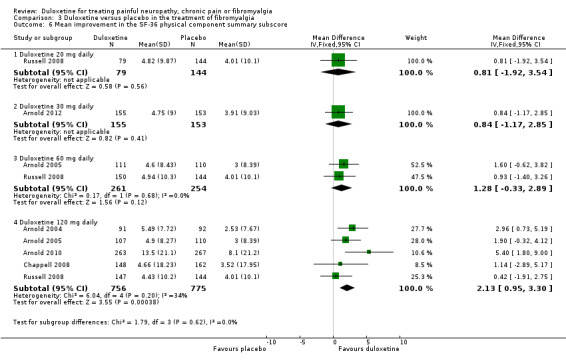
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 6 Mean improvement in the SF‐36 physical component summary subscore.
9.
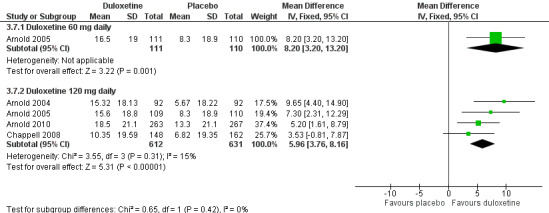
Duloxetine versus placebo in the treatment of fibromyalgia: SF‐36 bodily pain.
3.7. Analysis.
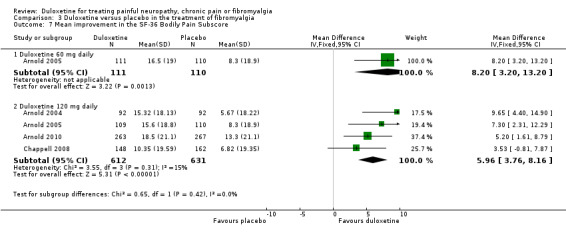
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 7 Mean improvement in the SF‐36 Bodily Pain Subscore.
Four studies reported the PGI‐I (Arnold 2005; Russell 2008; Chappell 2008; Arnold 2012), which was significantly in favour of duloxetine at the 20 mg, 30 mg, 60 mg and 120 mg doses (120 mg dose MD ‐0.44, 95% CI ‐0.66 to ‐0.23; 3 trials, 826 participants) (see Analysis 3.8). The magnitude of change at each dose was very similar, with no dose effect. However, the magnitude of change failed to reach a level considered to be clinically significant.
3.8. Analysis.
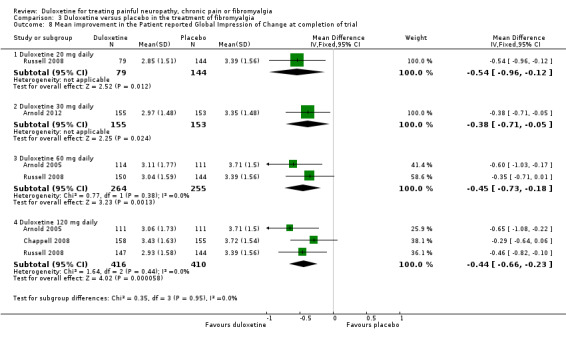
Comparison 3 Duloxetine versus placebo in the treatment of fibromyalgia, Outcome 8 Mean improvement in the Patient reported Global Impression of Change at completion of trial.
Sensitivity analysis
No data were suitable for the prespecified sensitivity analyses.
Trial sequential analysis in fibromyalgia
We performed TSA on the primary outcome data for ≥ 50% reduction in pain at ≤ 12 weeks with at least eight weeks of treatment with 60 mg duloxetine (the standard dose). WIth the data so far available, the Z‐score only just crossed the boundary of significance, although it was divergent from futility (Figure 10). The optimal information size is some way off and more trials and participants are required to make convincing statements about the efficacy of duloxetine for this indication at this dose.
10.

Trial sequential analysis of duloxetine 60 mg versus placebo for the 50% reduction in pain in fibromyalgia with at least 8 weeks treatment at 8‐12 weeks
Painful physical symptoms not explained by any known alternative diagnosis in the context of major depressive disorder ‐ duloxetine versus placebo
Primary outcome
Painful physical symptoms associated with major depressive disorder have been assessed in three studies each lasting eight weeks that used a duloxetine dose of 60 mg daily (Brecht 2007; Gaynor 2011a; Gaynor 2011b). The proportion of participants achieving ≥ 50% pain relief was greater with duloxetine than placebo in two studies (1023 participants) for which adequate information was available (RR 1.37, 95% CI 1.19 to 1.59) (see Analysis 4.1) and the magnitude of improvement was greater in those two studies (MD ‐0.55, 95% CI ‐0.75 to ‐0.35) (see Analysis 4.3). No data were available for Brecht 2007 in the comparison of magnitude as the report does not provide SDs, although the absolute magnitude of improvement was similar to the two studies of Gaynor.
4.1. Analysis.
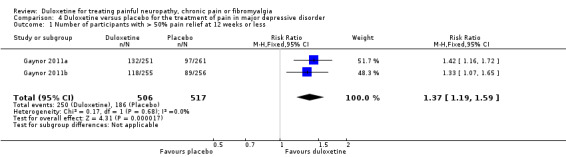
Comparison 4 Duloxetine versus placebo for the treatment of pain in major depressive disorder, Outcome 1 Number of participants with > 50% pain relief at 12 weeks or less.
4.3. Analysis.
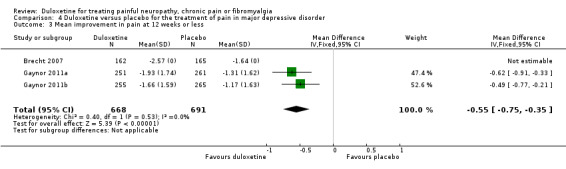
Comparison 4 Duloxetine versus placebo for the treatment of pain in major depressive disorder, Outcome 3 Mean improvement in pain at 12 weeks or less.
Secondary outcomes
More participants improved ≥ 30% in their levels of pain than with placebo at 60 mg duloxetine per day (RR 1.27, 95% CI 1.15 to 1.40; 3 studies, 1359 participants (see Analysis 4.2).
4.2. Analysis.
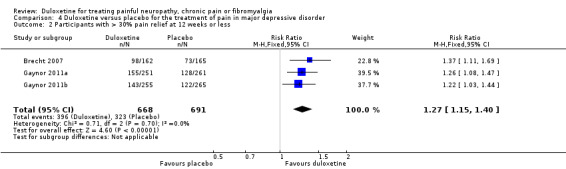
Comparison 4 Duloxetine versus placebo for the treatment of pain in major depressive disorder, Outcome 2 Participants with > 30% pain relief at 12 weeks or less.
No data were available for the subscores of the SF‐36 or to calculate a mean improvement in the PGI‐I scores.
Sensitivity analysis
No data were suitable for the prespecified sensitivity analyses.
Trial sequential analysis in painful physical symptoms in depression
TSA was performed on the primary outcome data for ≥ 50% reduction in pain at <12 weeks with at least 8 weeks of treatment with 60 mg duloxetine (the standard dose). The optimal information size was exceeded but there were not enough data to calculate an area of futility (Figure 11). However, there is convincing evidence from the small number of studies that duloxetine is efficacious.
11.

Trial Sequential Analysis of duloxetine 60 mg versus placebo in the treatment of painful physical symptoms in depression at less than 12 weeks with at least eight weeks of treatment
Central neuropathic pain ‐ duloxetine versus placebo
A single investigator‐led study with a low risk of bias but only 48 participants looked at the effect of duloxetine in people with central neuropathic pain (Vranken 2011). This was the only trial not sponsored or run by the company manufacturing duloxetine. However, it was the smallest of the studies and only one of three with no significant risk of bias.
There was no therapeutic effect of duloxetine on the neuropathic pain of the participants in this study on any of our pre‐defined outcome measures that the study reported (see Analysis 5.1; Analysis 5.2; Analysis 5.3; Analysis 5.4). There was a borderline effect in the bodily pain domain of the SF‐36 (MD 8.00, 95% CI ‐0.81 to 16.81), which did not reach significance, and the proportion of participants reporting improvement on the PGI‐I was just significant (RR 2.75, 95% CI 1.02 to 7.44) (see Analysis 5.4 and Analysis 5.5). The trial also reported a statistically significant improvement in the severity of dynamic and cold allodynia. Given that the trial was small, the trial authors recommended that more studies of central neuropathic pain are performed.
5.1. Analysis.
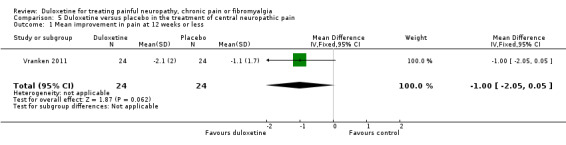
Comparison 5 Duloxetine versus placebo in the treatment of central neuropathic pain, Outcome 1 Mean improvement in pain at 12 weeks or less.
5.2. Analysis.
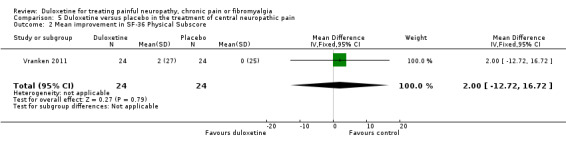
Comparison 5 Duloxetine versus placebo in the treatment of central neuropathic pain, Outcome 2 Mean improvement in SF‐36 Physical Subscore.
5.3. Analysis.
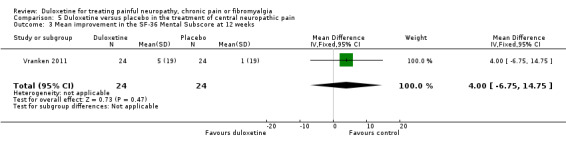
Comparison 5 Duloxetine versus placebo in the treatment of central neuropathic pain, Outcome 3 Mean improvement in the SF‐36 Mental Subscore at 12 weeks.
5.4. Analysis.
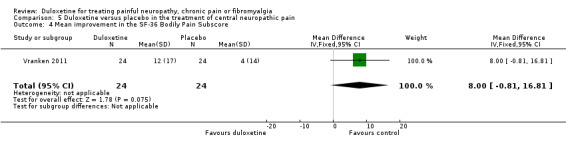
Comparison 5 Duloxetine versus placebo in the treatment of central neuropathic pain, Outcome 4 Mean improvement in the SF‐36 Bodily Pain Subscore.
5.5. Analysis.
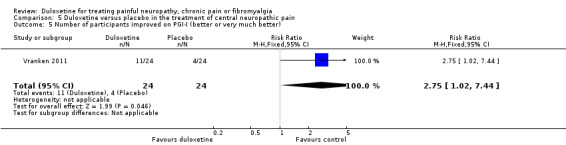
Comparison 5 Duloxetine versus placebo in the treatment of central neuropathic pain, Outcome 5 Number of participants improved on PGI‐I (better or very much better).
Sensitivity analysis
Vranken 2011 was the only trial of central neuropathic pain and no meta‐analysis or sensitivity analysis was therefore possible.
Trial sequential analysis ‐ central neuropathic pain
We did not perform TSA on the single study available.
Adverse events (all indications)
We analysed adverse events across all included studies (all indications).
Serious adverse events were uncommon and were no more frequent with duloxetine than placebo at any dose or when combining all doses together (42 events in 2785 duloxetine‐treated participants versus 39 events in 2191 placebo participants RR 0.81 (95% CI 0.53 to 1.25) (see Analysis 6.7).
6.7. Analysis.
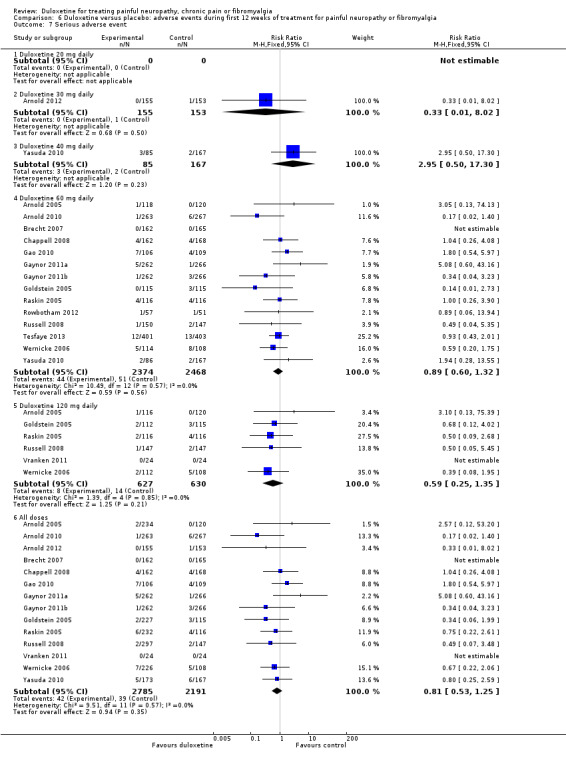
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 7 Serious adverse event.
Adverse events of any sort, however, were very common in all of the trials in both experimental and placebo groups.
The rate of any adverse event was high in both the treatment and placebo arms of all studies, with 1530 adverse events being reported in 2462 control participants and 2033 adverse events occurring in 2796 participants in the combined treatment arms covering all doses (RR 1.15, 95% CI 1.11 to 1.20) (see Analysis 6.1). Adverse events were significantly more common with duloxetine than with placebo especially in 60 mg (RR 1.15, 95% CI 1.10 to 1.20) and 120 mg (RR 1.19, 95% CI 1.09 to 1.30) duloxetine groups (Analysis 6.1). Doses of 60 mg and 120 mg duloxetine were also associated with a significantly greater risk of cessation compared to placebo (Analysis 6.6, Figure 12).
6.1. Analysis.
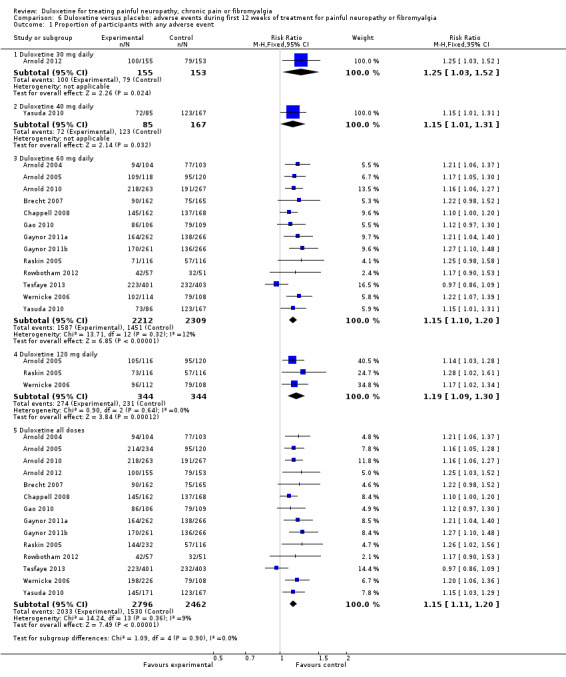
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 1 Proportion of participants with any adverse event.
6.6. Analysis.
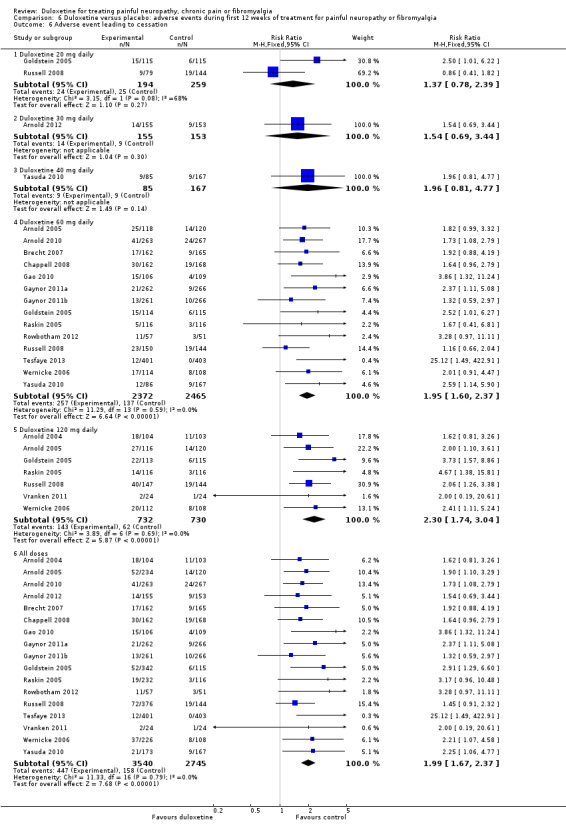
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 6 Adverse event leading to cessation.
12.
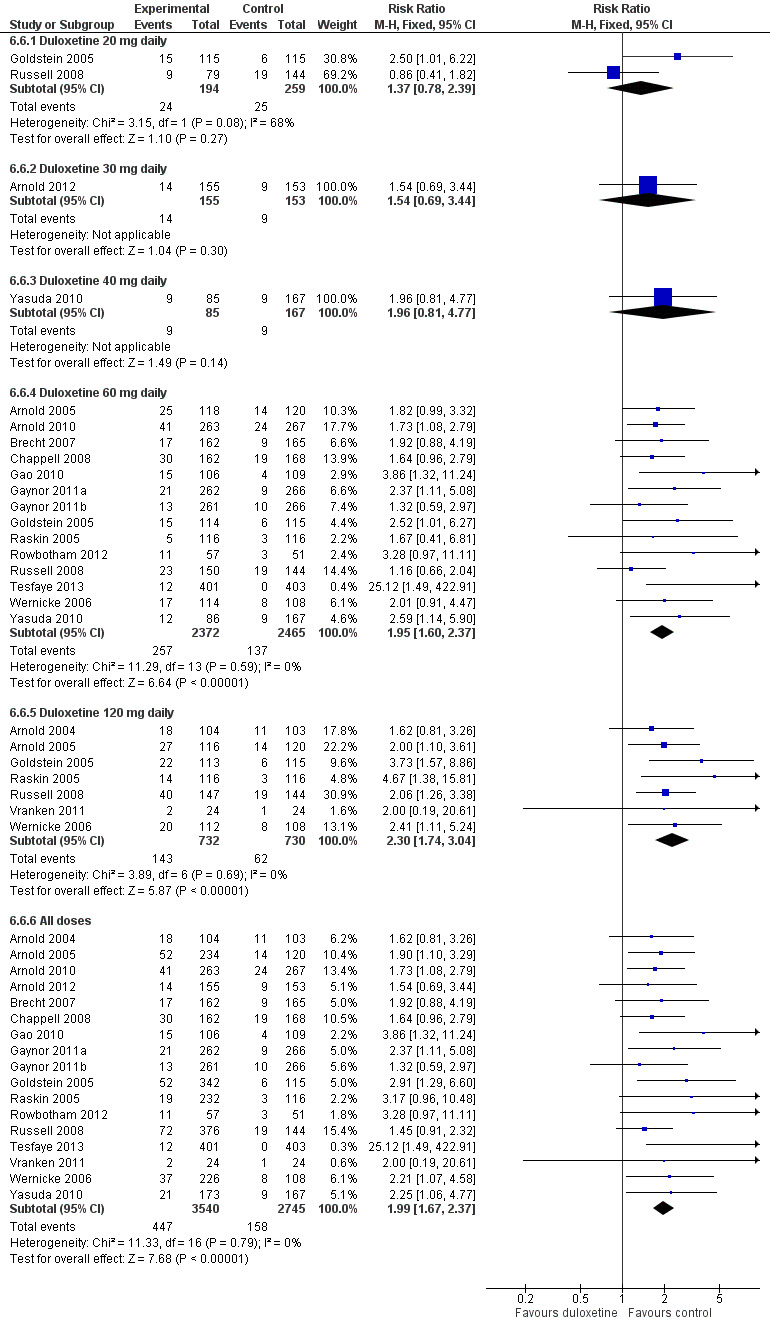
Adverse events leading to cessation of treatment.
The most common individual adverse events were nausea (Analysis 6.2), dry mouth (Analysis 6.3), dizziness (Analysis 6.4), somnolence (Analysis 6.5), fatigue, insomnia, constipation, decreased appetite, sweating and rhinitis. All had a dose dependency, with a greater frequency of side effects at 120 mg daily than 60 mg daily. No suicides were reported where suicide and risk were mentioned; this outcome was rarely specifically sought.
6.2. Analysis.
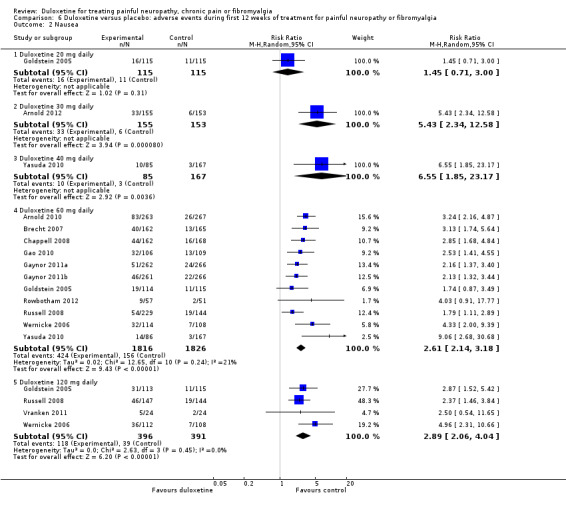
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 2 Nausea.
6.3. Analysis.
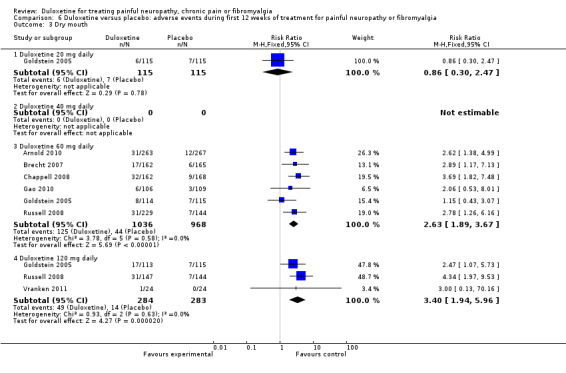
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 3 Dry mouth.
6.4. Analysis.
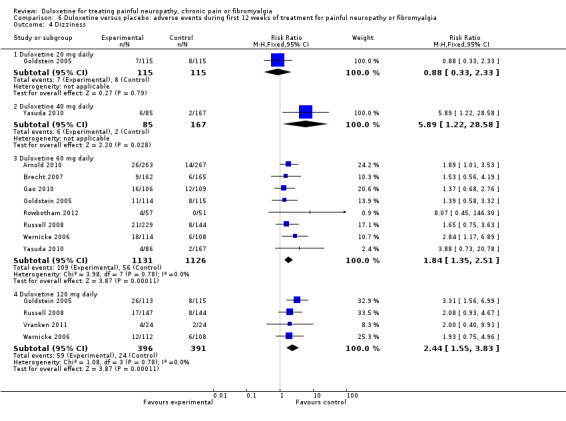
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 4 Dizziness.
6.5. Analysis.
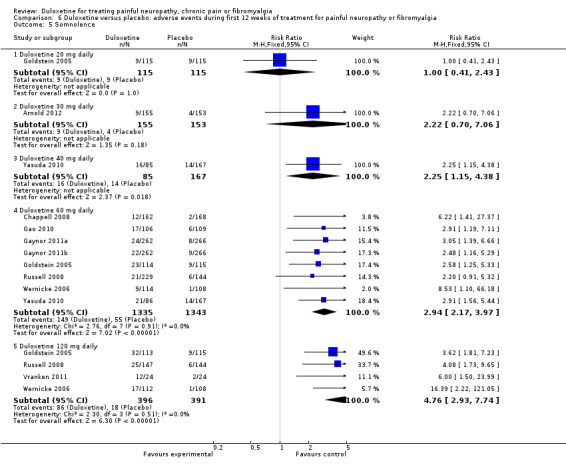
Comparison 6 Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia, Outcome 5 Somnolence.
Discussion
This updated Cochrane Systematic Review of duloxetine for the treatment of chronic pain and fibromyalgia identified 12 more studies than the original review that fitted the predefined quality criteria for inclusion, bringing the number of included studies to 18. These studies covered painful peripheral neuropathy (diabetic), central neuropathic pain, fibromyalgia, and painful physical symptoms in people with major depressive disorder and no underlying explanation for their pain. We excluded 25 studies identified in our searches for various reasons; many concerned pain from other conditions (for example, pelvic pain or osteoarthritis of the knee) that are the topics of other reviews. Some did not fulfil our criteria of treating participants for at least eight weeks (e.g. Brannan 2005), some were open label (e.g. Raskin 2006a and Raskin 2006b) and some not randomised or controlled (Canovas 2007). One trial comparing a novel agent ADL‐5859 to duloxetine and placebo has been completed but remained unpublished at the time of writing; no data were available from the company. Five studies are ongoing (NCT00457730; NCT00619983; NCT01179672; NCT01237587; NCT01552057) and the results of these are likely to become available in due course. Following extensive searches of ClinicalTrials.gov and reference databases, handsearching reference lists, cross‐correlating NCT codes, titles and abstracts, the authors did not identify any further trials. The Lilly Trials Database is freely available and contains extensive details of all trials. Unfortunately, neither the ClinicalTrials.gov NCT number nor the final published title of the research are published in the Lilly database, which make it very difficult to check whether all trials registered on ClinicalTrials.gov have been published. We have no reason to suspect extensive publication bias, certainly in the last few years, but we could not find publications corresponding to five ClinicalTrials.gov entries (Appendix 6).
We analysed the effects of duloxetine on painful diabetic neuropathy, central pain, fibromyalgia, and painful physical symptoms in depression separately. Each has a different pathogenesis and hence analysis of all conditions together would be meaningless. Furthermore, patients and caregivers are more likely to glean benefit from data presented for individual diseases. There is then the potential, should the need arise, to extrapolate such data as a guide to similar diseases where no evidence exists. In meta‐analyses, the magnitude of the benefit in terms of pain relief was similar in all the individual conditions.
For painful diabetic neuropathy, the RR of ≥ 50% reduction in pain at eight to 12 weeks at all doses of duloxetine versus placebo was 1.53 (95% CI 1.21 to 1.92) (NNTB 7 (5 to 10)). For the standard dosage of 60 mg duloxetine daily, this corresponds to an NNTB of 5 (95% CI 4 to 7) (Table 1). For ≥ 30% improvement of pain, the RR at 60 mg was 1.53 (95% CI 1.33 to 1.75), corresponding to an NNTB of 5 (95% CI 4 to 8) (Table 1). These NNTBs are of similar magnitude to the NNTB for tricyclic antidepressants for the achievement of moderate pain relief (NNTB 3.6, 95% CI 3 to 4.5) (Saarto 2007; Kajdasz 2007), and for amitriptyline for 'improvement' (NNTB) of 4.6 (95% CI 3.6 to 6.6) (Moore 2012). The mean difference in pain at a dose of 60 mg duloxetine daily was ‐0.96 points (95% CI ‐1.26 to ‐0.65 points) compared with placebo, on an 11‐point scale. A small dose‐effect may be present, but in most outcome measures, 20 mg to 30 mg is not clearly effective, and 40 mg to 120 mg is effective, with a very small efficacy increment as the dose is increased. There were improvements in the other prespecified secondary outcome measures at 40 mg, 60 mg, and 120 mg daily doses of duloxetine, again with slightly but not significantly greater improvements with the 120 mg dose. The PGIC secondary outcomes were statistically significant, but only border on magnitudes of change that are currently considered to be clinically significant (Dworkin 2008). We discovered no RCTs of the effect of duloxetine on painful diabetic neuropathy for periods longer than 12 weeks.
A number of studies exist presenting the minimal clinically important difference (MCID) for pain in various painful conditions on a 100 mm VAS, which can be roughly translated into an 11‐point VAS. Studies vary in design and inclusion criteria, and the MCID or minimal clinically important change varies with the position on the scale from which participants start. However, for people with pain measured in the middle of the VAS (moderate pain) at baseline, MCID is in the region of two to three points on an 11‐point scale (Mease 2011; Salaffi 2004). Changes of one point or less are unlikely to be clinically relevant. However, as with all pain studies, it is recognised that there is a U‐shaped response curve, with some people responding maximally and some not at all. This makes globalised linear measures of improvement moderately meaningless as the lack of response in some dilutes the effect that occurs in others. The proportion of people responding becomes a more useful measure.
For participants with fibromyalgia, the magnitude of improvement in pain at 12 weeks was similar to that seen in participants with diabetic peripheral neuropathy. There was no clear difference between the effects of duloxetine in fibromyalgia or diabetic peripheral neuropathy when assessed on the primary outcome measures, and so even if fibromyalgia is a "different sort of pain" (Dadabhoy 2006), it seems to respond to duloxetine in a similar way. However, the absolute risk reduction was marginally less than that for diabetic peripheral neuropathic pain and the corresponding NNTB for ≥ 50% or more pain relief at a duloxetine dose of 60 mg daily was 8 (95% CI 5 to 17) (Table 2). When we combined results for all doses, the NNTB was 9 (95% CI 7 to 13). Although there were fewer data for fibromyalgia, there again seemed to be a floor effect of dosage, where 30 mg was not effective but higher doses appeared to be. The ceiling effect (or lack of any additional therapeutic effect at 120 mg) was not evident as there were too few data.
It is notable that in fibromyalgia the magnitude of improvement in the SF‐36 mental subscore (MD, 120 mg dose, 4.22, 95% CI 2.43 to 6.02) was double that of peripheral neuropathic pain at the same dose (MD 2.23, 95% CI 0.69 to 3.77) or central neuropathic pain; whereas the magnitude of improvement in bodily pain scores in the neuropathic pain studies (MD 8.19, 95% CI 4.33 to 12.05) was 40% more than that in fibromyalgia (MD 5.96, 95% CI 3.76 to 8.16). As is noted above, the authors of Brecht 2007 comment on the difference in tempo of improvement of depression and pain scores, suggesting that different, but similar, mechanisms are responsible for the two phenomena. In Gaynor 2011a "..patients who met the >30% or >50% BPI response criteria at the 8 week LOCF endpoint had rates of [depression] remission (MADRS total scores…) that were higher compared to duloxetine treated patients who did not meet the BPI response criterion", and in Gaynor 2011b "..remission rates [of depression] were three times greater for patients taking duloxetine who reported at least 30% reduction in pain versus those who did not." However, in Raskin 2005, depression scales did not change despite similar pain scale improvements but the trial specifically excluded people with pre‐existing depression from entry. Russell 2008 used two regression models to separate the direct effect of duloxetine on pain and the indirect effect on the treatment of depression in the treatment of pain. The trial authors calculated that between 21% and 38% of the pain treatment effect was due to treatment of depressive symptoms. Fava 2004 estimated a 50% influence. This remains circumstantial evidence that the mechanisms of pain relief involve both mood and direct pain components and where mood is more involved there may be a greater influence on pain relief.
This new version of the review contains two large, new studies including people with major depressive symptoms and pain of unclear origin. Although these studies were generally well performed, limitations of allocation concealment, blinding and randomisation, along with the potential heterogeneous mix of causes of pain (including various pains of unknown origin), potentially leads to extensive imprecision and a lack of certainty about the results achieved. The mean improvement in pain was only ‐0.55 points on an 11‐point VAS (95% CI ‐0.75 to ‐0.35). The RR of people achieving 50% or more relief of pain at under 12 weeks was 1.37 (95% CI 1.19 to 1.59; NNTB 8, 95% CI 5 to 14) (Table 3), once again of a similar magnitude to the pain in fibromyalgia. Unfortunately, there were no data on the SF‐36 to make further comparisons.
This updated review also contains a single study independent from the makers of duloxetine exploring the effect of duloxetine versus placebo in the treatment of central neuropathic pain from spinal cord disorders or stroke. This single, high quality, independent and well‐performed study suffers from its small size and so effect estimates are associated with large CIs. The only result of statistical significance was the PGIC, with a RR favouring duloxetine that was just statistically significant at 2.75 (95% CI 1.02 to 7.44). The magnitude of improvement in pain was also similar to other conditions, although it was nonsignificant, with wide CIs.
A number of studies were not included in the meta‐analysis and are included here for completeness. Kaur 2011 performed a randomised cross‐over trial of amitriptyline and duloxetine. It was not included in a formal meta‐analysis because it was the only trial comparing those interventions. It did not meet our predefined inclusion criteria for length but we included it because it was the only one of its type. It had significant carryover between the cross‐over arms, which greatly affected quality. Duloxetine has a superior response to pregabalin in Tesfaye 2013, but the pregabalin response was at the level of a placebo response in other trials; other studies (for example, Moore 2009) suggest that pregabalin is effective and hence the comparative benefit of duloxetine over placebo remains unclear. Brannan 2005 performed a double‐blind RCT of duloxetine in major depressive disorders, in which they also measured the effect on pain. The outcomes were measured at seven weeks and hence it did not meet our eligibility criteria. Furthermore, the placebo and active arms of the trial were not balanced for depression as measured by the Hamilton Rating Scale for Depression at entry. However, depression scores did not change significantly and similar improvements were seen in pain scores as in the fibromyalgia and painful diabetic neuropathy trials. This compares favourably with the longer studies of Gaynor (Gaynor 2011a; Gaynor 2011b). The Raskin 2006a trial was a 52‐week extension phase of the prior 13‐week study (Raskin 2005) but although this trial was randomised, it was open, with a non‐standardised control group ("standard care"). Hence we excluded it. The only subscale outcomes of the SF‐36 eligible for this review from this trial did not change over one year, but duloxetine was safe and well‐tolerated over that period. Wernicke 2006b was a similar randomised but open extension phase study of Goldstein 2005. Again, safety and tolerability were the main focus. The SF‐36 bodily pain subscore improved significantly but by a much smaller magnitude than in the studies included in the meta‐analysis. Finally, the Goldstein 2004 study reported, with a dose of duloxetine of 80 mg, a median improvement in pain severity on a VAS for pain of ‐7.5% (interquartile range ‐25% to 1%), assessed at week eight. This was the only pain measure to show statistically significant improvement. Since pain was not an inclusion criterion for the trial (designed primarily to look at depression), the trial did not match our pre‐specified inclusion criteria. Hence it was not included in the analysis above but adds support to the therapeutic efficacy of duloxetine in the treatment of pain.
We included TSA in this review for the first time. These analyses are useful in a number of ways. There was not enough information to create a TSA for central pain. For the primary outcome in painful diabetic neuropathy, treatment with the standard dose of duloxetine had convincing evidence of non‐futility even though the optimal information size had not been reached. Within the predefined parameters, even after the first two studies the Z‐score had crossed the boundary line of efficacy. It remains that none of these trials was performed by an independent investigator, but the trials were generally of high to moderate quality and there was no reason to suspect that further independent trials would significantly skew the result to futility. In fibromyalgia, the optimal information size was some way off and further independent trials are recommended. For painful physical symptoms associated with depression, the optimal information size had been exceeded but there were insufficient data to calculate an area of futility. However, the efficacy in this indication is clear. As indicated above, the magnitude of improvement in terms of MD should be considered.
Adverse events were very common in these trials but were, in general, mild. The rates of any adverse event and adverse events leading to cessation of treatment were significantly greater with duloxetine than with placebo at the 60 mg and 120 mg doses, which are the doses used in clinical practice. However, withdrawals because of adverse events were relatively few (duloxetine, all doses combined, 12.6% versus placebo 5.8% (RR 1.99, 95% CI 1.67 to 2.37)). The NNTH for cessation of treatment was 17 (95% CI 13 to 26). These figures are in line with the retrospective analysis of Gahimer et al. in 23,983 patients in the duloxetine integrated exposures database (Gahimer 2007), where approximately 20% of patients withdrew because of adverse events. Adverse event rates were also dose related, being more common with the 120 mg than the 60 mg dose and with the 60 mg than the 20 mg or 30 mg doses, whether any adverse event or events leading to cessation are considered. The rates of serious adverse events were not greater with duloxetine than with placebo (RR 0.81, 95% CI 0.53 to 1.25). The adverse event profile was broadly in line with adverse events in the Cochrane Systematic Review of duloxetine in stress incontinence (Mariappan 2009). From observational studies, the most common side effects of duloxetine are quoted as nausea (37%), dry mouth (32%), dizziness (22%), somnolence (20%), insomnia (20%) and diarrhoea (14%) (Aronson 2007). In the studies included here, sweating (60 mg daily 6.5%, 120 mg daily 9.3%, versus control 0.88%) was also common, occurring at about the same frequency as reported by Gahimer et al. (6.2%) (Gahimer 2007). Tremor was also commonly reported, especially at the 120 mg dose in one trial (60 mg daily 3.3%, 120 mg daily 10.2%) (Russell 2008). There were small changes in blood pressure and heart rate in some but not all of the included studies, although we have not formally analysed these. However, in a review of the trials in depression, duloxetine was reported to produce a small, statistically significant rise in heart rate (2 beats per minute) and a sustained increase in systolic blood pressure versus placebo (1% rise versus 0.4%) (Aronson 2008). A company‐performed retrospective database review of 8504 participants in 42 placebo‐controlled studies of duloxetine covering five indications identified no significant cardiovascular risk (Wernicke 2007). There were no significant increases in suicide or suicidal thoughts in studies where these event were reported.
The studies included in this meta‐analysis contribute more than 6400 participants to meta‐analyses of the use of duloxetine in neuropathic pain, fibromyalgia and painful symptoms in depressive illness, and this is the most comprehensive assessment of any individual antidepressant drug for these indications of which we are aware. This provides a greater level of certainty to the conclusions of this review. Other tricyclic antidepressants have greater efficacy in terms of lower NNTBs than duloxetine, but the trials are small, many have methodological deficiencies and so efficacy may be overestimated. The European Federation of Neurological Societies (EFNS) guidelines for the pharmacological treatment of neuropathic pain recommend duloxetine as second line to tricyclic antidepressants and gabapentin or pregabalin, except where cardiovascular risk factors are present, when duloxetine is favoured (Attal 2006). The National Institue for Health and Clinical Excellence (NICE) recommends duloxetine as first line therapy in diabetic neuropathic pain in non‐specialist care settings but fails to mention it in the treatment of other painful neuropathies (http://publications.nice.org.uk/neuropathic‐pain‐cg96/guidance).
The cost effectiveness of duloxetine has not been formally investigated in RCTs. Four studies have addressed cost effectiveness (Wu 2006; Beard 2008; O'Connor 2008, Bellows 2012); the company manufacturing the drug performed two of them (Wu 2006; Beard 2008). The quality of the earlier studies was questionable but the latest independent study estimates similar levels of economic cost associated with duloxetine use. Wu 2006 performed an analysis of the cost effectiveness of duloxetine in participants completing the Goldstein 2005 trial in the USA. Using trial outcomes, the study authors compared the costs to healthcare, society and employers with those of the 'standard therapies' in the control group. Given that duloxetine was shown to be more effective than standard therapies, duloxetine was also shown to significantly reduce societal and employer costs with a trend towards cost effectiveness in medical costs when compared to standard treatment with other pain management therapies. Beard 2008 used a decision analytic model to represent the sequential management of people with diabetic peripheral neuropathic pain based upon current prescribing practice in the UK. Calculations in the model were based upon data for clinical efficacy that are good for duloxetine and pregabalin, but less robust for tricyclic antidepressants and gabapentin ('first line' drugs). However, recognising the inherent limitations, the authors calculated that duloxetine added as a second line therapy resulted in a predicted cost saving of GBP 77 per patient, on the basis that an additional 29 patients per 1000 achieved a full pain response compared to standard treatment. When duloxetine is added to the standard prescribing hierarchy as a second line therapy, the clinical benefit can also be expressed as adding an additional 0.0019 quality‐adjusted life years (QALYs) per patient. O'Connor 2008 also used a decision analytic model incorporating published and unpublished data from RCTs and cross‐sectional studies of duloxetine, gabapentin, pregabalin and desipramine. The QALY cost of duloxetine using this model varied from USD 47,700 to USD 867,000 per QALY depending upon the assumptions made in the analysis of the trial data. In the latest study, Bellows 2012 used a decision tree model in both clinical trial and real world studies comparing duloxetine to pregabalin. Duloxetine demonstrated an incremental cost of minus USD 187 per patient or incremental effectiveness of 0.011 QALYs. At a cost per QALY threshold of 50,000 USD (almost equivalent to the UK's GBP 30,000), duloxetine is more cost effective than pregabalin. This latter analysis presents the best case scenario, since although the cost effectiveness in people with good pain relief is high, the cost effectiveness can be overwhelmed by those patients in whom a drug does not work, and so cost effectiveness studies of this sort are questionable.
Authors' conclusions
Implications for practice.
There is moderate quality evidence from four studies performed by the manufacturers of duloxetine that doses of 60 mg and 120 mg daily are efficacious for treating pain in diabetic peripheral neuropathy but lower daily doses are not. In fibromyalgia, there is low to moderate quality evidence that duloxetine is effective at similar doses and with a similar magnitude of effect. That effect may be achieved through a greater improvement in mental symptomatology than somatic physical pain. There is also low to moderate quality evidence that pain relief is also achieved in pain associated with depressive symptoms but the NNTB of 8 in fibromyalgia and depression may not make this agent a first line choice if other more efficacious agents are available.
Minor adverse effects are common with duloxetine. They are more common with duloxetine 60 mg than placebo and certainly more common at doses of 120 mg daily. Adverse events were much less frequent at duloxetine 20 mg daily. Serious side effects are rare.
Implications for research.
Trial sequential analysis indicates that for painful diabetic neuropathy no further trials are needed to indicate the efficacy of duloxetine at 60 mg. More studies are required to convincingly demonstrate the efficacy of 60 mg of duloxetine in fibromyalgia, the painful physical symptoms of pain in depressive illness and in central pain. These trials should preferably be investigator led and independent of the company making the drug, as bias questions are always raised when the majority or all of the trials are drug company run or sponsored.
The trials were not designed to investigate mechanisms but there was some evidence that the effect on pain was independent from the effect on depression. Improved direct comparisons of duloxetine with other antidepressants and with other drugs, such as pregabalin, already shown to be efficacious in neuropathic pain would be appropriate. All trials should include unbiased economic analyses, although the economic analyses so far performed (including unbiased independent analysis) indicate that duloxetine is cost effective for treating neuropathic pain when tested in a number of models, at least in the US healthcare system.
Feedback
Duloxetine for treating painful neuropathy or chronic pain, 18 December 2009
Summary
Lunn et al (Lunn 2008) in their systematic review duloxetine for treating peripheral neuropathy for chronic pain came to the conclusion that “there is moderately strong evidence that duloxetine 60 mg and 120 mg daily are efficacious for treating pain in diabetic peripheral neuropathy and fibromyalgia but 20 mg is not.” However, we believe this claim may not be fully substantiated until the following issues are addressed.
Five of the six studies included in the review had a dropout rate of greater than 20%. It is not however specified what proportion of these dropouts were lost to follow‐up, or whether the dropouts were followed until completion of the study in question. Four of the five studies were listed as having used intention‐to‐treat or partial intention to treat analysis. (What was meant by partial intention to treat was further explained in the appendix but still did not address the issue as to when participants dropped out or whether they were lost to follow‐up). Bias caused by loss to follow‐up cannot be minimized by intention‐to‐treat analysis (Montori 2001). Once a patient is lost to follow‐up, investigators are unaware of their outcome. Even if intention‐to‐treat principle is performed, results may still be biased depending on what assumptions were made. For example, investigators may include those lost follow‐up in the denominators of study results (Montori 2001). This procedure assumes that those lost to follow‐up did not experience a target outcome, which may not be accurate. In the case of the duloxetine studies, results will depend on how the investigators imputed pain scores for those that were lost to follow‐up. One study was listed as using last observation carried forward analysis. Pain may fluctuate during the course of a patient's illness. Therefore using last observation carried forward data may result in bias in favor of duloxetine, if a patient dropped out after a pain reading which pain was not as severe as usual. Similarly, the bias could favor placebo if an individual dropped out after a pain reading which was unusually severe.
Pain improvement was taken to be an improvement of 50% or more from baseline on validated pain intensity scales. The effect of an improvement of 50% or more from baseline will vary significantly depending on the patient's initial pain severity scale. For example, a patient may present with a baseline VAS pain score on 60 out of 100 and after treatment achieve a score of 30 (a 50% reduction the their pain score). This could mean that the patient remained in the 'moderate pain' category despite receiving treatment (i.e. a VAS score of 60 and a VAS score of 30 both would be considered as 'moderate pain'). Conversely, a patient with a baseline score of 80 (severe pain) who achieves a 50% reduction in pain now would have a score of 40 (moderate pain). This scenario represents a more meaningful decrease in pain in our opinion. Since pain assessment measures are diverse and majority use standard subjective scales for pain intensity and/or pain relief, whichever pain scale used, one would expect changes in pain to trend towards the same direction. However, since a clinically meaningful difference in Patient Global Impression of Change (PGIC) is suggested as 1 point (Dworkin 2008), this may provide confirmation in addition to using pain improvement of 50% from baseline. According to this article, there were discrepancies in results when both PGIC and pain improvement of 50% from baseline were measured in the following 3 trials (Wernicke 2006, Raskin 2005, Goldstein 2005). When improvement of pain score compared with baseline was used, the combined data from all doses (20, 60, 120mg) from three trials together, showed a statistically significant result with a RR of 1.63 (95% CI 1.35 to 1.97) greater than placebo. One would expect that a clinically significant change in pain score would be accompanied by a significant change in PGIC (i.e. the patient's impression of the change in their clinical condition). However, a 50% reduction in pain score was not accompanied by a clinically significant improvement in pain in the opinion of the patients themselves (i.e. change in PGIC ‐0.59, 95% CI ‐0.78 to ‐0.41). One must ask how relevant is a 50% reduction in pain when measured by a pain scale when a patient is not able to perceive this as a clinical improvement?
Even though pooling data, such as in meta‐analyses, allows for detection of effects that might not be evident (due to being insufficiently powered) with individual small studies, there is a possibility of small‐study effect. Small‐study effect occurs when small treatment effects from multiple trials are pooled together leading to an exaggerated effect that may or may not reflect the true treatment effect size. Therefore, caution must be used. The sample sizes of the trials examined ranged between 200 and 400, which are considered relatively small. A way to mitigate small‐study effect has been suggested (Scott 2006): adopt a more stringent significance testing e.g. using a 99% CI rather than the traditional 95%. In the case of Duloxetine for treating painful neuropathy or chronic pain, the reduction of pain score by 50% seen in the short‐term treatment (12 weeks) of painful diabetic peripheral neuropathy had a RR of 1.65 (95% CI 1.34 to 2.03, NNT = 6) with duloxetine 60 mg daily may be exaggerated due to pooling multiple small studies together.
We look forward to hearing your response to our comments.
Sincerely,
Erica Lo Bsc. Pharm
Elisa Mok Bsc. Pharm
Elizabeth Monchesky Bsc. Pharm
Aaron M Tejani, BSc(Pharm), PharmD
References:
Dworkin 2008: Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar T, et al. Interpreting the Clinical Importance of Treatment Outcomes in Chronic Pain Clinical Trials: IMMPACT Recommendations. Journal of Pain 2008;9(2):105‐21.
Lunn 2008: Lunn MPT, Hughes RAC, Wiffen PJ. Duloxetine for treating painful neuropathy or chronic pain.Cochrane Database of Systematic Reviews 2009, Issue 4. Art. No.: CD007115. DOI: 10.1002/14651858.CD007115.pub2.
Montori 2001: Montori V, Guyatt GH. Intention‐to‐treat principle. CMAJ 2001; 165 (10): 1339‐1341.
Scott 2006: Scott I, Greenberg P, Poole P, Campbell D. Cautionary tales in the interpretation of systematic reviews of therapy trials. Internal Medicine Journal 2006; 36: 587‐99.
Reply
Dear Dr Tejani
Thank you very much for your feedback on our Cochrane review Duloxetine for treating painful neuropathy or chronic pain
We thank you for your thoughtful comment, and highlighting the issues which afflict Cochrane systematic reviews in general and this one in particular.
The methodology for performing a Cochrane systematic review is very clearly set out in the Cochrane Handbook. One of the reasons Cochrane reviews take so long to write is the stringent review procedures by content experts, statistical advisors and group statisticians and the editorial board. This takes place at all points in the process including title registration, protocol formulation, and review publication. Outcomes are all pre‐specified in the protocol, before the trials are searched. Inevitably the authors, being experienced in the field, have an idea of the outcomes that are likely to have been used in clinical trials in the area but those selected are done so on the basis that they may have some relevance to patients, health care providers, and clinicians. It is a difficult balance to strike. If one pre‐specifies derivative outcomes that would not necessarily be reported in the trials one runs the risk of coming up with no suitable evidence. However, even pre‐specifying commonly used outcomes leaves one open to risk.
I think some of your comments probably stem from the wording in the Abstract “There is moderately strong evidence that duloxetine 60 mg and 120 mg daily are efficacious for treating pain in diabetic peripheral neuropathy and fibromyalgia”. The “moderately strong” statement is derived from the GradePro terminology, a widely accepted system for reporting the quality of outcome measures. Under the GradePro system, meta‐analytical data from randomised controlled trials are all assumed to produce ‘high quality’ evidence. This is then downgraded as a result of bias and other methodological flaws qualitatively judged to affect the data across each outcome for all trials. Their application to each individual outcome is open to interpretation.
We very much recognise the problems with the high drop out rate, the difficulties with intention to treat analyses, the fluctuations in the clinical state of patients, and the difficulties with measuring pain. All of these studies had notable problems which we needed to take into account. However we have to commend Eli Lilly for their openness with their data. They provided us with all the data that were required to complete the study and indeed offered to forward the complete dataset for all the trials for re‐analysis (an offer which we did not take up given that the dataset is so massive). There has been no attempt on their part to obscure or withhold data from publication and we believe the trial represents ‘real life’ albeit not ideal.
Trials in patients with pain and associated mood disorders are notoriously difficult. We did not pre‐specify the exclusion of trials with a particular percentage drop out rate, but it is recognised that the levels of drop outs in these trials were substantial and could lead to bias. We had a number of long discussions about whether these should be excluded on the basis of the drop out rate, and a number of other issues. But when all the data are considered together we felt that these trials should be included. However as a result of these issues we felt the level of confidence in the evidence was reduced resulting in some but not substantial downgrading. As you point out in the latter part of your second paragraph, sometimes bias can decrease the apparent effect of an intervention as well as increase it. We have therefore we believe been open and transparent about the identified problems with the studies, and how these problems were used to downgrade the evidence as is available.
Patients in the studies all had their pain assessed prior to entry and were only included if they exceeded a baseline pain score. We excluded some trials of duloxetine which included pain scales as outcomes but where participants were not randomised with regards to pain status and where the origin and entry level of pain were not scored. A 50% reduction in pain for any patient with chronic pain is meaningful, and by many researchers considered to be a challenging target whatever level of chronic pain they start from, and we believe the 50% reduction in pain is an appropriate outcome.
Measurement scales that claim to measure the same metric are diverse and heterogeneous and clearly measure different aspects of the same thing, quantitatively and qualitatively. Although they frequently change in parallel no scale is perfect and hence there will always be differences in significance in outcome changes albeit with trends being in the same direction. Hence we appreciate that there are inconsistencies in the significance levels of these two outcomes.
We recognise that the studies may be relatively small in relation to some cardiology studies for instance. However they are still relatively large, larger than any of the other studies with similar pain agents and adequately powered to detect differences. They are also all the studies of the drug that have. We believe we have used caution in our meta‐analysis of these data, and do not think that using a more stringent, but still arbitrary, significance level of 99% rather than 95% as the confidence interval would improve the data.
It would of course be extremely nice to have huge, unbiased, beautifully performed randomised controlled trial studies, measuring the most meaningful outcomes and adverse events for every agent. Unfortunately real life, finance, ethics committees, and the vagaries of patients and their conditions do not allow such perfection. Systematic meta‐analysis of smaller trials undoubtedly has its problems, drawbacks, and critics. But as a means of extracting as much information from the historical studies which are available, and almost as importantly, pointing out the problems with those studies so that any future studies are better performed systematic reviews have their purpose.
Finally, two of the authors are Joint Co‐ordinating Editors of the Neuromuscular Group. We declare this as a conflict, but at each stage of the process the review was handled by an independent Contact Editor and peer review, editing and editorial decision making was carried out without the influence of the authors.
Dr Michael PT Lunn Joint Co‐ordinating Editor Cochrane Neuromuscular Disease Group.
Professor Richard AC Hughes Joint Co‐ordinating Editor Cochrane Neuromuscular Disease Group
Mr Philip J Wiffen Director of Training UK Cochrane Centre National Institute for Health Research Oxford
Contributors
Brian Dickie, Feedback Editor, Cochrane Neuromuscular Disease Group
Ruth Brassington, Managing Editor, Cochrane Neuromuscular Disease Group
Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia, 16 September 2015
Summary
Thank you for your systematic review on “Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia” by Lunn et al (2014). Clinicians depend on these systematic reviews to make treatment decisions as they are regarded as the highest standard in evidence‐based health care. However, we would caution against using this review in its current form to make such decisions.
After close appraisal of 5 of the studies included in the diabetic neuropathy analyses, we have concerns that the risks of biases in these trials were not adequately evaluated.
Goldstein et al (2005)
Blinding: Rated as low risk
We disagree with this assessment, as it was not explicitly stated in the study that the capsules looked identical in size and colour. It was unclear how blinding was protected in terms of the number of capsules received in each group as dosing for the 120mg duloxetine group was 60mg twice daily. As well, treatment‐emergent adverse events were higher in both the 60mg and 120mg duloxetine groups compared to placebo (nausea, somnolence, dizziness, constipation, dry mouth, and sweating were all statistically significantly different in the 120mg duloxetine group compared to placebo). It is possible that participants may have correctly anticipated which treatment they received (and personnel may have guessed what patients were receiving) and blinding may not have been maintained throughout the study. Therefore, we suggest the risk assessment to be rated as unclear.
Gao et al (2010)
Allocation concealment: Rated as low risk with comment of double‐blinding and study medication in capsules, or matching placebo.
We disagree with this assessment and the support for judgement. The term allocation concealment has been misinterpreted for blinding. Allocation concealment protects participants and investigators from foreknowledge of the intervention assignments, which was not clear in this study. We suggest the risk assessment to be rated as unclear.
Blinding: Rated as low
Again, it was unclear whether the different strengths of duloxetine were of the same size and colour. As in the Goldstein (2005) study, treatment emergent adverse events were higher in the duloxetine treated groups for nausea, somnolence, anorexia, and dysuria compared to placebo. It was unclear how blinding was maintained when doses were increased to 120mg for those participants that did not respond to 60mg based on the investigator’s judgement, and how the dose was returned to 60mg in patients who could not tolerate the increased dose. Therefore, we suggest the risk assessment to be rated as unclear.
Incomplete outcome data: Rated as low risk with comment that LOCF [last observation carried forward] and MMRM [mixed‐effect model repeated measure] used to minimize bias for a high dropout rate of 15.6% to 17.9%.
We disagree with this assessment. Using LOCF could have over or underestimated the effects of duloxetine, as it was unclear when the patients were lost to follow‐up and what impact this would have on the results. We suggest the risk assessment to be rated as unclear.
Other bias: Rated as low risk with comment that it was industry sponsored and that no adjustments were made for multiple comparisons.
We disagree with this assessment since multiple comparisons and therefore multiple testing renders a greater chance of type I errors. This is evident in the study’s post‐hoc analysis that found a statistically significant difference between the duloxetine 120mg group compared to placebo, which is inconsistent with their outcome data. A Cochrane review by Lundh et al (2013) suggests that industry funded studies lead to “more favourable results and conclusions” than non‐industry funded studies.
Raskin et al (2005)
Allocation concealment: We agree with the low risk assessment for allocation concealment, however, we do not agree with the support for judgement. The reason for low risk is that they used IVRS [interactive voice response system] not because capsules looked identical.
Blinding: Rated as low risk
We disagree with this assessment, as the mean average daily dose of concomitant acetaminophen was higher in the placebo group (202.53mg) compared to the duloxetine 60mg and 120mg groups (151.88, and 121.65mg). It is possible that participants and personnel may have correctly guessed what treatment group participants were in based on the amount of acetaminophen required. Again, blinding was compromised with treatment‐emergent adverse events being higher in both the 60mg and 120mg duloxetine groups compared to placebo; with nausea, somnolence, hyperhidrosis, and anorexia being significantly more frequent than placebo. We suggest the risk assessment to be rated as high risk.
Incomplete outcome data: Rated as low risk We disagree with this assessment. Although not explicitly stated in the study, “the last non‐missing observation after randomization” LOCF was used for the analyses. Although Raskin et al (2005) mentioned that most patients who discontinued treatment due to adverse events did so within the first 4 weeks, it is still unclear whether this would over or underestimate the results. We suggest the risk assessment to be rated as unclear.
Wernicke et al (2006)
Blinding: Rated as low risk
We disagree with this assessment due to the same reasons mentioned above in Raskin et al (2005) with acetaminophen doses being lower in the duloxetine groups and treatment emergent adverse events being higher in the duloxetine groups compared to placebo. We suggest the risk assessment to be rated as unclear.
Other bias: Rated as low risk
Wernicke et al (2006) stated that a 2‐point reduction of the Likert scale represented a clinically important difference. However, they powered the study to detect a treatment group difference of only 1.2 points which is inappropriate given what was defined as a clinically important difference. We therefore suggest the risk assessment to be rated as high.
Yasuda et al (2010)
Allocation concealment: We agree with the unclear risk assessment for allocation concealment, however, we do not agree with the “double‐blind” support for judgement. It was unclear because a stochastic minimization allocation was not explained.
Incomplete outcome data: Rated as low risk We would suggest the risk assessment to be rated as unclear for reasons as mentioned above for LOCF.
Reporting bias: Rated as low risk
Efficacy analyses of the primary and secondary end‐points were made by comparing the combined duloxetine groups compared to placebo which was not specified a priori. This is inappropriate and should be rated as high risk.
We disagree with many of the assessments made in the aforementioned 5 studies we have thus far reviewed and question whether the remainder of the included studies in this systematic review have been appropriately assessed. We suggest reviewing the assessments for the risk of biases.
Finally, we bring to your attention that numbers were flipped in Analysis 1.1 for “>50% improvement of pain at 12 weeks or less” on page 75 for “all doses”. Goldstein (2005) should have been 158/334 for favours control, and 29/101 for favours placebo. Raskin (2005) should have been 101/227 for favours control and 34/113 for favours placebo. The risk ratio remained the same with a slight change in the confidence interval [1.21, 1.92]. We suggest that this be corrected.
Sincerely,
Anna Maruyama Pharm D Student
Aaron M Tejani Pharm D
References:
1) Gao Y, Ning G, Jia WP, Zhou ZG, Xu ZR, Liu ZM, et al. Duloxetine versus placebo in the treatment of patients with diabetic neuropathic pain in China. Chin Med J (Engl) 2010 Nov;123(22):3184‐3192.
(2) Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain 2005 Jul;116(1‐2):109‐118.
(3) Lundh. Industry sponsorship and research outcome. Cochrane database of systematic reviews 2013(7).
(4) Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014 Jan 3;1:CD007115.
(5) Raskin J, Pritchett YL, Wang F, D'Souza DN, Waninger AL, Iyengar S, et al. A double‐blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med 2005 Sep‐Oct;6(5):346‐356.
(6) Wernicke JF, Pritchett YL, D'Souza DN, Waninger A, Tran P, Iyengar S, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology 2006 Oct 24;67(8):1411‐1420.
(7) Yasuda H, Hotta N, Nakao K, Kasuga M, Kashiwagi A, Kawamori R. Superiority of duloxetine to placebo in improving diabetic neuropathic pain: Results of a randomized controlled trial in Japan. J Diabetes Investig 2011 Apr 7;2(2):132‐139.
Reply
Dear Ms Maruyama and Dr Tejani
Thank you very much for submitting feedback on our review of `Duloxetine for the treatment of chronic pain and fibromyalgia'. We always appreciate constructive feedback of this sort as it assists with the review process. Despite the review having been actively written by all three of the authors, including many hours of discussion, analysis, writing and refinement, the possibility of human error remains and we are open to criticism and correction. The review has been updated twice and peer reviewed at least four times since its first publication. Furthermore Cochrane methodologies, including how risks of bias are judged, have changed.
We agree with some but not all of your points. Many of these concern subjective value judgements about risk of bias assessments. These were made independently by the CSR authors and then compared. Disagreements were few and we recorded conversations about these where there was disagreement. Some judgements were made following explanation by Eli Lilly investigators who confirmed that the methodologies of contemporaneous studies were the same, even though the reports were written independently and thus contain slightly different explanations of methodology. We do not think there has been any active obscuration of facts about allocation concealment and drug/placebo formulation for instance. We have made some changes in light of your comments and will indicate where we have made changes. For the most part we did not make changes and we have given the reasons why not. Your commentary is in italics, our answer below each point.
Blinding: Rated as low risk
We disagree with this assessment, as it was not explicitly stated in the study that the capsules looked identical in size and colour. It was unclear how blinding was protected in terms of the number of capsules received in each group as dosing for the 120mg duloxetine group was 60mg twice daily. As well, treatment‐emergent adverse events were higher in both the 60mg and 120mg duloxetine groups compared to placebo (nausea, somnolence, dizziness, constipation, dry mouth, and sweating were all statistically significantly different in the 120mg duloxetine group compared to placebo). It is possible that participants may have correctly anticipated which treatment they received (and personnel may have guessed what patients were receiving) and blinding may not have been maintained throughout the study. Therefore, we suggest the risk assessment to be rated as unclear.
We discussed this at the time of writing the review. MPL rated this as unclear and RACH as low risk. After discussion we decided to rate this as low risk. The reasoning for this was that although there is no explicit statement about size and colour, or the concealment of a twice daily dosage to 120mg, the treatments were given in pre‐assigned blister packs, against a placebo control, and that other studies run by Lilly at the same time explicitly state the twice daily 60mg was concealed with placebo for the 60mg once daily and 30mg once daily dosages.
We have therefore not changed this.
With regard to the treatment emergent side effects, these (especially those of the sort described) do not necessarily unblind patients, especially when there are significant numbers of adverse events in the placebo group. There are very few medications with no side‐effects and many side effects occur in placebo groups. We have not changed this on this basis here or below.
Allocation concealment: Rated as low risk with comment of double‐blinding and study medication in capsules, or matching placebo.
We disagree with this assessment and the support for judgement. The term allocation concealment has been misinterpreted for blinding. Allocation concealment protects participants and investigators from foreknowledge of the intervention assignments, which was not clear in this study. We suggest the risk assessment to be rated as unclear.
We are of course well aware of the difference between allocation concealment and double blinding and the importance of both. We have checked the files about this. MPL initially scored both blinding and allocation concealment as low risk and RACH scored allocation concealment as unclear and blinding as low risk. We wrote to the authors who assured us that the methods had been the same as in the other Eli Lilly sponsored studies in which we had concluded that allocation concealment was done properly. We accepted this reassurance and scored allocation concealment as low risk. The Cochrane Handbook states that ‘knowledge of who undertook the study can sometimes allow reasonable assumptions to be made about how the study was conducted…’.
Your commentary brings up further the difficulties of assessing blinding, allocation concealment and making judgements about these two areas from trial reports. We would not say that we have misinterpreted one for the other. Clearly patients, investigators and other trial personnel have to be blinded, but maintaining the blind requires allocation concealment.
Blinding: Rated as low – we have not changed this – see below.
Again, it was unclear whether the different strengths of duloxetine were of the same size and colour.
The study reports that “…capsules containing either 30mg or 60mg of duloxetine hydrochloride as enteric coated pellets or matching placebo. Throughout the study patients took one or two capsules in the morning or the evening [NB in figure legend to Figure 1 ‘randomly assigned to take …in the morning or the evening’]; however the full daily dose was contained in either the morning or the evening drug dose”. We think this is fairly clear.
As in the Goldstein (2005) study, treatment emergent adverse events were higher in the duloxetine treated groups for nausea, somnolence, anorexia, and dysuria compared to placebo.
As above
It was unclear how blinding was maintained when doses were increased to 120mg for those participants that did not respond to 60mg based on the investigator’s judgement, and how the dose was returned to 60mg in patients who could not tolerate the increased dose. Therefore, we suggest the risk assessment to be rated as unclear.
This we too found difficult at the time as there is no clear statement about pharmacists, treatment physicians, assessing physicians or other personnel, who was making the judgement and how it was actioned. The dose switch was complex and variable. We were unable to retrieve any data from the authors. However RACH and MPL discussed this and concluded that due to the complexity of the trial, the nature of the randomisation, blinding and allocation concealment that the blinding would have been maintained and is therefore low.
Incomplete outcome data: Rated as low risk with comment that LOCF and MMRM used to minimize bias for a high dropout rate of 15.6% to 17.9%.
We disagree with this assessment. Using LOCF could have over or underestimated the effects of duloxetine, as it was unclear when the patients were lost to follow‐up and what impact this would have on the results. We suggest the risk assessment to be rated as unclear.
We have been transparent about this in the text and the characteristics of included studies. Trials of pain medications frequently have very high dropout rates and <20% is quite low in this field. Using LOCF is not ideal, and there are significantly more dropouts in the treatment groups with adverse events than with placebo, but this might lead to an underestimate of effect. On the other hand, there aremore (not statistically different) dropouts from lack of efficacy in the placebo group which could overestimate the effect. We judged that this should remain low risk of bias.
Other bias: Rated as low risk with comment that it was industry sponsored and that no adjustments were made for multiple comparisons.
We disagree with this assessment since multiple comparisons and therefore multiple testing renders a greater chance of type I errors. This is evident in the study’s post‐hoc analysis that found a statistically significant difference between the duloxetine 120mg group compared to placebo, which is inconsistent with their outcome data. A Cochrane review by Lundh et al (2013) suggests that industry funded studies lead to “more favourable results and conclusions” than non‐industry funded studies.
Thank you for this comment. In general although ‘pharma are bad’, due to a number of high profile fraudulent studies, in general Cochrane does not consider the performance of a trial by a pharma company a risk of bias. Indeed many pharma studies are carried out exceptionally well and with far less risk of bias than single centre investigator led or academic studies. Eli Lilly have been quite forthcoming about their data, there is no evidence of publication bias that we can identify and patient level data have been offered. We have not changed this assessment.
Allocation concealment: We agree with the low risk assessment for allocation concealment, however, we do not agree with the support for judgement. The reason for low risk is that they used IVRS not because capsules looked identical.
Thank you for this observation. We have added this, as both are important to the allocation concealment.
Blinding: Rated as low risk
We disagree with this assessment, as the mean average daily dose of concomitant acetaminophen was higher in the placebo group (202.53mg) compared to the duloxetine 60mg and 120mg groups (151.88, and 121.65mg). It is possible that participants and personnel may have correctly guessed what treatment group participants were in based on the amount of acetaminophen required.
We feel this is extremely unlikely. Firstly as long as blinding was maintained the patients and investigators would not be able to identify this information. Secondly this dosage of acetaminophen is equivalent to 1/5 of one tablet per day and then difference equal to 1/10 of one tablet per day which is simply not clinically relevant or I suspect identifiable other than in statistics.
Again, blinding was compromised with treatment‐emergent adverse events being higher in both the 60mg and 120mg duloxetine groups compared to placebo; with nausea, somnolence, hyperhidrosis, and anorexia being significantly more frequent than placebo. We suggest the risk assessment to be rated as high risk.
We disagree with this statement also and doubt if this has any relevance to unblinding patients when rates of placebo and active adverse events are both significant. We have not changed this risk.
Incomplete outcome data: Rated as low risk We disagree with this assessment. Although not explicitly stated in the study, “the last non‐missing observation after randomization” LOCF was used for the analyses. Although Raskin et al (2005) mentioned that most patients who discontinued treatment due to adverse events did so within the first 4 weeks, it is still unclear whether this would over or underestimate the results. We suggest the risk assessment to be rated as unclear.
It is possible that using a LOCF methodology could over or underestimate the results; we do not know. Approximately equal numbers of patients withdrew from all groups, and reasons are specified for this. Drop outs were <20%. We do not think this is a significant bias and have not changed the risk of bias rating.
Blinding: Rated as low risk
We disagree with this assessment due to the same reasons mentioned above in Raskin et al (2005) with acetaminophen doses being lower in the duloxetine groups and treatment emergent adverse events being higher in the duloxetine groups compared to placebo. We suggest the risk assessment to be rated as unclear.
Unchanged as Raskin 2005 above
Other bias: Rated as low risk
Wernicke et al (2006) stated that a 2‐point reduction of the Likert scale represented a clinically important difference. However, they powered the study to detect a treatment group difference of only 1.2 points which is inappropriate given what was defined as a clinically important difference. We therefore suggest the risk assessment to be rated as high.
I think you have misinterpreted this. The study was ‘underpowered’ and yet still demonstrated a change in Likert Scale greater than the minimum clinically important difference and that for which the study was powered. We have not changed the risk assessment.
Allocation concealment: We agree with the unclear risk assessment for allocation concealment, however, we do not agree with the “double‐blind” support for judgement. It was unclear because a stochastic minimization allocation was not explained.
On analysis of the extraction files MPL did not change his data after discussion with RACH and ‘double blind’ was entered in both allocation concealment and blinding fields in error, whereas allocation concealment should be unclear with text ‘no clear explanation of methods’. Unclear remains.We have changed this explanation field.
Incomplete outcome data: Rated as low risk. We would suggest the risk assessment to be rated as unclear for reasons as mentioned above for LOCF.
As above. We disagree and have not changed this. LOCF was used. Losses were 17% with duloxetine and 10% with placebo with reasons explained; not ideal but within ‘acceptable’ limits. We have not changed this.
Reporting bias: Rated as low risk
Efficacy analyses of the primary and secondary end‐points were made by comparing the combined duloxetine groups compared to placebo which was not specified a priori. This is inappropriate and should be rated as high risk.
We agree that the study has reported a ‘combined dose’ outcome which narrows the confidence intervals. In the CSR we have taken the single dose outcomes in the meta analysis. We do not think that this single extra report (which we appreciate the authors made ‘headline’) constitutes a reason to make this high risk of bias. We have not changed this field.
We disagree with many of the assessments made in the aforementioned 5 studies we have thus far reviewed and question whether the remainder of the included studies in this systematic review have been appropriately assessed. We suggest reviewing the assessments for the risk of biases.
Thank you for this comment. However since we have disagreed with all but two of your comments, neither of which made any material difference to a risk of bias assessment, we respectfully decline to recheck the risk of bias assessments for all the other studies. If you wish to check them yourselves we will be pleased to consider any further comments.
Finally, we bring to your attention that numbers were flipped in Analysis 1.1 for “>50% improvement of pain at 12 weeks or less” on page 75 for “all doses”. Goldstein (2005) should have been 158/334 for favours control, and 29/101 for favours placebo. Raskin (2005) should have been 101/227 for favours control and 34/113 for favours placebo. The risk ratio remained the same with a slight change in the confidence interval [1.21, 1.92]. We suggest that this be corrected.
Thank you for pointing out this error which we acknowledge and have now corrected. As you indicate it makes no difference to the risk ratio, but we have corrected the CI in the text. Interestingly the table had incorrect headings at the top which we have now changed from ‘Favours control and placebo’ to ‘Duloxetine and placebo’. Thank you for drawing attention to this table.
Once again, thank you for your interest in this review. We always appreciate input and constructive criticism. While we have not accepted all your suggestions, we have made the changes indicated which enhance the accuracy and quality of the review.
On the wider note whether the medical community agree with the finding of the efficacy of duloxetine or not is a common debate. We are generally fairly sceptical in our approach to trials, but we have no reason to suspect that any of these studies were performed poorly or with biased methodology. None of us have a conflict of interest in these studies, and indeed our only interaction with the authors has been requesting clarifications of methodology and asking for additional data. The authors of the studies were helpful with their replies in general. Replies from some authors however were not forthcoming, although the queries were relatively minor. There is no evidence of publication bias in the duloxetine RCT literature or through checking trial reports. We have not found any substantial criticism of these trials and duloxetine does appear to have some efficacy, at least in painful diabetic neuropathy, compared with placebo.
We hope that this reply answers your criticisms adequately. We are always enthusiastic to engage with those with an interest in Evidence Based Medicine and the values that Cochrane expounds.
Yours sincerely
Michael Lunn, Richard Hughes and Phil Wiffen
Contributors
Brian Dickie, Feedback Editor, Cochrane Neuromuscular Disease Group
Ruth Brassington, Managing Editor, Cochrane Neuromuscular Disease Group
What's new
| Date | Event | Description |
|---|---|---|
| 21 September 2015 | Amended | Minor changes were made to Analysis 1.1 following feedback from the Cochrane Library site. No change to RR was made. Minor amendments to 'Risk of bias' assessments of no material difference to assessment grades. |
History
Protocol first published: Issue 2, 2008 Review first published: Issue 4, 2009
| Date | Event | Description |
|---|---|---|
| 5 February 2014 | Amended | Minor corrections to the following analyses and corresponding figures in text: Analysis 3.2 (Fibromyalgia: number of participants with ≥ 50% improvement of pain at more than 12 weeks, all doses), Analysis 6.6.6 (Adverse events leading to cessation, 'all doses') and Analysis 6.7.6 (Serious adverse events, 'all doses') |
| 17 January 2014 | Amended | Corrected the date assessed as up‐to‐date |
| 10 January 2014 | Amended | Minor editorial correction |
| 27 November 2013 | New search has been performed | Searches updated to November 2013 |
| 27 October 2013 | New citation required and conclusions have changed | Twelve new trials included. Scope extended to include fibromyalgia and other chronic pain conditions. Conclusions changed. Title change from 'Duloxetine for treating painful neuropathy or chronic pain' to 'Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia'. |
| 1 March 2011 | Amended | Contact details updated. |
| 9 February 2010 | Feedback has been incorporated | Feedback and authors' response added. |
| 14 April 2008 | Amended | Converted to new review format. |
Acknowledgements
MPTL is supported by the Biomedical Reseach Centre of UCLH Foundation Trust. The MRC Centre for Neuromuscular Disease hosts the Cochrane Neuromuscular Disease Group.
The Cochrane Neuromuscular Disease Group Trials Search Co‐ordinator, Angela Gunn, developed and ran searches.
Appendices
Appendix 1. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) <1946 to November Week 1 2013> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (389866) 2 controlled clinical trial.pt. (89904) 3 randomized.ab. (287333) 4 placebo.ab. (156850) 5 drug therapy.fs. (1767223) 6 randomly.ab. (199448) 7 trial.ab. (302482) 8 groups.ab. (1276425) 9 or/1‐8 (3299027) 10 exp animals/ not humans.sh. (4060470) 11 9 not 10 (2809295) 12 exp herpes zoster/ (9764) 13 herpes zoster.mp. (11326) 14 shingle$.mp. (867) 15 exp neuralgia, postherpetic/ (718) 16 postherpetic neuralgia.mp. (1429) 17 post‐herpetic neuralgia.mp. (574) 18 post‐herpetic pain.mp. (21) 19 postherpetic pain.mp. (52) 20 PHN.tw. (1202) 21 trigeminal neuralgia.mp. (6283) 22 exp Trigeminal Neuralgia/ (5589) 23 fibromyalgia.mp. (7494) 24 exp pain/ or (pain or painful).mp. (545148) 25 or/12‐24 (561051) 26 (duloxetine or cymbalta).mp. (1506) 27 11 and 25 and 26 (486) 28 remove duplicates from 27 (396)
Appendix 2. EMBASE (OvidSP) search strategy
Database: Embase <1980 to 2013 Week 46> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (38971) 2 double‐blind procedure.sh. (118651) 3 single‐blind procedure.sh. (18506) 4 randomized controlled trial.sh. (360008) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (1015543) 6 trial.ti. (155290) 7 or/1‐6 (1153429) 8 (animal/ or nonhuman/ or animal experiment/) and human/ (1321144) 9 animal/ or nonanimal/ or animal experiment/ (3494085) 10 9 not 8 (2877729) 11 7 not 10 (1057664) 12 limit 11 to embase (820767) 13 exp herpes zoster/ (16983) 14 herpes zoster.mp. (18732) 15 shingle$.mp. (1253) 16 exp postherpetic neuralgia/ (3802) 17 postherpetic neuralgia.mp. (4270) 18 post‐herpetic neuralgia.mp. (928) 19 post‐herpetic pain.mp. (30) 20 postherpetic pain.mp. (82) 21 PHN.tw. (1517) 22 trigeminal neuralgia.mp. (5471) 23 exp Trigeminus Neuralgia/ (8348) 24 painful neuropath$.mp. (826) 25 fibromyalgia.mp. (13627) 26 exp pain/dt or (pain or painful).tw,kw. (615267) 27 or/13‐26 (640712) 28 (duloxetine or cymbalta).mp. (6375) 29 12 and 27 and 28 (469) 30 remove duplicates from 29 (468)
Appendix 3. CENTRAL (DARE, HTA & NHSEED) search strategy
#1 MeSH descriptor: [Herpes Zoster] explode all trees #2 "herpes zoster" #3 shingle* #4 MeSH descriptor: [Neuralgia, Postherpetic] explode all trees #5 "postherpetic neuralgia" #6 "post‐herpetic neuralgia" #7 "post‐herpetic pain" #8 "postherpetic pain" #9 PHN #10 "trigeminal neuralgia" #11 fibromyalgia #12 MeSH descriptor: [Pain] explode all trees #13 pain or painful #14 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 #15 duloxetine or cymbalta #16 #14 and #15
Appendix 4. NMS Specialized Register (CRS) search strategy
#1 MeSH DESCRIPTOR Herpes Zoster Explode All [REFERENCE] [STANDARD] #2 "herpes zoster" or shingle* [REFERENCE] [STANDARD] #3 MeSH DESCRIPTOR Neuralgia, Postherpetic [REFERENCE] [STANDARD] #4 "postherpetic neuralgia" or "postherpetic pain" [REFERENCE] [STANDARD] #5 phn or "trigeminal neuralgia" or fibromyalgia [REFERENCE] [STANDARD] #6 MeSH DESCRIPTOR Trigeminal Neuralgia [REFERENCE] [STANDARD] #7 pain or painful [REFERENCE] [STANDARD] #8 MeSH DESCRIPTOR Pain Explode All [REFERENCE] [STANDARD] #9 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 [REFERENCE] [STANDARD] #10 duloxetine or cymbalta [REFERENCE] [STANDARD] #11 #9 and #10 [REFERENCE] [STANDARD] #12 (#9 and #10) AND (INREGISTER) [REFERENCE] [STANDARD]
Appendix 5. ClinicalTrials.gov
duloxetine AND pain
Appendix 6. Appendix table 1 ‐ identified trials completed without obvious study report
| www.clinicaltrials.gov reference | Title | Responsible body | Completed? | Last verified |
| NCT00489073 | Duloxetine versus placebo for fibromyalgia | Eli Lilly | Yes | June 2007 |
| NCT00233025 | Duloxetine versus placebo in the treatment of fibromyalgia syndrome | Eli Lilly | Yes | August 2007 |
| NCT00125892 | A study of duloxetine in the treatment of fibromyalgia | Eli Lilly | Yes | May 2007 |
| NCT00603265 | Safety and efficacy study of ADL5859 in subjects with neuropathic pain associated with diabetic peripheral neuropathy | Adolor Corporation | Yes | October 2008 |
| NCT01579279 | A study comparing the efficacy and safety of ABT‐652 to placebo in subjects with diabetic neuropathic pain | Abbott | Yes | May 2012 |
Data and analyses
Comparison 1. Duloxetine versus placebo in the treatment of painful diabetic neuropathy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants with ≥ 50% improvement of pain at 12 weeks or less | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Duloxetine 20 mg daily | 1 | 213 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.98, 2.09] |
| 1.2 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 1.91 [1.26, 2.87] |
| 1.3 Duloxetine 60 mg daily | 4 | 908 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [1.44, 2.08] |
| 1.4 Duloxetine 120 mg daily | 4 | 870 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [1.08, 1.97] |
| 1.5 All doses | 5 | 1655 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [1.21, 1.92] |
| 2 Mean improvement in pain at 12 weeks or less | 5 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Duloxetine 20 mg daily | 1 | 179 | Mean Difference (IV, Fixed, 95% CI) | ‐0.45 [‐1.05, 0.15] |
| 2.2 Duloxetine 60 mg daily | 4 | 722 | Mean Difference (IV, Fixed, 95% CI) | ‐0.96 [‐1.26, ‐0.65] |
| 2.3 Duloxetine 120 mg daily | 4 | 828 | Mean Difference (IV, Fixed, 95% CI) | ‐0.93 [‐1.21, ‐0.65] |
| 3 Number of participants with ≥ 30% improvement in pain at 12 weeks or less | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [1.18, 2.07] |
| 3.2 Duloxetine 60 mg daily | 4 | 799 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.53 [1.33, 1.75] |
| 3.3 Duloxetine 120 mg daily | 3 | 659 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.21, 1.58] |
| 3.4 All doses | 4 | 1220 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [1.30, 1.63] |
| 4 Mean improvement in SF‐36 Physical Subscore at 12 weeks or less | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Duloxetine 20 mg daily | 1 | 200 | Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐2.42, 1.88] |
| 4.2 Duloxetine 60 mg daily | 3 | 514 | Mean Difference (IV, Random, 95% CI) | 2.65 [1.38, 3.92] |
| 4.3 Duloxetine 120 mg daily | 2 | 409 | Mean Difference (IV, Random, 95% CI) | 2.80 [1.04, 4.55] |
| 5 Mean improvement in SF‐36 Mental Subscore at 12 weeks or less | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Duloxetine 20 mg daily | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 1.11 [‐0.98, 3.20] |
| 5.2 Duloxetine 60 mg daily | 3 | 514 | Mean Difference (IV, Fixed, 95% CI) | 1.08 [‐0.32, 2.48] |
| 5.3 Duloxetine 120 mg daily | 2 | 409 | Mean Difference (IV, Fixed, 95% CI) | 2.23 [0.69, 3.77] |
| 6 Mean improvement in SF‐36 Bodily Pain Subscore at 12 weeks or less | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Duloxetine 20 mg daily | 1 | 209 | Mean Difference (IV, Fixed, 95% CI) | 2.90 [‐2.37, 8.17] |
| 6.2 Duloxetine 60 mg daily | 2 | 421 | Mean Difference (IV, Fixed, 95% CI) | 5.58 [1.74, 9.42] |
| 6.3 Duloxetine 120 mg daily | 2 | 420 | Mean Difference (IV, Fixed, 95% CI) | 8.19 [4.33, 12.05] |
| 7 Mean improvement in Patient Reported Global Impression of Improvement at 12 weeks or less | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Duloxetine 20 mg daily | 1 | 219 | Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.56, 0.10] |
| 7.2 Duloxetine 40 mg daily | 1 | 252 | Mean Difference (IV, Random, 95% CI) | ‐0.65 [‐1.01, ‐0.29] |
| 7.3 Duloxetine 60 mg daily | 5 | 1018 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐0.77, ‐0.44] |
| 7.4 Duloxetine 120 mg daily | 4 | 870 | Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐0.73, ‐0.35] |
| 8 Mean improvement in BPI Severity ‐ average pain at 12 weeks or less | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Duloxetine 60 mg daily | 2 | 433 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐1.38, ‐0.57] |
| 8.2 Duloxetine 120 mg daily | 2 | 428 | Mean Difference (IV, Random, 95% CI) | ‐1.16 [‐1.91, ‐0.41] |
| 9 Mean improvement in pain at rest (night pain) at 12 weeks or less | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Duloxetine 20 mg daily | 1 | 222 | Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.90, 0.34] |
| 9.2 Duloxetine 60 mg daily | 3 | 664 | Mean Difference (IV, Random, 95% CI) | ‐0.92 [‐1.27, ‐0.57] |
| 9.3 Duloxetine 120 mg daily | 3 | 664 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐1.45, ‐0.75] |
Comparison 2. Duloxetine versus pregabalin in the treatment of painful diabetic neuropathy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants with ≥ 50% improvement in pain at 12 weeks or less | 1 | 804 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [1.19, 1.80] |
| 2 Mean improvement in pain at 12 weeks or less | 1 | 804 | Mean Difference (IV, Fixed, 95% CI) | ‐0.62 [‐0.92, ‐0.32] |
| 3 Number improved ≥ 30% at 12 weeks or less | 1 | 804 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [1.20, 1.68] |
Comparison 3. Duloxetine versus placebo in the treatment of fibromyalgia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants with ≥ 50% improvement of pain at 12 weeks or less | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Duloxetine 20 mg daily | 1 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.91, 2.14] |
| 1.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.75, 1.35] |
| 1.3 Duloxetine 60 mg daily | 2 | 528 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [1.20, 2.06] |
| 1.4 Duloxetine 120 mg daily | 4 | 1234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [1.40, 2.03] |
| 1.5 All doses | 5 | 1887 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.29, 1.75] |
| 2 Number of participants with ≥ 50% improvement of pain at more than 12 weeks | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Duloxetine 60 mg daily | 1 | 373 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [1.10, 2.27] |
| 2.2 Duloxetine 120 mg daily | 2 | 616 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.07, 1.79] |
| 2.3 Duloxetine all doses | 2 | 845 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [1.09, 1.79] |
| 3 Number of participants with ≥ 30% improvement of pain at 12 weeks or less | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Duloxetine 20 mg daily | 1 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.94, 1.79] |
| 3.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.89, 1.45] |
| 3.3 Duloxetine 60 mg daily | 2 | 528 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 [1.24, 1.85] |
| 3.4 Duloxetine 120 mg daily | 3 | 1020 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.46 [1.26, 1.69] |
| 3.5 All doses | 4 | 1673 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [1.22, 1.56] |
| 4 Mean improvement in pain at 12 weeks or less | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 4.1 Duloxetine 30 mg daily | 1 | 308 | Mean Difference (IV, Fixed, 95% CI) | ‐0.31 [‐0.86, 0.24] |
| 4.2 Duloxetine 120 mg daily | 1 | 507 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐1.35, ‐0.25] |
| 5 Mean improvement in the SF‐36 mental component summary subscore | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Duloxetine 20 mg daily | 1 | 223 | Mean Difference (IV, Random, 95% CI) | 0.81 [‐2.37, 3.99] |
| 5.2 Duloxetine 30 mg daily | 1 | 308 | Mean Difference (IV, Random, 95% CI) | 2.69 [0.31, 5.07] |
| 5.3 Duloxetine 60 mg daily | 2 | 515 | Mean Difference (IV, Random, 95% CI) | 3.31 [0.59, 6.02] |
| 5.4 Duloxetine 120 mg daily | 5 | 1531 | Mean Difference (IV, Random, 95% CI) | 4.22 [2.43, 6.02] |
| 6 Mean improvement in the SF‐36 physical component summary subscore | 6 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Duloxetine 20 mg daily | 1 | 223 | Mean Difference (IV, Fixed, 95% CI) | 0.81 [‐1.92, 3.54] |
| 6.2 Duloxetine 30 mg daily | 1 | 308 | Mean Difference (IV, Fixed, 95% CI) | 0.84 [‐1.17, 2.85] |
| 6.3 Duloxetine 60 mg daily | 2 | 515 | Mean Difference (IV, Fixed, 95% CI) | 1.28 [‐0.33, 2.89] |
| 6.4 Duloxetine 120 mg daily | 5 | 1531 | Mean Difference (IV, Fixed, 95% CI) | 2.13 [0.95, 3.30] |
| 7 Mean improvement in the SF‐36 Bodily Pain Subscore | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 Duloxetine 60 mg daily | 1 | 221 | Mean Difference (IV, Fixed, 95% CI) | 8.2 [3.20, 13.20] |
| 7.2 Duloxetine 120 mg daily | 4 | 1243 | Mean Difference (IV, Fixed, 95% CI) | 5.96 [3.76, 8.16] |
| 8 Mean improvement in the Patient reported Global Impression of Change at completion of trial | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 8.1 Duloxetine 20 mg daily | 1 | 223 | Mean Difference (IV, Fixed, 95% CI) | ‐0.54 [‐0.96, ‐0.12] |
| 8.2 Duloxetine 30 mg daily | 1 | 308 | Mean Difference (IV, Fixed, 95% CI) | ‐0.38 [‐0.71, ‐0.05] |
| 8.3 Duloxetine 60 mg daily | 2 | 519 | Mean Difference (IV, Fixed, 95% CI) | ‐0.45 [‐0.73, ‐0.18] |
| 8.4 Duloxetine 120 mg daily | 3 | 826 | Mean Difference (IV, Fixed, 95% CI) | ‐0.44 [‐0.66, ‐0.23] |
Comparison 4. Duloxetine versus placebo for the treatment of pain in major depressive disorder.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants with > 50% pain relief at 12 weeks or less | 2 | 1023 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [1.19, 1.59] |
| 2 Participants with > 30% pain relief at 12 weeks or less | 3 | 1359 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [1.15, 1.40] |
| 3 Mean improvement in pain at 12 weeks or less | 3 | 1359 | Mean Difference (IV, Fixed, 95% CI) | ‐0.55 [‐0.75, ‐0.35] |
Comparison 5. Duloxetine versus placebo in the treatment of central neuropathic pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean improvement in pain at 12 weeks or less | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | ‐1.0 [‐2.05, 0.05] |
| 2 Mean improvement in SF‐36 Physical Subscore | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 2.0 [‐12.72, 16.72] |
| 3 Mean improvement in the SF‐36 Mental Subscore at 12 weeks | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 4.0 [‐6.75, 14.75] |
| 4 Mean improvement in the SF‐36 Bodily Pain Subscore | 1 | 48 | Mean Difference (IV, Fixed, 95% CI) | 8.0 [‐0.81, 16.81] |
| 5 Number of participants improved on PGI‐I (better or very much better) | 1 | 48 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [1.02, 7.44] |
Comparison 6. Duloxetine versus placebo: adverse events during first 12 weeks of treatment for painful neuropathy or fibromyalgia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion of participants with any adverse event | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [1.03, 1.52] |
| 1.2 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.01, 1.31] |
| 1.3 Duloxetine 60 mg daily | 13 | 4521 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.10, 1.20] |
| 1.4 Duloxetine 120 mg daily | 3 | 688 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [1.09, 1.30] |
| 1.5 Duloxetine all doses | 14 | 5258 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [1.11, 1.20] |
| 2 Nausea | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Duloxetine 20 mg daily | 1 | 230 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [0.71, 3.00] |
| 2.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Random, 95% CI) | 5.43 [2.34, 12.58] |
| 2.3 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 6.55 [1.85, 23.17] |
| 2.4 Duloxetine 60 mg daily | 11 | 3642 | Risk Ratio (M‐H, Random, 95% CI) | 2.61 [2.14, 3.18] |
| 2.5 Duloxetine 120 mg daily | 4 | 787 | Risk Ratio (M‐H, Random, 95% CI) | 2.89 [2.06, 4.04] |
| 3 Dry mouth | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Duloxetine 20 mg daily | 1 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.30, 2.47] |
| 3.2 Duloxetine 40 mg daily | 0 | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 Duloxetine 60 mg daily | 6 | 2004 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.63 [1.89, 3.67] |
| 3.4 Duloxetine 120 mg daily | 3 | 567 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.40 [1.94, 5.96] |
| 4 Dizziness | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Duloxetine 20 mg daily | 1 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.33, 2.33] |
| 4.2 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.89 [1.22, 28.58] |
| 4.3 Duloxetine 60 mg daily | 8 | 2257 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.84 [1.35, 2.51] |
| 4.4 Duloxetine 120 mg daily | 4 | 787 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.44 [1.55, 3.83] |
| 5 Somnolence | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Duloxetine 20 mg daily | 1 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.41, 2.43] |
| 5.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.22 [0.70, 7.06] |
| 5.3 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.25 [1.15, 4.38] |
| 5.4 Duloxetine 60 mg daily | 8 | 2678 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.94 [2.17, 3.97] |
| 5.5 Duloxetine 120 mg daily | 4 | 787 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.76 [2.93, 7.74] |
| 6 Adverse event leading to cessation | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Duloxetine 20 mg daily | 2 | 453 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.78, 2.39] |
| 6.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.54 [0.69, 3.44] |
| 6.3 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.96 [0.81, 4.77] |
| 6.4 Duloxetine 60 mg daily | 14 | 4837 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.95 [1.60, 2.37] |
| 6.5 Duloxetine 120 mg daily | 7 | 1462 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [1.74, 3.04] |
| 6.6 All doses | 17 | 6285 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [1.67, 2.37] |
| 7 Serious adverse event | 16 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Duloxetine 20 mg daily | 0 | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.2 Duloxetine 30 mg daily | 1 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.02] |
| 7.3 Duloxetine 40 mg daily | 1 | 252 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.95 [0.50, 17.30] |
| 7.4 Duloxetine 60 mg daily | 14 | 4842 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.60, 1.32] |
| 7.5 Duloxetine 120 mg daily | 6 | 1257 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.25, 1.35] |
| 7.6 All doses | 14 | 4976 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.53, 1.25] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arnold 2004.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial of duloxetine in fibromyalgia | |
| Participants | 207 men or women over 18 years who fulfilled American College of Rheumatology criteria for fibromyalgia, and scoring 4 or more on the pain intensity item of the Fibromyalgia Impact Questionnaire (FIQ) | |
| Interventions | Duloxetine 60 mg twice daily versus placebo for 12 weeks with a 20‐day titration phase | |
| Outcomes | Follow‐up at 12 weeks Outcomes:
|
|
| Notes | Greater use of antidepressants in the placebo group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assignment to treatment groups was determined by a computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | Used an interactive voice response system |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind for all assessments in 12‐week therapy phase, Investigators adjusted the number of placebo capsules similarly to maintain the blinding. Single‐blind in run‐in phase |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 46/104 (44%) in duloxetine and 37/103 (36%) in placebo group discontinued treatment but all dropouts accounted for and LOCF |
| Selective reporting (reporting bias) | Unclear risk | As above in incomplete outcome data |
| Other bias | Low risk | More use of antidepressants in the placebo group but this would bias against the treatment arm |
Arnold 2005.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial of duloxetine in fibromyalgia | |
| Participants | 354 participants Women only, ≥ 18 years of age who met criteria for primary fibromyalgia as defined by the American College of Rheumatology, and had a score of ≥ 4 on the average pain severity item of the Brief Pain Inventory (BPI) at randomisation |
|
| Interventions | Duloxetine 60 mg daily, duloxetine 60 mg twice daily and placebo for 12 weeks | |
| Outcomes |
|
|
| Notes | Company sponsored and run trial. Fibromyalgia Impact Questionnaire abandoned in favour of BPI | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random assignment of women who met entry criteria following the screening phase to one of three treatment groups: duloxetine 60 mg daily, duloxetine 60 mg twice daily (forced titration from 60 mg daily for 3 days to 60 mg twice daily), or placebo, with randomisation in a 1:1:1 ratio. Random assignment of the participants to treatment groups occurred within two stratified groups, those with and those without current major depressive disorder |
| Allocation concealment (selection bias) | Unclear risk | Probably low risk of bias as previous trial used an adequate method |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High proportion of dropouts: 138 (39%) participants withdrew during the 12‐week therapy phase, 41 (35%) from the duloxetine 60 mg daily group, 45 (39%) from the duloxetine 60 mg twice daily group, and 52 (43%) from the placebo group (P = 0.407). Matched across groups but a high rate of loss "Partial intention to treat analysis". Efficacy analyses include all randomised participants with a baseline and at least one post‐baseline visit with efficacy data, while safety analyses included all randomised participants |
| Selective reporting (reporting bias) | Unclear risk | See incomplete outcome data above |
| Other bias | Low risk | Lilly study. No other bias identified |
Arnold 2010.
| Methods | Phase IV randomised, double‐blind (subject, caregiver, investigator, outcomes assessor), placebo‐controlled, parallel assignment safety and efficacy study of duloxetine in fibromyalgia | |
| Participants | Men or women
|
|
| Interventions | Duloxetine 60 to 120 mg daily for 24 weeks | |
| Outcomes | Time frame for all outcome measures 24 weeks Primary outcome
Secondary outcomes
|
|
| Notes | Completed and published | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned 1:1 in a double‐blind fashion to duloxetine 60 mg once daily or placebo by a computer‐generated random sequence using an IVRS |
| Allocation concealment (selection bias) | Low risk | "Double blind". "Variable transition to active treatment strategy…thereby blinding the onset of active treatment to reduce the patient's expectation of experiencing side effects" |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No comment on formulation of drug or placebo but almost certainly double blinded both in up and down titration. However, significantly more participants on duloxetine withdrew with adverse effects |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Accounted for as much as possible. High dropout rate (> 30%). Employs ITT ‐ use of a "restricted maximum likelihood‐based [mixed effects model repeated measures approach] analysis accounts for bias caused by non‐random missing data due to early discontinuation because of adverse events or lack of efficacy better than LOCF" |
| Selective reporting (reporting bias) | Low risk | "Patient Global Impression ‐ severity (PGI‐S) only assessed at baseline". Otherwise, paper reports all results |
| Other bias | Unclear risk | Lilly trial. 93.2% female participants, similar to all fibromyalgia studies |
Arnold 2012.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group study of duloxetine in fibromyalgia | |
| Participants | Women and men > 18 years of age who met the American College of Rheumatology 1990 criteria for primary fibromyalgia and had a score of > 4 on the average pain severity item of the Brief Pain Inventory (BPI)‐Modified Short Form. Patients with or without major depressive disorder or generalised anxiety disorder, as defined by the DSM‐IV and confirmed by the MINI were included. | |
| Interventions | Duloxetine 30 mg capsules or placebo for 12 weeks | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Lilly study. No other bias identified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation by a computer‐generated random sequence using an interactive voice response system (IVRS) |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The duloxetine and placebo capsules were identical in appearance to maintain the blinding. Participants and investigators were kept blinded to the rescue criteria and dose increase; site personnel entered the major depressive disorder status at baseline and the CGI‐I for Depression scores through IVRS at every visit |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 2 dropouts, both from duloxetine group and likely to have been of minimal significance |
| Selective reporting (reporting bias) | Low risk | Most outcomes presented except individual BPI severity items (worst pain, least pain, pain right now). However, no other outcomes with significant effect in completely negative trial |
| Other bias | Low risk | Lilly study. No other bias identified |
Brecht 2007.
| Methods | 8‐week, randomised, double‐blind, placebo‐controlled, parallel‐group efficacy and safety study of duloxetine in the treatment of pain of unknown aetiology in people with major depressive disorder | |
| Participants | Women or men > 18 with major depressive disorder defined by DSM‐IV. At baseline, depression score of > 20 on the MADRS and at least moderate pain on Brief Pain Inventory Short Form (BPI‐SF) ‐ 3 or higher for "24 hour average pain". Participants were also devoid of any other diagnosed pain syndrome as per a medical history | |
| Interventions | Duloxetine 60 mg versus placebo for 8 weeks | |
| Outcomes | Primary outcome
Seconday outcomes
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised" |
| Allocation concealment (selection bias) | Unclear risk | "Double blind" |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT using all participants with 1 dose of drug. 25% dropout rate |
| Selective reporting (reporting bias) | Low risk | None identified |
| Other bias | Low risk | Company sponsored trial |
Chappell 2008.
| Methods | Six‐month, randomised, double‐blind, placebo‐controlled, clinical trial of duloxetine in fibromyalgia | |
| Participants | Male and female outpatients were eligible for the study if they were ≥ 18 years of age, met criteria for fibromyalgia as defined by the American College of Rheumatology, with or without major depressive disorder No criteria for pain level at entry |
|
| Interventions | Duloxetine ‐ variable dose. Started at 60 mg (30 mg run in period over 1 week), randomised increase to 120 mg after 13 weeks if not > 50% reduction in pain on BPI average | |
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated random sequence within each study centre stratified by major depressive disorder |
| Allocation concealment (selection bias) | Low risk | Double‐blind |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Participants "blinded", but not clear how the study managed dose escalations and decreases and whether blinding was maintained |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 37.6% to 38.6% discontinuations, significantly different in lack of efficacy only. Investigators used LOCF and MMRM to correct for dropouts |
| Selective reporting (reporting bias) | Low risk | 30% improvement in BPI‐average added post hoc |
| Other bias | Unclear risk | Lilly sponsored trial Significant unexplained treatment by investigator interaction |
Gao 2010.
| Methods | Phase III randomised, double‐blind (subject, caregiver, investigator, outcomes assessor), placebo‐controlled, parallel assignment safety and efficacy study of duloxetine in painful diabetic neuropathy | |
| Participants | 215 participants Men or women, aged 18 to 75, pain due to bilateral peripheral neuropathy caused by type I or type II diabetes with the pain beginning in the feet, and present for at least 6 months. Score of 4 or greater on the Brief Pain Inventory (BPI) on the 24‐hour average pain item |
|
| Interventions | Duloxetine 60 mg daily for 12 weeks | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Closed and completed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned |
| Allocation concealment (selection bias) | Low risk | "Double blind". Study medication in capsules…or matching placebo' |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 15.6% to 17.9% dropout rate but ITT using LOCF and MMRM approach to minimise bias |
| Selective reporting (reporting bias) | Low risk | Selective use of MMRM or LOCF depending upon outcome. However, all measures reported |
| Other bias | Low risk | Lilly sponsored trial No adjustment for multiple comparisons |
Gaynor 2011a.
| Methods | Randomised, double‐blind, placebo‐controlled trial of duloxetine in people with major depressive disorder and painful physical symptoms | |
| Participants | Adult (18 years of age) male or female outpatients were eligible ...if they met all of the following: a current episode of major depressive disorder according to the DSM‐IV‐TR and confirmed by the MINI with a history of at least one separate, previous episode of depression, and at both the screening and randomisation visits a MADRS total score of 20, and at least moderate pain with a score of 3 on the Brief Pain Inventory Short Form (BPI) average pain item, and a Clinical Global Impression of Severity (CGI‐S) score 4. Painful symptoms were not allowed to have an identifiable underlying cause | |
| Interventions | Duloxetine 60 mg once daily orally for 8 weeks vs placebo | |
| Outcomes |
|
|
| Notes | Gaynor 2011b identical in design ‐ different patient group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No concerns |
| Selective reporting (reporting bias) | Low risk | None |
| Other bias | Low risk | None |
Gaynor 2011b.
| Methods | A randomised, double‐blind, placebo‐controlled trial of duloxetine in people with major depressive disorder and painful physical symptoms | |
| Participants | Adult (18 years of age) male or female outpatients were eligible ...if they met all of the following: a current episode of major depressive disorder according to the DSM‐IV‐TR and confirmed by the MINI with a history of at least one separate, previous episode of depression, and at both the screening and randomisation visits a MADRS total score of 20, and at least moderate pain with a score of 3 on the Brief Pain Inventory Short Form (BPI) average pain item, and a Clinical Global Impression of Severity (CGI‐S) score 4. Painful symptoms were not allowed to have an identifiable underlying cause | |
| Interventions | Duloxetine 60 mg once daily orally for 8 weeks vs placebo | |
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'Randomised' |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | 'Double Blind' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No concerns |
| Selective reporting (reporting bias) | Low risk | None |
| Other bias | Low risk | None identified |
Goldstein 2005.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group, trial of duloxetine in painful diabetic neuropathy | |
| Participants | 457 participants Participants, at least 18 years of age, had daily pain due to polyneuropathy caused by type 1 or type 2 diabetes mellitus, which was present for a minimum of 6 months. This pain had to have begun in the feet with relatively symmetrical onset. The diagnosis was confirmed by a score of at least 3 on the Michigan Neuropathy Screening Instrument (MNSI). Participants were required to have a minimum score of 4 on the 24‐hour average pain score rated on an 11‐point (0 to 10) Likert scale. |
|
| Interventions | Duloxetine 20 mg daily, 60 mg daily or 60 mg twice daily versus placebo for 8 weeks | |
| Outcomes |
|
|
| Notes | Company sponsored and run trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned in a 1:1:1:1 ratio by a computer generated random sequence |
| Allocation concealment (selection bias) | Low risk | Participant numbers were assigned consecutively at each study site. The interactive voice response system was used to assign blister cards containing the study drug to each participant confirmed through interactive voice response system entry of a confirmation number on the card |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | All analyses were undertaken as an ITT analysis. All participants were analysed in the safety analysis and all participants with at least one post entry data point were analysed in an ITT analysis. Dropout rate was 25% with significantly more in the higher dose treatment groups |
| Selective reporting (reporting bias) | Unclear risk | See above |
| Other bias | Low risk | Company sponsored and run trial |
Kaur 2011.
| Methods | Randomised, double‐blind, cross‐over clinical trial comparing amitriptyline and duloxetine in painful diabetic neuropathy | |
| Participants | 86 participants ‐ 65 randomised to treatment in 1st arm, 58 of whom completed both arms People of either sex with type 2 diabetes, aged between 18 and 75 years, who were on stable glucose‐lowering medications during the preceding month and who had painful diabetic neuropathy for at least 1 month were considered for the study. The study enrolled people who had a pain score of > 50%, as assessed by visual analogue scale (VAS). Painful diabetic neuropathy was confirmed by 1) medical history, 2) a diabetic neuropathy symptom (DNS) score of > 1 point (7), 3) a Diabetic Neuropathy Examination (DNE) score of > 3 points (8), 4) a modified neuropathy symptom score (mNSS) (9,10), and 5) increased thresholds on the vibration perception test and monofilament test |
|
| Interventions | Amitiyptyline 10, 25 or 50 mg once daily at night or duloxetine, 20, 40 or 60 mg once daily at night Intervention only 6 weeks before 2 week washout and cross‐over to alternate arm. Participants commenced on lowest dose and then increased every 2 weeks to next dose if required by treating physician; thus potentially only 2 weeks on maximum dose. 48% of amitriptyline and 65% of duloxetine participants reached the highest dose of drug. 17% vs 5% of the participants preferred higher dose duloxetine to amitriptyline |
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised (computer generated randomisation of blocks of 4) |
| Allocation concealment (selection bias) | Unclear risk | An independent person unrelated to the study carried out blinding and randomisation. Two separate companies provided medicines, so it is not clear that they were identical in appearance |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Blinded. Single physician assessment. "success of blinding was assessed by the accuracy of the physicians prediction at the end of the study" (34% correctly identified only) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No concerns |
| Selective reporting (reporting bias) | High risk | The primary end point of the study was the reduction in the median pain score from baseline, (patient’s global assessment of efficacy by VAS (0 to 100 points)). Secondary end points included the assessment of pain by the short‐form McGill Pain Questionnaire (11); an 11‐point Likert scale for pain (0 = no pain and 10 = excruciating pain); change in sleep pattern (increased, unchanged, or decreased); overall improvement by DNE score, DNS score, mNSS, and the 24‐point HAMD; and patient self evaluation of overall change on the basis of a 7‐point Patient Global Impression of Change (PGIC) scale not reported in analysis. |
| Other bias | High risk | Significant (but similar) carryover into period 2 despite 2 weeks' washout |
Raskin 2005.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial in diabetic peripheral neuropathic pain | |
| Participants | 348 participants Participants ≥ 18 years, with pain due to bilateral peripheral neuropathy caused by type 1 or type 2 diabetes mellitus. The pain had to begin in the feet with relatively symmetrical onset and be present for at least 6 months. Participants had to have a mean score of ≥ 4 when assessed for 24‐hour average pain severity on the Michigan Neuropathy Screening Instrument (MNSI) 11‐point Likert scale (from the patient diary prior to randomisation), and stable glycaemic control. Concomitant pain medications excluded. |
|
| Interventions | Duloxetine 60 mg daily or duloxetine 60 mg twice daily versus placebo for 12 weeks | |
| Outcomes |
|
|
| Notes | Company sponsored and run trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation performed at visit 3 in a 1:1:1 ratio. A computer‐generated random sequence determined assignment to treatment groups, using an IVRS |
| Allocation concealment (selection bias) | Low risk | Participants received either of (or a combination of, depending on their randomly assigned treatment) the following: 30 mg capsules of duloxetine hydrochloride or placebo capsules identical to duloxetine capsules. Participants randomly assigned to each treatment group were instructed to take two capsules (by mouth) every morning and every evening. Treatment was assigned using IVRS |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts were 52/340 (15%). Analysis was by ITT |
| Selective reporting (reporting bias) | Low risk | See above |
| Other bias | Low risk | Lilly study. No other bias identified |
Rowbotham 2012.
| Methods | Phase II, randomised, double‐blind, placebo‐controlled, single group assignment, safety and efficacy study comparing duloxetine, ABT‐894 and placebo in diabetic neuropathic pain | |
| Participants | 108 participants Men and women 18 to 75 Inclusion criteria:
|
|
| Interventions | Drug: ABT‐894, 1 mg, 2 mg, 4 mg twice daily
Drug: placebo
Drug: duloxetine 60 mg Duration 8 weeks |
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Published 2012 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants "were randomized 1:1 to each treatment arm via an interactive voice response system using a randomization schedule that was generated before study start" |
| Allocation concealment (selection bias) | Low risk | Careful attention to placebo and medication concealment noted |
| Blinding (performance bias and detection bias) All outcomes | Low risk | No concerns |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Almost 100% completed |
| Selective reporting (reporting bias) | Low risk | All reported |
| Other bias | Low risk | None identified |
Russell 2008.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial in fibromyalgia | |
| Participants | 520 participants Female and male outpatients ≥ 18 years of age who met criteria for fibromyalgia as defined by the American College of Rheumatology. Participants were required to have a score ≥ 4 on the average pain severity item (in the past 24 hours) of the Brief Pain Inventory (BPI‐modified Short Form at screening and at baseline. The study included people with or without current major depressive disorder and evaluated them for the presence of psychiatric disorders using the MINI. Prior to randomisation, the study required participants to discontinue any medications that might interfere with the evaluation of pain improvement, including analgesics (with the exception of up to 325 mg/day of aspirin for cardiac prophylaxis and paracetamol up to 2 g/day for pain), antidepressants, anticonvulsants, or other medications taken for fibromyalgia or pain |
|
| Interventions | Duloxetine 20 mg daily, 60 mg daily or 60 mg twice daily versus placebo for 6 months | |
| Outcomes |
|
|
| Notes | Company sponsored and run trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated random sequence determined assignment to treatment groups and the study randomly assigned each stratum (depressed and non‐depressed) within sites to achieve a relative balance across treatments |
| Allocation concealment (selection bias) | Unclear risk | Unclear although other trials from the same group have been adequate |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 35% to 40% dropout at the 3 month interim analysis phase and up to 46% dropout for the 6 month phase. "Intention‐to‐treat unless otherwise specified". Safety analyses in all participants and others with data for at least 1 measure |
| Selective reporting (reporting bias) | Unclear risk | See above |
| Other bias | Low risk | Lilly study. No other bias identified |
Tesfaye 2013.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group enrichment trials with three phases comparing duloxetine to pregabalin in painful diabetic neuropathy | |
| Participants | 401 participants treated with duloxetine and 403 with pregabalin Included participants had pain due to bilateral peripheral neuropathy (caused by type 1 or type 2 diabetes mellitus. Pain must have begun in the feet, with relatively symmetrical onset. Daily pain should have been present for more than 3 months (assessed by questioning the patient). • Score of at least 4 on the 24‐hour average pain severity score on an 11‐point Likert scale (on Brief Pain Inventory Modified Short Form (BPI‐mSF)) at screening and at randomisation • Participants not receiving treatment for diabetic peripheral neuropathic pain or received treatment for diabetic peripheral neuropathic pain, with a drug other than pregabalin or duloxetine, and completed the required washout • Participants never received treatment with duloxetine or pregabalin (short courses of less than 15 days of treatment, at any time previously, allowed) •Stable glycaemic control, as assessed by a physician investigator, and HbA1c less than or equal to 12% at screening |
|
| Interventions | Pregabalin titrated to 150 mg twice daily was compared to duloxetine titrated to 60 mg once daily (with placebo tablets to maintain blind between treatments) and treated in study phase II for 8 weeks. A third phase of non‐responding participants entered study phase III not included in this analysis | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised 1:1:1:1 in 4 parallel groups based on a computer generated sequence using IVRS |
| Allocation concealment (selection bias) | Unclear risk | Unclear ‐ although all drugs and placebo were similar and the allocation stratified by site, does not explicitly deal with concealment |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The trial maintained blinding by using over‐encapsulated duloxetine and pregabalin capsules, matching placebo and an identical dosing regimen for all groups in terms of numbers and timing of capsules |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropout in Phase II 17%, 9% with adverse events. All analyses performed on ITT (baseline + 1 measure for outcomes, all randomised for adverse events) with MMRM ‐ however no statement as to whether LOCF or BOCF used |
| Selective reporting (reporting bias) | High risk | Some partial reporting of outcomes (for example NPSI subscores not tabulated, PGI‐I and CGI‐I in figure form only and differences of reporting between phase II and phase III outcome reporting |
| Other bias | High risk | Lilly designed, interpreted, wrote and submitted. Ghost written by professional writer for company |
Vranken 2011.
| Methods | Stratified, randomised, double‐blind, placebo‐controlled, parallel group study of patients with severe central neuropathic pain of more than 6 months duration from cerebrovascular or spinal cord lesions | |
| Participants | 48 participants aged 18 years or older with > 6 month severe neuropathic pain from cord or cerebrovascular cause, > 6 on visual analogue scale (VAS) (10 points), which started after sustaining the lesion and with the distribution of pain concomitant with the somatosensory system involvement. The trial allowed other medication if doses were stable for 6 weeks, except other antidepressants, which had to be stopped more than 30 days prior to receiving study medication | |
| Interventions | Duloxetine or placebo for 8 weeks. Duloxetine 60 mg at start. Increased if participants did not meet criteria of > 1.8 points improvement on VAS. At week 8 and study end 15 participants on 120 mg and 8 participants on 60 mg | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Simple computerised random sampling (clorandm.exe) assigned study codes N = 1 to the placebo or duloxetine arm. Consecutive participants who met inclusion criteria were randomly assigned to treatment with flexible dose placebo or flexible dose duloxetine |
| Allocation concealment (selection bias) | Low risk | The association between type of treatment and study code was only known to the Department of Epidemiology, Biostatistics and Bioinformatics and the hospital pharmacy department |
| Blinding (performance bias and detection bias) All outcomes | Low risk | |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | |
| Selective reporting (reporting bias) | Low risk | |
| Other bias | Low risk | |
Wernicke 2006.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel group trial of duloxetine in diabetic peripheral neuropathic pain | |
| Participants | 334 participants Men or women ≥ 18 years and with > 6 months diabetic peripheral neuropathic pain secondary to type 1 or 2 diabetes (distal and symmetrical). At randomisation, score > 3 on Michigan Neuropathy Screening Instrument and average > 4 on 24 hour pain scale. Stable glucose control and HBA1c < 12. Multiple exclusions including other pain medications except paracetamol and aspirin |
|
| Interventions | Duloxetine 60 mg daily, 60 mg twice daily or placebo for 12 weeks | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Company sponsored and run trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed at the site level in that randomisation codes were assigned to sites in blocks, but there was no further stratification. Participants were randomly assigned to treatment in a 1:1:1 ratio. Assignment to a treatment group was determined by a computer‐generated random sequence using an IVRS |
| Allocation concealment (selection bias) | Low risk | The IVRS was used to assign blister cards containing study drug to each participant |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop outs were 29/114 (25%) in duloxetine 60 mg daily, 34/112 (30%) in duloxetine 60 mg twice daily and 23/108 (21.3%) in the placebo group |
| Selective reporting (reporting bias) | Unclear risk | An ITT principle was used in the analyses of all efficacy variables. For each efficacy variable, the analysis included all randomised participants with a baseline and at least one non‐missing observation after baseline |
| Other bias | Low risk | Lilly study. No other bias identified |
Yasuda 2010.
| Methods | Phase III randomised, double‐blind (subject, caregiver, investigator, outcomes assessor), placebo‐controlled, parallel assignment, safety and efficacy study of duloxetine in diabetic peripheral neuropathic pain | |
| Participants | 339 participants randomised Male or female outpatients aged 20 years or older but less than 80 years at the time of consent:
|
|
| Interventions | Duloxetine 40 mg or 60 mg orally daily versus placebo for 12 weeks | |
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Notes | Recruiting | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assigning table was prepared using Create Key Code 3.3. Participants were randomly assigned…' Stratified for pain, duration of diabetic peripheral neuropathy, diabetes type, study centre |
| Allocation concealment (selection bias) | Unclear risk | No clear explanation of methodology |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 10.2% to 16.9% dropout. All analyses using LOCF and MMRM |
| Selective reporting (reporting bias) | Low risk | None identified |
| Other bias | Low risk | Lilly sponsored |
DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, 4th ed HbA1c: haemoglobin A1c ITT: intention‐to‐treat BOCF: best observation carried forward LOCF: last observation carried forward MADRS: Montgomery–Åsberg Depression Rating Scale MINI: Mini International Neuropsychiatric Interview MMRM: mixed‐effect model repeated measure
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boyle 2012 | 28 days only |
| Brannan 2005 | 6 weeks of treatment only |
| Canovas 2007 | Not randomised or controlled |
| Chappell 2009 | Osteoarthritis of the knee ‐ likely to cross over with Cochrane Musculoskeletal Group |
| Chappell 2011 | Osteoarthritis of the knee ‐ likely to cross over with Cochrane Musculoskeletal Group |
| Goldstein 2004 | Trial of duloxetine in depression. Pain scales as secondary outcome measures only. It was not clear what sort of pain the participants had (for example musculoskeletal, neuropathic, headache) and the levels of pain at baseline were low compared to the included trials |
| Harrison 2013 | Four weeks treatment only in each group of 4 way crossover. Terminated early July 2013 |
| Lavoie Smith 2012 | Abstract publication of Smith 2013 |
| NCT00125892 | Open and then double‐blind study comparing 2 doses of duloxetine 60 mg and 120 mg |
| NCT00266643 | The first part of this cross‐over study was the only part of trial suitable for assessment (amitriptyline versus duloxetine) but only 4 weeks long ‐ thus excluded |
| NCT00385671 | Open label |
| NCT00425230 | Was registered in clinicaltrials.gov ‐ study terminated with no participants enrolled because no drug supplied |
| NCT00552682 | Open label duloxetine vs no treatment or pre‐existing antidepressant |
| NCT00641719 | Open label extension of Yasuda 2010 |
| NCT01451606 | Pelvic pain |
| Raskin 2005a | Not a randomised controlled study but a report of 3 trials included in this review |
| Raskin 2006a | Not a double‐blind trial |
| Raskin 2006b | Open label study with dosage control only |
| Russell 2006 | Summary report of 3 studies included in this review |
| Skljarevski 2008 | Back pain ‐ to be included in a Cochrane Back Group review ‐ Back Group informed. Published in full format in 2009 European Journal of Neurology 16: 1041‐8 |
| Skljarevski 2009 | Back pain ‐ to be included in a Cochrane Back Group review ‐ Back Group informed |
| Skljarevski 2009a | Open label extension |
| Skljarevski 2010 | Back pain ‐ to be included in a Cochrane Back Group review ‐ Back Group informed |
| Skljarevski 2010b | Back pain ‐ to be included in a Cochrane Back Group review ‐ Back Group informed |
| Smith 2013 | Duration of treatment only 4 weeks |
| Tanenberg 2011 | Open label ‐ non blinded study |
| Vollmer 2011 | Measured outcomes at durations of less than eight weeks |
| Wernicke 2006b | Not double‐blind ‐ extension of Goldstein 2005 |
| Wu 2006 | Open study, not blinded |
Characteristics of studies awaiting assessment [ordered by study ID]
NCT00603265.
| Methods | Phase II randomised, double blind, parallel assignment, safety and efficacy study |
| Participants | Male and female participants between 18 and 75 years of age Diabetes mellitus (type I or II) that is documented to be under stable glycaemic control over a period of at least 3 months, as indicated by a HbAIc of ≤ 12% and a stable dose of insulin or oral diabetic medication for 90 days prior to starting study medication. Evidence of symmetrical, bilateral pain in the lower extremities due to diabetic peripheral neuropathy. Presence of daily pain due to DPN for at least 3 months. Score ≥ 3 on the physical examination portion of the Michigan Neuropathy Screening Instrument (MNSI). Average weekly pain score of ≥ 4 on the numeric pain rating scale (NPRS) for symmetrical neuropathic pain in the feet and legs |
| Interventions | Drug: ADL5859 Drug: duloxetine Drug: placebo |
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
| Notes | Completed ‐ no reference in Pubmed ‐ no information on clinicaltrials.gov‐ e‐mail written to company with request for information September 2012. |
HbA1c: haemoglobin A1c
Characteristics of ongoing studies [ordered by study ID]
NCT00457730.
| Trial name or title | A randomised placebo controlled trial of duloxetine for central pain in multiple sclerosis |
| Methods | Randomised, double‐blind (caregiver, investigator), placebo‐controlled, parallel assignment safety/efficacy study |
| Participants | People with multiple sclerosis "who have central pain which is 4 or greater on a scale of 1‐10. Patients must have experienced pain for 2 months or longer prior to beginning the study." |
| Interventions | Duloxetine 30 mg (10 capsules) for 1 week, titrated up to 60 mg (40 capsules) for 5 weeks and titrated back down to 30 mg for 1 week Placebo for 7 weeks |
| Outcomes | Time frame for all outcomes, week 2 and week 6
|
| Starting date | January 2007 |
| Contact information | Brown, Theodore R., M.D., MPH Evergreen Healthcare Kirkland, Washington, United States, 98034 |
| Notes | NCT00457730 Lilly sponsored |
NCT00619983.
| Trial name or title | Three way interaction between gabapentin, duloxetine, and donepezil in patients with diabetic neuropathy |
| Methods | Randomised, double‐blind (subject, investigator, outcomes assessor), parallel assignment |
| Participants | Male or female. Diagnosis of diabetic neuropathy. Age 18 to 80 |
| Interventions | Group 1: donepezil 5 mg once per day for 12 weeks Group 2: duloxetine 30 mg twice a day for 12 weeks Group 3: combination of donepezil 2.5 mg and duloxetine 30 mg for 12 weeks Group 4: placebo pills. Gabapentin added to all groups at week 9 |
| Outcomes | Primary:
|
| Starting date | February 2008 to July 2010 |
| Contact information | Regina Curry, RN, CCRC 336‐716‐4294 recurry@wfubmc.edu Wake Forest University Baptist Medical Center Winston‐Salem, North Carolina, United States, 27157 |
| Notes | NCT00619983 Still recruiting 2013 ‐ estimated completion July 2013 |
NCT01179672.
| Trial name or title | Treatment of patients with diabetic peripheral neuropathic pain in China: duloxetine versus placebo |
| Methods | Randomized, double blind (subject, investigator), placebo‐controlled, parallel assignment, efficacy study |
| Participants | People over 18 years of age who present with pain due to bilateral diabetic peripheral neuropathy (type 1 or type 2 diabetes). Pain beginning in feet, relatively symmetrical onset, present daily for at least 6 months, confirmed by score of ≥ 3 on Michigan Neuropathy Screening Inventory |
| Interventions | Duloxetine 30 mg orally, once daily for 1 week; 60 mg once daily for next 11 weeks; 30 mg administered orally, once daily for 1 week during taper period Placebo once daily for 12 weeks, once daily for 1 week during taper period |
| Outcomes | Primary:
Secondary (changes measured from baseline to 12‐week endpoint):
|
| Starting date | April 2011 |
| Contact information | Eli Lilly and Company. Study director, tel: 1‐877‐CTLILLY (1‐877‐285‐4559) or 1‐317‐615‐4559 |
| Notes | NCT01179672 |
NCT01237587.
| Trial name or title | A study of duloxetine in adolescents with juvenile primary fibromyalgia syndrome |
| Methods | Phase III, randomised, double‐blind (subject, caregiver, investigator, outcomes assessor), parallel assignment, safety/efficacy study |
| Participants | Aged 13 to 17 years who meet criteria for primary juvenile primary fibromyalgia syndrome and have a score of greater than or equal to 4 on Brief Pain Inventory (BPI) average pain severity (Item 3) during screening |
| Interventions | Blinded period: 30 mg or 60 mg duloxetine or placebo once daily for 13 weeks Open label extension: 30 mg or 60 mg duloxetine once daily for 26 weeks |
| Outcomes | Primary:
Secondary:
|
| Starting date | February 2011 |
| Contact information | Eli Lilly and Company. Study Director: 1‐877‐CTLILLY (1‐877‐285‐4559) or 1‐317‐615‐4559 |
| Notes | NCT01237587 |
NCT01552057.
| Trial name or title | A phase III clinical trial of duloxetine in participants with fibromyalgia |
| Methods | Randomised, double‐blind (subject, investigator), placebo‐controlled, parallel assignment, safety/efficacy study |
| Participants | Participants with fibromyalgia aged 20 to 74 years Inclusion criteria:
|
| Interventions | Duloxetine hydrochloride orally 60 mg for 15 weeks or oral placebo for 15 weeks |
| Outcomes | Changes measured from baseline to 14 week endpoint Primary:
Secondary:
|
| Starting date | March 2012 |
| Contact information | Eli Lilly and Company, Shionogi. Tel: 1‐877‐CTLILLY (1‐877‐285‐4559) or 1‐317‐615‐4559 |
| Notes | NCT01552057 |
Differences between protocol and review
The review used the 'Risk of bias' table in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions instead of the previous methodological quality assessment and incorporated a 'Risk of bias' table. These methods were not available when the protocol was written.
We also included 'Summary of findings' tables for each comparison.
For this update we included trial sequential analyses.
We changed the title from 'Duloxetine for treating painful neuropathy or chronic pain' to 'Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia'
Contributions of authors
MPTL and RACH screened references, selected trials and extracted data independently. The first draft was written by MPTL and then revised and agreed by all three authors. The authors undertook the 2013 update in the same manner.
Sources of support
Internal sources
Institute of Neurology, University College London, UK.
-
National Institute for Health Research University College LondonBiomedical Research Centre, UK.
This Systematic Review Update was supported by researchers at the National Institute for Biomedical Research University College London Hospitals Biomedical Research Centre
External sources
None, UK.
Declarations of interest
RACH has no competing interests which affect his impartiality in preparing this review.
PJW: none known.
MPL has received honoraria for consultation from Baxter Pharmaceuticals, CSL Behring and LfB and a travel support grant from Grifols, all manufacturers of IVIg. He was a blinded investigator in the study of Comi et al. 2002.
MPL is one of two Joint Co‐ordinating Editors of the Cochrane Neuromuscular Disease Group and RACH is a member of the group's editorial board. Editorial decisions regarding the review were handled by other members of the editorial board without the influence of the review authors.
Edited (no change to conclusions)
References
References to studies included in this review
Arnold 2004 {published data only}
- Arnold LM, Lu Y, Crofford LJ, Wohlreich M, Detke MJ, Iyengar S, et al for the Duloxetine Fibromyalgia Trial Group. A double‐blind multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis and Rheumatism 2004;50(9):2974‐84. [PUBMED: 15457467] [DOI] [PubMed] [Google Scholar]
Arnold 2005 {published data only}
- Arnold LM, Rosen A, Pritchett YL, D'Souza DN, Goldstein DJ, Iyengar S, et al. A randomized double‐blind, placebo‐controlled trial of duloxetine in the treatment of women with fibromyalgia with or without major depressive disorder. Pain 2005;119(1‐3):5‐15. [PUBMED: 16298061] [DOI] [PubMed] [Google Scholar]
Arnold 2010 {published data only}
- Arnold LM, Clauw D, Wang F, Ahl J, Gaynor PJ, Wohlreich MM. Flexible dosed duloxetine in the treatment of fibromyalgia: a randomized double‐blind placebo‐controlled trial [A study comparing duloxetine and placebo in the treatment of fibromyalgia]. Journal of Rheumatology 2010;37(12):2578‐86. [PUBMED: 20843911] [DOI] [PubMed] [Google Scholar]
Arnold 2012 {published data only}
- Arnold LM, Zhang S, Pangallo BA. Efficacy and safety of duloxetine 30 mg/d in patients with fibromyalgia: a randomized, double‐blind, placebo‐controlled study. Clinical Journal of Pain 2012;28(9):775‐81. [PUBMED: 22971669] [DOI] [PubMed] [Google Scholar]
Brecht 2007 {published data only}
- Brecht S, Courtecuisse C, Debieuvre C, Croenlein J, Desaiah D, Raskin J, et al. Efficacy and safety of duloxetine 60 mg once daily in the treatment of pain in patients with major depressive disorder and at least moderate pain of unknown etiology: a randomized controlled trial. Journal of Clinical Psychiatry 2007;68(11):1707‐16. [PUBMED: 18052564] [DOI] [PubMed] [Google Scholar]
Chappell 2008 {published data only}
- Chappell AS, Bradley LA, Wiltse C, Detke MJ, D'Souza DN, Spaeth M. A six‐month double‐blind, placebo‐controlled, randomized clinical trial of duloxetine for the treatment of fibromyalgia. International Journal of General Medicine 2009;30(1):91‐102. [PUBMED: 20428412] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gao 2010 {published data only}
- Gao Y, Ning G, Jia WP, Zhou ZG, Xu ZR, Liu ZM, et al. Duloxetine versus placebo in the treatment of patients with diabetic peripheral neuropathic pain in China. Chinese Medical Journal 2010;123(22):3184‐92. [CTG: NCT00408993; PUBMED: 21163113] [PubMed] [Google Scholar]
Gaynor 2011a {published data only}
- Gaynor PJ, Gopal M, Zheng W, Martinez JM, Robinson MJ, Hann D, Marangell LB. Duloxetine versus placebo in the treatment of major depressive disorder and associated painful physical symptoms: a replication study. Current Medical Research and Opinion 2011;27(10):1859‐67. [PUBMED: 21838410] [DOI] [PubMed] [Google Scholar]
Gaynor 2011b {published data only}
- Gaynor PJ, Gopal M, Zheng W, Martinez JM, Robinson MJ, Marangell LB. A randomized placebo‐controlled trial of duloxetine in patients with major depressive disorder and associated painful physical symptoms. Current Medical Research and Opinion 2011;27(10):1849‐58. [PUBMED: 21838411] [DOI] [PubMed] [Google Scholar]
Goldstein 2005 {published data only}
- Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain 2005;116(1‐2):109‐18. [PUBMED: 15927394] [DOI] [PubMed] [Google Scholar]
Kaur 2011 {published data only}
- Kaur H, Hota D, Bhansali A, Dutta P, Bansal D, Chakrabarti A. A comparative evaluation of amitriptyline and duloxetine in painful diabetic neuropathy: a randomized, double‐blind, cross‐over clinical trial. Diabetes Care 2011;34(4):818‐22. [PUBMED: 21355098] [DOI] [PMC free article] [PubMed] [Google Scholar]
Raskin 2005 {published data only}
- Raskin J, Pritchett YL, Wang F, D'Souza DN, Waninger AL, Iyengar S, et al. A double‐blind, randomized multicentre trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Medicine 2005;6(5):346‐56. [PUBMED: 16266355] [DOI] [PubMed] [Google Scholar]
Rowbotham 2012 {published data only}
- Rowbotham MC, Arslanian A, Nothaft W, Duan WR, Best AE, Pritchett Y, et al. Efficacy and safety of the α4β2 neuronal nicotinic receptor agonist ABT‐894 in patients with diabetic peripheral neuropathic pain [Safety and efficacy study in subjects with diabetic neuropathic pain]. Pain 2012;153(4):862‐8. [CTG: NCT00507936; PUBMED: 22386472] [DOI] [PubMed] [Google Scholar]
Russell 2008 {published data only}
- Russell IJ, Mease PJ, Smith TR, Kajdasz DK, Wohlreich MM, Detke MJ, et al. Efficacy and safety of duloxetine for the treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6‐month randomized double‐blind, placebo‐controlled, fixed‐dose trial. Pain 2008;136(3):432‐44. [PUBMED: 18395345] [DOI] [PubMed] [Google Scholar]
Tesfaye 2013 {published and unpublished data}
- Tesfaye S, Wilhelm S, Lledo A, Schacht A, Tölle T, Bouhassira D, et al. Duloxetine and pregabalin: high‐dose monotherapy or their combination? The “COMBO‐DN study” – a multinational, randomized, double‐blind, parallel‐group study in patients with diabetic peripheral neuropathic pain [Use of duloxetine or pregabalin in monotherapy versus combination therapy of both drugs in patients with painful diabetic neuropathy "The COMBO ‐ DN (COmbination vs Monotherapy of pregaBalin and dulOxetine in Diabetic Neuropathy) Study"]. Pain 2013 May 31 [Epub ahead of print]. [CTG: NCT01089556; PUBMED: 23732189] [DOI] [PubMed] [Google Scholar]
Vranken 2011 {published data only}
- Vranken JH, Hollmann MW, Vegt MH, Kruis MR, Heesen M, Vos K, et al. Duloxetine in patients with central neuropathic pain caused by spinal cord injury or stroke: a randomized, double‐blind, placebo‐controlled trial. Pain 2011;152(2):267‐73. [DOI] [PubMed] [Google Scholar]
Wernicke 2006 {published data only}
- Wernicke JF, Pritchett YL, D'Souza DN, Waninger A, Tran P, Iyengar S, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology 2006;67(8):1411‐20. [PUBMED: 17060567] [DOI] [PubMed] [Google Scholar]
Yasuda 2010 {published and unpublished data}
- Yasuda H, Hotta N, Nakao K, Kasuga M, Kashiwagi A, Kawamori R. Superiority of duloxetine to placebo in improving painful diabetic neuropathic pain: results of a randomized controlled trial in Japan [A superiority study of LY248686 versus placebo in the treatment of patients with diabetic peripheral neuropathic pain]. Journal of Diabetes Investigation 2011;2(2):132‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Boyle 2012 {published data only}
- Boyle J, Eriksson ME, Gribble L, Gouni R, Johnsen S, Coppini DV, Kerr D. Randomized, placebo‐controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: impact on pain, polysomnographic sleep, daytime functioning, and quality of life. [Effects of pregabalin, duloxetine & amitriptyline on pain & sleep (NCT00370656)]. Diabetes Care. 2012;35(12):2451‐8. [DOI: 10.2337/dc12-0656] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brannan 2005 {published data only}
- Brannan SK, Mallinckrodt CH, Brown EB, Wohlreich MM, Watkin JG, Schatzberg AF. Duloxetine 60mg once daily in the treatment of painful physical symptoms in patients with major depressive disorder. Journal of Psychiatric Research 2005;39(1):43‐53. [PUBMED: 15504423] [DOI] [PubMed] [Google Scholar]
Canovas 2007 {published data only}
- Cánovas L, Illodo G, Castro M, Mouriz L, Vázquez‐Martínez A, Centeno J, et al. Effects of duloxetin and amitriptilin in neuropathic pain: study in 180 patients [Efectos de duloxetina y amitriptilina en el dolor neuropatico: estudio en 180 casos]. Revista de la Sociedad Espanola del Dolor 2007;14(8):568‐73. [Google Scholar]
Chappell 2009 {published data only}
- Chappell AS, Ossanna MJ, Liu‐Seifert H, Iyengar S, Skljarevski V, Li LC, et al. Duloxetine, a centrally acting analgesic, in the treatment of patients with osteoarthritis knee pain: a 13‐week, randomized, placebo‐controlled trial. Pain 2009;146(3):253‐60. [PUBMED: 19625125] [DOI] [PubMed] [Google Scholar]
Chappell 2011 {published data only}
- Chappell AS, Desaiah D, Liu‐Seifert H, Zhang S, Skljarevski V, Belenkov Y, et al. A double‐blind, randomized, placebo‐controlled study of the efficacy and safety of duloxetine for the treatment of chronic pain due to osteoarthritis of the knee. Pain Practice 2011;11(1):33‐41. [PUBMED: 20602715] [DOI] [PubMed] [Google Scholar]
Goldstein 2004 {published data only}
- Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C, Demitrack MA. Duloxetine in the treatment of depression: a double‐blind placebo‐controlled comparison with paroxetine. Journal of Clinical Psychopharmacology 2004;24(4):389‐99. [PUBMED: 15232330] [DOI] [PubMed] [Google Scholar]
Harrison 2013 {published data only}
- Harrison T, Miyahara S, Lee A, Evans S, Bastow B, Simpson D, et al. ACTG A5252 Team. Experience and challenges presented by a multicenter crossover study of combination analgesic therapy for the treatment of painful HIV‐associated polyneuropathies [Combination Pain Therapy in HIV Neuropathy]. Pain Medicine 2013;14(7):1039‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lavoie Smith 2012 {published data only}
- Lavoie Smith EM, Pang H, Cirrincione C, Fleishman SB, Paskett ED, Fadul CE, et al. CALGB 170601: A phase III double blind trial of duloxetine to treat painful chemotherapy‐induced peripheral neuropathy (CIPN). Journal of clinical oncology 2012;Conference:32676. [Google Scholar]
NCT00125892 {published data only}
- NCT00125892. A study of duloxetine in the treatment of fibromyalgia ‐ [A 1‐year safety study of duloxetine in patients with fibromyalgia; www.lillytrials.com/results/Cymbalta.pdf]. clinicaltrials.gov/show/NCT00125892; (Accessed 7 November 2013). [Lilly Clinical Trials Register Reference Summary #: 9075]
NCT00266643 {published data only}
- NCT00266643. Tolerability of switching to duloxetine for the management of diabetic nerve pain (NCT00266643) [A comparison of strategies for switching patients from amitriptyline to duloxetine for the management of diabetic peripheral neuropathic pain (Lilly trial reference Summary ID# 8952).]. clinicaltrials.gov/show/NCT00266643; www.lillytrials.com/results/Cymbalta.pdf (accessed 7 November 2013). [Lilly trial reference Summary ID#: 8952]
NCT00385671 {published data only}
- NCT00385671. An open‐label comparison of duloxetine to other alternatives for the management of diabetic peripheral neuropathic pain. www.clinicaltrials.gov/show/NCT00385671 (Accessed 7 November 2013).
NCT00425230 {published data only}
- NCT00425230. Open label study of duloxetine for the treatment of phantom limb pain [Pilot study of use of duloxetine for the treatment of phantom limb pain]. www.clinicaltrials.org/show/NCT00425230 (accessed 7 November 2013).
NCT00552682 {unpublished data only}
- NCT00552682. Pilot, opened, randomized clinical trial to assess the efficacy of duloxetine in the treatment of fibromyalgia in patients with infection by HIV 1+. www.clinicaltrials.gov\show\NCT00552682 (Accessed 7 November).
NCT00641719 {published data only}
- NCT00641719. A long‐term study for the treatment of painful diabetic neuropathy. www.clinicaltrials.gov/show/NCT00641719 (Accessed 7 November 2013).
NCT01451606 {unpublished data only}
- NCT01451606. Duloxetine for the treatment of chronic pelvic pain. http://clinicaltrials.gov/ct2/show/NCT01451606 (Accessed 7 November 2013). [CTG: NCT01451606]
Raskin 2005a {published data only}
- Raskin J, Pritchett YL, Chappell AS, D‐Souza DN, Wong CK, Ferdinand SJ, et al. Duloxetine in the treatment of diabetic peripheral neuropathic pain ‐ results from three clinical trials. European Journal of Neurology 2005;12(s2):221. [Google Scholar]
Raskin 2006a {published data only}
- Raskin J, Smith TR, Wong K, Pritchett YL, D'Souza DN, Iyengar S, et al. Duloxetine versus routine care in the long‐term management of diabetic neuropathic pain. Journal of Palliative Medicine 2006;9(1):29‐40. [PUBMED: 16430342] [DOI] [PubMed] [Google Scholar]
Raskin 2006b {published data only}
- Raskin J, Wang F, Pritchett YL, Goldstein DJ. Duloxetine for patients with diabetic peripheral neuropathic pain: a 6‐month open‐label safety study. Pain Medicine 2006;7(5):373‐85. [PUBMED: 17014595] [DOI] [PubMed] [Google Scholar]
Russell 2006 {published data only}
- Russell JW. Does duloxetine safely and effectively reduce the severity of diabetic peripheral neuropathic pain?. Nature Clinical Practice Neurology 2006;2(1):18‐9. [PUBMED: 16932514] [DOI] [PubMed] [Google Scholar]
Skljarevski 2008 {published data only}
- Skljarevski V, Desaiah D, Liu‐Siefert H, Zhang Q, Chappell AS, Detke MJ, et al. Efficacy of duloxetine in low back pain. European Journal of Neurology 2008;15(Suppl 3):320. [Google Scholar]
Skljarevski 2009 {published data only}
- Skljarevski V, Ossanna M, Liu‐Seifert H, Zhang Q, Chappell A, Iyengar S, et al. A double‐blind, randomized trial of duloxetine versus placebo in the management of chronic low back pain. European Journal of Neurology 2009;16(9):1041‐8. [PUBMED: 19469829] [DOI] [PubMed] [Google Scholar]
Skljarevski 2009a {published data only}
- Skljarevski V, Desaiah D, Zhang Q, Chappell AS, Detke MJ, Gross JL, et al. Evaluating the maintenance of effect of duloxetine in patients with diabetic peripheral neuropathic pain [Maintenance of effect of duloxetine in patients with diabetic peripheral neuropathic pain]. Diabetes/Metabolism Research and Reviews 2009;25(7):623‐31. [CTG: NCT00322621; PUBMED: 19637208] [DOI] [PubMed] [Google Scholar]
Skljarevski 2010 {published data only}
- Skljarevski V, Desaiah D, Liu‐Seifert H, Zhang Q, Chappell AS, Detke MJ, et al. Efficacy and safety of duloxetine in patients with chronic low back pain. Spine 2010;35(13):E578‐85. [PUBMED: 20461028] [DOI] [PubMed] [Google Scholar]
Skljarevski 2010b {published data only}
- Skljarevski V, Zhang S, Desaiah D, Alaka KJ, Palacios S, Miazgowski T, et al. Duloxetine versus placebo in patients with chronic low back pain: a 12‐week, fixed‐dose, randomized, double‐blind trial. Journal of Pain 2010;11(12):1282‐90. [DOI] [PubMed] [Google Scholar]
Smith 2013 {published data only}
- Smith EML, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy‐induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013;309(13):1359‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanenberg 2011 {published data only}
- Tanenberg RJ, Irving GA, Risser RC, Ahl J, Robinson MJ, Skljarevski V, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open‐label, randomized, noninferiority comparison. Mayo Clinic Proceedings 2011;86(7):615‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vollmer 2011 {published data only}
- Vollmer T, Robinson M, Risser R, Malcolm SK, Carlson J. A randomised, double‐blind, placebo‐controlled trial of duloxetine for the treatment of central neuropathic pain associated with multiple sclerosis. Multiple Sclerosis Journal 2011;17 (10 suppl):P1044. [DOI] [PubMed] [Google Scholar]
Wernicke 2006b {published data only}
- Wernicke JF, Raskin J, Rosen A, Pritchett YL, D'Souza DN, Iyengar S, et al. Duloxetine in the long‐term management of diabetic neuropathic pain: an open‐label, 52‐week extension of a randomized controlled clinical trial. Current Therapeutic Research 2006;67(5):283‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2006 {published data only}
- Wu EQ, Birnbaum HJ, Mareva MN, Le TK, Robinson RL, Rosen A, et al. Cost‐effectiveness of duloxetine versus routine treatment for U.S. patients with diabetic peripheral neuropathic pain. The Journal of Pain 2006;7(6):399‐407. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT00603265 {published data only}
- NCT00603265. A phase 2a, randomized, double‐blind, placebo‐ and active‐controlled, parallel‐group, multicenter study to assess the safety and efficacy of ADL5859 100 mg BID in subjects with neuropathic pain associated with diabetic peripheral neuropathy [Safety and efficacy study of ADL5859 in subjects with neuropathic pain associated with diabetic peripheral neuropathy]. www.clinicaltrials.gov/show/NCT00603265 (Accessed 7 November 2013). [CTG: NCT00603265]
References to ongoing studies
NCT00457730 {unpublished data only}
- NCT00457730. A randomized placebo controlled trial of duloxetine for central pain in multiple sclerosis [A study to test the use of duloxetine for pain in MS]. http://clinicaltrials.gov/show/NCT00457730 Accessed 19 December 2013.
NCT00619983 {published data only}
- NCT00619983. Three way interaction between gabapentin, duloxetine, and donepezil in patients with diabetic neuropathy. http://clinicaltrials.gov/show/NCT00619983 Accessed 19 December 2013.
NCT01179672 {unpublished data only}
- NCT01179672. A study in patients with diabetic peripheral neuropathic pain in China. http://clinicaltrials.gov/show/NCT01179672 (Accessed 19 December 2013).
NCT01237587 {unpublished data only}
- NCT01237587. Effect of duloxetine 30/60 mg once daily versus placebo in adolescents with juvenile primary fibromyalgia syndrome [A study of duloxetine in adolescents with juvenile primary fibromyalgia syndrome]. http://clinicaltrials.gov/show/NCT01237587 Accessed 19 December 2013.
NCT01552057 {unpublished data only}
- NCT01552057. A phase III clinical trial of duloxetine in participants with fibromyalgia [A study of duloxetine in fibromyalgia]. http://clinicaltrials.gov/show/NCT01552057 Accessed 19 December 2013.
Additional references
Aronson 2007
- Aronson JK. Chapter 2 Antidepressant Drugs. In: Aronson JK editor(s). Side Effects of Drugs. Vol. 29, Amsterdam: Elsevier, 2007:23. [Google Scholar]
Aronson 2008
- Aronson JK. Chapter 2 Antidepressant Drugs. In: Aronson JK editor(s). Side Effects of Drugs. Vol. 30, Amsterdam: Elsevier, 2008:19. [Google Scholar]
Attal 2006
- Attal N, Cruccu G, Haanpää M, Hansson P, Jensen TS, Nurmikko T, et al. EFNS Task Force. EFNS guidelines on pharmacological treatment of neuropathic pain. European Journal of Neurology 2006;13(11):1153‐69. [DOI] [PubMed] [Google Scholar]
Beard 2008
- Beard SM, McCrink L, Le TK, Garcia‐Cebrian A, Monz B, Malik RA. Cost effectiveness of duloxetine in the treatment of diabetic peripheral neuropathic pain in the UK. Current Medical Research and Opinion 2008;24(2):385‐99. [DOI] [PubMed] [Google Scholar]
Bellows 2012
- Bellows BK, Dahal A, Jiao T, Biskupiak J. A cost‐utility analysis of pregabalin versus duloxetine for the treatment of painful diabetic neuropathy. Journal of Pain & Palliative Care Pharmacotherapy 2012;26(2):153–64. [DOI] [PubMed] [Google Scholar]
Breivik 2006
- Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. European Journal of Pain 2006;10(4):287‐333. [DOI] [PubMed] [Google Scholar]
Burckhardt 1991
- Burckhardt CS, Clark SR, Bennett RM. The fibromyalgia impact questionnaire: development and validation. Journal of Rheumatology 1991;18(5):728‐33. [PubMed] [Google Scholar]
Bymaster 2001
- Bymaster FP, Dreshfield‐Ahmad LJ, Threlkeld PG, Shaw JL, Thompson L, Nelson DL, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology 2001;25(6):871‐80. [DOI] [PubMed] [Google Scholar]
Bymaster 2005
- Bymaster FP, Lee TC, Knadler MP, Detke MJ, Iyengar S. The dual transporter inhibitor duloxetine: a review of its preclinical pharmacology, pharmacokinetic profile, and clinical results in depression. Current Pharmaceutical Design 2005;11(12):1475‐93. [DOI] [PubMed] [Google Scholar]
Dadabhoy 2006
- Dadabhoy D, Clauw DJ. Therapy Insight: fibromyalgia ‐ a different type of pain needing a different type of treatment. Nature Clinical Practice Rheumatology 2006;2(7):364‐72. [DOI] [PubMed] [Google Scholar]
Daousi 2004
- Daousi C, MacFarlane IA, Woodward A, Nurmikko TJ, Bundred PE, Benbow SJ. Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabetic Medicine 2004;21(9):976‐82. [DOI] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fava 2004
- Fava M, Mallinckrodt CH, Detke MJ, Watkin JG, Wohlreich MM. The effect of duloxetine on painful physical symptoms in depressed patients: do improvements in these symptoms result in higher remission rates?. Journal of Clinical Psychiatry 2004;65(4):521‐30. [DOI] [PubMed] [Google Scholar]
Gahimer 2007
- Gahimer J, Wernicke J, Yalcin I, Ossanna MJ, Wulster‐Radcliffe M, Viktrup L. A retrospective polled analysis of duloxetine safety in 23,983 subjects. Current Medical Research Opinion 2007;23(1):175‐84. [DOI] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- Jan Brozek, Andrew Oxman, Holger Schünemann. GRADEpro. Version Version 3.2 for Windows. Jan Brozek, Andrew Oxman, Holger Schünemann, 2008.
Hall 2006
- Hall GC, Carroll D, Parry D, McQuay HJ. Epidemiology and treatment of neuropathic pain: the UK primary care perspective. Pain 2006;122(1‐2):156‐62. [DOI] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley & Sons Ltd, 2008:187‐244. [MEDLINE: ] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kajdasz 2007
- Kajdasz DK, Iyengar S, Desaiah D, Backonja MM, Farrar JT, Fishbain DA, et al. Duloxetine for the management of diabetic peripheral neuropathic pain: evidence‐based findings from post hoc analysis of three multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group studies. Clinical Therapeutics 2007;29(Supplement):2536‐46. [DOI] [PubMed] [Google Scholar]
Mariappan 2009
- Mariappan P, Alhasso AA, Grant A, N'Dow JMO. Serotonin and noradrenaline reuptake inhibitors (SNRI) for stress urinary incontinence in adults. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD004742.pub2] [DOI] [PubMed] [Google Scholar]
Mease 2011
- Mease PJ, Spaeth M, Clauw DJ, Arnold LM, Bradley LA, Russell IJ, et al. Estimation of minimum clinically important difference for pain in fibromyalgia. Arthritis Care & Research (Hoboken). 2011;63(8):821‐6. [DOI] [PubMed] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2012
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [CENTRAL: CD008242; DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Nagda 2004
- Nagda J, Bajwa ZH. Definitions and Classification of Pain. Principles and practice of pain management. New York: McGraw Hill, 2004:51‐4. [Google Scholar]
Nose 2007
- Nose M, Cipriani A, Furukawa TA, Omori I, Churchill R, McGuire H et al for the Meta‐Analysis of New Generation Antidepressants (MANGA) Study Group. Duloxetine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD005454.pub2] [DOI] [Google Scholar]
O'Connor 2008
- O'Connor AB, Noyes K, Holloway RG. A cost‐utility comparison of four first‐line medications in painful diabetic neuropathy. Pharmacoeconomics 2008;26(12):1045‐64. [DOI] [PubMed] [Google Scholar]
Perahia 2006
- Perahia DG, Pritchett YL, Desaiah D, Raskin J. Efficacy of duloxetine in painful symptoms: analgesic or antidepressant effect?. International Clinical Psychopharmacology 2006;21(6):311‐7. [DOI] [PubMed] [Google Scholar]
Saarto 2007
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD005454] [DOI] [PMC free article] [PubMed] [Google Scholar]
Salaffi 2004
- Salaffi F, Stancati A, Silvestri CA, Ciapetti A, Grassi W. Minimal clinically important changes in chronic musculoskeletal pain intensity measured on a numerical rating scale. European Journal of Pain 2004;8(4):283‐91. [DOI] [PubMed] [Google Scholar]
Schuessler 2006
- Scheussler B. What do we know about duloxetine's mode of action? Evidence from animals to humans. British Journal of Obstetrics and Gynaecology 2006;113 Suppl 1:5‐9. [DOI] [PubMed] [Google Scholar]
Thorlund 2011 [Computer program]
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. Trial Sequential Analysis. Version Version 0.9 beta. Copenhagen: Copenhagen Trials Unit, 2011.
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐35. [DOI] [PubMed] [Google Scholar]
Wernicke 2007
- Wernicke J, Lledo A, Raskin J, Kajdasz DK, Wang F. An evaluation of the cardiovascular safety profile of duloxetine. Drug Safety 2007;30(5):437‐55. [DOI] [PubMed] [Google Scholar]
Wiffen 2005
- Wiffen PJ, McQuay HJ, Edwards JE, Moore RA. Gabapentin for acute and chronic pain. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005452] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Lunn 2008
- Lunn MPT, Hughes RAC, Wiffen PJ. Duloxetine for treating painful neuropathy or chronic pain. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD007115] [DOI] [Google Scholar]
Lunn 2009
- Lunn MPT, Hughes RAC, Wiffen PJ. Duloxetine for treating painful neuropathy or chronic pain. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007115.pub2] [DOI] [PubMed] [Google Scholar]