

Abstract
Connective tissue diseases (CTDs) can be associated with various forms of pulmonary hypertension, including pulmonary arterial hypertension (PAH), pulmonary veno‐occlusive disease, pulmonary venous hypertension, interstitial lung disease‐associated pulmonary hypertension, chronic thromboembolic pulmonary hypertension, and sometimes a combination of several processes. The prevalence of PAH varies among the different CTDs, with systemic sclerosis (SSc) having the highest at 8%–12%. The most recent European Society of Cardiology/European Respiratory Society guidelines recommend routine annual screening for PAH in SSc and CTDs with SSc features. As CTDs can be associated with a myriad of presentations of pulmonary hypertension, a thorough evaluation to include a right heart catheterization to clearly delineate the hemodynamic profile is essential in developing an appropriate treatment plan. Treatment strategies will depend on the predominant phenotype of pulmonary vasculopathy. In general, management approach to CTD‐PAH mirrors that of idiopathic PAH. Despite this, outcomes of CTD‐PAH are inferior to those of idiopathic PAH, with those of SSc‐PAH being particularly poor. Reasons for this may include extrapulmonary manifestations of CTDs, including renal disease and gastrointestinal involvement, concurrent interstitial lung disease, and differences in the innate response of the right ventricle to increased pulmonary vascular resistance. Early referral for lung transplant evaluation of patients with CTD‐PAH, particularly SSc‐PAH, is recommended. It is hoped that in the near future, additional therapies may be added to the armamentarium of effective treatments for CTD‐PAH. Ultimately, a better understanding of the pathogenesis of CTD‐PAH will be required to develop targeted therapies for this morbid condition.
Keywords: connective tissue disease, pulmonary arterial hypertension, pulmonary hypertension, scleroderma, systemic lupus erythematosus
INTRODUCTION
Connective tissue diseases (CTDs) are a heterogeneous group of systemic autoimmune rheumatic diseases that can affect multiple organ systems. 1 The CTDs include multiple disorders, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), polymyositis/dermatomyositis (PM/DM), mixed connective tissue disease (MCTD), Sjogren's syndrome, and various systemic vasculitides. 2 These disorders are characterized by immune dysregulation and resultant disease‐specific autoantibody production. 1 CTDs frequently affect the cardiopulmonary system, with respiratory manifestations being the most common cause of death in many of these disorders. 2 Patients with CTD are at a substantially higher risk for the development of pulmonary hypertension (PH) than the general population, including pulmonary arterial hypertension (PAH), a rare progressive disorder that affects pre‐capillary vasculature and can lead to right heart failure and death. 3 Despite being included in Group 1 PAH classification, patients with CTD‐PH may have processes other than primary pulmonary vasculopathy contributing to the pathogenesis of disease, including interstitial lung disease (ILD), diastolic heart failure and chronic thromboembolic disease (CTEPH), which can affect approach to treatment. Furthermore, the histopathologic manifestations, prevalence and prognosis of PH, and approach to treatment differ amongst the various CTDs. Moreover, gastrointestinal involvement in some CTDs can impact the tolerability of pulmonary vasodilator therapy and impact treatment choices. In this review, we hope to shed light on the nuances of evaluating and treating CTD‐PH in all its various forms.
HOW IS PULMONARY HYPERTENSION DEFINED AND CATEGORIZED? WHERE DO CTDs FIT INTO THE CATEGORIZATION?
PH is defined and categorized based on hemodynamic profiles obtained by right heart catheterization (RHC). For many years, a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg was the threshold for diagnosing PH; however, in the last decade, this definition has evolved. 4 In 2018, the sixth World Symposium on Pulmonary Hypertension revised the definition of PH to a mPAP > 20 mmHg. 5 Additionally, a requirement of a pulmonary vascular resistance (PVR) ≥ 3 Wood units to establish a diagnosis of “pre‐capillary” PH was added. 5 More recently, the European Society of Cardiology (ESC)/European Respiratory Society (ERS) has released guidelines on the diagnosis and treatment of PH which have further modified the definition of pre‐capillary PH by decreasing the PVR threshold to ≥2 Wood units. 6 In addition, the ESC/ERS guidelines provide a hemodynamic definition for exercise‐induced PH (mPAP/cardiac output slope >3 mmHg/L/min from rest to exercise; Table 1). 6 Whether the upcoming seventh World Symposium on Pulmonary Hypertension will uphold this definition is unknown.
Table 1.
Updated hemodynamic definitions of pulmonary hypertension according to the 2022 ESC/ERS pulmonary hypertension guidelines.
| Definition | Characteristics |
|---|---|
| Pulmonary hypertension | mPAP > 20 mmHg |
| Pre‐capillary PH |
mPAP > 20 mmHg PVR > 2 WU PAWP ≤ 15 mmHg |
| Isolated post‐capillary PH |
mPAP > 20 mmHg PVR < 2 WU PAWP > 15 mmHg |
| Combined pre‐ and post‐capillary PH |
mPAP > 20 mmHg PVR > 2 WU PAWP > 15 mmHg |
| Exercise PH | mPAP/CO slope between rest and exercise > 3 mmHG/L/min |
Abbreviations: CO, cardiac output; ESC, European Society of Cardiology; ERS, European Respiratory Society; mPAP, mean pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; WU, Wood units.
The WHO organizes PH into five clinical subgroups in an attempt to combine conditions with similar pathophysiologic mechanisms, hemodynamic profiles, clinical presentations, and treatment strategies. 5 The categories are as follows: Group 1—PAH; Group 2—PH due to left heart disease (LHD); Group 3—PH due to lung diseases and/or hypoxia; Group 4—PH due to pulmonary artery obstructions; and Group 5—PH with unclear and/or multifactorial mechanisms. 5 Patients with CTDs can develop PH that falls into any of the five WHO Groups, although the propensity to cause a particular type of PH varies among individual CTDs. For instance, SSc is commonly associated with WHO Group 1 PAH, but can also be associated with PH due to LHD (Group 2), PH due to lung disease (Group 3), PH due to CTEPH (Group 4), or PH from end‐stage renal disease (Group 5), or a combination of above.
Although all forms of CTD‐PAH are lumped together in the WHO PH classification, it is worth recognizing that the individual conditions vary widely in their predilection to Group 1 PAH, propensity for concurrent ILD, response to immunosuppression, and prognosis. SSc, in particular, is phenotypically different from other CTDs. 7
One could reasonably argue that SSc‐PAH merits a dedicated category of its own within the WHO definition.
IS CTD‐PAH COMMON? WHAT IS THE PREVALENCE OF PAH AMONGST THE VARIOUS CTDS?
PAH is a relatively rare disorder, with an estimated prevalence of 5–15 cases per one million adults. 8 , 9 , 10 Amongst this rare disorder, CTD‐PAH is commonly encountered, however, accounting for approximately one quarter of cases. 8 , 10 PAH may develop in any of the CTDs, but the prevalence appears to vary widely between disorders. Data regarding the prevalence of PAH development in the various CTDs is limited. Estimates vary substantially depending on the specific population studied, the duration of time they were followed, and whether RHC was required for diagnosis. SSc accounts for the majority of cases of CTD‐PAH in Europe and North America despite the lower prevalence of SSc compared to other CTDs. PAH is estimated to occur in 8‐12% of patients with SSc. 11 , 12 , 13 , 14 Patients with limited cutaneous SSc (lcSSc) have a higher incidence of PAH than those with diffuse cutaneous SSc (dcSSc). 15 In Asia, SLE‐PAH appears to be more common than SSc‐PAH. 15 This is in part due to a higher prevalence of SLE than SSc in Asia, but also seems to be related to a predilection of Asians with SLE to develop SLE‐PAH. 15 Estimates from Western countries report SLE‐PAH rates of approximately 0.1%, 7 , 16 , 17 which is in contrast to a rate of 2.06% observed in 11,735 SLE patients from an Asian registry. 14 Development of PAH is thought to be much less common in other CTDs. The best available data come from a national health insurance database in Taiwan which reported the development of PAH in 0.22% of 17,316 Sjogren's syndrome patients, 0.33% of 1811 myositis patients, and 0.04% of 32,296 RA patients. 14 MCTD patients were not included in this report; however, a nationwide multicenter cohort of patients with MCTD from Norway reported PAH in 3.4% of 147 patients. 18 Prevalence of PAH in MCTD is difficult to elucidate due to misclassification of patients with “overlap syndromes” as MCTD. Patients with MCTD with prominent features of SSc have a higher prevalence of PAH than “pure” ribonucleoprotein (RNP)‐positive MCTD. 15 How the new definition of PAH will affect the prevalence of CTD‐PAH remains to be determined. Two cohort studies found that less than 2% of patients with SSc were reclassified as having PAH following the decrease in mPAP to ≥20 mmHg proposed by the sixth World Symposium. 19 , 20 However, a substantial number of patients in these studies would be reclassified as having PAH if the new ESC/ERS definition incorporating a PVR > 2 Wood units is applied. 17 , 18 Table 2 summarizes the epidemiology of PAH in the various CTDs.
Table 2.
Characteristics of various connective tissue diseases with regard to pulmonary hypertension.
| SSc | SLE | MCTD | Sjogren's | Myositis | RA | |
|---|---|---|---|---|---|---|
|
Estimated prevalence of RHC confirmed PAH |
8%–14% | 0.1%–2% (Higher in Asian cohorts) | 1%–3% | 0.2% | 0.3% | 0.04% |
| Other associated WHO PH groups |
1 (PVOD) 2, 3, 4, 5 |
2, 3, 4, 5 | 3 | 3 | 2, 3 | 2, 3 |
| Predilection to ILD |
lSSC: + dSSC: ++ |
+ | ++ | ++ | +++ | + |
Abbreviations: dSSc, diffuse systemic sclerosis; ILD, interstitial lung disease; lSSC, limited systemic sclerosis; MCTD, mixed connective tissue disease; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; PVOD, pulmonary veno‐occlusive disease; RA, rheumatoid arthritis; SS, Sjogren's syndrome; SLE, systemic lupus erythematosus; SSc, systemic sclerosis; WHO, World Health Organization.
IS THE PROGNOSIS OF CTD‐PAH DIFFERENT FROM IDIOPATHIC PAH? IF SO, WHY?
Numerous studies have established that CTD‐PAH is associated with a worse prognosis than idiopathic PAH (iPAH). 7 , 16 , 21 Patients with SSc constitute a majority of CTD patients included in several drug trials and registries. An analysis of the REVEAL registry compared the outcomes of 1251 patients with iPAH to 641 patients with CTD‐PAH. 7 Despite having more favorable hemodynamic profiles, patients with CTD‐PAH had lower 6‐min walk test distances, higher B‐type natriuretic peptide levels, and lower diffusing capacity of carbon monoxide (DLCO) levels. One‐year survival and freedom from hospitalization were significantly lower in the CTD‐PAH population than in the iPAH population (86% vs. 93%, p < 0.0001; 67% vs. 73%, p < 0.03, respectively). 7 A recent meta‐analysis compared outcomes in CTD‐PAH versus iPAH from 11 randomized controlled trials of pulmonary vasodilator therapy. 22 The authors found that while pulmonary vasodilator therapy was effective in improving outcomes in CTD‐PAH patients, survival in CTD‐PAH remained inferior to iPAH (62% vs. 72% 3‐year survival). Despite the inferior outcomes noted in CTD‐PAH patients, it was encouraging to see that CTD‐PAH patients from trials after 2010 had improved outcomes. 22 A similar analysis of RCT data found patients with CTD‐PAH treated with pulmonary vasodilator therapy had less improvement in 6MWT distance and a higher rate of clinical worsening compared to iPAH patients. 23
Among the various CTDs, SSc is consistently associated with the worst outcomes. Data from the REVEAL registry demonstrated a 1‐year survival rate of only 82% for SSc‐PAH compared to 94% for SLE‐PAH and 96% for RA‐PAH. 7 Epidemiologic data from a cohort of Taiwanese patients demonstrated inferior 5‐year survival in SSc‐PAH (61.9%) compared to SLE‐PAH (69.4%) and Sjogren's‐PAH (80.6%). 14 Outcomes of SLE‐PAH vary regionally with better outcomes in Western compared to Asian cohorts. 14 , 17 The lone exception to the superior outcomes of other CTDs in comparison to SSc may be MCTD which was associated with a 1‐year survival of 88% in the REVEAL registry, similar to the 1‐year survival of 82% in SSc. 7 Additionally, data from the United Kingdom registry showed poor 3‐year survival of 63% for MCTD‐PAH, which is markedly worse than other forms of CTD‐PAH. 16
PAH is not the only form of PH associated with adverse outcomes. Bourji and colleagues reported that patients with SSc who had WHO Group 2 PH due to heart failure with a preserved ejection fraction (PH‐HFpEF) had a similar median survival to patients with SSc‐PAH. 24 Furthermore, this group of patients had a twofold increased risk of death when controlling for hemodynamics. Dong et al. found similar results among a group of 202 patients with different types of CTDs, of whom only approximately 10% had SSc. CTD patients with WHO Group 2 PH had worse short‐term survival but comparable 5‐year survival to those with CTD‐PAH. 25 WHO Group 3 PH due to ILD is also associated with poor outcomes. In fact, patients with PH associated with severe SSc‐ILD have a dismal 3‐year survival rate of 39%, which is worse than patients with SSc‐PAH without ILD. 11 A retrospective study of 75 patients with various forms of CTD‐ILD found PH to be the most important predictor of mortality (odds ratio [OR] 14.4, p = 0.006). 26
The reason for the disparity in outcomes between CTD‐PAH and iPAH is incompletely understood but many potential reasons have been proposed. Perhaps extrapulmonary manifestations of CTD, including gastrointestinal disease and renal insufficiency, contribute to morbidity or lead to decreased tolerance of PAH‐targeted therapy. Concurrent ILD may also have impacted outcomes in the CTD‐PAH population as it is well associated with reduced survival. 11 Pulmonary veno‐occlusive disease (PVOD) can complicate various forms of CTD and is commonly reported in autopsy data from SSc‐PAH patients. 27 Additionally, mixed pre‐ and post‐capillary PH due to HFpEF can complicate CTDs and affect response to pulmonary vasodilator therapy. 24 While the above factors may contribute in part to the inferior outcomes observed in CTD‐PAH, it seems likely that phenomena inherent to CTD are also at play. It has been hypothesized that patients with CTD‐PAH have reduced endothelial metabolic function and a reduced functional capillary surface area comparison to iPAH patients. 28 There also seem to be differences in RV adaptation to increases in pulmonary vascular resistance. Despite controlling for confounders, N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) levels are higher in SSc‐PAH compared with iPAH. 29 In fact, functional and structural alterations of the RV, including collagen deposition and fibrosis, are observed in patients with SSc even before the onset of PAH. 30 In summary, there are multiple potential reasons for the increased mortality observed in CTD‐PAH, some of which are attributable to differences in the pulmonary vascular and right ventricular response to pulmonary hypertension.
WHAT CAUSES PULMONARY HYPERTENSION TO DEVELOP IN CTDS?
The mechanisms leading to the development of PH in CTDs are complex and incompletely understood. As in iPAH, endothelial dysfunction results in reduced production of pulmonary vasodilators, increased production of pulmonary vasoconstrictors, and upregulation of proliferative mediators which increase vascular tone and cause remodeling of vessels. 31 Inflammation and autoimmunity likely contribute as well; this is supported by a histopathologic study that demonstrated deposition of immunoglobulins and complement in pulmonary arterial walls from patients with SLE‐PAH. 32 Immune dysregulation due to diminished regulatory T‐cell activity may lead to the activation of autoreactive B and T cells with resultant pathogenic autoantibody production. 33 The role of inflammation appears to be less important in the pathogenesis of SSc‐PAH and possibly MCTD‐PAH. Biopsy specimens from these conditions show fibrinous intimal thickening of medium‐sized arteries and small vessels. This is in contrast to SLE‐PAH, which is characterized by plexiform lesions similar to those found in iPAH. 32 These differences in the degree of inflammation seen in SSc‐PAH may explain why immunosuppressive therapies have shown little success in SSc‐PAH. Genetic abnormalities appear to be less common in CTD‐PAH than in iPAH; however, they may have a role in some cases. In a recent analysis of 79 patients with CTD‐PAH assessed for a PAH‐specific panel of 35 genes, abnormalities were found in 9 patients (11.4%). 34 LV dysfunction, which commonly occurs in CTDs, may lead to pulmonary venous hypertension. Pulmonary veno‐occlusive lesions have been described in a variety of CTDs and are fairly commonly encountered in autopsies of SSc‐PAH patients. 27 Finally, ILD may result in obliteration of the pulmonary vascular bed and hypoxemic pulmonary vasoconstriction. It should be noted, however, that these mechanisms alone do not adequately explain the development of WHO Group 3 PH. It is likely that endothelial dysfunction, autoimmunity, and inflammation are integral to the development of Group 3 PH as well. 35
WHAT ARE THE CHARACTERISTICS OF THE VARIOUS CTDS? WHAT TYPES OF PH COMMONLY MANIFEST IN EACH?
Systemic sclerosis or scleroderma
SSc is a systemic autoimmune rheumatic disease characterized by fibrosis, vasculopathy, and inflammation. It is classified into three subtypes based on the presence and extent of skin involvement. 36 Patients with lcSSchave skin thickening of the fingers, and in some cases, the face and distal extremities. Patients with dcSSc by definition have skin thickening that involves the upper arms, thighs, chest, and/or abdomen. They also commonly have skin thickening distal to the elbows and distal to the knees. Facial skin thickening can be present in both lcSSc and dcSSc. SSc patients without skin thickening are classified as “scleroderma sine scleroderma.” 36 Raynaud phenomenon affects greater than 95% of SSc patients and can precede the development of other SSc manifestations by months to years. 37 SSc can also “overlap” with other rheumatologic diseases, including SLE, RA, myositis, and Sjogren's. ILD is most common in patients with the diffuse cutaneous subtype and in those who are positive for the anti‐topoisomerase I antibody (also known as the anti‐Scl‐70 antibody). Some degree of ILD is present in greater than 50% of patients with SSc, but only 30% develop progressive pulmonary fibrosis (PPF). 38 PAH is more commonly associated with the limited cutaneous subtype than the diffuse cutaneous subtype and is more likely to develop in those who are positive for the anti‐centromere antibody; however, PAH can develop in both subtypes and in those who are negative for the anti‐centromere antibody. 36
Numerous etiologies can lead to the development of PH in SSc. In fact, all five WHO PH groups have been implicated in the development of SSc‐PH. The epidemiology of WHO Group 1 PAH has been discussed extensively above. PVOD is a rare form of PAH caused by occlusive thickening of pulmonary veins and venules. PVOD lesions have been described in up to 75% of autopsies from patients with SSc‐PAH, but overt, clinically manifest PVOD is much less common. 27 Despite the identical hemodynamic profile between PVOD and PAH, patients with PVOD develop hypoxemia due to pulmonary edema when treated with pulmonary vasodilator therapy. PVOD should be considered in any patient with suspected SSc‐PAH who clinically deteriorates when pulmonary vasodilator therapy is initiated. Given the poor prognosis of SSC‐PVOD, early lung transplant referral in this population is warranted. 39
Cardiac involvement is common in SSc, with an estimated prevalence of clinically overt disease of 7%–39%. 40 , 41 Myocardial disease may develop from SSc‐related fibrosis or microvascular disease as well as from traditional risk factors, including coronary artery disease and systemic hypertension. 39 Echocardiographic studies found that approximately 25% of SSc patients had diastolic dysfunction and approximately 5% had a reduced ejection fraction. 42 , 43 Avouac and colleagues performed RHC on 206 patients with suspected SSc‐PH. Among 83 patients with hemodynamically confirmed PH, 17 (8.3%) had Group 2 disease from LHD. 44 It should be noted that patients with SSc‐PAH may have concurrent PH‐LHD. A retrospective study comparing SSc‐PAH to iPAH found concurrent diastolic dysfunction in 33% of SSc‐PAH patients compared to 10% of iPAH patients. 45 SSc‐PH‐LHD may also masquerade as SSc‐PAH. Fox et al. found that following a provocative fluid challenge during RHC, 11 of the 29 patients originally diagnosed with SSc‐PAH were reclassified as SSc‐PH‐LHD. 46
PH‐ILD is also frequently encountered in the SSc population. As mentioned previously, the majority of patients with SSc have some degree of ILD and 30% go on to develop PPF. 38 Risk factors for development of SSc‐ILD include male sex, older age at disease onset, Black race, anti‐topoisomerase‐I antibody positivity, and the diffuse cutaneous subtype. 39 Differentiation between SSc‐PAH and SSc‐PH‐ILD is somewhat arbitrary as both disease states have precapillary hemodynamic profiles. The prevalence of SSc‐PH‐ILD is poorly characterized. One large cohort study found that of 93 patients with SSc‐ILD, 29 (31.2%) had RHC‐proven PH. 47 SSc‐PH‐ILD is associated with very poor survival. A meta‐analysis reported a 3‐year survival of only 35% for SSc‐PH‐ILD compared to 56% in SSc‐PAH. 48
CTEPH, that is, WHO Group 4 PH, can develop in patients with SSc and should be excluded in all patients with SSc‐PH as part of their evaluation. Patients with scleroderma renal crisis can progress to end‐stage renal disease and develop WHO Group 5 PH. Both processes are relatively uncommon but round out the myriad potential etiologies of PH complicating SSc.
Systemic lupus erythematosus
SLE is a systemic autoimmune rheumatic disease that is associated with multiple autoantibodies, can have variable clinical presentations, and can affect virtually any organ system. Constitutional symptoms, arthralgia and arthritis, and rash are common. Diagnosis can be challenging given the myriad and protean clinical manifestations which can be present. The 2019 European Alliance of Associations of Rheumatology (EULAR)/American College of Rheumatology (ACR) classification criteria require the presence of a positive antinuclear antibody (ANA) in combination with weighted clinical domains to establish a diagnosis. 49 Although pleural involvement in SLE is frequent, other forms of lung involvement, such as acute pneumonitis, ILD, PH, shrinking lung, and hemorrhage, are much less common. 50 Much like SSc, PH can manifest in SLE in a variety of ways that span the five WHO PH groups. As mentioned earlier in the manuscript, SLE‐PAH can be observed—albeit rarely—in Western populations and is more frequent in Asian populations. 7 , 14 , 16 , 17 Not surprisingly, the development of SLE‐PAH is associated with decreased survival in comparison to SLE patients without PAH. 17 , 51
Cardiovascular disease commonly complicates SLE and may lead to WHO Group 2 PH. Atherosclerotic coronary artery disease, myocarditis, pericarditis, valvular disease, and heart failure can all be observed. 52 The estimated prevalence of heart failure in SLE ranges from 1% to 10%. 53 , 54 , 55 SLE‐associated ILD occurs in 3%–9% of patients. 50 Risk factors for the development of ILD include older age, longer duration of disease, RNP positivity, and clinical overlap with features of SSc including Raynaud phenomenon, abnormal nailfold capillaries, and sclerodactyly. 50 , 56 , 57 SLE‐PH‐ILD can develop, but the incidence of this is not well known. CTEPH (WHO Group 4) may occur in patients with secondary antiphospholipid antibody syndrome, which markedly increases the predilection to thromboembolism. Patients with SLE‐PH with or without secondary antiphospholipid antibody syndrome should be screened for CTEPH. Testing for antiphospholipid antibody syndrome should be checked for in all patients with unprovoked venous thromboembolism or CTEPH. 58 Finally, SLE patients can develop lupus nephritis which can result in end‐stage renal disease that could lead to development of WHO Group 5 PH.
Mixed connective tissue disease
MCTD is a systemic autoimmune rheumatic disease characterized by a positive U1‐RNP antibody, some combination of Raynaud phenomenon, puffy hands, sclerodactyly, synovitis, and myositis, and overlapping features of SLE, SSc, RA, and idiopathic inflammatory myopathy. 59 Given the variable presentation as well as multiple existing and somewhat contradictory diagnostic criteria, the diagnosis can be elusive. 60 , 61 It should be noted while MCTD may have features of SLE, SSc, RA, or idiopathic inflammatory myopathy, it is a distinct clinical entity. Nearly any organ system may be involved, although severe renal disease and severe central nervous system involvement are uncommon. 62 Joint involvement is common and may be erosive. 63 Esophageal dysmotility is also frequently present. 64 As mentioned above, large nationwide series report that MCTD‐PAH is relatively uncommon with an estimated prevalence of around 3%. 18 Other series estimate it to be more common with longer follow‐up time. For example, a cohort study from Hungary found that 18% of patients with MCTD developed PAH after 14.5 years of follow‐up. 65 PH is an important cause of mortality in patients with, although studies have shown that patients with CTD‐PAH who are U1‐RNP positive have better survival than those who are U1‐RNP negative. 66 , 67 ILD commonly complicates MCTD with an estimated 50%–70% having CT evidence of parenchymal lung involvement. 59 Patients with MCTD‐ILD may develop Group 3 PH, although the prevalence is poorly defined. Symptomatic cardiac disease can develop in approximately one third of MCTD patients. 68 Rates of diastolic dysfunction in patients with MCTD are higher than in age‐ and sex‐matched controls, so Group 2 PH is a potential complication but so far has not been frequently reported. 68
Sjogren's syndrome
Sjogren's is a systemic autoimmune rheumatic disease characterized by ocular and oral dryness due to lacrimal and salivary gland dysfunction, respectively. 69 Sjogren's may occur as a primary disorder or secondary to another CTD. In addition to experiencing keratoconjunctivitis sicca and xerostomia from exocrine gland involvement, patients may also have symptoms related to extraglandular organ involvement. 69 For example, they may develop arthralgias, xerosis with resultant pruritis, peripheral neuropathy, tubulointerstitial nephritis, and lymphoma. Cardiac involvement is rare. ILD occurs in approximately 20% of patients and is associated with substantial morbidity and mortality. 70 PH complicating Sjogren's primarily manifests as either PAH or Group 3 PH. Sjogren's‐PAH is rare, with a reported prevalence of <1%. 14 The prevalence of Group 3 PH in the context of Sjogren's‐ILD is not well known.
Polymyositis/dermatomyositis
Polymyositis and dermatomyositis are types of idiopathic inflammatory myopathies, a heterogeneous group of autoimmune disorders characterized by muscle inflammation. 71 , 72 , 73 Patients with dermatomyositis, unlike those with polymyositis, have skin involvement. In addition, some patients with dermatomyositis have no clinically apparent muscle involvement (so‐called clinically amyopathic dermatomyositis). There are several different myositis‐specific and myositis‐associated antibodies that have particular clinical associations. For example, the anti‐synthetase antibodies (Jo‐1, PL‐7, PL‐12, EJ, OJ, KS, Ha, and Zo) and anti‐MDA5 antibody are associated with ILD; anti‐SRP with treatment‐resistant necrotizing myopathy; and anti‐TIF‐1gamma with malignancy. 74 ILD occurs in up to 40% of patients with polymyositis or dermatomyositis and in some cases may be rapidly progressive. Group 3 PH can complicate myositis‐associated ILD, but the prevalence is poorly delineated. Based on the published literature PAH seems to be rare, and it is unclear what proportion of patients diagnosed with PAH actually instead have Group 3 PH due to ILD. 14 Approximately 10% of patients with idiopathic inflammatory myopathies develop clinically evident cardiac disease. 75 Both replacement cardiac fibrosis and myocarditis may occur and could potentially result in Group 2 PH from LHD. 75
Rheumatoid arthritis
RA is a systemic autoimmune rheumatic disease characterized by arthralgia, synovitis, and morning‐predominant stiffness in the joints. It is relatively common, affecting 1.0% of the adult US population. 76 Symmetric arthritis of the small joints of the hands, wrists, and feet is the typical pattern, although medium and large joints can be involved as well. Patients typically have elevated inflammatory markers as well as a positive rheumatoid factor and/or anti‐cyclic citrullinated peptide antibody. PH can manifest in RA in several ways. RA is associated with both ischemic heart disease and HFpEF and can therefore cause Group 2 PH. 77 ILD complicates between 7.7% and 67% of cases of RA and is a leading cause of mortality and the second leading cause of death in patients with RA. 78 Group 3 PH due to RA‐ILD is likely the most common cause of PH encountered in RA given the prevalence of ILD. PAH secondary to RA has been reported but the incidence is low. 14 , 79
SHOULD YOU SCREEN FOR PAH IN CTD? IF SO, HOW?
Given the varying risk for the development of PAH in CTD, recommendations on screening for PAH in CTD also vary based on the underlying disease process. The morbidity and mortality of PAH in SSc and SSc spectrum disorders (i.e., other CTDs with scleroderma features) is high and multiple studies have demonstrated that systematic screening in these patients has resulted in earlier diagnosis and improved survival. 6 , 80 , 81 , 82 The most recent WSPH and ESC/ERS guidelines recommend screening all asymptomatic patients with SSc spectrum diseases for PAH. 5 , 6
Several different screening algorithms have been studied, with most international guidelines recommending the use of the DETECT algorithm in patients with a diagnosis of SSc for >3 years, a DLCO > 60%, and an FVC ≥ 40%. The DETECT algorithm is a two‐step diagnostic algorithm in which the total score determines the need for RHC. Step 1 of DETECT incorporates FVC/DLCO ratio, anti‐centromere antibody status, presence/absence of telangiectasias, serum urate, NTproBNP, and right axis deviation on ECG to determine the need for an echocardiogram. Step 2 incorporates the echocardiographic variables of right atrial enlargement and TR jet velocity to then determine the need for RHC. The DETECT screening algorithm demonstrated a much lower false negative rate of PAH diagnosis of 4% compared to 29% with the use of the 2015 ESC/ERS guidelines alone. Notably, the DETECT study also demonstrated that the use of the TR jet velocity or presence of dyspnea could not reliably discriminate between the absence or presence of PAH in SSc patients. 83
Another screening algorithm was developed by the Australian Scleroderma Interest Group (ASIG) which utilizes NT‐proBNP, DLCO, and FVC/DLCO for determining the need for echocardiogram and RHC. 82 , 84 A comparison study by Hao et al. demonstrated that both the DETECT and ASIG algorithms had equivalent sensitivity and negative predictive value for detecting SSc‐PAH and both algorithms outperformed the 2009 ESC/ERS guidelines. 85 Although the simplicity of the ASIG algorithm is attractive, the decreased systematic use of RHC and the smaller size of the ASIG study cohort make it a less robustly validated tool compared to the DETECT algorithm. 86 As these DETECT and ASIG screening algorithms were validated using an mPAP cutoff of ≥25 and PVR > 3, it is unknown if they will remain effective in patients in the context of the recently updated diagnostic criteria. 6 , 87 Figure 1 shows the DETECT and ASIG algorithms.
Figure 1.
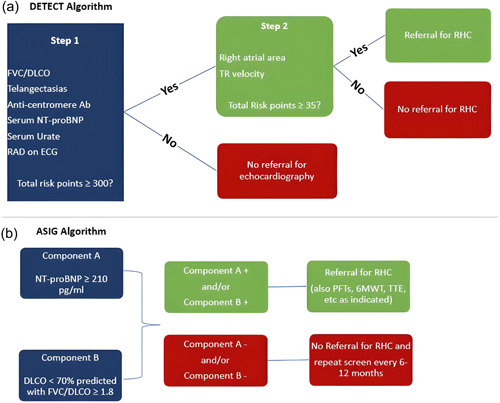
DETECT versus ASIG algorithms for screening for CTD‐PAH. ASIG, Australian Scleroderma Interest Group; CTD, connective tissue disease; PAH, pulmonary arterial hypertension.
A study by Huang et al. also identified several risk factors for PAH development in SLE including the presence of ILD, absence of a skin rash, presence of a pericardial effusion, positive anti‐SSA and anti‐RNP antibodies, low disease activity and ESR, and high serum uric acid. 88 MCTD accounts for the second most common form of CTD‐PAH; however, there remains little data on risk factors for the development of PAH in this population. More studies are needed to recommend screening in asymptomatic patients with forms of CTD other than SSc and SSc spectrum disease. Given the elevated risk of PH development in those with CTD‐ILD, it is our practice to also screen all patients with CTD‐ILD for the presence of PH. 89
HOW DO YOU DIAGNOSE PULMONARY HYPERTENSION IN PATIENTS WITH CTD? WHAT TESTS ARE RECOMMENDED?
The evaluation of PH in patients with CTD should focus on confirmation of the presence of PH, ascertainment of the dominant etiology and classification, and assessment of PH severity.
The initial evaluation should include a thorough physical examination and history, with a particular focus on identifying risk factors for pulmonary hypertension (e.g., family history of PH, methamphetamine use, VTE, etc). Additionally, the utilization of electrocardiogram (ECG), laboratory testing, echocardiogram, pulmonary function testing, and 6‐min walk tests may help identify, screen, and risk stratify patients with CTD‐PH.
Electrocardiogram
ECG is important to evaluate for dysrhythmias and ischemic heart disease; the presence of right atrial enlargement and right ventricular strain and hypertrophy may suggest the presence of PH. A small study of 61 PAH patients demonstrated that 87% had abnormal ECGs, most often demonstrating RV strain, hypertrophy, and right axis deviation. 90
Laboratory
Presuming the patient has already had autoantibody testing for CTD, additional diagnostic testing should include liver function tests, drug toxicology screening, basic metabolic panel, complete blood count, human immunodeficiency virus testing, and BNP/NT‐proBNP. Patients with certain CTDs are at a particularly elevated risk for VTE, so a hypercoagulability evaluation (e.g., antiphospholipid antibodies) should be considered if clinically indicated. 91 Although nonspecific biomarkers, BNP/NT‐proBNP are often elevated in patients with PH and correlate with disease severity and prognosis. In one study, BNP levels greater than 150 pg/mL at the time of diagnosis and persistent elevations greater than 180 pg/mL despite pulmonary vasodilatory therapy were associated with worse outcomes. 92 In patients with SSc specifically, an elevated NT‐proBNP (≥395 pg/mL) predicted the presence of PH (sensitivity of 56% and specificity of 95%) and correlated with increased mortality and PH severity. 29 , 93 , 94 , 95
Pulmonary function testing and 6‐min walk test
Pulmonary function testing should be performed in all patients with suspected PH and concomitant lung disease. An isolated reduction in the DLCO or DLCO decreased out of proportion to lung volumes or FVC may suggest the presence of PH. In patients with SSc, a ratio of FVC to DLCO > 1.6 or a DLCO < 60% of predicted was associated with an increased likelihood of PH. 96 , 97 The 6‐min walk test is another simple, easy‐to‐perform test with established prognostic significance in patients with PAH. 6 , 98 , 99 Patients with CTD frequently have extrapulmonary manifestations that may limit their walk distance (e.g., muscle weakness, arthralgias, and joint contractures), thus changes in walk distance in these patients should be interpreted in the appropriate clinical context. In CTD patients with stable ILD, a decrease in 6‐min walk distance, oxygen saturation nadir (SaO2), and heart rate response should all raise suspicion for the presence of PH. 89 Table 3 lists signs suggestive of development of PH in ILD patients.
Table 3.
Diagnostic signs of potential PH in ILD.
| Test | Finding |
|---|---|
| PFTs |
|
| 6MWT |
|
| CT Scanning |
|
| Echocardiography |
|
| Laboratory testing |
|
Abbreviations: 3‐D, 3‐dimensional; 6MWT, 6‐min walk test; BNP, brain natriuretic peptide; DLCO, ¼ diffusing capacity of the lung for carbon monoxide; ILD, interstitial lung disease; LV, left ventricular; PA:A, pulmonary artery to aorta; PFT, pulmonary function test; RV, right ventricular; RVOT, RV outflow tract; RVSP, RV systolic pressure; TAPSE, tricuspid annular plane systolic excursion.
Echocardiogram
Echocardiography remains the primary screening tool for suspected PH. Current guidelines recommend using an echocardiogram‐derived peak tricuspid regurgitation jet velocity (TRV) > 2.8 m/s, rather than an estimated systolic pulmonary artery pressure (sPAP), as the primary variable suggestive of PH. 6 This is due to the unreliability of estimated RA pressures via echocardiography. The TRV alone cannot reliably identify the presence or absence of PH, however contextualizing and combining it with risk factors for PH and additional echocardiographic assessments of the right heart, PA, and IVC may help further stratify the probability of PH (Table 4). 100 Attention should also be given to the presence of LV hypertrophy, diastolic dysfunction, and LA size to evaluate for the signs of Group 2 PH, especially in patients with risk factors such as obesity, systemic hypertension, obstructive sleep apnea and diabetes. This is particularly the case in patients with SSc, in which diastolic dysfunction is common. 101 , 102 In patients with a high probability of PH via echocardiogram, right heart catheterization should be pursued to confirm the diagnosis.
Table 4.
Additional echocardiographic signs of PH.
| Right Atrium and IVC |
|
| Right Ventricle |
|
| Pulmonary Artery |
|
| Pericardium |
|
Abbreviations: AcT, acceleration time; IVS, intraventricular septum; IVC, inferior vena cava; LV, left ventricle; PA, pulmonary artery; PR, pulmonary regurgitation; PW, pulse‐wave; RA, right atrium; RV, right ventricle; RVOT, right ventricular outflow tract; TAPSE, tricuspid annular plane systolic excursion.
Cardiac MR
Cardiac magnetic resonance (CMR) imaging has multiple advantages over echocardiography and its use for the assessment of PAH and RV dysfunction has increased over the years. In addition to its increased diagnostic accuracy for PH compared to echocardiography, it may be particularly useful in patients with CTD, who often have additional cardiac pathologies. 103 , 104 CMR allows for a more detailed evaluation of biventricular function, vessels, the myocardium, and the pericardium all in a single study and may additionally allow for the identification of early, subclinical disease. 105 , 106 Additionally, in both CTD‐PAH and PAH not due to CTD, CMR has been shown to be an effective tool for prognostication. 107 , 108 Despite its numerous advantages, the use of CMR remains limited by its availability, cost, and need for expert readers.
Right heart catheterization
The diagnosis of PH must ultimately be confirmed via right heart catheterization. In patients with suspected Group 2 PH or unexplained dyspnea with normal resting hemodynamics, exercise RHC or fluid loading should be considered. Exercise PH is now defined in the current guidelines as an mPAP/CO slope > 3 with a PAWP/CO slope < 2. 6 An elevated PAWP/CO slope (>2) and/or PAWP > 25 mmHg with supine exercise is diagnostic for the presence of HFpEF. Given variability in exercise protocols and lack of uniform availability of exercise testing, a fluid challenge during RHC is recommended in patients with suspected Group 2 PH who have a normal resting PAWP. A rise in PAWP ≥ 18 mmHg with rapid infusion of approximately 500 mL of intravenous fluids is suggestive of HFpEF. 6 If the accuracy of the PAWP is in question, a left ventricular end‐diastolic pressure (LVEDP) measurement should be obtained. Interestingly, a recent study demonstrated that an increase in PAWP to above 11 and 19 mmHg during passive leg raise (PLR) demonstrated a 100% sensitivity and specificity, respectively, for occult‐HFpEF. 109 In suspected HFpEF patients in whom exercise or fluid administration challenge cannot be performed, the use of PLR during RHC can be considered. 109
Imaging (CT/DECT/VQ/MRI)
Once the diagnosis of PH is confirmed, additional testing may be indicated to allow for accurate identification and classification of PH. Given the increased prevalence of venous thromboembolism and interstitial lung disease in patients with CTD, studies to evaluate for these should be performed in all patients with suspected or confirmed PH. 91 Ventilation‐perfusion scans remain the gold standard diagnostic test for CTEPH; however, newer imaging modalities such as dual‐energy CT, iodine subtraction mapping CT, and MRI perfusion scans appear promising and may be considered in the future. 6 Despite potential advantages over V/Q scans, these newer modalities lack large, multi‐center data and are often limited by availability and cost. High‐resolution chest CT scans should be performed in these patients to evaluate for possible interstitial lung disease and thus Group 3 PH.
Cardiopulmonary exercise testing
Cardiopulmonary exercise testing (CPET) allows for a robust assessment of functional capacity and the identification of the etiology of exercise limitation. A recent study by Pezzuto et al. demonstrated that the combination of peak VO2 and PETCO2 correlated with the degree of hemodynamic impairment in patients with PAH and multiple previous studies have demonstrated the prognostic utility of peak VO2 and VE/VCO2. 110 , 111 , 112 , 113 Notably, patients with CTD‐PH tend to perform worse on CPET compared to iPAH patients. In a study by Zhang et al., CTD‐PAH patients had more impaired ventilation, cardiac function, and muscular strength than iPAH patients, despite comparable hemodynamic profiles, WHO FC, NT‐proBNP, and 6MWD. 114 It remains unclear, however, if the routine use of CPET has much added value to the current diagnostic paradigm.
HOW SHOULD CTD‐PH BE TREATED?
Careful consideration of the hemodynamic profile of the patient and the presence or absence of ILD and other co‐morbidities is required when developing a treatment plan for CTD‐PH. Clinicians must also be aware that not all CTD patients will fit neatly into a specific WHO Group. Rather, many patients with CTD may also have features of Group 2, 3, or 4 PH as well.
WHAT IS THE ROLE FOR PULMONARY VASODILATOR TREATMENT IN CTD‐PAH?
The most current ESC/ERS guidelines include CTD‐PAH and iPAH under the same treatment umbrella protocol and use risk stratification to dictate the initial choice of drugs, with upfront dual therapy for patients in low‐ and intermediate‐risk status, and upfront triple therapy for patients in high‐risk status (Figure 2). 6 Escalation to triple drug therapy with a parenteral prostanoid is warranted in patients on dual therapy who either remain in the intermediate‐high risk group or progress into the high‐risk group at follow up assessment. 6 In patients who stratify as intermediate‐low risk at follow‐up, therapy augmentation with the addition of a nonparenteral prostanoid and/or a switch from PDE5i to a sGC stimulator is recommended. 6 Most large RCTs for PAH‐specific therapies included CTD‐PAH patients (20%–30%), and the relative response to therapy appears to be largely equivalent to those with iPAH. In a recent meta‐analysis of 11 RCTs and 19 registries, Khanna et al. found that patients with CTD‐PAH and iPAH had similar relative risk reduction in clinical morbidity/mortality events compared with control subjects; however, survival in CTD‐PAH patients was lower than in iPAH patients. 22 Efficacy of multiple pulmonary vasodilator medications in CTD‐PAH has been established in their respective clinical trials. A prospective study of upfront ambrisentan and tadalafil in SSc‐PAH by Hassoun et al. demonstrated significantly improved RV function, hemodynamics, 6MWD, and BNP. 115 Additionally, a sub‐group analysis of the landmark AMBITION study confirmed the benefit of upfront dual therapy with tadalafil and ambrisentan in CTD‐PAH patients. 116 In the GRIPHON trial, addition of selexipag to background treatment of one to two pulmonary vasodilator treatments demonstrated a similar reduction in composite morbidity/mortality of 40% and 41% in both the overall cohort and CTD‐PAH subgroup, respectively. 117 Moreover, the subgroup analysis of CTD‐PAH patients treated with selexipag versus placebo demonstrated a risk reduction of 44% and 35% in SSc‐PAH and SLE‐PAH patients, respectively.
Figure 2.
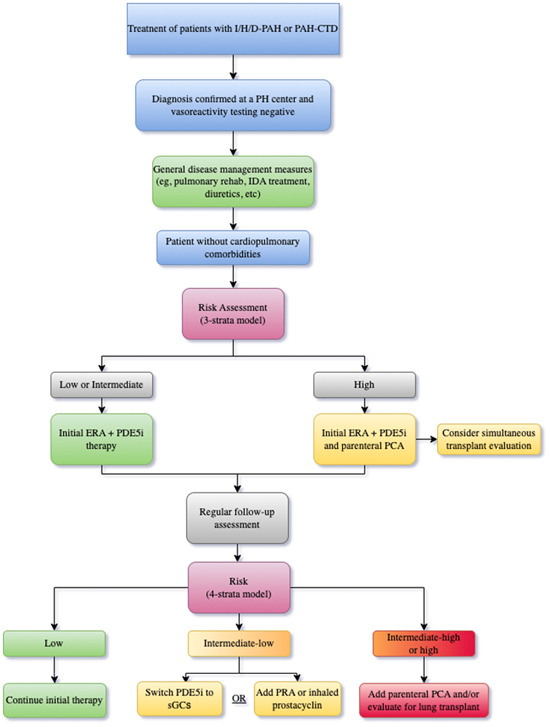
Recommended treatment algorithm for PAH from the 2022 ESC/ERS guidelines. ESC/ERS, European Society of Cardiology/European Respiratory Society; PAH, pulmonary arterial hypertension.
WHAT ARE POTENTIAL PITFALLS WHEN USING PULMONARY VASODILATORS IN PATIENTS WITH CTD‐PH?
In contrast to iPAH, CTD‐PAH presents several unique challenges given the potential for overlapping and evolving contributory PH pathologies (i.e., Groups 1, 2, 3, and 4) that often occur concurrently. As detailed earlier, patients with CTD‐PAH, particularly those with SSc and MCTD, have an increased prevalence of diastolic dysfunction as compared to iPAH. In these patients with combined pre‐/post‐capillary PH, the use of PAH‐targeted therapies may not be tolerated due to worsening pulmonary edema, dyspnea, and/or hypoxemia. Therefore, achieving optimal therapy for pre‐capillary PAH may be limited in some patients, even after volume optimization. Furthermore, patients with SSc‐PAH may have PVOD lesions that may result in pulmonary edema and worsening hypoxemia following initiation of pulmonary vasodilator therapy. Thus, careful phenotyping of patients with particular attention to the echocardiographic findings and other risk factors for HFpEF should be performed before treatment is initiated. Following therapy initiation, patients with CTD‐PH require close monitoring for unmasking of these complications.
DOES ONE APPROACH TREATMENT OF PH DIFFERENTLY IN PATIENTS WITH ILD?
The prevalence of ILD in CTD varies substantially depending on the underlying CTD, with reports of over 50% in patients with SSc. 47 , 118 Distinguishing between Group 3 PH and Group 1 PAH can be particularly challenging in patients with CTD who can present with various severities of PH and ILD, respectively (Table 5). Features favoring Group 3 PH include significant lung disease manifested by greater impairment of pulmonary function testing, extensive radiographic involvement, and milder hemodynamic impairment. Conversely, Group 1 PAH is suggested by milder underlying pulmonary disease and more severe hemodynamic impairment. To date, no firm diagnostic criteria exist that clearly delineate this distinction, and categorization of Group 3 versus Group 1 PH is left to the discretion of the clinician.
Table 5.
Factors favoring Group 1 versus Group 3 PH in connective tissue diseases.
| Favors Group 1 maladaptive phenotype | Favors Group 3 adaptive phenotype | |
|---|---|---|
| PFT |
|
|
| Imaging |
|
|
| Echocardiography |
|
|
| Hemodynamics |
|
|
Abbreviations: DLCO, Diffusing capacity of carbon monoxide; PAP, pulmonary artery pressure; PFT, Pulmonary function test; PH, Pulmonary hypertension; RV, Right ventricle.
Although it makes sense that treating the underlying lung disease should impact the trajectory of associated PH, this concept has not been studied in great depth. Given this, the authors advocate for appropriate treatment of the underlying parenchymal process. It is possible that in patients with ILD responsive to immunosuppression, treatment of the parenchymal process may result in improved pulmonary hemodynamics. Further study on this important topic is needed.
Until recently, there were no FDA‐approved therapies for Group 3 PH. Neither ERAs nor riociguat is recommended in treatment of patients with ILD‐PH due to safety concerns. 119 , 120 Although patients with CTD‐ILD were not included in these trials, it appears prudent to avoid these agents in patients with significant underlying ILD.
Clinical trial data for sildenafil in ILDs is somewhat mixed and primarily originates from studies in patients with idiopathic pulmonary fibrosis (IPF). 121 , 122 , 123 , 124 , 125 , 126 A few studies of sildenafil in IPF and fibrotic ILD with associated PH have shown clinical improvements in 6MWD and BNP/NT‐proBNP, but these data are limited by the lack of placebo arms and small sample sizes. The majority of these studies showed that sildenafil was well tolerated without negative effects on oxygenation or systemic blood pressure. 127
The INCREASE trial, which included 22% of patients with CTD‐ILD, provided data supporting the use of inhaled treprostinil for ILD‐PH. 128 The trial met its primary endpoint of change in 6MWD at week 16, as well as secondary endpoints including change in NT‐proBNP and time to clinical worsening. Post‐hoc analyses of the INCREASE trial have suggested that treatment with inhaled treprostinil has additional clinical benefits. One analysis demonstrated that all subgroups of patients, including those with CTD‐ILD, who received inhaled treprostinil were less likely to experience multiple disease progression events compared to those who received placebo. 128
Patients with mild ILD and significant PH are often treated according to the Group 1 treatment algorithm. 80 Patients with significant ILD present a bigger treatment challenge as they historically have been excluded from clinical trials. For patients with severe ILD and mild PH, based on the INCREASE data, our current approach is to use inhaled treprostinil as first‐line therapy. Although there are no data to support this practice, consideration can be given to the addition of sildenafil in patients with more severe hemodynamic impairment on an individual basis and following appropriate discussion with the patient. Similarly, patients with very severe PH and significant ILD can be managed with a combination of parenteral prostanoid therapy and sildenafil. It is recommended that pulmonary vasodilator therapy initiation be performed by experienced centers with careful observation of the patients to ensure safety.
IS THERE A ROLE FOR IMMUNOSUPPRESSION IN THE TREATMENT OF CTD‐PAH?
The use of immunosuppressive therapy for the treatment of CTD‐PAH is limited but may play a role in some patients. Although no randomized controlled trials have been performed in non‐SSc CTD‐PAH, limited data from small case series, observational studies, and registry analyses have demonstrated hemodynamic and clinical improvements with immunosuppressive therapies. 17 , 129 , 130 , 131 , 132 The role of immunosuppression in SSC‐PAH is less clear. No response has been demonstrated with corticosteroids or Cytoxan. The pathophysiologic differences in SSc‐PAH and other forms of CTD‐PAH likely account for the observed difference in response to immunosuppression. A recent study by Zamanian et al. evaluating the use of rituximab for SSc‐PAH failed to reach its primary endpoint of change in 6MWD at 24 weeks, but there was a trend toward improvement. 133 The authors posited that low levels of RF, IL‐2, and IL‐17 are predictive of response to rituximab. Based on the limited available data, the addition of immunosuppressive therapy (e.g., glucocorticoids, mycophenolate) as part of the PH treatment armamentarium can be considered in patients with non‐SSc CTD‐PAH, particularly if they have noncardiopulmonary manifestations which would benefit from treatment. The role of rituximab treatment for select patients with SSc‐PAH requires further study.
WHAT ADJUNCTIVE CARE SHOULD BE PROVIDED WHEN TREATING CTD‐PAH?
In addition to PAH‐targeted therapies, adjunctive supportive measures remain vital to the management of CTD‐PAH. The use of diuretics and salt/fluid restrictions are a cornerstone of PAH therapy to prevent volume overload and worsening right ventricular failure. Although there are no data on the benefit of long‐term oxygen use in PAH, we support the guideline recommendations for the utilization of supplemental oxygen when the PaO2 is <8 kPa (60 mmHg; alternatively, SaO2 < 92%) at rest or with exertion. Iron deficiency in PAH is associated with increased mortality and myocardial dysfunction; however, the long‐term clinical benefit of iron supplementation in PAH remains unclear. 134 , 135 , 136 , 137 As iron supplementation appears safe in PAH, the most recent ERS guidelines recommend repletion and regular monitoring of iron status in PAH patients with iron deficiency anemia. 138 , 139 , 140 In both iPAH and CTD‐PAH, the completion of a formal exercise program, such as pulmonary rehabilitation, was found to improve quality of life, functional capacity, and even pulmonary artery pressures. 141 , 142 The benefit of anticoagulation in PAH remains unclear and data on CTD‐PAH patients in particular are lacking. Of the available data, there appears to be particular harm with the use of anticoagulation in patients with SSc‐PAH. 143 , 144 As such, in the absence of other indications (e.g., atrial fibrillation, VTE, antiphospholipid antibody syndrome, etc.), the use of anticoagulation in CTD‐PAH is currently not recommended.
WHEN AND HOW SHOULD ONE REASSESS THE RESPONSE TO TREATMENT?
As touched upon in the treatment algorithm from the latest ESC/ERS guidelines, both initial and follow‐up risk assessments are fundamental for prognostication and guiding therapy decisions. Ongoing risk assessment allows clinicians to assess both treatment response as well as the possible need for therapy augmentation and/or lung transplant consideration. Numerous risk assessment tools available have demonstrated efficacy in PAH, including but not limited to REVEAL 2.0, REVEAL Lite 2.0, COMPERA 2.0, and the latest ESC/ERS guideline risk stratification model. 6 , 145 , 146 , 147 Based on the new COMPERA 2.0 model, the 2022 ESC/ERS models have moved from a three‐strata assessment to a more simplified four‐strata assessment at follow‐up, now dividing the intermediate‐risk strata into intermediate‐low and intermediate‐high risk categories (Figure 3). The current ESC/ERS guidelines continue to recommend the use of their three‐strata risk model at the initial assessment and the utilization of the simplified four‐risk strata model for follow‐up assessments. Data on the use of this new four‐strata risk assessment model from both the COMPERA and French PH registries demonstrated improved risk prediction, particularly for those in the intermediate risk category, compared to the previous three‐strata model. 145 , 146 The COMPERA 2.0 study additionally validated the four‐risk strata model in patients with CTD‐PAH. Although the external validation study of the COMPERA 2.0 model in the French PH registry did not specifically validate the model for any particular PAH sub‐groups, 27% of the cohort had CTD‐PAH. The REVEAL 2.0 risk calculator uses a score based on 13 weighted variables to categorize patients into either low, intermediate, or high‐risk strata. It has also been validated in PAH, in contrast to the aforementioned four‐risk strata models and assigns a negative prognostic value for CTD‐PAH in its model. 148 As previously mentioned, frequent risk reassessment is essential in the management of PAH; however, the need for invasively derived variables often limits the practicality and utility of the four‐risk strata and REVEAL 2.0 models at follow‐up. To that end, the REVEAL Lite 2.0 score uses only six noninvasive variables and has demonstrated good risk discrimination in both iPAH and CTD‐PAH with c‐indexes of 0.74 (95% confidence interval [CI], 0.71–0.77) and 0.76 (95% CI, 0.73–‐0.79), respectively. 147
Figure 3.
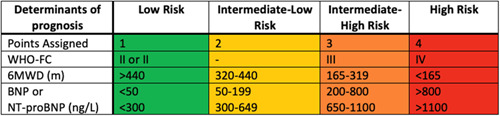
ESC/ERS PAH Guideline 4‐stratum risk assessment. ESC/ERS, European Society of Cardiology/European Respiratory Society; PAH, pulmonary arterial hypertension.
WHAT IS THE ROLE OF LUNG TRANSPLANTATION IN CTD‐PAH?
Lung transplantation remains an important therapeutic option in patients with severe CTD‐PAH and an insufficient response to medical therapy. In addition to an inadequate response to therapy, those with intermediate‐high or high‐risk of death (based on stratification tools), on parenteral prostanoid therapy, and/or rapidly progressive disease should undergo early referral for lung transplantation. 6 Given the unpredictable course of CTD‐PAH, early and timely identification of patients in need of referral for lung transplantation and bridging support therapies (i.e., ECMO) is critical, as delays in referral may result in poor outcomes. 149 , 150 Patients should have continued reassessments and further optimization of their CTD‐PAH while undergoing a transplant evaluation and while actively listed. We recommend that given the high preoperative mortality and complexity of management, CTD‐PAH transplant candidates should be referred to centers with a large volume of experience and expertise. Bilateral lung transplant remains the procedure of choice in patients with PAH, as right ventricular recovery occurs relatively rapidly after transplantation, even in those with severe RV dysfunction. 151 , 152 , 153 Heart‐ung transplantation is not usually indicated in patients with CTD‐PAH unless there is concurrent left heart dysfunction or congenital heart disease. The use of peri‐operative ECMO in severe PAH is associated with improved short‐ and long‐term outcomes. 154 , 155 , 156 , 157 In patients with severe CTD‐PAH who are not already being bridged with mechanical circulatory support, the initiation of awake ECMO before intubation and anesthetic induction should be considered.
CONCLUSION
PH can complicate the course of a variety of CTDs. While many only think of PAH in the context of CTDs, PH due to any of the WHO etiologic groups can occur. The most likely cause of PH can vary depending on the specific CTD. Development of an appropriate treatment plan for CTD‐PH requires consideration of numerous factors, including the specific CTD involved, the hemodynamic profile of the patient, and the presence of ILD. Despite CTD‐PAH being treated in a similar fashion to other causes of PAH, outcomes remain inferior. It is hoped that further study will improve our understanding of CTD‐PAH, allowing for improved outcomes in these complex conditions.
AUTHOR CONTRIBUTIONS
Vikramjit Khangoora, Christopher S. King, and Oksana A Shlobin: Development of concept, manuscript writing, editing.
CONFLICT OF INTEREST STATEMENT
Christopher S. King—Speaker: United Therapeutics, Actelion, Altavant; Consulting—UT, Merck. Oksana A Shlobin—Speaker B: UT, Janssen; Consulting: UT, Janssen, Aerovate, Aerami, Renzyvant, Merck; Educational: Total CME, Medscape. Vikramjit Khangoora—Consulting: Janssen. Elana J. Bernstein—Consulting: Boehringer Ingelheim; Contracted research: Boehringer Ingelheim, Pfizer, Kadmon.
ETHICS STATEMENT
The ethics statement is not available.
ACKNOWLEDGMENTS
EJB's work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant K23‐AR‐075112), the National Heart, Lung, and Blood Institute (grant R01‐HL‐164758), and the Department of Defense (grant W81XWH2210163).
Khangoora V, Bernstein EJ, King CS, Shlobin OA. Connective tissue disease‐associated pulmonary hypertension: a comprehensive review. Pulm Circ. 2023;13:e12276. 10.1002/pul2.12276
REFERENCES
- 1. Mulhearn B, Tansley SL, McHugh NJ. Autoantibodies in connective tissue disease. Best Pract Res Clin Rheumatol. 2020;34(1):101462. 10.1016/j.berh.2019.101462 [DOI] [PubMed] [Google Scholar]
- 2. Kondoh Y, Makino S, Ogura T, Suda T, Tomioka H, Amano H, Anraku M, Enomoto N, Fujii T, Fujisawa T, Gono T, Harigai M, Ichiyasu H, Inoue Y, Johkoh T, Kameda H, Kataoka K, Katsumata Y, Kawaguchi Y, Kawakami A, Kitamura H, Kitamura N, Koga T, Kurasawa K, Nakamura Y, Nakashima R, Nishioka Y, Nishiyama O, Okamoto M, Sakai F, Sakamoto S, Sato S, Shimizu T, Takayanagi N, Takei R, Takemura T, Takeuchi T, Toyoda Y, Yamada H, Yamakawa H, Yamano Y, Yamasaki Y, Kuwana M. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir Investig. 2021;59(6):709–740. 10.1016/j.resinv.2021.04.011 [DOI] [PubMed] [Google Scholar]
- 3. Yang X, Mardekian J, Sanders KN, Mychaskiw MA, Thomas J. Prevalence of pulmonary arterial hypertension in patients with connective tissue diseases: a systematic review of the literature. Clin Rheumatol. 2013;32(10):1519–1531. 10.1007/s10067-013-2307-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hatano S, Strasser T, World Health Organization . Primary Pulmonary Hypertension. Report on a WHO Meeting. World Health Organization. 1975.
- 5. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, Schwerzmann M, Dinh‐Xuan AT, Bush A, Abdelhamid M, Aboyans V, Arbustini E, Asteggiano R, Barberà JA, Beghetti M, Čelutkienė J, Cikes M, Condliffe R, de Man F, Falk V, Fauchier L, Gaine S, Galié N, Gin‐Sing W, Granton J, Grünig E, Hassoun PM, Hellemons M, Jaarsma T, Kjellström B, Klok FA, Konradi A, Koskinas KC, Kotecha D, Lang I, Lewis BS, Linhart A, Lip GYH, Løchen ML, Mathioudakis AG, Mindham R, Moledina S, Naeije R, Nielsen JC, Olschewski H, Opitz I, Petersen SE, Prescott E, Rakisheva A, Reis A, Ristić AD, Roche N, Rodrigues R, Selton‐Suty C, Souza R, Swift AJ, Touyz RM, Ulrich S, Wilkins MR, Wort SJ. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731. 10.1093/eurheartj/ehac237 [DOI] [PubMed] [Google Scholar]
- 7. Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, Miller DP, Nicolls MR, Zamanian RT. Characterization of connective tissue disease‐associated pulmonary arterial hypertension from REVEAL. Chest. 2010;138(6):1383–1394. 10.1378/chest.10-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud‐Gaubert M, Haloun A, Laurent M, Hachulla E, Simonneau G. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–1030. 10.1164/rccm.200510-1668OC [DOI] [PubMed] [Google Scholar]
- 9. Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JSR, Howard LS, Pepke‐Zaba J, Sheares KKK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186(8):790–796. 10.1164/rccm.201203-0383OC [DOI] [PubMed] [Google Scholar]
- 10. McGoon MD, Benza RL, Escribano‐Subias P, Jiang X, Miller DP, Peacock AJ, Pepke‐Zaba J, Pulido T, Rich S, Rosenkranz S, Suissa S, Humbert M. Pulmonary arterial hypertension. JACC. 2013;62(25 Suppl):D51–D59. 10.1016/j.jacc.2013.10.023 [DOI] [PubMed] [Google Scholar]
- 11. Mathai SC, Hummers LK, Champion HC, Wigley FM, Zaiman A, Hassoun PM, Girgis RE. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis Rheum. 2009;60(2):569–577. 10.1002/art.24267 [DOI] [PubMed] [Google Scholar]
- 12. Mukerjee D. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2003;62(11):1088–1093. 10.1136/ard.62.11.1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hachulla E, de Groote P, Gressin V, Sibilia J, Diot E, Carpentier P, Mouthon L, Hatron PY, Jego P, Allanore Y, Tiev KP, Agard C, Cosnes A, Cirstea D, Constans J, Farge D, Viallard JF, Harle JR, Patat F, Imbert B, Kahan A, Cabane J, Clerson P, Guillevin L, Humbert M. The three‐year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in France. Arthritis Rheum. 2009;60(6):1831–1839. 10.1002/art.24525 [DOI] [PubMed] [Google Scholar]
- 14. Lin CY, Ko CH, Hsu CY, Chen HA. Epidemiology and mortality of connective tissue disease‐associated pulmonary arterial hypertension: a national cohort study in Taiwan. Semin Arthritis Rheum. 2020;50(5):957–962. 10.1016/j.semarthrit.2020.06.005 [DOI] [PubMed] [Google Scholar]
- 15. Fayed H, Coghlan JG. Pulmonary hypertension associated with connective tissue disease. Semin Respir Crit Care Med. 2019;40(2):173–183. 10.1055/s-0039-1685214 [DOI] [PubMed] [Google Scholar]
- 16. Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JSR, Vrapi F, Das C, Elliot CA, Johnson M, DeSoyza J, Torpy C, Goldsmith K, Hodgkins D, Hughes RJ, Pepke‐Zaba J, Coghlan JG. Connective tissue disease‐associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179(2):151–157. 10.1164/rccm.200806-953OC [DOI] [PubMed] [Google Scholar]
- 17. Hachulla E, Jais X, Cinquetti G, Clerson P, Rottat L, Launay D, Cottin V, Habib G, Prevot G, Chabanne C, Foïs E, Amoura Z, Mouthon L, Le Guern V, Montani D, Simonneau G, Humbert M, Sobanski V, Sitbon O, Balquet MH, Ziza JM, Clauvel JP, Brouet JC, Pison C, Chabot JF, Velly JF, Dos Santos PD, Meurice JC, Fauchais AL, Guillevin L, Cadranel J, Traclet J, Mornex JF, Mabo P, Didier A. Pulmonary arterial hypertension associated with systemic lupus erythematosus. Chest. 2018;153(1):143–151. 10.1016/j.chest.2017.08.014 [DOI] [PubMed] [Google Scholar]
- 18. Gunnarsson R, Andreassen AK, Molberg O, Lexberg AS, Time K, Dhainaut ASS, Bertelsen LT, Palm O, Irgens K, Becker‐Merok A, Nordeide JL, Johnsen V, Pedersen S, Proven A, Garabet LSN, Garen T, Aalokken TM, Gilboe IM, Gran JT. Prevalence of pulmonary hypertension in an unselected, mixed connective tissue disease cohort: results of a nationwide, Norwegian cross‐sectional multicentre study and review of current literature. Rheumatology. 2013;52(7):1208–1213. 10.1093/rheumatology/kes430 [DOI] [PubMed] [Google Scholar]
- 19. Xanthouli P, Jordan S, Milde N, Marra A, Blank N, Egenlauf B, Gorenflo M, Harutyunova S, Lorenz HM, Nagel C, Theobald V, Lichtblau M, Berlier C, Ulrich S, Grünig E, Benjamin N, Distler O. Haemodynamic phenotypes and survival in patients with systemic sclerosis: the impact of the new definition of pulmonary arterial hypertension. Ann Rheum Dis. 2020;79(3):370–378. 10.1136/annrheumdis-2019-216476 [DOI] [PubMed] [Google Scholar]
- 20. Jaafar S, Visovatti S, Young A, Huang S, Cronin P, Vummidi D, McLaughlin V, Khanna D. Impact of the revised haemodynamic definition on the diagnosis of pulmonary hypertension in patients with systemic sclerosis. Eur Respir J. 2019;54(2):1900586. 10.1183/13993003.00586-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ, Billings C, Lawrie A, Sabroe I, Akil M, O'Toole L, Kiely DG. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a REferral centre. Eur Respir J. 2012;39(4):945–955. 10.1183/09031936.00078411 [DOI] [PubMed] [Google Scholar]
- 22. Khanna D, Zhao C, Saggar R, Mathai SC, Chung L, Coghlan JG, Shah M, Hartney J, McLaughlin V. Long‐term outcomes in patients with connective tissue disease‐associated pulmonary arterial hypertension in the modern treatment era: meta‐analyses of randomized, controlled trials and observational registries. Arthritis Rheum. 2021;73(5):837–847. 10.1002/art.41669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rhee RL, Gabler NB, Sangani S, Praestgaard A, Merkel PA, Kawut SM. Comparison of treatment response in idiopathic and connective tissue disease‐associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(9):1111–1117. 10.1164/rccm.201507-1456OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bourji KI, Kelemen BW, Mathai SC, Damico RL, Kolb TM, Mercurio V, Cozzi F, Tedford RJ, Hassoun PM. Poor survival in patients with scleroderma and pulmonary hypertension due to heart failure with preserved ejection fraction. Pulm Circ. 2017;7(2):409–420. 10.1177/2045893217700438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong X, Shi Y, Xia Y, Zhang X, Qian J, Zhao J, Peng J, Wang Q, Weng L, Li M, Du B, Zeng X. Diversity of hemodynamic types in connective tissue disease associated pulmonary hypertension: more than a subgroup of pulmonary arterial hypertension. BMC Pulm Med. 2022;22(1):295. 10.1186/s12890-022-02081-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliveira RP, Ribeiro R, Melo L, Grima B, Oliveira S, Alves JD. Connective tissue disease‐associated interstitial lung disease. Pulmonology. 2022;28(2):113–118. 10.1016/j.pulmoe.2020.01.004 [DOI] [PubMed] [Google Scholar]
- 27. Dorfmüller P, Humbert M, Perros F, Sanchez O, Simonneau G, Müller KM, Capron F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38(6):893–902. 10.1016/j.humpath.2006.11.022 [DOI] [PubMed] [Google Scholar]
- 28. Langleben D, Orfanos SE, Giovinazzo M, Hirsch A, Baron M, Senécal JL, Armaganidis A, Catravas JD. Pulmonary capillary endothelial metabolic dysfunction: severity in pulmonary arterial hypertension related to connective tissue disease versus idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2008;58(4):1156–1164. 10.1002/art.23405 [DOI] [PubMed] [Google Scholar]
- 29. Mathai SC, Bueso M, Hummers LK, Boyce D, Lechtzin N, Le Pavec J, Campo A, Champion HC, Housten T, Forfia PR, Zaiman AL, Wigley FM, Girgis RE, Hassoun PM. Disproportionate elevation of N‐terminal pro‐brain natriuretic peptide in scleroderma‐related pulmonary hypertension. Eur Respir J. 2010;35(1):95–104. 10.1183/09031936.00074309 [DOI] [PubMed] [Google Scholar]
- 30. Cucuruzac R, Muntean I, Benedek I, Mester A, Rat N, Mitre A, Chitu M, Benedek T. Right ventricle remodeling and function in scleroderma patients. BioMed Res Int. 2018;2018:1–9. 10.1155/2018/4528148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zanatta E, Polito P, Famoso G, Larosa M, De Zorzi E, Scarpieri E, Cozzi F, Doria A. Pulmonary arterial hypertension in connective tissue disorders: pathophysiology and treatment. Exp Biol Med. 2019;244(2):120–131. 10.1177/1535370218824101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sasaki N, Kamataki A, Sawai T. A histopathological study of pulmonary hypertension in connective tissue disease. Allergol Int. 2011;60(4):411–417. 10.2332/allergolint.11-RAI-0337 [DOI] [PubMed] [Google Scholar]
- 33. Nicolls MR. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–1118. 10.1183/09031936.05.00045705 [DOI] [PubMed] [Google Scholar]
- 34. Hernandez‐Gonzalez I, Tenorio‐Castano J, Ochoa‐Parra N, Gallego N, Pérez‐Olivares C, Lago‐Docampo M, Palomino Doza J, Valverde D, Lapunzina P, Escribano‐Subias P. Novel genetic and molecular pathways in pulmonary arterial hypertension associated with connective tissue disease. Cells. 2021;10(6):1488. 10.3390/cells10061488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh N, Dorfmüller P, Shlobin OA, Ventetuolo CE. Group 3 pulmonary hypertension: from bench to bedside. Circ Res. 2022;130(9):1404–1422. 10.1161/CIRCRESAHA.121.319970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. 10.1016/S0140-6736(17)30933-9 [DOI] [PubMed] [Google Scholar]
- 37. Walker UA, Tyndall A, Czirjak L, Denton C, Farge‐Bancel D, Kowal‐Bielecka O, Muller‐Ladner U, Bocelli‐Tyndall C, Matucci‐Cerinic M. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis. 2007;66(6):754–763. 10.1136/ard.2006.062901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khanna D, Nagaraja V, Tseng C, Abtin F, Suh R, Kim G, Wells A, Furst DE, Clements PJ, Roth MD, Tashkin DP, Goldin J. Predictors of lung function decline in scleroderma‐related interstitial lung disease based on high‐resolution computed tomography: implications for cohort enrichment in systemic sclerosis‐associated interstitial lung disease trials. Arthritis Res Ther. 2015;17:372. 10.1186/s13075-015-0872-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haque A, Kiely DG, Kovacs G, Thompson AAR, Condliffe R. Pulmonary hypertension phenotypes in patients with systemic sclerosis. Eur Respir Rev. 2021;30(161):210053. 10.1183/16000617.0053-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bissell LA, Md Yusof MY, Buch MH. Primary myocardial disease in scleroderma‐a comprehensive review of the literature to inform the UK Systemic Sclerosis Study Group cardiac working group. Rheumatology. 2016;56(6):kew364. 10.1093/rheumatology/kew364 [DOI] [PubMed] [Google Scholar]
- 41. Fernández‐Codina A, Simeón‐Aznar CP, Pinal‐Fernandez I, Rodríguez‐Palomares J, Pizzi MN, Hidalgo CE, Del Castillo AG, Prado‐Galbarro FJ, Sarria‐Santamera A, Fonollosa‐Plà V, Vilardell‐Tarrés M. Cardiac involvement in systemic sclerosis: differences between clinical subsets and influence on survival. Rheumatol Int. 2017;37(1):75–84. 10.1007/s00296-015-3382-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tennøe AH, Murbræch K, Andreassen JC, Fretheim H, Garen T, Gude E, Andreassen A, Aakhus S, Molberg Ø, Hoffmann‐Vold AM. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. JACC. 2018;72(15):1804–1813. 10.1016/j.jacc.2018.07.068 [DOI] [PubMed] [Google Scholar]
- 43. Allanore Y, Meune C, Vonk MC, Airo P, Hachulla E, Caramaschi P, Riemekasten G, Cozzi F, Beretta L, Derk CT, Komócsi A, Farge D, Balbir A, Riccieri V, Distler O, Chialà A, Papa ND, Simic KP, Ghio M, Stamenkovic B, Rednic S, Host N, Pellerito R, Zegers E, Kahan A, Walker UA, Matucci‐Cerinic M. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis. 2010;69(1):218–221. 10.1136/ard.2008.103382 [DOI] [PubMed] [Google Scholar]
- 44. Avouac J, Airò P, Meune C, Beretta L, Dieude P, Caramaschi P, Tiev K, Cappelli S, Diot E, Vacca A, Cracowski JL, Sibilia J, Kahan A, Matucci‐cerinic M, Allanore Y. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J Rheumatol. 2010;37(11):2290–2298. 10.3899/jrheum.100245 [DOI] [PubMed] [Google Scholar]
- 45. Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten‐Harris T, Hummers L, Krishnan JA, Wigley F, Hassoun PM. Clinical differences between idiopathic and scleroderma‐related pulmonary hypertension. Arthritis Rheum. 2006;54(9):3043–3050. 10.1002/art.22069 [DOI] [PubMed] [Google Scholar]
- 46. Fox BD, Shimony A, Langleben D, Hirsch A, Rudski L, Schlesinger R, Eisenberg MJ, Joyal D, Hudson M, Boutet K, Serban A, Masetto A, Baron M. High prevalence of occult left heart disease in scleroderma‐pulmonary hypertension. Eur Respir J. 2013;42(4):1083–1091. 10.1183/09031936.00091212 [DOI] [PubMed] [Google Scholar]
- 47. Young A, Vummidi D, Visovatti S, Homer K, Wilhalme H, White ES, Flaherty K, McLaughlin V, Khanna D. Prevalence, treatment, and outcomes of coexistent pulmonary hypertension and interstitial lung disease in systemic sclerosis. Arthritis Rheum. 2019;71(8):1339–1349. 10.1002/art.40862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lefèvre G, Dauchet L, Hachulla E, Montani D, Sobanski V, Lambert M, Hatron PY, Humbert M, Launay D. Survival and prognostic factors in systemic sclerosis‐associated pulmonary hypertension: a systematic review and meta‐analysis. Arthritis Rheum. 2013;65(9):2412–2423. 10.1002/art.38029 [DOI] [PubMed] [Google Scholar]
- 49. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey‐Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, Jayne D, Cervera R, Costedoat‐Chalumeau N, Diamond B, Gladman DD, Hahn B, Hiepe F, Jacobsen S, Khanna D, Lerstrøm K, Massarotti E, McCune J, Ruiz‐Irastorza G, Sanchez‐Guerrero J, Schneider M, Urowitz M, Bertsias G, Hoyer BF, Leuchten N, Tani C, Tedeschi SK, Touma Z, Schmajuk G, Anic B, Assan F, Chan TM, Clarke AE, Crow MK, Czirják L, Doria A, Graninger W, Halda‐Kiss B, Hasni S, Izmirly PM, Jung M, Kumánovics G, Mariette X, Padjen I, Pego‐Reigosa JM, Romero‐Diaz J, Rúa‐Figueroa Fernández Í, Seror R, Stummvoll GH, Tanaka Y, Tektonidou MG, Vasconcelos C, Vital EM, Wallace DJ, Yavuz S, Meroni PL, Fritzler MJ, Naden R, Dörner T, Johnson SR. 2019 European League against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2019;71(9):1400–1412. 10.1002/art.40930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Amarnani R, Yeoh SA, Denneny EK, Wincup C. Lupus and the lungs: the assessment and management of pulmonary manifestations of systemic lupus erythematosus. Front Med. 2021;7:610257. 10.3389/fmed.2020.610257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hannah JR, D'Cruz DP. Pulmonary complications of systemic lupus erythematosus. Semin Respir Crit Care Med. 2019;40(2):227–234. 10.1055/s-0039-1685537 [DOI] [PubMed] [Google Scholar]
- 52. Dhakal BP, Kim CH, Al‐Kindi SG, Oliveira GH. Heart failure in systemic lupus erythematosus. Trends Cardiovasc Med. 2018;28(3):187–197. 10.1016/j.tcm.2017.08.015 [DOI] [PubMed] [Google Scholar]
- 53. Kim CH, Al‐Kindi SG, Jandali B, Askari AD, Zacharias M, Oliveira GH. Incidence and risk of heart failure in systemic lupus erythematosus. Heart. 2017;103(3):227–233. 10.1136/heartjnl-2016-309561 [DOI] [PubMed] [Google Scholar]
- 54. Bartels CM, Buhr KA, Goldberg JW, Bell CL, Visekruna M, Nekkanti S, Greenlee RT. Mortality and cardiovascular burden of systemic lupus erythematosus in a US population‐based cohort. J Rheumatol. 2014;41(4):680–687. 10.3899/jrheum.130874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fernández‐Nebro A, Rúa‐Figueroa Í, López‐Longo FJ, Galindo‐Izquierdo M, Calvo‐Alén J, Olivé‐Marqués A, Ordóñez‐Cañizares C, Martín‐Martínez MA, Blanco R, Melero‐González R, Ibáñez‐Rúan J, Bernal‐Vidal JA, Tomero‐Muriel E, Uriarte‐Isacelaya E, Horcada‐Rubio L, Freire‐González M, Narváez J, Boteanu AL, Santos‐Soler G, Andreu JL, Pego‐Reigosa JM. Cardiovascular events in systemic lupus erythematosus: a nationwide study in Spain from the RELESSER registry. Medicine. 2015;94(29):e1183. 10.1097/MD.0000000000001183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eisenberg H. Diffuse interstitial lung disease in systemic lupus erythematosus. Ann Intern Med. 1973;79(1):37–45. 10.7326/0003-4819-79-1-37 [DOI] [PubMed] [Google Scholar]
- 57. ter Borg EJ, Groen H, Horst G, Limburg PC, Wouda AA, Kallenberg CGM. Clinical associations of antiribonucleoprotein antibodies in patients with systemic lupus erythematosus. Semin Arthritis Rheum. 1990;20(3):164–173. 10.1016/0049-0172(90)90057-m [DOI] [PubMed] [Google Scholar]
- 58. Xourgia E, Tektonidou MG. An update on antiphospholipid syndrome. Curr Rheumatol Rep. 2022;23(12):84. 10.1007/s11926-021-01051-5 [DOI] [PubMed] [Google Scholar]
- 59. Perelas A, Arrossi AV, Highland KB. Pulmonary manifestations of systemic sclerosis and mixed connective tissue disease. Clin Chest Med. 2019;40(3):501–518. 10.1016/j.ccm.2019.05.001 [DOI] [PubMed] [Google Scholar]
- 60. Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52(2):148–159. 10.1016/0002-9343(72)90064-2 [DOI] [PubMed] [Google Scholar]
- 61. Alarcón‐Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16(3):328–334. https://www.ncbi.nlm.nih.gov/pubmed/2724251 [PubMed] [Google Scholar]
- 62. Tanaka Y, Kuwana M, Fujii T, Kameda H, Muro Y, Fujio K, Itoh Y, Yasuoka H, Fukaya S, Ashihara K, Hirano D, Ohmura K, Tabuchi Y, Hasegawa H, Matsumiya R, Shirai Y, Ogura T, Tsuchida Y, Ogawa‐Momohara M, Narazaki H, Inoue Y, Miyagawa I, Nakano K, Hirata S, Mori M. 2019 Diagnostic criteria for mixed connective tissue disease (MCTD): from the Japan research committee of the ministry of health, labor, and welfare for systemic autoimmune diseases. Mod Rheumatol. 2021;31(1):29–33. 10.1080/14397595.2019.1709944 [DOI] [PubMed] [Google Scholar]
- 63. Bennett RM, O'Connell DJ. The arthritis of mixed connective tissue disease. Ann Rheum Dis. 1978;37(5):397–403. 10.1136/ard.37.5.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gunnarsson R, Hetlevik SO, Lilleby V, Molberg Ø. Mixed connective tissue disease. Best Pract Res Clin Rheumatol. 2016;30(1):95–111. 10.1016/j.berh.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 65. Hajas A, Szodoray P, Nakken B, Gaal J, Zöld E, Laczik R, Demeter N, Nagy G, Szekanecz Z, Zeher M, Szegedi G, Bodolay E. Clinical course, prognosis, and causes of death in mixed connective tissue disease. J Rheumatol. 2013;40(7):1134–1142. 10.3899/jrheum.121272 [DOI] [PubMed] [Google Scholar]
- 66. Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long‐term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum. 1999;42(5):899–909. [DOI] [PubMed] [Google Scholar]
- 67. Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, Handler CE, Denton CP, Coghlan JG. Characteristics and survival of anti‐U1 RNP antibody‐positive patients with connective tissue disease‐associated pulmonary arterial hypertension. Arthritis Rheum. 2016;68(2):484–493. 10.1002/art.39432 [DOI] [PubMed] [Google Scholar]
- 68. Ungprasert P, Wannarong T, Panichsillapakit T, Cheungpasitporn W, Thongprayoon C, Ahmed S, Raddatz DA. Cardiac involvement in mixed connective tissue disease: a systematic review. Int J Cardiol. 2014;171(3):326–330. 10.1016/j.ijcard.2013.12.079 [DOI] [PubMed] [Google Scholar]
- 69. Negrini S, Emmi G, Greco M, Borro M, Sardanelli F, Murdaca G, Indiveri F, Puppo F. Sjögren's syndrome: a systemic autoimmune disease. Clin Exp Med. 2022;22(1):9–25. 10.1007/s10238-021-00728-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Luppi F, Sebastiani M, Silva M, Sverzellati N, Cavazza A, Salvarani C, Manfredi A. Interstitial lung disease in Sjögren's syndrome: a clinical review. Clin Exp Rheumatol. 2020;38 Suppl 126(4):291–300. https://www.ncbi.nlm.nih.gov/pubmed/33095142 [PubMed] [Google Scholar]
- 71. Fujisawa T. Management of myositis‐associated interstitial lung disease. Medicina. 2021;57(4):347. 10.3390/medicina57040347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baig S, Paik JJ. Inflammatory muscle disease—an update. Best Pract Res Clin Rheumatol. 2020;34(1):101484. 10.1016/j.berh.2019.101484 [DOI] [PubMed] [Google Scholar]
- 73. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971–982. 10.1016/S0140-6736(03)14368-1 [DOI] [PubMed] [Google Scholar]
- 74. Basuita M, Fidler LM. Myositis antibodies and interstitial lung disease. J Appl Lab Med. 2022;7(1):240–258. 10.1093/jalm/jfab108 [DOI] [PubMed] [Google Scholar]
- 75. Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher‐Stine L, Mammen AL, Miller FW. Idiopathic inflammatory myopathies. Nat Rev Dis Primers. 2021;7(1):86. 10.1038/s41572-021-00321-x [DOI] [PubMed] [Google Scholar]
- 76. Sparks JA. Rheumatoid arthritis. Ann Intern Med. 2019;170(1):ITC1–ITC16. 10.7326/AITC201901010 [DOI] [PubMed] [Google Scholar]
- 77. Chen J, Norling LV, Cooper D. Cardiac dysfunction in rheumatoid arthritis: the role of inflammation. Cells. 2021;10(4):881. 10.3390/cells10040881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dai Y, Wang W, Yu Y, Hu S. Rheumatoid arthritis‐associated interstitial lung disease: an overview of epidemiology, pathogenesis and management. Clin Rheumatol. 2021;40(4):1211–1220. 10.1007/s10067-020-05320-z [DOI] [PubMed] [Google Scholar]
- 79. Panagiotidou E, Sourla E, Kotoulas SX, Akritidou S, Bikos V, Bagalas V, Stanopoulos I, Pitsiou G. Rheumatoid arthritis associated pulmonary hypertension: clinical challenges reflecting the diversity of pathophysiology. Respir Med Case Rep. 2017;20:164–167. 10.1016/j.rmcr.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Morrisroe K, Stevens W, Sahhar J, Rabusa C, Nikpour M, Proudman S. Epidemiology and disease characteristics of systemic sclerosis‐related pulmonary arterial hypertension: results from a real‐life screening programme. Arthritis Res Ther. 2017;19(1):42. 10.1186/s13075-017-1250-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Humbert M, Yaici A, de Groote P, Montani D, Sitbon O, Launay D, Gressin V, Guillevin L, Clerson P, Simonneau G, Hachulla E. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long‐term survival. Arthritis Rheum. 2011;63(11):3522–3530. 10.1002/art.30541 [DOI] [PubMed] [Google Scholar]
- 82. Thakkar V, Stevens W, Prior D, Youssef P, Liew D, Gabbay E, Roddy J, Walker JG, Zochling J, Sahhar J, Nash P, Lester S, Rischmueller M, Proudman SM, Nikpour M. The inclusion of N‐terminal pro‐brain natriuretic peptide in a sensitive screening strategy for systemic sclerosis‐related pulmonary arterial hypertension: a cohort study. Arthritis Res Ther. 2013;15(6):R193. 10.1186/ar4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Coghlan JG, Denton CP, Grünig E, Bonderman D, Distler O, Khanna D, Müller‐Ladner U, Pope JE, Vonk MC, Doelberg M, Chadha‐Boreham H, Heinzl H, Rosenberg DM, McLaughlin VV, Seibold JR. Evidence‐based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis. 2014;73(7):1340–1349. 10.1136/annrheumdis-2013-203301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thakkar V, Stevens WM, Prior D, Moore OA, Byron J, Liew D, Patterson K, Hissaria P, Roddy J, Zochling J, Sahhar J, Nash P, Tymms K, Celermajer D, Gabbay E, Youssef P, Proudman SM, Nikpour M. N‐terminal pro‐brain natriuretic peptide in a novel screening algorithm for pulmonary arterial hypertension in systemic sclerosis: a case‐control study. Arthritis Res Ther. 2012;14(3):R143. 10.1186/ar3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hao Y, Thakkar V, Stevens W, Morrisroe K, Prior D, Rabusa C, Youssef P, Gabbay E, Roddy J, Walker J, Zochling J, Sahhar J, Nash P, Lester S, Rischmueller M, Proudman SM, Nikpour M. A comparison of the predictive accuracy of three screening models for pulmonary arterial hypertension in systemic sclerosis. Arthritis Res Ther. 2015;17(1):7. 10.1186/s13075-015-0517-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Weatherald J, Montani D, Jevnikar M, Jaïs X, Savale L, Humbert M. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev. 2019;28(153):190023. 10.1183/16000617.0023-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th world symposium on pulmonary hypertension. Eur Respir J. 2019;53(1):1802148. 10.1183/13993003.02148-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang C, Li M, Liu Y, Wang Q, Guo X, Zhao J, Lai J, Tian Z, Zhao Y, Zeng X. Baseline characteristics and risk factors of pulmonary arterial hypertension in systemic lupus erythematosus patients. Medicine. 2016;95(10):e2761. 10.1097/MD.0000000000002761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. King CS, Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease. Chest. 2020;158(4):1651–1664. 10.1016/j.chest.2020.04.046 [DOI] [PubMed] [Google Scholar]
- 90. Ahearn GS, Tapson VF, Rebeiz A, Greenfield JC. Electrocardiography to define clinical status in primary pulmonary hypertension and pulmonary arterial hypertension secondary to collagen vascular disease. Chest. 2002;122(2):524–527. 10.1378/chest.122.2.524 [DOI] [PubMed] [Google Scholar]
- 91. Lee JJ, Pope JE. A meta‐analysis of the risk of venous thromboembolism in inflammatory rheumatic diseases. Arthritis Res Ther. 2014;16(5):435. 10.1186/s13075-014-0435-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, Kakishita M, Fukushima K, Okano Y, Nakanishi N, Miyatake K, Kangawa K. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102(8):865–870. 10.1161/01.cir.102.8.865 [DOI] [PubMed] [Google Scholar]
- 93. Fijalkowska A, Kurzyna M, Torbicki A, Szewczyk G, Florczyk M, Pruszczyk P, Szturmowicz M. Serum N‐terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest. 2006;129(5):1313–1321. 10.1378/chest.129.5.1313 [DOI] [PubMed] [Google Scholar]
- 94. Andreassen AK, Wergeland R, Simonsen S, Geiran O, Guevara C, Ueland T. N‐terminal pro‐B‐type natriuretic peptide as an indicator of disease severity in a heterogeneous group of patients with chronic precapillary pulmonary hypertension. Am J Cardiol. 2006;98(4):525–529. 10.1016/j.amjcard.2006.02.061 [DOI] [PubMed] [Google Scholar]
- 95. Williams MH, Handler CE, Akram R, Smith CJ, Das C, Smee J, Nair D, Denton CP, Black CM, Coghlan JG. Role of N‐terminal brain natriuretic peptide (N‐TproBNP) in scleroderma‐associated pulmonary arterial hypertension. Eur Heart J. 2006;27(12):1485–1494. 10.1093/eurheartj/ehi891 [DOI] [PubMed] [Google Scholar]
- 96. Schreiber BE, Valerio CJ, Keir GJ, Handler C, Wells AU, Denton CP, Coghlan JG. Improving the detection of pulmonary hypertension in systemic sclerosis using pulmonary function tests. Arthritis Rheum. 2011;63(11):3531–3539. 10.1002/art.30535 [DOI] [PubMed] [Google Scholar]
- 97. Hachulla E, Gressin V, Guillevin L, Carpentier P, Diot E, Sibilia J, Kahan A, Cabane J, Francès C, Launay D, Mouthon L, Allanore Y, Tiev KP, Clerson P, Groote P, Humbert M. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum. 2005;52(12):3792–3800. 10.1002/art.21433 [DOI] [PubMed] [Google Scholar]
- 98. Vachiery JL, Yerly P, Huez S. How to detect disease progression in pulmonary arterial hypertension. Eur Respir Rev. 2012;21(123):40–47. 10.1183/09059180.00009011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Benza RL, Miller DP, Gomberg‐Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long‐Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–172. 10.1161/CIRCULATIONAHA.109.898122 [DOI] [PubMed] [Google Scholar]
- 100. D'Alto M, Di Maio M, Romeo E, Argiento P, Blasi E, Di Vilio A, Rea G, D'Andrea A, Golino P, Naeije R. Echocardiographic probability of pulmonary hypertension: a validation study. Eur Respir J. 2022;60(2):2102548. 10.1183/13993003.02548-2021 [DOI] [PubMed] [Google Scholar]
- 101. de Groote P, Gressin V, Hachulla E, Carpentier P, Guillevin L, Kahan A, Cabane J, Frances C, Lamblin N, Diot E, Patat F, Sibilia J, Petit H, Cracowski JL, Clerson P, Humbert M. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):31–36. 10.1136/ard.2006.057760 [DOI] [PubMed] [Google Scholar]
- 102. Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014;6(9):993–1005. 10.4330/wjc.v6.i9.993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ullah W, Minalyan A, Saleem S, Nadeem N, Abdullah HM, Abdalla A, Chan V, Saeed R, Khan M, Collins S, Mukhtar M, Grover H, Sattar Y, Panchal A, Narayana Gowda S, Khwaja U, Lashari B, Fischman DL. Comparative accuracy of non‐invasive imaging versus right heart catheterization for the diagnosis of pulmonary hypertension: a systematic review and meta‐analysis. IJC Heart Vasc. 2020;29:100568. 10.1016/j.ijcha.2020.100568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mavrogeni S, Pepe A, Nijveldt R, Ntusi N, Sierra‐Galan LM, Bratis K, Wei J, Mukherjee M, Markousis‐Mavrogenis G, Gargani L, Sade LE, Ajmone‐Marsan N, Seferovic P, Donal E, Nurmohamed M, Cerinic MM, Sfikakis P, Kitas G, Schwitter J, Lima JAC, Dawson D, Dweck M, Haugaa KH, Keenan N, Moon J, Stankovic I, Donal E, Cosyns B. Cardiovascular magnetic resonance in autoimmune rheumatic diseases: a clinical consensus document by the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2022;23(9):e308–e322. 10.1093/ehjci/jeac134 [DOI] [PubMed] [Google Scholar]
- 105. Mavrogeni S, Markousis‐Mavrogenis G, Koutsogeorgopoulou L, Dimitroulas T, Bratis K, Kitas GD, Sfikakis P, Tektonidou M, Karabela G, Stavropoulos E, Katsifis G, Boki KA, Kitsiou A, Filaditaki V, Gialafos E, Plastiras S, Vartela V, Kolovou G. Cardiovascular magnetic resonance imaging pattern at the time of diagnosis of treatment naïve patients with connective tissue diseases. Int J Cardiol. 2017;236:151–156. 10.1016/j.ijcard.2017.01.104 [DOI] [PubMed] [Google Scholar]
- 106. Bezante GP, Rollando D, Sessarego M, Panico N, Setti M, Filaci G, Molinari G, Balbi M, Cutolo M, Barsotti A, Indiveri F, Ghio M. Cardiac magnetic resonance imaging detects subclinical right ventricular impairment in systemic sclerosis. J Rheumatol. 2007;34(12):2431–2437. https://www.ncbi.nlm.nih.gov/pubmed/17985401 [PubMed] [Google Scholar]
- 107. Alabed S, Garg P, Johns CS, Alandejani F, Shahin Y, Dwivedi K, Zafar H, Wild JM, Kiely DG, Swift AJ. Cardiac magnetic resonance in pulmonary hypertension—an update. Curr Cardiovasc Imaging Rep. 2020;13(12):30. 10.1007/s12410-020-09550-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hagger D, Condliffe R, Woodhouse N, Elliot CA, Armstrong IJ, Davies C, Hill C, Akil M, Wild JM, Kiely DG. Ventricular mass index correlates with pulmonary artery pressure and predicts survival in suspected systemic sclerosis‐associated pulmonary arterial hypertension. Rheumatology. 2009;48(9):1137–1142. 10.1093/rheumatology/kep187 [DOI] [PubMed] [Google Scholar]
- 109. van de Bovenkamp AA, Wijkstra N, Oosterveer FPT, Vonk Noordegraaf A, Bogaard HJ, van Rossum AC, de Man FS, Borlaug BA, Handoko ML. The value of passive leg raise during right heart catheterization in diagnosing heart failure with preserved ejection fraction. Circ Heart Fail. 2022;15(4):e008935. 10.1161/CIRCHEARTFAILURE.121.008935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pezzuto B, Badagliacca R, Muratori M, Farina S, Bussotti M, Correale M, Bonomi A, Vignati C, Sciomer S, Papa S, Palazzo Adriano E, Agostoni P. Role of cardiopulmonary exercise test in the prediction of hemodynamic impairment in patients with pulmonary arterial hypertension. Pulm Circ. 2022;12(1):e12044. 10.1002/pul2.12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Badagliacca R, Papa S, Poscia R, Valli G, Pezzuto B, Manzi G, Torre R, Gianfrilli D, Sciomer S, Palange P, Naeije R, Fedele F, Vizza CD. The added value of cardiopulmonary exercise testing in the follow‐up of pulmonary arterial hypertension. J Heart Lung Transplant. 2019;38(3):306–314. 10.1016/j.healun.2018.11.015 [DOI] [PubMed] [Google Scholar]
- 112. Deboeck G, Scoditti C, Huez S, Vachiéry JL, Lamotte M, Sharples L, Melot C, Naeije R. Exercise testing to predict outcome in idiopathic versus associated pulmonary arterial hypertension. Eur Respir J. 2012;40(6):1410–1419. 10.1183/09031936.00217911 [DOI] [PubMed] [Google Scholar]
- 113. Wensel R, Francis DP, Meyer FJ, Opitz CF, Bruch L, Halank M, Winkler J, Seyfarth HJ, Gläser S, Blumberg F, Obst A, Dandel M, Hetzer R, Ewert R. Incremental prognostic value of cardiopulmonary exercise testing and resting haemodynamics in pulmonary arterial hypertension. Int J Cardiol. 2013;167(4):1193–1198. 10.1016/j.ijcard.2012.03.135 [DOI] [PubMed] [Google Scholar]
- 114. Zhang Y, Jin Q, Zhao Z, Zhao Q, Yu X, Yan L, Li X, An C, Ma X, Xiong C, Luo Q, Liu Z. Exercise pathophysiology differs between connective tissue diseases‐associated pulmonary arterial hypertension and idiopathic pulmonary arterial hypertension. Clin Exp Rheumatol. 2021;39(5):1063–1070. 10.55563/clinexprheumatol/z0i6ys [DOI] [PubMed] [Google Scholar]
- 115. Hassoun PM, Zamanian RT, Damico R, Lechtzin N, Khair R, Kolb TM, Tedford RJ, Hulme OL, Housten T, Pisanello C, Sato T, Pullins EH, Corona‐Villalobos CP, Zimmerman SL, Gashouta MA, Minai OA, Torres F, Girgis RE, Chin K, Mathai SC. Ambrisentan and tadalafil up‐front combination therapy in scleroderma‐associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(9):1102–1110. 10.1164/rccm.201507-1398OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kuwana M, Blair C, Takahashi T, Langley J, Coghlan JG. Initial combination therapy of ambrisentan and tadalafil in connective tissue disease‐associated pulmonary arterial hypertension (CTD‐PAH) in the modified intention‐to‐treat population of the AMBITION study: post hoc analysis. Ann Rheum Dis. 2020;79(5):626–634. 10.1136/annrheumdis-2019-216274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galiè N, Ghofrani HA, Lang IM, McLaughlin V, Preiss R, Rubin LJ, Simonneau G, Sitbon O, Tapson VF, Hoeper MM. Selexipag for the treatment of connective tissue disease‐associated pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1602493. 10.1183/13993003.02493-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bergamasco A, Hartmann N, Wallace L, Verpillat P. Epidemiology of systemic sclerosis and systemic sclerosis‐associated interstitial lung disease. Clin Epidemiol. 2019;11:257–273. 10.2147/CLEP.S191418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Nathan SD, Behr J, Collard HR, Cottin V, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Boateng F, Wells AU. Riociguat for idiopathic interstitial pneumonia‐associated pulmonary hypertension (RISE‐IIP): a randomised, placebo‐controlled phase 2b study. Lancet Respir Med. 2019;7(9):780–790. 10.1016/S2213-2600(19)30250-4 [DOI] [PubMed] [Google Scholar]
- 120. Raghu G. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med. 2013;158(9):641–649. 10.7326/0003-4819-158-9-201305070-00003 [DOI] [PubMed] [Google Scholar]
- 121. Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131(3):897–899. 10.1378/chest.06-2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zisman DA, Schwarz M, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network , A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–628. 10.1056/NEJMoa1002110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Han MK, Bach DS, Hagan PG, Yow E, Flaherty KR, Toews GB, Anstrom KJ, Martinez FJ. Sildenafil preserves exercise capacity in patients with idiopathic pulmonary fibrosis and right‐sided ventricular dysfunction. Chest. 2013;143(6):1699–1708. 10.1378/chest.12-1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ghofrani HA, Wiedemann R, Rose F, Schermuly RT, Olschewski H, Weissmann N, Gunther A, Walmrath D, Seeger W, Grimminger F. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360(9337):895–900. 10.1016/S0140-6736(02)11024-5 [DOI] [PubMed] [Google Scholar]
- 125. Corte TJ, Gatzoulis MA, Parfitt L, Harries C, Wells AU, Wort SJ. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010;15(8):1226–1232. 10.1111/j.1440-1843.2010.01860.x [DOI] [PubMed] [Google Scholar]
- 126. Chapman TH, Wilde M, Sheth A, Madden BP. Sildenafil therapy in secondary pulmonary hypertension: is there benefit in prolonged use? Vascul Pharmacol. 2009;51(2–3):90–95. 10.1016/j.vph.2009.04.002 [DOI] [PubMed] [Google Scholar]
- 127. Dawes TJW, McCabe C, Dimopoulos K, Stewart I, Bax S, Harries C, Samaranayake CB, Kempny A, Molyneaux PL, Seitler S, Semple T, Li W, George PM, Kouranos V, Chua F, Renzoni EA, Kokosi M, Jenkins G, Wells AU, Wort SJ, Price LC. Phosphodiesterase 5 inhibitor treatment and survival in interstitial lung disease pulmonary hypertension: a Bayesian retrospective observational cohort study. Respirology. 2023;28(3):262–272. 10.1111/resp.14378 [DOI] [PubMed] [Google Scholar]
- 128. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–334. 10.1056/NEJMoa2008470 [DOI] [PubMed] [Google Scholar]
- 129. Sanchez O, Sitbon O, Jaïs X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases‐associated pulmonary arterial hypertension. Chest. 2006;130(1):182–189. 10.1378/chest.130.1.182 [DOI] [PubMed] [Google Scholar]
- 130. Kato M, Kataoka H, Odani T, Fujieda Y, Otomo K, Oku K, Horita T, Yasuda S, Atsumi T, Ohira H, Tsujino I, Nishimura M, Koike T. The short‐term role of corticosteroid therapy for pulmonary arterial hypertension associated with connective tissue diseases: report of five cases and a literature review. Lupus. 2011;20(10):1047–1056. 10.1177/0961203311403347 [DOI] [PubMed] [Google Scholar]
- 131. Miyamichi‐Yamamoto S, Fukumoto Y, Sugimura K, Ishii T, Satoh K, Miura Y, Tatebe S, Nochioka K, Aoki T, Do.e Z, Shimokawa H. Intensive immunosuppressive therapy improves pulmonary hemodynamics and long‐term prognosis in patients with pulmonary arterial hypertension associated with connective tissue disease. Circ J. 2011;75(11):2668–2674. 10.1253/circj.cj-11-0473 [DOI] [PubMed] [Google Scholar]
- 132. Jais X, Launay D, Yaici A, Le Pavec J, Tchérakian C, Sitbon O, Simonneau G, Humbert M. Immunosuppressive therapy in lupus‐ and mixed connective tissue disease‐associated pulmonary arterial hypertension: a retrospective analysis of twenty‐three cases. Arthritis Rheum. 2008;58(2):521–531. 10.1002/art.23303 [DOI] [PubMed] [Google Scholar]
- 133. Zamanian RT, Badesch D, Chung L, Domsic RT, Medsger T, Pinckney A, Keyes‐Elstein L, D'Aveta C, Spychala M, White RJ, Hassoun PM, Torres F, Sweatt AJ, Molitor JA, Khanna D, Maecker H, Welch B, Goldmuntz E, Nicolls MR. Safety and efficacy of B‐cell depletion with rituximab for the treatment of systemic sclerosis‐associated pulmonary arterial hypertension: a multicenter, double‐blind, randomized, placebo‐controlled trial. Am J Respir Crit Care Med. 2021;204(2):209–221. 10.1164/rccm.202009-3481OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rhodes CJ, Wharton J, Howard L, Gibbs JSR, Vonk‐Noordegraaf A, Wilkins MR. Iron deficiency in pulmonary arterial hypertension: a potential therapeutic target. Eur Respir J. 2011;38(6):1453–1460. 10.1183/09031936.00037711 [DOI] [PubMed] [Google Scholar]
- 135. Ruiter G, Lanser IJ, de Man FS, van der Laarse WJ, Wharton J, Wilkins MR, Howard LS, Vonk‐Noordegraaf A, Voskuyl AE. Iron deficiency in systemic sclerosis patients with and without pulmonary hypertension. Rheumatology. 2014;53(2):285–292. 10.1093/rheumatology/ket331 [DOI] [PubMed] [Google Scholar]
- 136. Van De Bruaene A, Delcroix M, Pasquet A, De Backer J, De Pauw M, Naeije R, Vachiery JL, Paelinck B, Morissens M, Budts W. Iron deficiency is associated with adverse outcome in Eisenmenger patients. Eur Heart J. 2011;32(22):2790–2799. 10.1093/eurheartj/ehr130 [DOI] [PubMed] [Google Scholar]
- 137. Sonnweber T, Nairz M, Theurl I, Petzer V, Tymoszuk P, Haschka D, Rieger E, Kaessmann B, Deri M, Watzinger K, Steringer‐Mascherbauer R, Tancevski I, Weiss G, Löffler‐Ragg J. The crucial impact of iron deficiency definition for the course of precapillary pulmonary hypertension. PLoS One. 2018;13(8):e0203396. 10.1371/journal.pone.0203396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kramer T, Wissmüller M, Natsina K, Gerhardt F, Freyhaus H, Dumitrescu D, Viethen T, Hellmich M, Baldus S, Rosenkranz S. Ferric carboxymaltose in patients with pulmonary arterial hypertension and iron deficiency: a long‐term study. J Cachexia Sarcopenia Muscle. 2021;12(6):1501–1512. 10.1002/jcsm.12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Olsson KM, Fuge J, Brod T, Kamp JC, Schmitto J, Kempf T, Bauersachs J, Hoeper MM. Oral iron supplementation with ferric maltol in patients with pulmonary hypertension. Eur Respir J. 2020;56(5):2000616. 10.1183/13993003.00616-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Howard LSGE, He J, Watson GMJ, Huang L, Wharton J, Luo Q, Kiely DG, Condliffe R, Pepke‐Zaba J, Morrell NW, Sheares KK, Ulrich A, Quan R, Zhao Z, Jing X, An C, Liu Z, Xiong C, Robbins PA, Dawes T, de Marvao A, Rhodes CJ, Richter MJ, Gall H, Ghofrani HA, Zhao L, Huson L, Wilkins MR. Supplementation with iron in pulmonary arterial hypertension. Two randomized crossover trials. Ann Am Thorac Soc. 2021;18(6):981–988. 10.1513/AnnalsATS.202009-1131OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Grünig E, Maier F, Ehlken N, Fischer C, Lichtblau M, Blank N, Fiehn C, Stöckl F, Prange F, Staehler G, Reichenberger F, Tiede H, Halank M, Seyfarth HJ, Wagner S, Nagel C. Exercise training in pulmonary arterial hypertension associated with connective tissue diseases. Arthritis Res Ther. 2012;14(3):R148. 10.1186/ar3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Pandey A, Garg S, Khunger M, Garg S, Kumbhani DJ, Chin KM, Berry JD. Efficacy and safety of exercise training in chronic pulmonary hypertension: systematic review and meta‐analysis. Circ Heart Fail. 2015;8(6):1032–1043. 10.1161/CIRCHEARTFAILURE.115.002130 [DOI] [PubMed] [Google Scholar]
- 143. Preston IR, Roberts KE, Miller DP, Sen GP, Selej M, Benton WW, Hill NS, Farber HW. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the Registry to Evaluate Early and Long‐Term PAH Disease Management (REVEAL). Circulation. 2015;132(25):2403–2411. 10.1161/CIRCULATIONAHA.115.018435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang P, Hu L, Yin Y, Yan D, Zheng H, Zhang J, Li Y. Can anticoagulants improve the survival rate for patients with idiopathic pulmonary arterial hypertension? A systematic review and meta‐analysis. Thromb Res. 2020;196:251–256. 10.1016/j.thromres.2020.08.024 [DOI] [PubMed] [Google Scholar]
- 145. Hoeper MM, Pausch C, Olsson KM, Huscher D, Pittrow D, Grünig E, Staehler G, Vizza CD, Gall H, Distler O, Opitz C, Gibbs JSR, Delcroix M, Ghofrani HA, Park DH, Ewert R, Kaemmerer H, Kabitz HJ, Skowasch D, Behr J, Milger K, Halank M, Wilkens H, Seyfarth HJ, Held M, Dumitrescu D, Tsangaris I, Vonk‐Noordegraaf A, Ulrich S, Klose H, Claussen M, Lange TJ, Rosenkranz S. COMPERA 2.0: a refined four‐stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J. 2022;60(1):2102311. 10.1183/13993003.02311-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Boucly A, Weatherald J, Savale L, de Groote P, Cottin V, Prévot G, Chaouat A, Picard F, Horeau‐Langlard D, Bourdin A, Jutant EM, Beurnier A, Jevnikar M, Jaïs X, Simonneau G, Montani D, Sitbon O, Humbert M. External validation of a refined four‐stratum risk assessment score from the French pulmonary hypertension registry. Eur Respir J. 2022;59(6):2102419. 10.1183/13993003.02419-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Benza RL, Kanwar MK, Raina A, Scott JV, Zhao CL, Selej M, Elliott CG, Farber HW. Development and validation of an abridged version of the REVEAL 2.0 risk score calculator, REVEAL Lite 2, for use in patients with pulmonary arterial hypertension. Chest. 2021;159(1):337–346. 10.1016/j.chest.2020.08.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Benza RL, Gomberg‐Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, McGoon MD, Pasta DJ, Selej M, Burger CD, Frantz RP. Predicting survival in patients with pulmonary arterial hypertension. Chest. 2019;156(2):323–337. 10.1016/j.chest.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 149. de Perrot M, Granton JT, McRae K, Pierre AF, Singer LG, Waddell TK, Keshavjee S. Outcome of patients with pulmonary arterial hypertension referred for lung transplantation: a 14‐year single‐center experience. J Thorac Cardiovasc Surg. 2012;143(4):910–918. 10.1016/j.jtcvs.2011.08.055 [DOI] [PubMed] [Google Scholar]
- 150. Khangoora VS, King CS, Shlobin OA. Managing pulmonary arterial hypertension: how to select and facilitate successful transplantation. Curr Opin Organ Transplant. 2022;27(3):169–176. 10.1097/MOT.0000000000000980 [DOI] [PubMed] [Google Scholar]
- 151. Gorter TM, Verschuuren EAM, van Veldhuisen DJ, Hoendermis ES, Erasmus ME, Bogaard HJ, Vonk Noordegraaf A, Berger RMF, van Melle JP, Willems TP. Right ventricular recovery after bilateral lung transplantation for pulmonary arterial hypertension†. Interact Cardiovasc Thorac Surg. 2017;24(6):890–897. 10.1093/icvts/ivx025 [DOI] [PubMed] [Google Scholar]
- 152. Kasimir M. Reverse cardiac remodelling in patients with primary pulmonary hypertension after isolated lung transplantation. Eur J Cardiothorac Surg. 2004;26(4):776–781. 10.1016/j.ejcts.2004.05.057 [DOI] [PubMed] [Google Scholar]
- 153. Schuba B, Michel S, Guenther S, Weig T, Emser J, Schneider C, Kneidinger N, Strueven A, Sisic A, Hagl C, Schramm R. Lung transplantation in patients with severe pulmonary hypertension—focus on right ventricular remodelling. Clin Transplant. 2019;33(6):e13586. 10.1111/ctr.13586 [DOI] [PubMed] [Google Scholar]
- 154. Moser B, Jaksch P, Taghavi S, Muraközy G, Lang G, Hager H, Krenn C, Roth G, Faybik P, Bacher A, Aigner C, Matilla JR, Hoetzenecker K, Hacker P, Lang I, Klepetko W. Lung transplantation for idiopathic pulmonary arterial hypertension on intraoperative and postoperatively prolonged extracorporeal membrane oxygenation provides optimally controlled reperfusion and excellent outcome. Eur J Cardiothorac Surg. 2018;53(1):178–185. 10.1093/ejcts/ezx212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tudorache I, Sommer W, Kühn C, Wiesner O, Hadem J, Fühner T, Ius F, Avsar M, Schwerk N, Böthig D, Gottlieb J, Welte T, Bara C, Haverich A, Hoeper MM, Warnecke G. Lung transplantation for severe pulmonary hypertension—awake extracorporeal membrane oxygenation for postoperative left ventricular remodelling. Transplantation. 2015;99(2):451–458. 10.1097/TP.0000000000000348 [DOI] [PubMed] [Google Scholar]
- 156. Salman J, Ius F, Sommer W, Siemeni T, Kuehn C, Avsar M, Boethig D, Molitoris U, Bara C, Gottlieb J, Welte T, Haverich A, Hoeper MM, Warnecke G, Tudorache I. Mid‐term results of bilateral lung transplant with postoperatively extended intraoperative extracorporeal membrane oxygenation for severe pulmonary hypertension. Eur J Cardiothorac Surg. 2017;52(1):163–170. 10.1093/ejcts/ezx047 [DOI] [PubMed] [Google Scholar]
- 157. Stącel T, Antończyk R, Latos M, Nęcki M, Przybyłowski P, Zembala M, Ochman M, Urlik M. Extracorporeal membrane oxygenation as a postoperative left ventricle conditioning tool after lung transplantation in patients with primary pulmonary artery hypertension: first polish experience. Transplant Proc. 2020;52(7):2113–2117. 10.1016/j.transproceed.2020.02.113 [DOI] [PubMed] [Google Scholar]