

Abstract
Multiple sclerosis (MS) is characterized by the chronic inflammatory destruction of myelinated axons in the central nervous system. Several ideas have been put forward to clarify the roles of the peripheral immune system and neurodegenerative events in such destruction. Yet, none of the resulting models appears to be consistent with all the experimental evidence. They also do not answer the question of why MS is exclusively seen in humans, how Epstein-Barr virus contributes to its development but does not immediately trigger it, and why optic neuritis is such a frequent early manifestation in MS.
Here we describe a scenario for the development of MS that unifies existing experimental evidence as well as answers the above questions. We propose that all manifestations of MS are caused by a series of unfortunate events that usually unfold over a longer period of time after a primary EBV infection and involve periodic weakening of the blood–brain barrier, antibody-mediated CNS disturbances, accumulation of the oligodendrocyte stress protein αB-crystallin and self-sustaining inflammatory damage.
Keywords: multiple sclerosis, EBV, MOG, αB-crystallin
Here we describe a scenario for the development of multiple sclerosis (MS) that unifies existing experimental evidence as well as answers the above questions. We propose a hypothesis that manifestations of MS are caused by a series of unfortunate events that usually unfold over a longer period of time after a primary EBV infection and involve a periodic weakening of the blood-brain barrier, antibody-mediated CNS disturbances, accumulation of the oligodendrocyte stress protein aB-crystallin and self-sustaining inflammatory damage.
Graphical Abstract
Graphical Abstract.
Introduction
In this paper, we present a hypothesis for the pathogenesis of multiple sclerosis (MS) that explains many of its key features. These include the fact that MS only occurs in humans, that a prior infection with Epstein-Barr virus (EBV) is an essential prerequisite for MS, that it is linked to optic neuritis as a frequent early clinical manifestation, and those peripheral immunological parameters in people with MS are not fundamentally different from those in healthy subjects. While the risk to develop MS is influenced by genetic factors, it is not a classic genetic disorder. In most populations, the gene encoding the human leukocyte antigen DRB1*1501 increases the MS risk approximately 4-fold while dozens of other genes have a minor additional effect on this risk [1]. Collectively, the genetic evidence supports the notion that the immune system is closely involved in disease development, but it does not offer any specific clues as to its pathogenesis.
While the immune system is clearly involved, there is no convincing evidence that MS is caused by a primary attack on myelin by peripherally activated myelin-reactive T cells, as already emphazised above. This scenario is often found in textbook explanations of MS, inspired by the chain of events that drives experimental MS-like disease in laboratory animals. In people with MS, however, no disease-specific and persistent myelin-reactivity of peripheral T cells has ever been found, despite decades of research on the subject [2, 3]. Over the years, many reports have suggested differences exist between MS patients and control subjects in this context, but none of these have stood the test of time. We still do not have a blood test for MS based on any such difference. In addition, an attack by peripheral T cells as the primary cause of disease would not explain how in normal-appearing brain tissue, foci of cellular stress and innate immune activation emerge during MS in the absence of any lymphocytes having entered the tissue, as further explained below. Another concept, sometimes referred to as the ‘inside-out’ model assumes a neurodegenerative process to be the primary cause of MS. Again, the experimental evidence does not support this idea either. The causative role of EBV, the enhanced MS risk that is conferred by certain HLA alleles, the appearance of oligoclonal antibodies in the cerebrospinal fluid (CSF), and the impact of therapeutic elimination of B cells all argue against it. Both the immune system and the central nervous system (CNS) are clearly involved, but the evidence indicates that neither plays a role as a sole driver. Here, we present a hybrid scenario for the development of MS that involves a series of unfortunate events in the interplay between an essentially normal immune system, a common virus, and events within the CNS.
Event 1: EBV infection and a normal immune system
Many studies have presented compelling epidemiological evidence that infection with EBV is a prerequisite for the development of MS [4], the latest one describing the clinical history of over 10 million young adults [5]. The collective data conclusively demonstrate that an EBV infection greatly increases the risk of developing MS and that MS is essentially never found in EBV seronegative people. Consistent with a key role of EBV, the single gene that is closely linked to the MS risk in most populations, HLA-DRB1*1501, facilitates EBV infection and—along with complement receptors—leads to increased viral load [6, 7].
A simple explanation for the role of EBV could be that persistent infection of the CNS itself would trigger the inflammatory reactions that cause lesions. Yet, pathological studies have shown that most brain samples from MS patients do not contain the virus [8]. While a few EBV-infected B cells might well escape detection during routine pathology, current evidence does not support the idea that EBV infection of the CNS is a prominent feature of MS. Other pathogenic mechanisms have therefore been proposed. One concept is that EBV induces immune responses that are cross-reactive to myelin antigens, so-called molecular mimicry at the level of either T cells or antibodies. As an example, the latest report on this phenomenon documents cross-reactivity at both levels between the viral nucleoprotein 1 and the myelin-associated protein αB-crystallin, which is discussed in more detail below [9]. Several other protein targets of cross-reactivity have been described as well. However, any EBV-triggered pathogenic immune response that would be powerful and persistent enough to cause MS should distinguish MS patients from healthy subjects, and as stated before, there is no evidence for such MS-specific autoreactive T-cell responses. This alone renders it highly unlikely that molecular mimicry is the key pathogenic mechanism. The biggest problem with molecular mimicry as a proposed cause for MS, however, is that cross-reactivity between pathogen sequences and self-proteins at the level of single T or B cells is nothing special and will likely develop in response to any pathogen. This is because any individual T-cell receptor can respond to as many as a million different antigenic sequences and likewise, any single antibody can bind to hundreds of target sequences [10, 11]. This degeneracy in the specificity of individual T- and B-cell receptors is a necessity for the immune system with only finite number of leukocytes to be able to respond to a near infinite number of amino acid sequences in pathogenic microorganisms and to do so in an optimized manner [12, 13]. Conversely, this implies that each pathogen-derived epitope will be able to engage an equally large number of clonotypes of T or B cells. With dozens of potential epitope sequences contained within the collective set of EBV proteins [14], the total number of clonotypes involved in a polyclonal T-cell or antibody response triggered by an EBV infection will be enormous. Given their high level of receptor degeneracy, cross-reactivity to a host protein sequence can probably be found for many of such individual clonotypes, yet, each time such reactivity will be directed toward another host sequence, functionally and anatomically unrelated to host sequences recognized by other clonotypes. In contrast to the shared and thus, focused EBV-specific reactivity of all clonotypes together, the diffuse cross-reactivities of individual clonotypes toward a wide range of unrelated host sequences will generally remain irrelevant.
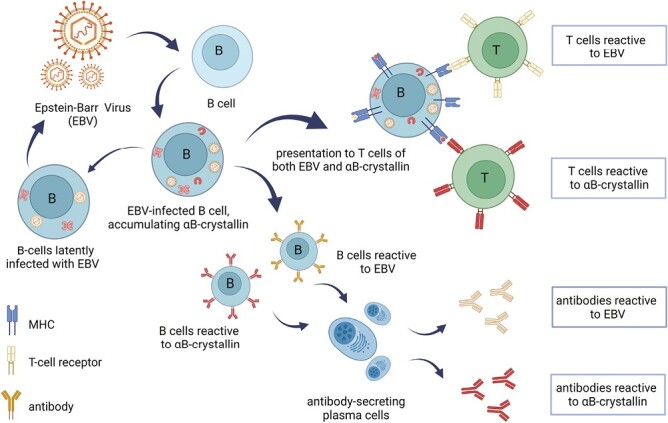
In our view, a more relevant effect of an EBV infection in the context of MS is the induction of the small heat shock protein αB-crystallin in infected B cells ( [15]; Figs. 1 and 2). As an anti-apoptotic molecular chaperone, αB-crystallin is induced in EBV-infected cells to counteract virus-induced cell death, along with several other well-known anti-apoptotic factors [16, 17]. As illustrated in Figs. 1 and 2, EBV induces not only an accumulation of αB-crystallin in B cells but also its subsequent presentation to T cells. Signaling by the viral latency membrane protein-1 as well as by EBV-induced expression of CD40 and CD ligand contributes to the establishment of a potent cytotoxic CD4+ and CD8+ memory T-cell repertoire against infected B cells [18] which at some point may involve as many as 50% of all peripheral B cells [19]. This response is not only directed against EBV antigens but also against αB-crystallin.
Figure 1.
EBV infection of B cells leads to the establishment of an αB-crystallin-reactive immune repertoire in humans. EBV infection of B cells leads to the intracellular accumulation of the stress protein αB-crystallin that counteracts virus-induced apoptosis. When EBV-derived antigens are subsequently presented to T cells, αB-crystallin-derived epitopes are presented by B cells along with viral epitopes. Since the protein is not expressed in human thymus, potentially reactive T cells that have escaped deletion become activated and form an αB-crystallin-reactive memory T-cell repertoire. They also provide help to B cells that mature into plasma cells which produce antibodies not only against EBV antigens but also against αB-crystallin. Periodic reactivation of the latent EBV infection supports this memory immune repertoire for life (for further details see ref 15; Figure created with Biorender).
Figure 2.
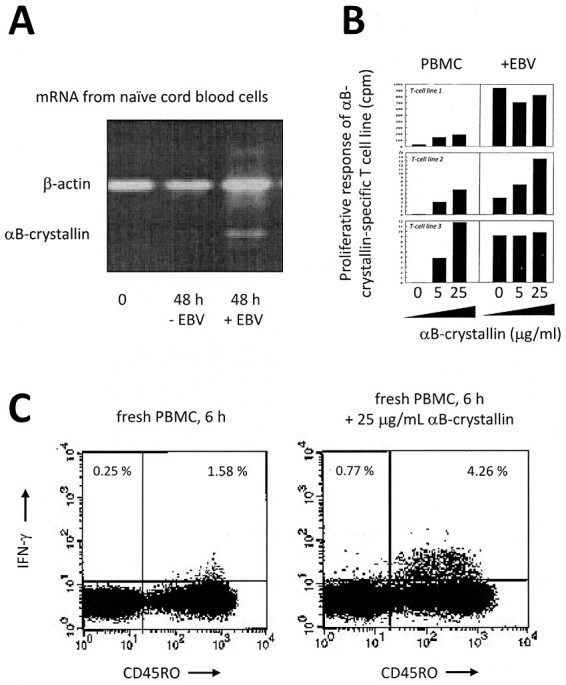
Key elements in the induction of αB-crystallin immune reactivity by EBV. By inducing de novo expression of αB-crystallin in B cells (A) and its subsequent HLA-DR-restricted presentation to T cells. EBV not only activates an immune repertoire against its own viral antigens but also against αB-crystallin. Note how different αB-crystallin-specific T cell lines respond to EBV-infected B cells even before their specific antigen has been exogenously added to the culture (B). Human peripheral blood mononuclear cells frequently contain up to 5% of CD45RO memory T cells that release IFN-γ in response to αB-crystallin (C). This population contains both CD4+ and CD8+ T cells in variable ratios. Originally published in The Journal of Immunology. Van Sechel et al 1997. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol. 1999 Jan 1;162(1):129-35. PMID: 9886378. Copyright © [1997] The American Association of Immunologists, Inc.
Different from other mammals, humans do not express αB-crystallin transcripts in the thymus. This allows αB-crystallin-reactive T cells to escape negative selection [15]. The collective data, including those of Phase I clinical study including 76 healthy subjects, indicate that frequently up to around 5% of all human CD45RO+ memory T cells (including both CD4+ and CD8+ T cells in variable ratios) proliferate in response to αB-crystallin while releasing significant amounts of IFN-γ [20, 21]. In addition, readily detectable levels of IgG against αB-crystallin are routinely found in the serum of adults [22]. Clearly, αB-crystallin is not tolerated as ‘self’ by the human immune system but provokes substantial immune reactions in humans. Given the persistence of EBV in the human body and periodic reactivation of the virus [23], a memory immune repertoire against αB-crystallin will be maintained for life. Importantly, this is a uniquely human trait. In rodents and many other mammals, robust thymic expression of αB-crystallin leads to tolerance, even preventing the induction of experimental disease with αB-crystallin in these animals [24, 25]. This first unfortunate event may well explain why MS only occurs in humans.
The establishment of the αB-crystallin reactive immune repertoire in humans as the result of an EBV infection represents the first in a series of unfortunate events that may eventually culminate in the development of MS. As illustrated by the fact that most seropositive humans do not immediately develop MS following an EBV infection, however, it only sets the stage. The subsequent occurrence of other events will be essential for the actual development of MS, often only years later.
As explained further below, we propose that the second event involves the emergence of myelin-reactive serum antibodies and their entry into the CNS. The presence of myelin-reactive antibodies is perfectly normal in adult humans and does not distinguish MS patients from healthy subjects [22, 26–34]. This appears to be at odds with the recently propagated use of such antibodies as a diagnostic criterion to distinguish myelin-oligodendrocyte glycoprotein antibody disease (MOGAD) from neuromyelitis optica (NMO) and MS [35]. These three conditions are collectively referred to as NMO spectrum disorders (NMOSD). Serum antibodies to the astrocyte surface protein aquaporin-4 (AQP4) are currently used to diagnose NMO, while MOG-reactive antibodies in serum are taken to be indicative of MOGAD. The pathogenicity of these antibodies has been confirmed in animal models but as further explained below, the idea that they selectively accumulate in people with certain NMOSD variants in our opinion results from the clinical application of highly selective antibody assays. In our view, this has led to some misconceptions with regard to anti-myelin serum antibodies.
Event 2: Myelin-reactive serum antibodies and peripheral B cells that produce them
It is important to appreciate the technical difficulties in evaluating antibodies against membrane antigens such as AQP4 and especially MOG. As membrane-embedded proteins, almost all myelin antigens are very hydrophobic and extremely difficult to purify to homogeneity in aqueous media using current technologies. Not only does their hydrophobic character render this problematic but also the fact that many of these proteins exist as a collection of various isoforms and post-translationally modified variants [36, 37]. The name ‘MOG’, for example, does not refer to a single protein but to a group of structurally related MOG variants. Thus, the surrogate antigens commonly used in antibody assays such as synthetic peptides, recombinant constructs, or cell lines transfected with a fusion protein inevitably fall short in adequately representing the biologically relevant dynamic collection of antigens that exist within the CNS. This likely explains the widely different results that have been obtained using different surrogate antigens and varying types of assays [35, 38, 39].
As an example, the presence of MOG-specific serum antibodies as evaluated by western blotting was claimed to be indicative of the conversion of clinically isolated syndrome to MS about 20 years ago [30]. This report was already at odds with the results of earlier studies using enzyme-linked immunosorbent assays (ELISA) which demonstrated anti-MOG antibodies in the serum of about 80% of MS patients and 70% of healthy controls [29]. After new cell-based antibody assays were introduced, serum antibodies to MOG are currently considered as a core diagnostic criterion for MOGAD, a much rarer condition than MS. However, even when using the recommended assay for antibody detection the consistency in results from different centers is only adequate with high-titer samples [35]. Furthermore, a significant minority of 20–30% of NMO patients are still AQP4-seronegative, not all MOGAD patients have MOG-reactive antibodies, and AQP4/MOG double seropositive patients also exist [40]. Despite refinements in clinical testing protocols up to 30% of patients with NMOSD remain seronegative for either MOG or AQP4 [41, 42]. To complicate matters further, serum titers of anti-MOG antibodies vary markedly over time and seropositive subjects can even convert to full seronegativity [43].
Especially in the case of myelin antigens, therefore, the experience is unfortunately that the outcome of an antibody assay is largely determined by the way the assay is performed. Furthermore and as clarified in the case of MOG-reactive antibodies, the isotype is relevant for an antibody’s ability to fix complement and consequently, for its pathogenic potential [44]. As a rule, this feature of anti-myelin serum antibodies is ignored in diagnostic approaches. Thus, the field of antibody specificities and antibody titers in people with NMOSD contains significant pitfalls and regularly produces poorly consistent data sets that are strongly biased by technical factors. The results of more comprehensive methods such as approaches that have evaluated serum antibody reactivity to the entire collection of human myelin proteins following their solubilization in strongly organic solvents [45] clarify that such antibodies are a normal feature of an adult human immune system, also in perfectly healthy subjects.
The above technical limitations also apply to studies of CSF antibodies or more specifically, oligoclonal bands (OCB) of IgG or IgM that are typically found in the CSF of MS patients [46]. Also, their specific target(s) have been the subject of several studies with often poorly corresponding results. More recent studies have identified a number of linear peptide sequences that can be bound by CSF antibodies but no study has yet shed real light on the specificity of OCB in MS patients [47, 48]. It is truly difficult to define the specificity of antibodies that can be identified as ‘oligoclonal’ only by a migration pattern during iso-electric focusing. An additional problem in studying the specificity of OCB is that the target(s) may be so abundant within the CNS that the vast majority of reactive antibodies are sequestered in the CNS and no longer remain available for detection in the CSF. Despite this, and largely based on the presence of myelin-reactive antibodies in the CSF (see further below), it is reasonable to assume that at least some CSF antibodies react to CNS antigens including myelin antigens, similar to serum antibodies.
CNS-reactive antibodies in serum or CSF are unlikely to be products of B cells that enter the CNS from the skull and then exit into the circulation [49, 50]. After all, such B cells are at least in part tolerized for local CNS antigens on site before they exit the CNS [51]. As a more likely alternative, deep cervical or lumbar lymph nodes containing antigens drained from the CNS following tissue turnover or injury in the CNS may be the source of peripheral B cells against these CNS antigens, including myelin-derived antigens ([52–55]; see Fig. 3). Since a certain level of tissue turnover and repair is likely a normal feature of the CNS as it is of other organs, such a mechanism would readily explain the lifelong persistence of CNS-reactive peripheral B cells and serum antibodies, if not their gradual accumulation with age. For the same reason, if any CSF-borne antibody—including OCBs—would have a specificity against CNS antigens, they will likely be the product of peripheral B cells that have entered the CNS and persisted. That traffic of B cells occurs between the CNS and the circulation has been convincingly demonstrated [56–63]. Yet, still much has to be learned on this particular issue, including the question of how B cells may persist within the CNS and how so-called ectopic follicles within the CNS are formed [64, 65].
Figure 3.
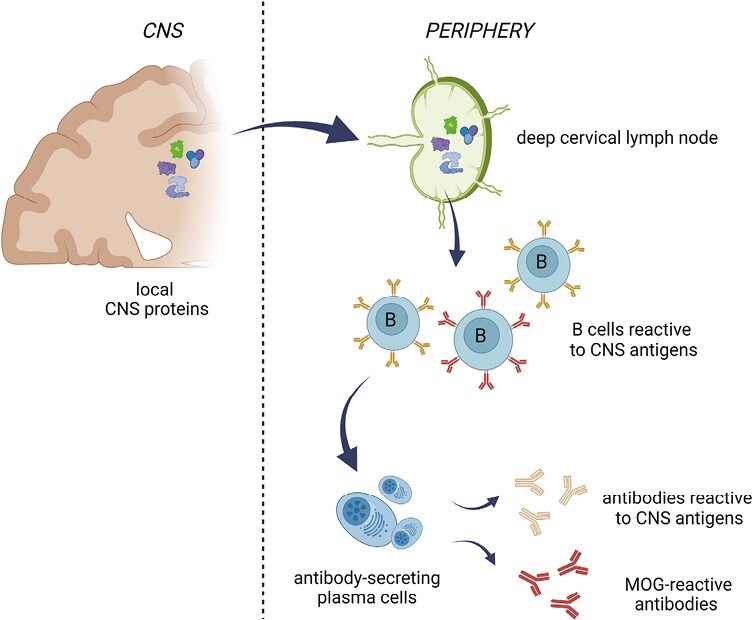
Drainage of CNS antigens to deep cervical or lumbar lymph nodes is the most likely pathway for the generation of the peripheral anti-CNS antibodies that are found in all adult humans. When cells or tissues within the CNS are subjected to turnover or damage, molecular debris is carried off by the lymphatic system and collected in deep cervical or lumbar lymph nodes. CNS debris in these secondary lymphoid organs will activate resident B and T cells. This likely leads to the accumulation over time of self-reactive T and B cells as well as self-reactive antibodies in the periphery. The immune repertoire thus generated contains antibodies against ubiquitous cellular proteins as well as proteins that are selectively expressed within the CNS (Figure created with Biorender).
Event 3. Entry of serum-borne antibodies and B cells into the CNS
One of the striking features of MS is its close association with optic neuritis. The majority of MS patients show signs of ON during disease and intriguingly, over 25% of MS patients even present with ON as their first clinical symptom. Following an initial episode of acute ON, about 50% of people develop MS within 15 years [66]. This number may even be an underestimation given that ON can be mild and transient and could easily come and go unnoticed. This is emphasized by the finding that post-mortem neuropathological abnormalities are present in the optic nerves of MS patients in whom visual disturbances were not reported [67]. Of the 154 cases reviewed in this latter study, no less than 75% showed optic nerve pathology while only one of those nerves was actually examined per case. The diagnostic use of optical coherence tomography further illustrates the fact that inflammatory damage to the optic and retinal nerve is a consistent early event during the development of MS, and even found to precede the onset of clinical signs [68, 69] in line with several other lines of evidence [70]. Furthermore, damage to the optic nerve is well known to allow anterograde trans-synaptic spreading of neurodegeneration to other regions of the CNS [71, 72], which is also consistent with the anterior visual pathway being a site of primary disturbance during the development of MS.
A possible explanation for the striking predominance of ON in MS may be the relative vulnerability of the optic nerve itself, or that visual disturbances are more easily recognized by patients than other symptoms. Still, a more intriguing consideration is the finding that the blood–brain barrier (BBB) at the optic nerve head is relatively permeable [73–78]. While this feature is shared with a few other regions in the CNS including, e.g., the choroid plexus and the area postrema [79], the strong relationship between ON and MS still points to the possibility that variations in BBB permeability are highly relevant to the development of both ON and MS since it controls the traffic of antibodies and leucocytes into the CNS. A normal healthy BBB already allows between 0.1% and 0.2% of peripherally administered antibodies to enter the CNS [80, 81]. Yet, when vascular endothelial cells of the BBB are exposed to cytokines such as IL-1β, TNF-α, IL-6, and IFN-γ, BBB permeability markedly increases [82]. This likely has the strongest impact in CNS regions that already have a weak BBB. Given that increased levels of the above-mentioned serum cytokines are found during peripheral viral infection, including EBV [83–86], a temporary increase in BBB permeability around the optic nerve and choroid plexus and intensified antibody and leucocyte trafficking under such conditions is a likely consequence. In support of this notion, many case reports have documented the development of ON as the direct result of an infection with peripheral pathogens including varicella zoster virus, influenza virus, herpes simplex virus, SARS-CoV2, Lyme disease, and EBV ( [87] and references therein). In addition, peripheral respiratory tract infections during MS are known to trigger exacerbations [88–90], as does the experimental systemic administration of IFN-γ [91].
In our view, therefore, the frequent involvement of the optic nerve in MS and the impact of peripheral infections on its disease course strongly suggests that a temporarily permeable BBB—also in other regions of the CNS—plays a critical role in the development of MS. Over the period following a primary EBV infection and up to the point of the first clinical symptoms of MS, a periodically permeable BBB may well be instrumental in allowing potentially damaging antibodies and the B cells that produce such antibodies to enter and accumulate within the CNS (Figs. 4 and 6). The time it takes to gradually build up pathogenic levels of such antibodies may sometimes be quite short, as in the case of pediatric MS, but could routinely also take many years [5]. It is clear, however, that the development of MS does not start at the time of the first clinical symptoms, but well before that.
Figure 4.
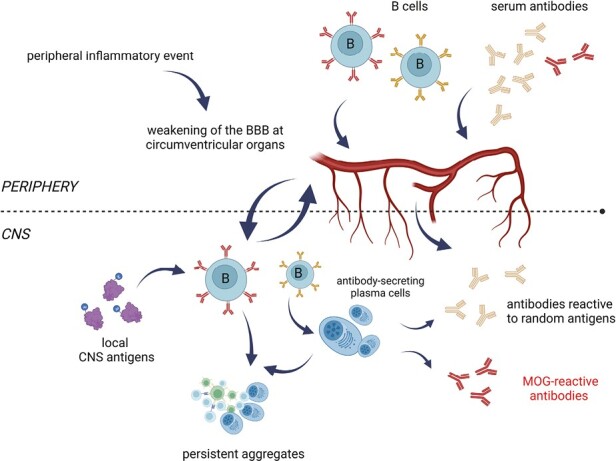
By inducing release of inflammatory cytokines, peripheral inflammatory events may temporarily weaken the blood-brain barrier at certain locations. This will promote trafficking into the CNS of CNS-reactive antibodies and B cells. Peripheral inflammatory events including viral infections lead to temporarily increased serum levels of cytokines such as IFN-γ, TNF-α, and IL-6 that will increase the permeability of the BBB. Especially in regions where the BBB is already relatively permeable under normal conditions, serum antibodies and the B cells that can produce them will thus gain easier access to the CNS than normal. The latter can even go on to form aggregates that persist in the CNS (see also Fig. 6). Among the antibodies entering the CNS will be myelin-reactive ones such as MOG-reactive antibodies (Figure created with Biorender).
Figure 6.
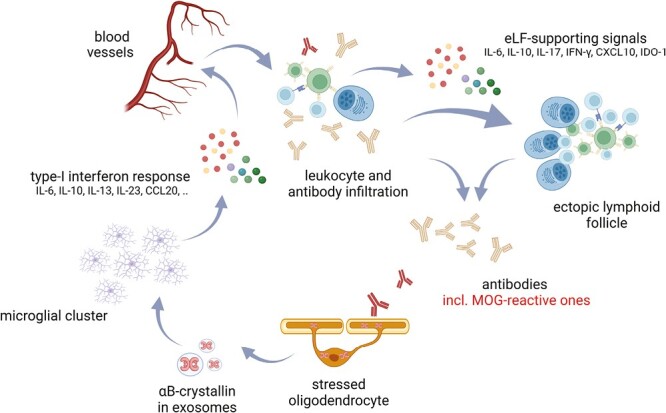
Myelin-reactive antibodies induce oligodendroglial αB-crystallin which, in turn, triggers a protective and tolerizing type I interferon-like response in microglia. Exosomes containing αB-crystallin that are released by antibody-stressed oligodendrocyte activate surrounding microglia via CD14 and TLR2. The protective type I interferon-like response this induces will cause microglia to cluster and it promotes repair and immunological tolerance. It also leads to the release of chemokines that stimulate leucocyte recruitment. Collectively, the unique cocktail of mediators produced by activated microglia and infiltrated leucocytes will promote the formation of B-cell aggregates and ectopic follicles that thus become a persistent local source of antibodies including OCBs (Figure created with Biorender).
Event 4. Anti-myelin antibodies, oligodendrocyte stress, and the small heat shock protein αB-crystallin
As explained above, we propose that the periodically enhanced influx of serum antibodies and leucocytes into the CNS via a temporarily permeable BBB initiates a chain of events that ultimately culminates in MS. The idea that antibodies in general are key players in its pathogenesis is supported by the fact that elevated levels of IgG in the CNS and the presence of OCB of IgG and IgM have long been acknowledged as the single most important distinguishing trait of MS patients. More recent pathological evidence has added significant refinement to this notion in showing that myelin damage during MS is generally initiated by the binding of antibodies and complement to myelin sheaths, followed by the formation of small myelin deformations and spreading of damage to the oligodendrocyte cell body [92, 93]. Animal model data support a co-pathogenic role for myelin-reactive antibodies but they also emphasize that antibodies alone are usually insufficient to trigger chronic demyelination as seen in MS [44]. While it may be argued that antibodies could be generally important in the inflammatory process irrespective of their antigenic specificity [94], other pathogenic factors are clearly needed too [95].
Several reports have illustrated that the above scenario for the initial development of antibody-mediated damage during MS can be reiterated by MOG-reactive antibodies both in cell culture and in animal models. When they bind to cultured oligodendrocytes such antibodies induce cellular stress [96, 97]. As one of the earliest events in the development of experimental optic neuritis in rats, serum-borne MOG-reactive antibodies enter the CNS at the optic nerve head and induce oligodendrocyte stress [78, 98]. In the above cases, oligodendrocyte stress was demonstrated by the accumulation of the small heat shock protein αB-crystallin, which is of particular relevance, as explained below. While several other studies have confirmed a pathogenic role for MOG-reactive antibodies in promoting oligodendrocyte perturbations and complement-mediated demyelination [99–103], it is likely that other CNS-reactive antibodies can have such effects too [79]. The above findings are just well-documented examples that clarify the link between anti-myelin antibodies and the induction of oligodendrocyte stress as evidenced by the accumulation of αB-crystallin (Fig. 5). While anti-myelin antibodies binding to their target is an early event in the development of an MS lesion, so is the accumulation of αB-crystallin in oligodendrocytes [104]. In contrast, accumulation of αB-crystallin in astrocytes is only observed at later stages of lesion development [104]. Clearly, αB-crystallin accumulation is not a secondary response by oligodendrocytes to ongoing inflammatory damage during MS. Instead, it is a critical factor that accompanies early antibody-induced disturbances of oligodendrocytes that eventually lead to such damage.
Figure 5.
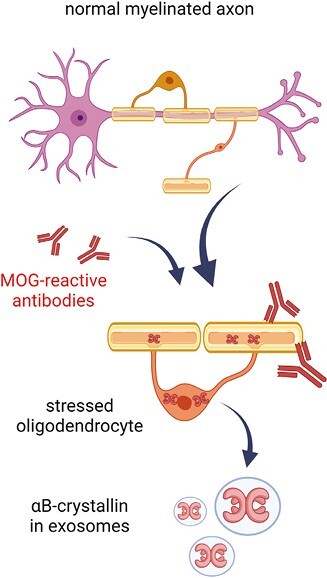
Myelin-reactive antibodies such as those against MOG trigger oligodendrocyte stress. This subsequently results in the release of exosomes containing the stress protein αB-crystallin. Antibodies that bind to the oligodendroglial transmembrane protein MOG trigger membrane perturbations and intracellular changes including stress. To counteract apoptosis, oligodendrocytes accumulate intracellular αB-crystallin which is subsequently also secreted as exosome cargo (Figure created with Biorender).
Event 5. αB-crystallin activates protective microglial responses
In this section, we discuss the impact of the accumulation of αB-crystallin in oligodendrocytes, a next event in the development of MS. Apart from a role as an intracellular anti-apoptotic protectant of oligodendrocytes [105, 106] αB-crystallin also serves an extracellular signaling role following its secretion as exosome cargo [107–109]. Via the innate toll-like receptor heterodimer TLR1/TLR2 and using CD14 as co-receptor, it activates a response by surrounding microglia and macrophages following its uptake by macropinocytosis [110, 111]. In both cell types, this leads to the production of anti-inflammatory mediators dominated by IL-10, IL-6, IL-13, many chemokines, and several tolerance-inducing factors that will control pro-inflammatory T-cell responses [110–112]. That this signaling effect of αB-crystallin is beneficial and serves to suppress inflammatory damage has been documented in a wide range of experimental models of inflammatory disease in which exogenously supplied αB-crystallin consistently suppresses clinical symptoms (summarized in Refs [20]. and [113]).
It may be somewhat surprising that TLR2, which normally responds to lipoproteins and lipopeptides, also responds to a protein such as αB-crystallin that contains no lipid moieties. Yet, things become clearer when it is considered that αB-crystallin contains hydrophobic stretches of amino acids that play a crucial role in its function as molecular chaperone (reviewed in Refs. [114, 115]). Under normal conditions, such a stretch is paired with the corresponding ‘sticky’ stretch of another subunit within the 24- to 32-subunit complexes that αB-crystallin forms. In this way, relatively stable dimers are paired in larger oligomeric complexes. Yet, individual subunits dissociate continuously from this complex as a single protein chain, after which they are rapidly absorbed by another αB-crystallin complex. This offers a temporary opportunity for the hydrophobic part of such a traveling subunit to capture the exposed hydrophobic part of partially unfolded other proteins. This is the way αB-crystallin acts as a molecular chaperone, interferes with pro-apoptotic pathways, and indeed, binds to TLR2.
The tell-tale signs of αB-crystallin-mediated activation of microglia during MS are obvious in so-called ‘normal-appearing’ white matter as clusters of mildly activated microglia that are referred to as ‘preactive MS lesions’ [116–119], alternatively described as ‘microglial nodules’ or ‘newly-forming MS lesions’ [120, 121]. They are generally considered to represent the very first stage of a localized disturbance that can develop into an MS lesion. These microglial clusters exist in the absence of overt demyelination, BBB or leucocyte infiltration [117] but they do co-localize with oligodendrocytes in which αB-crystallin expression is prominent [111]. We have previously discussed in detail the possible drivers behind their emergence and their possible relevance to MS [122]. The appearance of preactive MS lesions in chronic disease is not an isolated phenomenon but part of a much wider manifestation of tissue abnormalities that develop in so-called ‘normal-appearing’ white as well as grey matter CNS regions in people with ongoing MS. Prominent features of these abnormalities include antibody (IgG and IgM) deposition, oxidative stress and oligodendrocyte apoptosis [123, 124]. It is likely that these are all associated with the presence of abnormal levels of myelin- or otherwise CNS-reactive antibodies.
Microglia in preactive MS lesions contain both CD14 and TLR2 as joint receptors for αB-crystallin and they express HLA-DR along with several chemokines and cytokines as signs of cellular activation. A comparison of their molecular activation profile with that of human microglia activated with purified αB-crystallin in culture revealed striking similarities [111]. This strongly suggests that microglial activation in preactive MS lesions is indeed the result of exposure to oligodendrocyte-derived αB-crystallin. This microglial response as evaluated by microarray transcript profiling is reminiscent of an antiviral type I interferon response [111, 112]. It leads to induction of chemokines such as CCL4 and CCL5 that fully explain microglial aggregation, and of beneficial cytokines including IL-10 and IL-13. While IL-13 is a key driver of reparative (‘alternative’) macrophage activation [125], IL-10 regulates immunological activity within the CNS and limits tissue damage [126]. Importantly, it also promotes B cell proliferation, differentiation, and class switch [127] and may thus well play a role in controlling IgG production within the CNS, including oligoclonal band formation [128]. Indeed, IL-10 levels in the CSF of MS patients are generally high [129]. The broad protective qualities of the microglial mediators induced by αB-crystallin in preactive MS lesions along with the widespread appearance of these lesions strongly suggest that most of them will resolve without causing any further damage. They simply reflect the successful response by the CNS to counteract antibody-mediated oligodendrocyte stress by actively deploying microglia to help resolve inflammatory damage (Fig. 6). Yet, there is a risk that events take a different course when the local concentrations of individual factors start to exceed a safe threshold and a memory T-cell response is locally triggered.
Event 6. The process derails; recruitment of leucocytes subverts repair and triggers destruction
As stated above, microglial mediators induced by αB-crystallin include chemokines such as CCL4 and CCL5 that are likely instrumental in creating the microglial aggregates that form preactive MS lesions [111]. Additional chemokines that are induced such as CCL20 and CXCL10, however, are also able to promote influx of potentially pathogenic T and B cells from the circulation [130, 131]. This is especially likely to happen when at the same time, peripheral viral infections lead to BBB weakening by inducing serum cytokines such as IFN-γ and IL-6, as discussed above. Recruitment of leucocytes represents a risk factor for subversion of the local repair process when memory T cells come into play, as further explained below. While the critical role of B cells in MS has become widely recognized over the past 10 years, this still does not eliminate T cells as additional critical players in its pathogenesis.
Previously, the central role of T cells in MS has inspired many studies addressing the question which of the many different myelin antigens play a role in activating pathogenic T cells. In this context, we previously screened the reactivity of peripheral T-cells against the complete collection of human myelin proteins following their high-resolution fractionation by high-performance liquid chromatography [20]. Authentic myelin-derived proteins were purified from post-mortem brains of MS patients or healthy controls using a rigorous protocol based on extraction with organic solvents. Peripheral blood T cells were isolated from both test groups as well and their response to the fractionated myelin proteins was examined in a crossover design: T cells from MS patients or healthy controls were tested against myelin proteins isolated from both groups. The results showed no discernible difference between the T-cell response profiles of either MS patients or healthy subjects, emphasizing again the lack of any obvious disease-specific peripheral T-cell autoreactivity in MS patients. Instead, a single protein fraction from MS-affected brain white matter consistently triggered much stronger responses from both sets of T cells than the corresponding protein fraction isolated from control brains. This striking difference was due to the presence of much higher levels of αB-crystallin in this protein fraction [20]. Follow-up data confirmed that oligodendrocytes and myelin in and around active MS lesions contain unusually high levels of αB-crystallin [104, 132]. Together, these data indicate that when the concentration of αB-crystallin produced by oligodendrocytes exceeds a critical level, it can activate the human memory T-cell repertoire that had been established by an EBV infection years before.
Importantly, however, this will only happen when B cells act as antigen-presenting cells. When αB-crystallin is presented to T cells by microglia or macrophages, TLR-mediated signaling leads to production of factors including indoleamine 2,3-dioxygenase (IDO-1), programmed cell death ligand-1 (PD-1L; CD274) and the adenosine receptor 2A. These are all powerful inhibitors of T-cell responses and they will promote tolerance [133–135]. Yet, the situation becomes fundamentally different when B cells act as local antigen-presenting cells [136]. Different from microglia and macrophages, B cells lack the co-receptor CD14 that is essential for αB-crystallin to act as an agonist for TLR2. B cells will therefore do not develop such a protective TLR2-mediated response but simply present αB-crystallin as an antigen, thus allowing the development of a full-blown pro-inflammatory memory T-cell response against it. This critical role of B cells is fully in line with the reports that B-cell targeting therapeutic interventions in MS are more effective than previously expected (reviewed in Ref. [137]).
T-cell activation of memory T cells by αB-crystallin within the CNS poses a serious risk. As illustrated in Fig. 2C, we have frequently found that up to 5% of these cells release IFN-γ in response to such activation. When sufficient amounts of IFN-γ are released locally, a dramatic change will occur in the way microglia and macrophages respond to the αB-crystallin cargo of oligodendroglial exosomes. The TLR-mediated protective response changes into a destructive one (Fig. 7). This is because IFN-γ inactivates the inhibitory mechanisms that are mediated by IL-10 and signal transducer and activator of transcription (STAT)3. Instead, it shifts TLR-mediated responses toward pro-inflammatory STAT1-dominated responses that activate a destructive macrophage program [138–140]. Indeed, the addition of IFN-γ to cultured microglia and macrophages that are already exposed to αB-crystallin abrogates IL-10 induction and causes a release of strongly increased levels of TNF-α, IL-6, IL-12, IL-1β along with reactive oxygen and nitrogen species [141]. The molecular markers of such double-activated microglia and macrophages are consistently found in actively demyelinating MS lesions, emphasizing the relevance of this phenomenon. In addition, CXCL9, CXCL10, and CXL11 are induced, which are key signals for T-cell recruitment into the CNS [142]. In this way, a self-amplifying destructive process is ignited by the local accumulation of αB-crystallin and subsequent release of IFN-γ by memory T cells that respond to it. It is important to note that this subversion of the originally protective microglial response only occurs when all of the local factors involved exceed a quantitative threshold that controls the process [21]. Local concentrations of myelin-reactive antibodies will have to exceed a certain threshold, as will local concentrations of αB-crystallin. Also, the local numbers of B cells will have to reach a certain minimum, as well as the numbers of αB-crystallin-reactive T cells. Such accumulation is another unfortunate event in the pathogenesis of MS.
Figure 7.
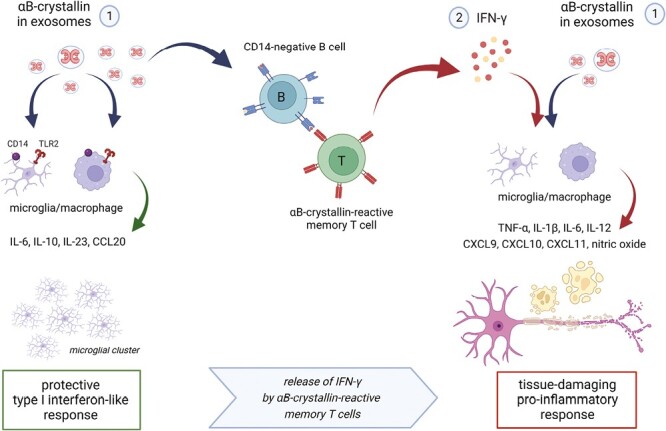
Memory T cells against αB-crystallin cause derailment of the process. When sufficient numbers of αB-crystallin-reactive T cells encounter B cells within the CNS that present their target antigen at a sufficiently high concentration, things will go wrong. B cells lack CD14 and, therefore, fail to mount the regulatory and immune-suppressive response to αB-crystallin as seen in microglia and macrophages. Instead, they will allow the development of a substantial IFN-γ response by T cells (see Fig. 2C). This response changes everything since IFN-γ reprograms TLR signaling pathways in microglia and macrophages. Their originally protective response to αB-crystallin now changes into a full-blown destructive response, leading to tissue damage. Furthermore, it will cause substantial destabilization of the BBB and active recruitment of leucocytes, adding more fuel to the fire (Figure created with Biorender).
Due to the accumulation of cytokines and chemokines that destabilize the BBB and promote leucocyte recruitment under the above conditions, more pathogenic factors come into play including IL-17 that will be produced by some T-cells. These products will be mixed with locally produced IL-10, IL-6, CXCL10, and IDO-1 as well as local antigens that all promote the persistence of B cells in localized niches within the CNS and antibody production [143–147] including the development of ectopic follicles [65]. While much remains to be clarified on this particular point, the resulting cocktail of soluble factors that is thus generated by the local inflammatory response likely contributes to persistent local production of potentially pathogenic antibodies including OCB. T cells within ectopic follicles may similarly become a persistent source of IFN-γ. Since IFN-γ may subsequently diffuse from its production site to adjacent regions within the CNS where it will promote destructive macrophage activity in the way described above, this could well lead to a gradually expanding zone of destruction at later stages of MS, as is seen in cases of cortical demyelination in MS. A self-sustaining or even self-amplifying process of inflammatory activity that develops in this way within the CNS readily explains the chronic nature of MS.
Therapeutic intervention with αB-crystallin
A wealth of data has clarified that repeated systemic administration of αB-crystallin counteracts neuronal and glial cell apoptosis, mitigates inflammation, reduces tissue damage, and promotes recovery in a variety of animal models for neuroinflammatory disorders including autoimmune encephalomyelitis, stroke, spinal cord injury, and optic nerve damage (summarized in Refs. [21] and [105]). These effects have been documented by various groups following repeated intravenous or intraperitoneal administration of purified αB-crystallin. As explained above, this leads to the activation of relatively short-lived TLR2-mediated protective microglia/macrophage responses. Such a therapeutic strategy based on daily intravenous administrations is difficult to apply in MS not only for practical reasons but also since in this case, the long-term inhibition of a specific memory T-cell response to αB-crystallin would be the goal. If this could be achieved, it would provide strong support for the model presented here.
While intravenous administration of protein antigens is a time-honored method to induce tolerance at the level of naïve T cells, things are more complicated when memory T cells are the target. Both deletion of existing memory T cells and activation of peripheral regulatory T cells may help establish functional tolerance. For deletion of T cells in vivo, at least two intravenous doses of antigen generally appear to be required [148]. However, considering that reactivation of EBV will inevitably occur at some point in time, newly generated T cells against αB-crystallin are likely to reappear. For activation of antigen-specific peripheral regulatory T cells as an alternative way to suppress the target response, sub-immunogenic doses of antigen are required. Again, the impact of such an intervention will be limited given the finite life span of peripheral regulatory T cells [149, 150]. Any effect of tolerance induction is therefore likely to be only temporary and will require periodic reiteration.
In randomized placebo-controlled clinical Phase I and IIa studies the possibilities were explored to use repeated intravenous administration of highly purified αB-crystallin at sub-immunogenic doses as a strategy to suppress specific memory T-cell responses to the protein [21]. At these sub-immunogenic doses, peak serum concentrations of αB-crystallin remain below the threshold for triggering IFN-γ release by pathogenic T cells but still become sufficiently high to activate beneficial macrophage responses that promote tolerance. After all, these distinct responses are mediated by different cell types that use different receptor complexes. As illustrated in Fig. 8B, peak serum concentrations between 5 and 10 μg/ml should achieve this separation of the beneficial from the pathogenic impact of αB-crystallin. As a main goal of these first-in-man studies, the safety of the intervention was firmly established in both healthy subjects and MS patients. Furthermore, the collective data on 76 healthy subjects involved in the Phase I study and 32 MS patients involved in the Phase IIa study confirmed that essentially all of them had serum antibodies against αB-cystallin which were readily detectable by ELISA, accompanied by marked levels of memory T-cell reactivity. Unsurprisingly, antibody titers were variable, as were levels of T-cell reactivity against the protein. Yet, consistent with earlier findings [15], up to around 5% of all memory T cells were frequently found to be responsive to αB-crystallin prior to treatment, and such T cells consistently released IFN-γ (Fig. 8).
Figure 8.
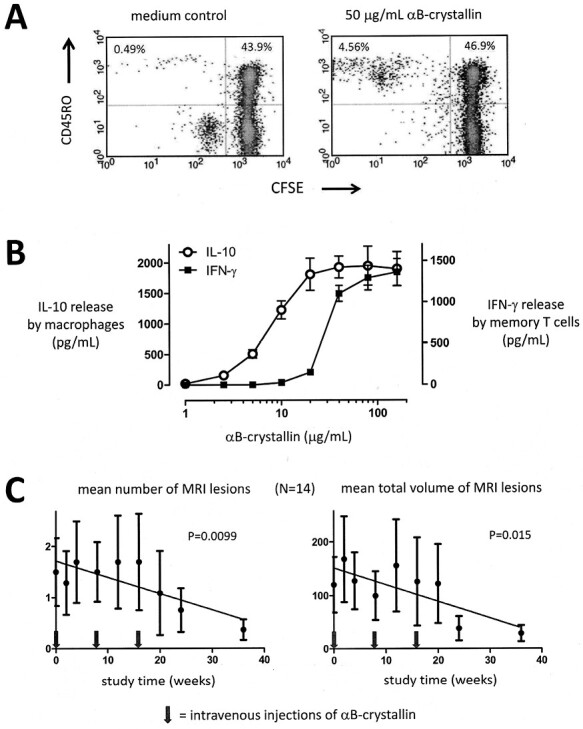
Therapeutic intervention in MS with αB-crystallin. A significant memory T-cell response against αB-crystallin is a normal part of the human adult immune repertoire. This is illustrated by the proliferative response of CD45RO + memory T cells as measured by dilution of the cellular marker carboxyfluorescein succinimidyl ester (CFSE) in an example assay using fresh blood from a normal healthy adult (A). As many as 3–5% of all memory T cells frequently respond to αB-crystallin in this way. This T-cell response is associated with the production of IFN-γ, as illustrated by another example assay in panel B. Yet, approximately 10-fold higher concentrations of αB-crystallin are required to trigger this T-cell response as compared to the TLR2-mediated response by macrophages that induces IL-10 production. This difference offers a therapeutic concentration window for tolerance induction. As long as the maximum serum concentration of intravenously administered αB-crystallin remains below the threshold for T-cell activation of around 10 μg/mL, benefit can be taken from the tolerizing macrophage response while avoiding the pathogenic IFN-γ response by T cells. Intravenous doses of either 7.5 or 12.5 mg αB-crystallin lead to sub-immunogenic peak serum concentrations of well below 5 μg/mL. After three bimonthly doses at these levels the total number as well as total volume of gadolinium-enhancing MRI lesions in MS patients are significantly suppressed by about 75% (previously published in part by van Noort et al. 2015, Ref. 21).
Using intravenous dose levels of αB-crystallin that remained below the threshold for T-cell activation, three bimonthly injections led to a significant reduction in the number as well as the total volume of gadolinium-enhancing magnetic resonance imaging (MRI) lesions in the brains of MS patients in this study, but only during a limited time window (Fig. 8C). In line with the above considerations, the reduction in MRI lesion load became apparent after three consecutive injections and was sustained for about 20 weeks. At the end of this response period, both total numbers and total volumes of MRI brain lesions were significantly reduced by 75% relative to pre-treatment values. No such change was observed in a higher dose group, or the placebo group who received intravenous phosphate-buffered saline. At the end of the study period at 48 weeks, MRI lesion load tended to increase again despite the observation that at that point in time, no clinical relapses were recorded in the entire treatment population anymore. Unfortunately, technical limitations led to unreliable data collected in the T-cell assays that were performed to monitor actual T-cell responsiveness. This requires an analysis of freshly isolated peripheral blood mononuclear cells rather than frozen cells, posing significant logistic demands that could not be adequately met during the multicenter study in Bulgaria.
In summary, the outcome of the Phase IIa study did reveal a significant reduction in MRI lesions and the complete suppression of clinical relapses. Yet, these effects were limited to a 20-week period toward the end of the study and no data could be obtained to demonstrate a direct association with antigen-specific tolerance at that stage. While these data therefore fell somewhat short of providing conclusive evidence for the model presented here, they do support it. Further refinement of the tolerizing approach and assays to quantitate memory T-cell responsiveness in patients may finally clarify whether or not the specific elimination of αB-crystallin-reactive memory T cells will be enough to halt MS.
A final note: is MS a single disease or just part of a spectrum of demyelinating CNS disorders?
The debate over the issue of whether or not MS is a single disease is ongoing [151]. Should we consider the possibility that separate pathogenic pathways exist for the clinically diverse forms of MS? These different forms may be variable and they are associated with different responses to certain pharmaceutical interventions. It is understandable that clinicians, researchers, and MS patients interested in treatment options and prognosis attach value to these variations at the clinical level. On the other hand, the key biological features of MS do not differ between people with different forms of MS. The pathological picture, epidemiology, genetic factors, and immunology of MS all support the idea that MS is one disease. We therefore prefer the idea that the variable clinical manifestations are caused by differences in age, gender, disease duration, genetic make-up, infection history, lifestyle, diet, vascular architecture, the antigen specificity, and isotype of anti-myelin antibodies involved, and many other biological confounders. After all, such factors are well-known to similarly influence the course of experimental autoimmune encephalomyelitis in laboratory animals, even when such disease is induced using the same immunogen [152–156]. In our view and in the absence of compelling evidence to indicate otherwise, we should give priority to the simplest explanation that all manifestations of MS reflect the outcome of the single pathogenic pathway described here.
As an extension of this issue, this pathogenic pathway may perhaps not be very different from the one that causes the pathologically and clinically similar conditions that are collectively referred to as NMOSD [157]. NMO, for example, has long been considered as a variant of MS and was only classified as a different disorder after pathogenic serum antibodies against AQP4 were found in the majority of patients. More recently, the term MOGAD was introduced for similar reasons [158, 159]. Yet, the clinical, immunological, and pathological characteristics of NMO and MOGAD are strongly overlapping with each other as well as with MS, and all three conditions are equally linked to ON as a frequent first symptom [42, 160–162]. Only levels of serum antibody reactivities to either MOG or AQP4 may be different between subjects with the above conditions, but as explained above, the outcome of the antibody assays used in this context is confounded by significant technical issues. This is a relevant final point since it clarifies that an exclusively diagnostic focus may distract from the cause of a disease, in this case by suggesting an association with levels of certain serum antibodies that is far from straightforward. It may well be more productive for our understanding of NMOSD including MS to focus on the common biological parameters that unify clinical variations rather than aiming at the reduction of a complex biological problem to ever smaller parts, and getting lost in the process. A more holistic approach will likely be more helpful to further understand the way these disorders develop and to define new ways to treat them.
Acknowledgments
Not applicable.
Glossary
Abbreviations:
- AQP4
aquaporin-4
- BBB
blood–brain barrier
- CNS
central nervous system
- CSF
cerebrospinal fluid
- EBV
Epstein-Barr virus
- ELISA
enzyme-linked immunosorbent assays
- HLA-DR
human leukocyte antigen-DR
- IDO-1
indoleamine 2;3-dioxygenase
- IFN-γ
interferon gamma
- MOG
myelin-oligodendrocyte glycoprotein
- MOGAD
MOG antibody disease
- magnetic resonance imaging
MRI
- MS
multiple sclerosis
- NMOSD
neuromyelitis optica spectrum disorder
- OCB
oligoclonal bands
- ON
optic neuritis
- PD-1L
programmed cell death ligand-1
- STAT-3
signal transducer and activator of transcription 3
- TLR
toll like receptor.
Contributor Information
Johannes M van Noort, Department of Pathology, Amsterdam UMC, Location VUmc, Amsterdam, The Netherlands.
David Baker, Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK.
Markus Kipp, Institute of Anatomy, Rostock University Medical Center, Rostock, Germany.
Sandra Amor, Department of Pathology, Amsterdam UMC, Location VUmc, Amsterdam, The Netherlands; Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK; Institute of Anatomy, Rostock University Medical Center, Rostock, Germany.
Ethical Approval
Not applicable.
Conflict of Interests
The authors declare no conflict of interest.
Funding
No funding to report.
Data Availability
All data submitted is available.
Author Contributions
All authors contributed equally.
Permission to Reproduce
Not applicable.
References
- 1. Compston A, Coles A.. Multiple sclerosis. Lancet 2008, 372, 1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2. Lisak RP, Zweiman B, Burns JB, Rostami A, Silberberg DH.. Immune responses to myelin antigens in multiple sclerosis. Ann N Y Acad Sci 1984, 436, 221–30. doi: 10.1111/j.1749-6632.1984.tb14793.x. [DOI] [PubMed] [Google Scholar]
- 3. McFarland HF, Martin R.. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 2007, 89, 913–9. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 4. Soldan SS, Lieberman PM.. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol 2023, 21, 51–64. doi: 10.1038/s41579-022-00770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. doi: 10.1126/science.abj8222. [DOI] [PubMed] [Google Scholar]
- 6. Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, et al. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med 2018, 16, 80. doi: 10.1186/s12967-018-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baker D, Pryce G, Amor S, Giovannoni G, Schmierer K.. Learning from other autoimmunities to understand targeting of B cells to control multiple sclerosis. Brain 2018, 141, 2834–47. doi: 10.1093/brain/awy239. [DOI] [PubMed] [Google Scholar]
- 8. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM; NeuroproMiSe EBV Working Group. NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue--report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain 2011, 134, 2772–86. doi: 10.1093/brain/awr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas OG, Bronge M, Tengvall K, Akpinar B, Nilsson OB, Holmgren E, et al. Cross-reactive EBNA1 immunity targets alpha-crystallin B and is associated with multiple sclerosis. Sci Adv 2023, 9, eadg3032. doi: 10.1126/sciadv.adg3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wucherpfennig KW. T cell receptor cross-reactivity as a general property of T cell recognition. Mol Immunol 2004, 40, 1009–17. doi: 10.1016/j.molimm.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 11. Kramer A, Keitel T, Winkler K, Stöcklein W, Höhne W, Schneider-Mergener J.. Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell 1997, 91, 799–809. doi: 10.1016/s0092-8674(00)80468-7. [DOI] [PubMed] [Google Scholar]
- 12. Mason D. A very high level of cross-reactivity is an essential feature of the T-cell receptor. Immunol Today 1998, 19, 395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 13. Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol 2012, 12, 669–77. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olotu FA, Soliman MES.. Immunoinformatics prediction of potential B-cell and T-cell epitopes as effective vaccine candidates for eliciting immunogenic responses against Epstein-Barr virus. Biomed J 2021, 44, 317–37. doi: 10.1016/j.bj.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM.. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol 1999, 162, 129–35. [PubMed] [Google Scholar]
- 16. Altmann M, Hammerschmidt W.. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 2005, 3, e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wyżewski Z, Mielcarska MB, Gregorczyk-Zboroch KP, Myszka A.. Virus-mediated inhibition of apoptosis in the context of EBV-associated diseases: molecular mechanisms and therapeutic perspectives. Int J Mol Sci 2022, 23, 7265. doi: 10.3390/ijms23137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Imadome K, Shirakata M, Shimizu N, Nonoyama S, Yamanashi Y.. CD40 ligand is a critical effector of Epstein-Barr virus in host cell survival and transformation. Proc Natl Acad Sci USA 2003, 100, 7836–40. doi: 10.1073/pnas.1231363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA.. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J Virol 2004, 78, 5194–204. doi: 10.1128/jvi.78.10.5194-5204.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Noort JM, van Sechel AC, Bajramovic JJ, el Ouagmiri M, Polman CH, Lassmann H, et al. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature 1995, 375, 798–801. doi: 10.1038/375798a0. [DOI] [PubMed] [Google Scholar]
- 21. van Noort JM, Bsibsi M, Nacken PJ, Verbeek R, Venneker EH.. Therapeutic intervention in multiple sclerosis with alpha B-crystallin: a randomized controlled phase IIa trial. PLoS One 2015, 10, e0143366. doi: 10.1371/journal.pone.0143366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Noort JM, Verbeek R, Meilof JF, Polman CH, Amor S.. Autoantibodies against alpha B-crystallin, a candidate autoantigen in multiple sclerosis, are part of a normal human immune repertoire. Mult Scler 2006, 12, 287–93. doi: 10.1191/135248506ms1271oa. [DOI] [PubMed] [Google Scholar]
- 23. Thorley-Lawson DA. EBV Persistence–Introducing the Virus. Curr Top Microbiol Immunol 2015, 390, 151–209. doi: 10.1007/978-3-319-22822-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Stipdonk MJ, Willems AA, Plomp AC, van Noort JM, Boog CJ.. Tolerance controls encephalitogenicity of alpha B-crystallin in the Lewis rat. J Neuroimmunol 2000, 103, 103–11. doi: 10.1016/s0165-5728(99)00171-x. [DOI] [PubMed] [Google Scholar]
- 25. van Stipdonk MJ, Willems AA, Verbeek R, Boog CJ, van Noort JM.. T- and B-cell non-responsiveness to self-alpha B-crystallin in SJL mice prevents the induction of experimental allergic encephalomyelitis. Cell Immunol 2000, 204, 128–34. doi: 10.1006/cimm.2000.1698. [DOI] [PubMed] [Google Scholar]
- 26. Traugott U, Scheinberg LC, Raine CS.. Multiple sclerosis: circulating antigen-reactive lymphocytes. Ann Neurol 1979, 6, 425–9. doi: 10.1002/ana.410060509. [DOI] [PubMed] [Google Scholar]
- 27. Paterson PY, Day ED, Whitacre CC, Berenberg RA, Harter DH.. Endogenous myelin basic protein-serum factors (MBP-SFs) and anti-MBP antibodies in humans. Occurrence in sera of clinically well subjects and patients with multiple sclerosis. J Neurol Sci 1981, 52, 37–51. doi: 10.1016/0022-510x(81)90132-5. [DOI] [PubMed] [Google Scholar]
- 28. Hellings N, Barée M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res 2001, 63, 290–302. doi:. [DOI] [PubMed] [Google Scholar]
- 29. Tejada-Simon MV, Hong J, Rivera VM, Zhang JZ.. Skewed autoantibody reactivity to the extracellular domain of myelin oligodendrocyte glycoprotein in multiple sclerosis. Immunology 2002, 107, 403–10. doi: 10.1046/j.1365-2567.2002.01533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med 2003, 349, 139–45. doi: 10.1056/NEJMoa022328. [DOI] [PubMed] [Google Scholar]
- 31. Rauer S, Euler B, Reindl M, Berger T.. Antimyelin antibodies and the risk of relapse in patients with a primary demyelinating event. J Neurol Neurosurg Psychiatry 2006, 77, 739–42. doi: 10.1136/jnnp.2005.077784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Munger KL, Reindl M, O’Reilly EJ, Levin LI, Berger T, et al. Myelin oligodendrocyte glycoprotein antibodies and multiple sclerosis in healthy young adults. Neurology 2008, 71, 1142–6. doi: 10.1212/01.wnl.0000316195.52001.e1. [DOI] [PubMed] [Google Scholar]
- 33. Chan A, Decard BF, Franke C, Grummel V, Zhou D, Schottstedt V, et al. Serum antibodies to conformational and linear epitopes of myelin oligodendrocyte glycoprotein are not elevated in the preclinical phase of multiple sclerosis. Mult Scler 2010, 16, 1189–92. doi: 10.1177/1352458510376406. [DOI] [PubMed] [Google Scholar]
- 34. Prineas JW, Parratt JDE.. Multiple sclerosis: serum anti-CNS autoantibodies. Mult Scler 2018, 24, 610–22. doi: 10.1177/1352458517706037. [DOI] [PubMed] [Google Scholar]
- 35. Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm 2020, 7, e674. doi: 10.1212/NXI.0000000000000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campagnoni AT, Macklin WB.. Cellular and molecular aspects of myelin protein gene expression. Mol Neurobiol 1988, 2, 41–89. doi: 10.1007/BF02935632. [DOI] [PubMed] [Google Scholar]
- 37. Taylor CM, Marta CB, Claycomb RJ, Han DK, Rasband MN, Coetzee T, et al. Proteomic mapping provides powerful insights into functional myelin biology. Proc Natl Acad Sci USA 2004, 101, 4643–8. doi: 10.1073/pnas.0400922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Menge T, von Büdingen HC, Lalive PH, Genain CP.. Relevant antibody subsets against MOG recognize conformational epitopes exclusively exposed in solid-phase ELISA. Eur J Immuno 2007, 37, 3229–39. doi: 10.1002/eji.200737249. [DOI] [PubMed] [Google Scholar]
- 39. Schanda K, Peschl P, Lerch M, Seebacher B, Mindorf S, Ritter N, et al. Differential binding of autoantibodies to MOG isoforms in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm 2021, 8, e1027. doi: 10.1212/NXI.0000000000001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spiezia AL, Carotenuto A, Iovino A, Moccia M, Gastaldi M, Iodice R, et al. AQP4-MOG double-positive neuromyelitis optica spectrum disorder: case report with central and peripheral nervous system involvement and review of literature. Int J Mol Sci 2022, 23, 14559. doi: 10.3390/ijms232314559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dinoto A, Sechi E, Flanagan EP, Ferrari S, Solla P, Mariotto S, et al. Serum and cerebrospinal fluid biomarkers in neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disease. Front Neurol 2022, 13, 866824. doi: 10.3389/fneur.2022.866824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fadda G, Flanagan EP, Cacciaguerra L, Jitprapaikulsan J, Solla P, Zara P, et al. Myelitis features and outcomes in CNS demyelinating disorders: comparison between multiple sclerosis, MOGAD, and AQP4-IgG-positive NMOSD. Front Neurol 2022, 13, 1011579. doi: 10.3389/fneur.2022.1011579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wendel EM, Thonke HS, Bertolini A, Baumann M, Blaschek A, Merkenschlager A, et al.; BIOMARKER Study Group. Temporal dynamics of MOG antibodies in children with acquired demyelinating syndrome. Neurol Neuroimmunol Neuroinflamm 2022, 9, e200035. doi: 10.1212/NXI.0000000000200035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morris-Downes MM, Smith PA, Rundle JL, Piddlesden SJ, Baker D, Pham-Dinh D, et al. Pathological and regulatory effects of anti-myelin antibodies in experimental allergic encephalomyelitis in mice. J Neuroimmunol 2002, 125, 114–24. doi: 10.1016/s0165-5728(02)00040-1. [DOI] [PubMed] [Google Scholar]
- 45. van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, et al. Alpha B-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 2010, 69, 694–703. doi: 10.1097/NEN.0b013e3181e4939c. [DOI] [PubMed] [Google Scholar]
- 46. Brändle SM, Obermeier B, Senel M, Bruder J, Mentele R, Khademi M, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc Natl Acad Sci USA 2016, 113, 7864–9. doi: 10.1073/pnas.1522730113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Graner M, Pointon T, Manton S, Green M, Dennison K, Davis M, et al. Oligoclonal IgG antibodies in multiple sclerosis target patient-specific peptides. PLoS One 2020, 15, e0228883. doi: 10.1371/journal.pone.0228883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Z, Kennedy PG, Dupree C, Wang M, Lee C, Pointon T, et al. Antibodies from multiple sclerosis brain identified Epstein-Barr virus nuclear antigen 1 & 2 epitopes which are recognized by oligoclonal bands. J Neuroimmune Pharmacol 2021, 16, 567–80. doi: 10.1007/s11481-020-09948-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 2021, 373, eabf7844. doi: 10.1126/science.abf7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 2021, 373, eabf9277. doi: 10.1126/science.abf9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y, Chen D, Xu D, Huang C, Xing R, He D, et al. Early developing B cells undergo negative selection by central nervous system-specific antigens in the meninges. Immunity 2021, 54, 2784–2794.e6. doi: 10.1016/j.immuni.2021.09.016. [DOI] [PubMed] [Google Scholar]
- 52. van Zwam M, Huizinga R, Melief MJ, Wierenga-Wolf AF, van Meurs M, Voerman JS, et al. Brain antigens in functionally distinct antigen-presenting cell populations in cervical lymph nodes in MS and EAE. J Mol Med (Berl) 2009, 87, 273–86. doi: 10.1007/s00109-008-0421-4. [DOI] [PubMed] [Google Scholar]
- 53. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 2014, 6, 248ra–107. doi: 10.1126/scitranslmed.3008879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salvador F, Deramoudt L, Leprêtre F, Figeac M, Guerrier T, Boucher J, et al. Spontaneous model of experimental autoimmune encephalomyelitis provides evidence of MOG-Specific B cell recruitment and clonal expansion. Front Immunol 2022, 13, 755900. doi: 10.3389/fimmu.2022.755900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schiefenhövel F, Immig K, Prodinger C, Bechmann I.. Indications for cellular migration from the central nervous system to its draining lymph nodes in CD11c-GFP+ bone-marrow chimeras following EAE. Exp Brain Res 2017, 235, 2151–66. doi: 10.1007/s00221-017-4956-x. [DOI] [PubMed] [Google Scholar]
- 56. Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Börsch AL, et al. Single-cell profiling of CNS border compartment leucocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat Neurosci 2021, 24, 1225–34. doi: 10.1038/s41593-021-00880-y. [DOI] [PubMed] [Google Scholar]
- 57. Pollok K, Mothes R, Ulbricht C, Liebheit A, Gerken JD, Uhlmann S, et al. The chronically inflamed central nervous system provides niches for long-lived plasma cells. Acta Neuropathol Commun 2017, 5, 88. doi: 10.1186/s40478-017-0487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tomescu-Baciu A, Johansen JN, Holmøy T, Greiff V, Stensland M, de Souza GA, et al. Persistence of intrathecal oligoclonal B cells and IgG in multiple sclerosis. J Neuroimmunol 2019, 333, 576966. doi: 10.1016/j.jneuroim.2019.576966. [DOI] [PubMed] [Google Scholar]
- 59. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med 2014, 6, 248ra–106. doi: 10.1126/scitranslmed.3008930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol 2014, 75, 266–76. doi: 10.1002/ana.24088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. von Büdingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest 2012, 122, 4533–43. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA.. B cells in multiple sclerosis—from targeted depletion to immune reconstitution therapies. Nat Rev Neurol 2021, 17, 399–414. doi: 10.1038/s41582-021-00498-5. [DOI] [PubMed] [Google Scholar]
- 63. Soldan SS, Su C, Lamontagne RJ, Grams N, Lu F, Zhang Y, et al. Epigenetic plasticity enables CNS-trafficking of EBV-infected B lymphocytes. PLoS Pathog 2021, 17, e1009618. doi: 10.1371/journal.ppat.1009618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F.. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004, 14, 164–74. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Negron A, Stüve O, Forsthuber TG.. Ectopic lymphoid follicles in multiple sclerosis: centers for disease control? Front Neurol 2020, 11, 607766. doi: 10.3389/fneur.2020.607766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008, 65, 727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fernández Blanco L, Marzin M, Leistra A, van der Valk P, Nutma E, Amor S.. Immunopathology of the optic nerve in multiple sclerosis. Clin Exp Immunol 2022, 209, 236–46. doi: 10.1093/cei/uxac063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Knier B, Berthele A, Buck D, Schmidt P, Zimmer C, Mühlau M, et al. Optical coherence tomography indicates disease activity prior to clinical onset of central nervous system demyelination. Mult Scler 2016, 22, 893–900. doi: 10.1177/1352458515604496. [DOI] [PubMed] [Google Scholar]
- 69. Saidha S, Syc SB, Ibrahim MA, Eckstein C, Warner CV, Farrell SK, et al. Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain 2011, 134, 518–33. doi: 10.1093/brain/awq346. [DOI] [PubMed] [Google Scholar]
- 70. Balcer LJ. Optic neuritis. N Engl J Med 2006, 354, 1273–80. doi: 10.1056/NEJMcp053247. [DOI] [PubMed] [Google Scholar]
- 71. Tur C, Goodkin O, Altmann DR, Jenkins TM, Miszkiel K, Mirigliani A, et al. Longitudinal evidence for anterograde trans-synaptic degeneration after optic neuritis. Brain 2016, 139, 816–28. doi: 10.1093/brain/awv396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. You Y, Joseph C, Wang C, Gupta V, Liu S, Yiannikas C, et al. Demyelination precedes axonal loss in the transneuronal spread of human neurodegenerative disease. Brain 2019, 142, 426–42. doi: 10.1093/brain/awy338. [DOI] [PubMed] [Google Scholar]
- 73. Flage T. A defect in the blood-retina barrier in the optic nerve head region in the rabbit and the monkey. Acta Ophthalmol (Copenh) 1980, 58, 645–51. doi: 10.1111/j.1755-3768.1980.tb08307.x. [DOI] [PubMed] [Google Scholar]
- 74. Guy J, Rao NA.. Acute and chronic experimental optic neuritis. Alteration in the blood-optic nerve barrier. Arch Ophthalmol 1984, 102, 450–4. doi: 10.1001/archopht.1984.01040030364039. [DOI] [PubMed] [Google Scholar]
- 75. Tso MO, Shih CY, McLeans IW.. Is there a blood-brain barrier at the optic nerve head? Arch Ophthalmol 1975, 93, 815–25. [DOI] [PubMed] [Google Scholar]
- 76. Hofman P, Hoyng P, vanderWerf F, Vrensen GF, Schlingemann RO.. Lack of blood-brain barrier properties in microvessels of the prelaminar optic nerve head. Invest Ophthalmol Vis Sci 2001, 42, 895–901. [PubMed] [Google Scholar]
- 77. Grieshaber MC, Flammer J.. Does the blood-brain barrier play a role in Glaucoma? Surv Ophthalmol 2007, 52 Suppl 2, S115–21. doi: 10.1016/j.survophthal.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 78. Stojic A, Bojcevski J, Williams SK, Bas-Orth C, Nessler S, Linington C, et al. Preclinical stress originates in the rat optic nerve head during development of autoimmune optic neuritis. Glia 2019, 67, 512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hillebrand S, Schanda K, Nigritinou M, Tsymala I, Böhm D, Peschl P, et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol 2019, 137, 467–85. doi: 10.1007/s00401-018-1950-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chang HY, Morrow K, Bonacquisti E, Zhang W, Shah DK.. Antibody pharmacokinetics in rat brain determined using microdialysis. MAbs 2018, 10, 843–53. doi: 10.1080/19420862.2018.1473910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Q, Delva L, Weinreb PH, Pepinsky RB, Graham D, Veizaj E, et al. Monoclonal antibody exposure in rat and cynomolgus monkey cerebrospinal fluid following systemic administration. Fluids Barriers CNS 2018, 15, 10. doi: 10.1186/s12987-018-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang J, Ran M, Li H, Lin Y, Ma K, Yang Y, et al. New insight into neurological degeneration: Inflammatory cytokines and blood-brain barrier. Front Mol Neurosci 2022, 15, 1013933. doi: 10.3389/fnmol.2022.1013933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Attarbaschi T, Willheim M, Ramharter M, Hofmann A, Wahl K, Winkler H, et al. T cell cytokine profile during primary Epstein-Barr virus infection (infectious mononucleosis). Eur Cytokine Netw 2003, 14, 34–9. [PubMed] [Google Scholar]
- 84. Wada T, Muraoka M, Yokoyama T, Toma T, Kanegane H, Yachie A.. Cytokine profiles in children with primary Epstein-Barr virus infection. Pediatr Blood Cancer 2013, 60, E46–8. doi: 10.1002/pbc.24480. [DOI] [PubMed] [Google Scholar]
- 85. Chai Q, He WQ, Zhou M, Lu H, Fu ZF.. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol 2014, 88, 4698–710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, et al. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm 2020, 7, e841. doi: 10.1212/NXI.0000000000000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Spencer C, Bhat NN, Bindiganavile S, Lee AG.. Postinfectious optic neuritis after hand-foot-mouth disease. J Neuroophthalmol 2021, 41, e351–3. [DOI] [PubMed] [Google Scholar]
- 88. Steelman AJ. Infection as an environmental trigger of multiple sclerosis disease exacerbation. Front Immunol 2015, 6, 520. doi: 10.3389/fimmu.2015.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. McKay KA, Jahanfar S, Duggan T, Tkachuk S, Tremlett H.. Factors associated with onset, relapses or progression in multiple sclerosis: a systematic review. Neurotoxicology 2017, 61, 189–212. doi: 10.1016/j.neuro.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 90. Marrodan M, Alessandro L, Farez MF, Correale J.. The role of infections in multiple sclerosis. Mult Scler 2019, 25, 891–901. doi: 10.1177/1352458518823940. [DOI] [PubMed] [Google Scholar]
- 91. Panitch HS, Hirsch RL, Haley AS, Johnson KP.. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–5. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- 92. Romanelli E, Merkler D, Mezydlo A, Weil MT, Weber MS, Nikić I, et al. Myelinosome formation represents an early stage of oligodendrocyte damage in multiple sclerosis and its animal model. Nat Commun 2016, 7, 13275. doi: 10.1038/ncomms13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Höftberger R, Lassmann H, Berger T, Reindl M.. Pathogenic autoantibodies in multiple sclerosis - from a simple idea to a complex concept. Nat Rev Neurol 2022, 18, 681–8. doi: 10.1038/s41582-022-00700-2. [DOI] [PubMed] [Google Scholar]
- 94. Pryce G, Baker D.. Oligoclonal bands in multiple sclerosis; functional significance and therapeutic implications. Does the specificity matter? Mult Scler Relat Disord 2018, 25, 131–7. doi: 10.1016/j.msard.2018.07.030. [DOI] [PubMed] [Google Scholar]
- 95. Bradl M, Reindl M, Lassmann H.. Mechanisms for lesion localization in neuromyelitis optica spectrum disorders. Curr Opin Neurol 2018, 31, 325–33. doi: 10.1097/WCO.0000000000000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Duvanel CB, Monnet-Tschudi F, Braissant O, Matthieu JM, Honegger P.. Tumor necrosis factor-alpha and alpha B-crystallin up-regulation during antibody-mediated demyelination in vitro: a putative protective mechanism in oligodendrocytes. J Neurosci Res 2004, 78, 711–22. doi: 10.1002/jnr.20310. [DOI] [PubMed] [Google Scholar]
- 97. Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH.. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci USA 2005, 102, 13992–7. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fairless R, Williams SK, Hoffmann DB, Stojic A, Hochmeister S, Schmitz F, et al. Preclinical retinal neurodegeneration in a model of multiple sclerosis. J Neurosci 2012, 32, 5585–97. doi: 10.1523/JNEUROSCI.5705-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhou D, Srivastava R, Nessler S, Grummel V, Sommer N, Brück W, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci USA 2006, 103, 19057–62. doi: 10.1073/pnas.0607242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Peschl P, Schanda K, Zeka B, Given K, Böhm D, Ruprecht K, et al. Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation 2017, 14, 208. doi: 10.1186/s12974-017-0984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Blauth K, Soltys J, Matschulat A, Reiter CR, Ritchie A, Baird NL, et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid cause demyelination of spinal cord explants. Acta Neuropathol 2015, 130, 765–81. doi: 10.1007/s00401-015-1500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liu Y, Given KS, Harlow DE, Matschulat AM, Macklin WB, Bennett JL, et al. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol Commun 2017, 5, 25. doi: 10.1186/s40478-017-0428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. von Büdingen HC, Harrer MD, Kuenzle S, Meier M, Goebels N.. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. Eur J Immunol 2008, 38, 2014–23. doi: 10.1002/eji.200737784. [DOI] [PubMed] [Google Scholar]
- 104. Bajramović JJ, Lassmann H, van Noort JM.. Expression of alpha B-crystallin in glia cells during lesional development in multiple sclerosis. J Neuroimmunol 1997, 78, 143–51. doi: 10.1016/s0165-5728(97)00092-1. [DOI] [PubMed] [Google Scholar]
- 105. Boelens WC. Cell biological roles of αB-crystallin. Prog Biophys Mol Biol 2014, 115, 3–10. doi: 10.1016/j.pbiomolbio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 106. Kim JY, Kim CH, Lee EY, Seo JH.. Alpha B-crystallin overexpression protects oligodendrocyte precursor cells against oxidative stress-induced apoptosis through the Akt pathway. J Mol Neurosci 2020, 70, 751–8. doi: 10.1007/s12031-020-01485-z. [DOI] [PubMed] [Google Scholar]
- 107. Guo YS, Liang PZ, Lu SZ, Chen R, Yin YQ, Zhou JW.. Extracellular αB-crystallin modulates the inflammatory responses. Biochem Biophys Res Commun 2019, 508, 282–8. doi: 10.1016/j.bbrc.2018.11.024. [DOI] [PubMed] [Google Scholar]
- 108. Reddy VS, Madala SK, Trinath J, Reddy GB.. Extracellular small heat shock proteins: exosomal biogenesis and function. Cell Stress Chaperones 2018, 23, 441–54. doi: 10.1007/s12192-017-0856-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci 2011, 124, 447–58. doi: 10.1242/jcs.074088. [DOI] [PubMed] [Google Scholar]
- 110. van Noort JM, Bsibsi M, Nacken PJ, Gerritsen WH, Amor S, Holtman IR, et al. Activation of an immune-regulatory macrophage response and inhibition of lung inflammation in a mouse model of COPD using heat-shock protein alpha B-crystallin-loaded PLGA microparticles. Biomaterials 2013, 34, 831–40. doi: 10.1016/j.biomaterials.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 111. Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJ, Boddeke E, van der Valk P, et al. Alpha-B-crystallin induces an immune-regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 2013, 72, 970–9. doi: 10.1097/NEN.0b013e3182a776bf. [DOI] [PubMed] [Google Scholar]
- 112. Holtman IR, Bsibsi M, Gerritsen WH, Boddeke HW, Eggen BJ, van der Valk P, et al. Identification of highly connected hub genes in the protective response program of human macrophages and microglia activated by alpha B-crystallin. Glia 2017, 65, 460–73. doi: 10.1002/glia.23104. [DOI] [PubMed] [Google Scholar]
- 113. Zhu Z, Reiser G.. The small heat shock proteins, especially HspB4 and HspB5 are promising protectants in neurodegenerative diseases. Neurochem Int 2018, 115, 69–79. doi: 10.1016/j.neuint.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 114. Hayashi J, Carver JA.. The multifaceted nature of αB-crystallin. Cell Stress Chaperones 2020, 25, 639–54. doi: 10.1007/s12192-020-01098-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Inoue R, Takata T, Fujii N, Ishii K, Uchiyama S, Sato N, et al. New insight into the dynamical system of αB-crystallin oligomers. Sci Rep 2016, 6, 29208. doi: 10.1038/srep29208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. De Groot CJ, Bergers E, Kamphorst W, Ravid R, Polman CH, Barkhof F, et al. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: increased yield of active demyelinating and (p)reactive lesions. Brain 2001, 124, 1635–45. doi: 10.1093/brain/124.8.1635. [DOI] [PubMed] [Google Scholar]
- 117. van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation 2012, 9, 156. doi: 10.1186/1742-2094-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. van der Valk P, Amor S.. Preactive lesions in multiple sclerosis. Curr Opin Neurol 2009, 22, 207–13. doi: 10.1097/WCO.0b013e32832b4c76. [DOI] [PubMed] [Google Scholar]
- 119. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W.. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013, 125, 595–608. doi: 10.1007/s00401-013-1082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Henderson AD, Barnett MH, Parratt JDE, Prineas JW.. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009, 66, 739–53. doi: 10.1002/ana.21800. [DOI] [PubMed] [Google Scholar]
- 121. Sato F, Martinez NE, Stewart EC, Omura S, Alexander JS, Tsunoda I.. “Microglial nodules” and “newly forming lesions” may be a Janus face of early MS lesions; implications from virus-induced demyelination, the inside-out model. BMC Neurol 2015, 15, 219. doi: 10.1186/s12883-015-0478-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. van Noort JM, van den Elsen PJ, van Horssen J, Geurts JJ, van der Valk P, Amor S.. Preactive multiple sclerosis lesions offer novel clues for neuroprotective therapeutic strategies. CNS Neurol Disord Drug Targets 2011, 10, 68–81. doi: 10.2174/187152711794488566. [DOI] [PubMed] [Google Scholar]
- 123. Muñoz U, Sebal C, Escudero E, Esiri M, Tzartos J, Sloan C, et al. Main role of antibodies in demyelination and axonal damage in multiple sclerosis. Cell Mol Neurobiol 2022, 42, 1809–27. doi: 10.1007/s10571-021-01059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N.. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol 2003, 13, 554–73. doi: 10.1111/j.1750-3639.2003.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hu Q, Wu G, Wang R, Ma H, Zhang Z, Xue Q.. Cutting edges and therapeutic opportunities on tumor-associated macrophages in lung cancer. Front Immunol 2022, 13, 1007812. doi: 10.3389/fimmu.2022.1007812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Savarin C, Bergmann CC.. Fine tuning the cytokine storm by IFN and IL-10 following neurotropic coronavirus encephalomyelitis. Front Immunol 2018, 9, 3022. doi: 10.3389/fimmu.2018.03022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Itoh K, Hirohata S.. The role of IL-10 in human B-cell activation, proliferation, and differentiation. J Immunol 1995, 154, 4341–50. [PubMed] [Google Scholar]
- 128. Nakashima I, Fujihara K, Misu T, Okita N, Takase S, Itoyama Y.. Significant correlation between IL-10 levels and IgG indices in the cerebrospinal fluid of patients with multiple sclerosis. Neuroimmunol 2000, 111, 64–7. [DOI] [PubMed] [Google Scholar]
- 129. Talbot J, Højsgaard Chow H, Mahler M, Buhelt S, Holm Hansen R, Lundell H, et al. Relationship between cerebrospinal fluid biomarkers of inflammation and tissue damage in primary progressive multiple sclerosis. Mult Scler Relat Disord 2022, 68, 104209. doi: 10.1016/j.msard.2022.104209. [DOI] [PubMed] [Google Scholar]
- 130. Heng AHS, Han CW, Abbott C, McColl SR, Comerford I.. Chemokine-driven migration of pro-inflammatory CD4+ T cells in CNS autoimmune disease. Front Immunol 2022, 13, 817473. doi: 10.3389/fimmu.2022.817473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE.. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol 2013, 87, 2420–9. doi: 10.1128/JVI.01769-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001, 294, 1731–5. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 133. Katz JB, Muller AJ, Prendergast GC.. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev 2008, 222, 206–21. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 134. Burke KP, Patterson DG, Liang D, Sharpe AH.. Immune checkpoint receptors in autoimmunity. Curr Opin Immunol 2023, 80, 102283. doi: 10.1016/j.coi.2023.102283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun 2021, 12, 3236. doi: 10.1038/s41467-021-23331-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jain RW, Yong W.. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol 2022, 22, 513–24. doi: 10.1038/s41577-021-00652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Baker D, Jacobs BM, Gnanapavan S, Schmierer K, Giovannoni G.. Plasma cell and B cell-targeted treatments for use in advanced multiple sclerosis. Mult Scler Relat Disord 2019, 35, 19–25. doi: 10.1016/j.msard.2019.06.030. [DOI] [PubMed] [Google Scholar]
- 138. Hu X, Chakravarty SD, Ivashkiv LB.. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev 2008, 226, 41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity 2013, 39, 454–69. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kann O, Almouhanna F, Chausse B.. Interferon γ: a master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci 2022, 45, 913–27. doi: 10.1016/j.tins.2022.10.007. [DOI] [PubMed] [Google Scholar]
- 141. Bsibsi M, Peferoen LA, Holtman IR, Nacken PJ, Gerritsen WH, Witte ME, et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-γ and alpha B-crystallin. Acta Neuropathol 2014, 128, 215–29. doi: 10.1007/s00401-014-1317-8. [DOI] [PubMed] [Google Scholar]
- 142. Sørensen TL, Trebst C, Kivisäkk P, Klaege KL, Majmudar A, Ravid R, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol 2002, 127, 59–68. doi: 10.1016/s0165-5728(02)00097-8. [DOI] [PubMed] [Google Scholar]
- 143. Lai YH, Mosmann TR.. Mouse IL-13 enhances antibody production in vivo and acts directly on B cells in vitro to increase survival and hence antibody production. J Immunol 1999, 162, 78–87. [PubMed] [Google Scholar]
- 144. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R, et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med 2009, 206, 69–78. doi: 10.1084/jem.20081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lightman SM, Peresie JL, Carlson LM, Holling GA, Honikel MM, Chavel CA, et al. Indoleamine 2,3-dioxygenase 1 is essential for sustaining durable antibody responses. Immunity 2021, 54, 2772–2783.e5. doi: 10.1016/j.immuni.2021.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Dale RC, Morovat A.. Interleukin-6 and oligoclonal IgG synthesis in children with acute disseminated encephalomyelitis. Neuropediatrics 2003, 34, 141–5. doi: 10.1055/s-2003-41281. [DOI] [PubMed] [Google Scholar]
- 147. Agrafiotis A, Dizerens R, Vincenti I, Wagner I, Kuhn R, Shlesinger D, et al. Persistent virus-specific and clonally expanded antibody-secreting cells respond to induced self-antigen in the CNS. Acta Neuropathol 2023, 145, 335–55. doi: 10.1007/s00401-023-02537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. David A, Crawford F, Garside P, Kappler JW, Marrack P, MacLeod M.. Tolerance induction in memory CD4 T cells requires two rounds of antigen-specific activation. Proc Natl Acad Sci USA 2014, 111, 7735–40. doi: 10.1073/pnas.1406218111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Akbar AN, Vukmanovic-Stejic M, Taams LS, Macallan DC.. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat Rev Immunol 2007, 7, 231–7. doi: 10.1038/nri2037. [DOI] [PubMed] [Google Scholar]
- 150. Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 2016, 351, 858–63. doi: 10.1126/science.aac5560. [DOI] [PubMed] [Google Scholar]
- 151. Kuhlmann T, Moccia M, Coetzee T, Cohen JA, Correale J, Graves J, et al.; International Advisory Committee on Clinical Trials in Multiple Sclerosis. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol 2023, 22, 78–88. doi: 10.1016/S1474-4422(22)00289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zuo M, Ramaglia V, Gommerman JL.. Age-related changes in multiple sclerosis and experimental autoimmune encephalomyelitis. Semin Immunol 2022, 59, 101631. doi: 10.1016/j.smim.2022.101631. [DOI] [PubMed] [Google Scholar]
- 153. Jensen SN, Cady NM, Shahi SK, Peterson SR, Gupta A, Gibson-Corley KN, et al. Isoflavone diet ameliorates experimental autoimmune encephalomyelitis through modulation of gut bacteria depleted in patients with multiple sclerosis. Sci Ad. 2021, 7, eabd4595. doi: 10.1126/sciadv.abd4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Reddy J, Waldner H, Zhang X, Illes Z, Wucherpfennig KW, Sobel RA, et al. Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J Immunol 2005, 175, 5591–5. doi: 10.4049/jimmunol.175.9.5591. [DOI] [PubMed] [Google Scholar]
- 155. Adzemovic MZ, Zeitelhofer M, Hochmeister S, Gustafsson SA, Jagodic M.. Efficacy of vitamin D in treating multiple sclerosis-like neuroinflammation depends on developmental stage. Exp Neurol 2013, 249, 39–48. doi: 10.1016/j.expneurol.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 156. La Flamme AC, Ruddenklau K, Bäckström BT.. Schistosomiasis decreases central nervous system inflammation and alters the progression of experimental autoimmune encephalomyelitis. Infect Immun 2003, 71, 4996–5004. doi: 10.1128/IAI.71.9.4996-5004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–89. doi: 10.1212/WNL.0000000000001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Marignier R, Hacohen Y, Cobo-Calvo A, Pröbstel AK, Aktas O, Alexopoulos H, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol 2021, 20, 762–72. doi: 10.1016/S1474-4422(21)00218-0. [DOI] [PubMed] [Google Scholar]
- 159. Sechi E, Cacciaguerra L, Chen JJ, Mariotto S, Fadda G, Dinoto A, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): a review of clinical and MRI features, diagnosis, and management. Front Neurol 2022, 13, 885218. doi: 10.3389/fneur.2022.885218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sechi E, Krecke KN, Messina SA, Buciuc M, Pittock SJ, Chen JJ, et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology 2021, 97, e1097–109. doi: 10.1212/WNL.0000000000012467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rempe T, Tarhan B, Rodriguez E, Viswanathan VT, Gyang TV, Carlson A, et al. Anti-MOG associated disorder—clinical and radiological characteristics compared to AQP4-IgG+ NMOSD-A single-center experience. Mult Scler Relat Disord 2021, 48, 102718. doi: 10.1016/j.msard.2020.102718. [DOI] [PubMed] [Google Scholar]
- 162. Benard-Seguin E, Costello F.. Optic neuritis: current challenges in diagnosis and management. Curr Opin Neurol 2023, 36, 10–8. doi: 10.1097/WCO.0000000000001128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data submitted is available.