

Summary
Metformin prevents progression of non-alcoholic fatty liver disease (NAFLD). However, the potential mechanism is not entirely understood. Ferroptosis, a recently recognized nonapoptotic form of regulated cell death, has been reported to be involved in the pathogenesis of NAFLD. Here, we investigated the effects of metformin on ferroptosis and its potential mechanism in NAFLD. We found that metformin prevented the progression of NAFLD, and alleviated hepatic iron overload (HIO), ferroptosis and upregulated ferroportin (FPN) expression in vivo and in vitro. Mechanically, metformin reduced the lysosomal degradation pathway of FPN through activation AMPK, thus upregulated the expression of FPN protein, alleviated HIO and ferroptosis, and prevented progression of NAFLD. These findings discover a mechanism of metformin, suggesting that targeting FPN may have the therapeutic potential for treating NAFLD and related disorders.
Subject areas: Pharmacology, Biological sciences, Biochemistry, Physiology, Cell biology
Graphical abstract
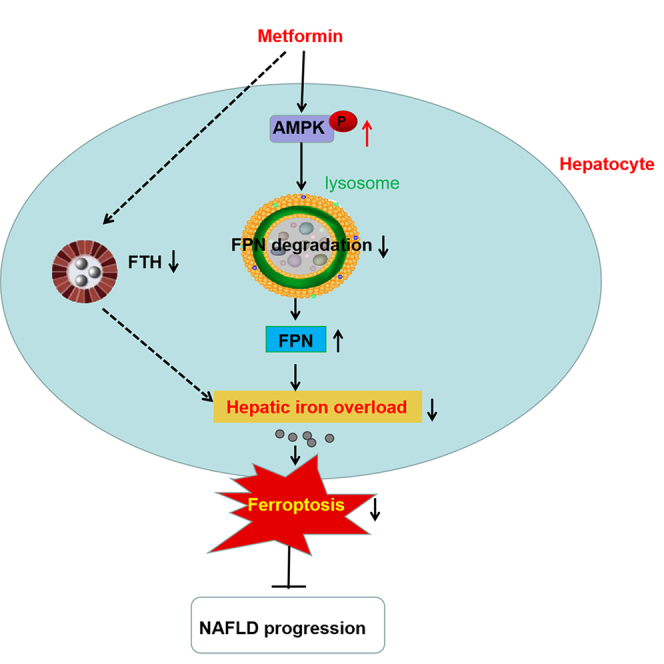
Highlights
-
•
Metformin alleviates HIO and ferroptosis in HFD-induced NAFLD
-
•
FPN is involved in the molecular mechanism of metformin on HIO in HFD-induced NAFLD
-
•
Metformin upregulates FPN expression by reducing lysosomal ubiquitination degradation
Pharmacology; Biological sciences; Biochemistry; Physiology; Cell biology
Introduction
Non-alcoholic fatty liver disease (NAFLD), the hepatic manifestation of metabolic syndrome, is emerging to be the most common chronic liver disease with a global prevalence of 20∼30%.1 The majority of patients with NAFLD remain asymptomatic, but 20% progress and develop chronic hepatic inflammation, i.e., non-alcoholic steatohepatitis (NASH), which in turn may lead to hepatic fibrosis, cirrhosis, hepatocellular carcinoma (HCC) and increased mortality.2 Presently, no pharmacotherapies have been approved yet for NAFLD treatment.3
Metformin is the first-line pharmacologic treatment in most clinical guidelines for type 2 diabetes with low cost, robust efficacy, and good tolerability.4 In animal experiment, metformin has been shown to improve hepatic steatosis and inflammation and inhibit NAFLD-associated HCC.5,6 Clinically, even though metformin as a treatment for NAFLD/NASH is not recommended at this time, several cohort studies have observed an intriguing association of metformin use with decreased liver volume and alanine aminotransferase (ALT) concentration, reduced hepatic fibrosis and HCC risk or mortality in NASH patients, highlighting that preventive effects of metformin on the progression of NAFLD.7,8 However, the mechanism of metformin in NAFLD is still not fully understood. In-depth investigation of the underlying molecular mechanism may pave the way for developing a promising therapeutic strategy.
Ferroptosis, a nonapoptotic form of regulated cell death that is driven by iron-dependent lipid peroxidation, was identified as a distinct phenomenon and named a decade ago.9 Recent studies have implicated that ferroptosis plays an important role as the trigger for initiating inflammation in steatohepatitis and affect the progression of NASH.10,11 Ferroptosis inducers, e.g., erastin, exacerbate hepatic steatosis and inflammation in NAFLD mice, and treatment with ferroptosis inhibitors significantly decreases the severity of NASH.12 Taken together, these results suggest that ferroptosis may serve as a potential therapeutic target for NAFLD.
Iron accumulation is necessary for ferroptosis to occur.13 Liver is the master regulating organ for iron homeostasis by facilitating both its systemic regulation and the storage of excess iron in case of iron overload.14 Also, hepatic iron overload (HIO) has been reported in patients with NAFLD and NASH, and has been associated with increased severity and progression of NAFLD.15
Thus, in the present study, we sought to evaluate the effects of metformin on HIO and ferroptosis in an HFD-induced NAFLD model and to provide mechanistic insight into its effects. We show here that metformin upregulates the expression of ferroportin (FPN), the only known mammalian cellular iron exporter,16 through AMPK-dependent lysosomal degradation pathway, resulting in the alleviation of HIO and ferroptosis and prevents progression of NAFLD.
Results
Metformin prevented the progression of NAFLD in HFD rats
HFD-C rats had increased body weight, liver weight, serum ALT and AST than normal control (NC) rats at the end of the experiment (p < 0.05). Metformin treatment significantly decreased body weight, liver weight, serum ALT and AST levels in high-fat diet (HFD) rats (Figures 1A–1D). Histologically, HFD led to severe steatosis, hepatocellular ballooning, lobular inflammation, liver fibrosis, elevated NAFLD activity score (NAS) and liver fibrosis score vs. NC rats, and metformin improved all these pathological changes (Figures 1E–1H). The data indicated that metformin prevented the progression of NAFLD in HFD rats.
Figure 1.

Metformin prevented the progression of NAFLD in HFD rats
(A and B) Body weight and liver weight of rats fed with normal diet (ND) or high-fat diet (HFD) without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks. Data are represented as mean ± SEM (n = 6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C and D) Serum ALT and AST of rats fed with normal diet (ND) or high-fat diet (HFD) without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks. Data are represented as mean ± SEM (n = 6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
(E) Liver histopathology assessed by H&E staining (magnification, 200×), scale bar: 100 μm.
(F) NAFLD active score. Data are represented as mean ± SEM (n = 6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, ∗∗∗p < 0.001.
(G) Liver fibrosis determined by Masson staining (magnification, 200×), scale bar: 100 μm.
(H) Liver fibrosis score. Data are represented as mean ± SEM (n = 6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗∗p < 0.001. NC, normal control; HFD-C, high fat diet control; Met, metformin; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Metformin ameliorated HIO and ferroptosis in HFD-fed rats
Serum ferritin, a commonly used marker for iron stores,17 was significantly increased in HFD-C group vs. NC group, and was significantly decreased in HFD+Met 300 group vs. HFD-C group (Figure 2A). As revealed by Prussian blue staining and liver iron measurements, a significant increase in hepatic iron concentration was observed in HFD-C group vs. NC group. Compared with the HFD-C group, iron concentrations were significantly decreased in liver tissue of the HFD+Met 150 group (Figures 2B and 2C). Correlation analyses showed significant positive correlations between hepatic iron and serum IL-6 (r = 0.4613, p < 0.05), NAS (r = 0.6071, p < 0.01) and liver fibrosis score (r = 0.4450, p < 0.05) (Figures 2D–2F).
Figure 2.

Metformin ameliorated hepatic iron overload and ferroptosis in HFD-fed rats
(A and B) Serum ferritin and hepatic iron content of rats fed with ND or HFD without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks. Data are represented as mean ± SEM (n = 5–6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, ∗∗∗p < 0.001.
(C) Hepatic iron deposition assessed by Prussian blue staining (magnification, 200×), scale bar: 100 μm.
(D) Pearson correlation analysis between hepatic iron and serum IL-6.
(E) Spearman correlation analysis between hepatic iron and NAS.
(F) Spearman correlation analysis between hepatic iron and liver fibrosis score.
(G) MDA concentrations in liver tissue in rats fed with ND or HFD without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks. Data are represented as mean ± SEM (n = 5–6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05.
(H) qPCR analysis of liver GPX4 in rats fed with ND or HFD without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks. Data are represented as mean ± SEM (n = 5–6 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05.
(I) Western blotting of hepatic GPX4, FTH, and FPN in rats fed with ND or HFD without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks, GAPDH was used as the loading control.
(J) Representative immunofluorescence staining image of hepatic GPX4 of rats fed with ND or HFD without or with metformin (150 mg/kg or 300 mg/kg) intragastrically for 8 weeks (magnification, 200×), scale bar: 100 μm. IL-6, interleukin-6; NAFLD, nonalcoholic fatty liver disease; MDA, malondialdehyde; FPN, ferroportin; FTH, ferritin heavy chain; GPX4, glutathione peroxidase 4; DAPI, 4′,6-diamidino-2-phenylindole.
Lipid peroxidation and GPX4 (a key membrane lipid repair enzyme) are involved in ferroptosis.18 Malondialdehyde (MDA), the final product of lipid peroxidation,19 significantly increased in liver tissues of HFD-C rats vs. NC rats. Metformin significantly reduced hepatic MDA in HFD rats (Figure 2G). Compared with NC rats, liver GPX4 protein expression was decreased in HFD-C rats, and HFD+Met300 rats had increased liver expressions of GPX4 mRNA and protein than the HFD-C rats (Figures 2H–2J).
Metformin ameliorated iron overload and ferroptosis in PA-treated WRL68 cells
We then examined effects of metformin on iron overload and ferroptosis in PA-treated WRL68 cells, an in vitro NAFLD model. According to CCK-8 cell viability assay, cell survival was decreased with 0.4 mM palmitic acid (PA) treatment, and treatment with 2.5 mM metformin significantly increased cell survival in PA-treated WRL68 cells (Figures 3A and 3B). Thus, we selected 0.4 mM PA, and 2.5 mM metformin for the following experiments.
Figure 3.

Metformin ameliorated iron overload and ferroptosis in PA-treated WRL68 cells
(A) Cell viability assessed with a CCK-8 assay after 24 h or 48 h of pretreatment with different concentrations of PA (0.1 mM–0.6 mM). Data are represented as mean ± SEM (n = 4 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, vs. control group; #p < 0.05, vs. PA 0.1 mM group; $p < 0.05, vs. PA 0.2 mM group; &p < 0.05, vs. PA 0.3 mM group.
(B) Cell survival assessed by CCK-8 after 24 h of pretreatment with different concentrations of metformin (0.01 mM–25 mM) and PA (0.4 mM) for 24 h. Data are represented as mean ± SEM (n = 5 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, vs. control group; #p < 0.05, vs. PA 0.4 mM group.
(C–F) Intracellular levels of TG, MDA, GSH, and T-SOD in WRL68 cells treated with PA (0.4 mM) without or with metformin (0.5 mM, 1.0 mM, and 2.5 mM) or AICAR (1.0 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; #p < 0.05, ##p < 0.01, vs. PA 0.4 mM group; &&p < 0.01, vs. PA + Metformin 0.5 mM group.
(G) Distribution of iron between cells and supernatants in WRL68 cells treated with PA (0.4 mM), FAC (0.1 mM) without or with metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; $$p < 0.05, vs. PA + FAC group.
(H) Representative images of Oil Red O staining of WRL68 cells treated with PA (0.4 mM) in the absence or presence of Fer-1 (4 μM) or metformin (2.5 mM) for 24 h (magnification, 200×), scale bar: 100 μm.
(I) Western blotting of AMPKα, P-AMPKα, and GPX4 after 24 h of PA (0.4 mM) treatment in the absence or presence of Fer-1 (4 μM) or metformin (2.5 mM) in WRL68 cells. GAPDH was used as the loading control.
(J) Cell viability assessed by CCK-8 after 24 h of pretreatment with erastin (20 μM) without or with metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, ∗∗∗p < 0.001.
(K–M) Intracellular levels of MDA, GSH and T-SOD in WRL68 cells treated with erastin (20 μM) without or with metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; $$p < 0.01, vs. erastin group.
(N) Analysis of total iron, Fe2+and Fe3+ in pellets of WRL68 cells after erastin (20 μM) treatment for 24 h without or with metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; ##p < 0.01, vs. erastin group.
(O) Total iron, Fe2+ and Fe3+ in supernatants of WRL68 cells after erastin (20 μM) treatment for 24 h without or with metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group. PA, palmitic acid; Met, metformin; TG, triglyceride; MDA, malondialdehyde; GSH, glutathione; T-SOD, total superoxide dismutase; Fer-1, ferrostatin-1.
The intracellular TG assay revealed that PA incubation increased lipid accumulation, and metformin decreased lipid accumulation in PA-treated WRL68 cells (Figure 3C). Enhanced lipid peroxidation, increases in iron levels in the pellets of WRL68 cells and the accompanied decreases in iron levels in the supernatants of WRL68 cells, decreased intracellular glutathione (GSH) levels were observed in PA-treated WRL68 cells, and these changes were remarkably ameliorated by metformin treatment (Figures 3D, 3E, and 3G). Ferrostatin-1 (Fer-1), a ferroptosis inhibitor, reversed lipid accumulation and enhanced the protein expression of GPX4 in PA-treated WRL68 cells (Figures 3H and 3I).
Effects of metformin on iron overload and ferroptosis in vitro
To determine whether metformin could directly affect ferroptosis, WRL68 cells were treated with 20 μM erastin, a classical inducer of ferroptosis, in the presence or absence of metformin for 24 h. We found that metformin mitigated erastin-induced cell inviability, iron accumulation (total iron, and Fe2+) in the pellets of WRL68 cells (Figures 3J and 3N). A decreased in intracellular T-SOD levels was observed in erastin-treated WRL68 cells, which was significantly reversed by metformin treatment (Figure 3M). In erastin-treated WRL68 cells, P-AMPKα was downregulated, and metformin significantly upregulated protein expression of P-AMPKα (Figure S1).
We then explored the effect of metformin on iron overload in vitro. In WRL68 cells, to which ferric ammonium citrate (FAC) (an iron source) was supplied at 0.1 mM, metformin intervention led to decreases in total iron, Fe2+, and Fe3+ of pellets in PA+FAC-treated WRL68 cells (Figure 4A). AICAR, an AMPK agonist, reduced total iron, Fe2+, and Fe3+ of pellets in PA+FAC-treated WRL68 cells, suggesting that metformin might attenuate liver iron overload partial through AMPK pathway.
Figure 4.

Effects of metformin on iron overload in vitro NAFLD model
(A) Concentrations of total iron, Fe2+and Fe3+ in pellets of WRL68 cells treated with PA (0.4 mM), and FAC (0.1 mM) without or with metformin (2.5 mM) or AICAR (1.0 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; $p < 0.05, $$p < 0.01, vs. PA + FAC group.
(B) Concentrations of total iron, Fe2+ and Fe3+ in supernatants of WRL68 cells treated with PA (0.4 mM), and FAC (0.1 mM) without or with metformin (2.5 mM) or AICAR (1.0 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗p < 0.05, ∗∗p < 0.01, vs. control group; $p < 0.05, $$p < 0.01, vs. PA + FAC group; &&p < 0.01, vs. PA+FAC+Metformin group.
(C and D) Intracellular levels of TG and TC in WRL68 cells treated with FAC (0.1 mM, 0.2 mM) without or with PA (0.4 mM) in the absence or presence of metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; ##p < 0.01, vs. FAC 0.2 mM group; $$p < 0.01, vs. PA + FAC 0.1 mM group; &&p < 0.01, vs. PA + FAC 0.2 mM group.
(E–G) Intracellular levels of MDA, GSH and T-SOD in WRL68 cells treated with FAC (0.1 mM, 0.2 mM) without or with PA (0.4 mM) in the absence or presence of metformin (2.5 mM). Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were assessed by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; $$p < 0.01, vs. PA + FAC group; &&p < 0.01, vs. PA+FAC+Metformin group. TG, triglyceride; TC, total cholesterol; FAC, ferric ammonium citrate; MDA, malondialdehyde; GSH, glutathione; T-SOD, total superoxide dismutase.
In WRL68 cells, FAC alone led to lipid accumulation, and co-treatment of PA with FAC augmented lipid accumulation. Metformin reduced lipid accumulation in PA + FAC co-treated WRL68 cells (Figures 4C and 4D). An increase in MDA level, and decreases in GSH, T-SOD levels were observed in PA + FAC group vs. control group (p < 0.05), and metformin significantly decreased intracellular MDA levels, and increased intracellular GSH, T-SOD levels in PA+FAC-treated WRL68 cells (p < 0.05). AICAR, an AMPK agonist, also led to a decline in intracellular MDA level and an elevation in intracellular T-SOD level in PA+FAC-treated WRL68 cells (Figures 4E–4G).
Metformin improved HIO through upregulation of FPN
To further explore the mechanisms by which metformin alleviated HIO, we detected the expression of iron regulatory proteins, such as FPN and ferritin heavy chain (FTH), which were responsible for iron output and iron storage, respectively.20 The expression of FPN was unchanged with metformin treatment in WRL68 cells (Figure S2), while in liver tissues of HFD-C rats or PA-treated WRL68 cells, FPN was downregulated and FTH was upregulated, and metformin significantly upregulated protein expression of FPN and downregulated the protein expression of FTH (Figures 2I and 5B).
Figure 5.

Metformin upregulates the expression of FPN by reducing its lysosomal ubiquitination degradation in an AMPK-dependent manner
(A) Metformin upregulates FPN mRNA expression in PA-treated WRL68 cells. Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were determined by one-way ANOVA analysis. ∗∗∗p < 0.001.
(B) Metformin upregulates FPN protein expression and downregulates FTH protein expression in PA-treated WRL68 cells.
(C) WRL68 cells were transfected with siRNA-NC or siRNA-FPNs (siRNA-FPN1, siRNA-FPN2, and siRNA-FPN3) for 48 h, and the relative mRNA expression of FPN was determined by RT-PCR. Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were determined by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; $$p < 0.01, vs. SiRNA-NC group; &&p < 0.01, vs. SiRNA-FPN2 group.
(D) WRL68 cells were transfected with siRNA-FPN3 or siRNA-NC for 48 h. FPN knockdown was confirmed through western blotting.
(E–G) Intracellular total iron, Fe2+ and Fe3+ levels. WRL68 cells were transfected with siRNA-FPN or siRNA-NC for 48 h, then treated with PA (0.4 mM) without or with metformin (2.5 mM) for 24 h, and intracellular total iron, Fe2+ and Fe3+ levels in WRL68 cells were measured. Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were determined by one-way ANOVA analysis. ∗p < 0.05, ∗∗p < 0.01, vs. control+siRNA-NC group; #p < 0.05, ##p < 0.01, vs. PA+siRNA-NC group; $p < 0.05, vs. siRNA-FPN group; &p < 0.05, vs. PA+siRNA-FPN group.
(H) PA-treated WRL68 cells were treated without or with metformin (2.5 mM), were then incubated with 100 μM CQ (a lysosome inhibitor) for 2 h, and the expression of FPN protein was assessed by western blotting.
(I) PA-treated WRL68 cells were treated without or with metformin (2.5 mM), were then incubated with 10 μM MG132 (a proteasome inhibitor) for 8 h, and the expression of FPN protein was assessed by western blotting.
(J) Ubiquitin/FPN co-immunoprecipitation of cells extracts of WRL68 cells transfected with GFP-tagged human overexpression FPN plasmid. WRL68 cells were divided into four groups: (1) GFP-FPN+CQ group; (2) GFP-FPN+CQ + PA group; (3) GFP-FPN+CQ + PA + Met group, and (4) GFP-FPN+CQ + PA + AICAR group. Cells were then lysed and immunoprecipitated with anti-FPN antibody or IgG as negative control, and immunocomplexes were immunoblotted by anti-ubiquitin or anti-FPN antibody (output). Also, the expressions of FPN in cell lysates before immunoprecipitation were assessed (input).
(K) Short hairpin RNAs (shRNA) for transient silencing of AMPKα gene (sh-AMPKα) or non-targeting control shRNA (sh-NC) were constructed. The one with the highest silencing efficiency (sh-AMPKα-2) was selected for the experiment. WRL68 cells were transfected with GFP-tagged human overexpression FPN plasmid. The cells were then divided into four groups: (1) GFP-FPN+CQ+sh-NC;( 2) GFP-FPN+CQ + PA+sh-NC; (3) GFP-FPN +CQ + PA+Met+ sh-NC; and (4) GFP-FPN+CQ + PA+Met+sh-AMPKα-2. Ubiquitin/FPN was assessed by co-immunoprecipitation. PA, palmitic acid; FPN, ferroportin; FTH, ferritin heavy chain; GPX4, glutathione peroxidase 4; CQ, chloroquine.
To confirm the involvement of FPN in the metformin-mediated improvement of liver iron overload in NAFLD, three different siRNAs targeting FPN gene were constructed and transfected into WRL68 cells (Figure 5C). We selected siRNA-FPN3 construct, which showed the strongest knockdown efficiency, as evidenced by RT-PCR and western blotting (Figures 5C and 5D). We found that FPN knockdown by siRNA-FPN3 construct prevented the reversal of metformin-mediated decreases in iron level in pellets of PA-treated WRL68 cells (Figures 5E–5G).
Metformin ameliorated iron overload through AMPK/FPN pathway
GFP-tagged human FPN (pcDNA 3.1-GFP) plasmid was transfected into WRL68 cells. And the efficiency of overexpressing GFP/FPN was assessed by western blotting (Figure 6A). Metformin reduced total iron, Fe2+ levels in the pellets of WRL68 cells, similar results were also found in cells with overexpression of FPN (Figures 6B and 6C). Compound C, an AMPK inhibitor, prevented the decrease of iron levels of cell pellets by metformin or GFP/FPN overexpression in WRL68 cells (Figures 6B–6D). The results indicated that metformin ameliorated iron overload through AMPK/FPN pathway.
Figure 6.

Metformin ameliorated iron overload through AMPK/FPN pathway in PA+FAC-induced WRL68 cells
(A) WRL68 cells were transfected with GFP-tagged human overexpression FPN plasmid. The protein expressions of FPN were confirmed by western blotting.
(B–D) Intracellular total iron, Fe2+ and Fe3+ levels. WRL68 cells were transfected with GFP-tagged human overexpression FPN plasmid or empty vector plasmid, then cells were treated with PA (0.4 mM), FAC (0.1 mM) without or with metformin (2.5 mM) or Compound C (15 μM), and intracellular total iron, Fe2+ and Fe3+ levels in WRL68 cells were measured. Data are represented as mean ± SEM (n = 3 in each group). Differences among groups were determined by one-way ANOVA analysis. ∗∗p < 0.01, vs. control group; ##p < 0.01, vs. PA+FAC+empty vector group; &&p < 0.01, vs. PA+FAC+empty vector +Met +Compound C group. PA, palmitic acid; FPN, ferroportin; FAC, ferric ammonium citrate.
Metformin upregulates the expression of FPN by reducing its lysosomal ubiquitination degradation in an AMPK-dependent manner
It has been reported that the degradation of FPN is mainly initiated by ligand-induced ubiquitination in lysosome and proteasome pathways.21 In PA-induced WRL68 cells, chloroquine (CQ, a lysosomal protease inhibitor) prevented the decrease in FPN protein expression, while proteasome inhibitor MG132 did not alter the expressions of FPN (Figures 5H and 5I). PA induced ubiquitination degradation of FPN in WRL68 cells overexpressing GFP/FPN, and metformin, AICAR decreased ubiquitination degradation of FPN (Figure 5J). We then established sh-AMPKα constructs, and selected the one with the highest silencing efficiency (sh-AMPKα-2) for the following experiments (Figure S3). We found that AMPKα knockdown by sh-AMPKα-2 construct prevented the reversal of metformin-mediated decreases in ubiquitination degradation of FPN in WRL68 cells overexpressing GFP/FPN (Figure 5K). These data suggested that metformin upregulated protein expression of FPN in part by reducing its lysosomal ubiquitination degradation in an AMPK-dependent manner.
Discussion
The present study confirmed the preventive effects of metformin on the progression of NAFLD in the HFD-induced model, which is consistent with previous observations.22,23 However, the effects of metformin on HIO and ferroptosis in NAFLD has not been well-documented. Here, we found that metformin alleviated HIO and ferroptosis in both in vivo and in vitro NAFLD models. More importantly, the study provided new molecular mechanisms that metformin mitigates HIO through activation of AMPK-FPN pathway, eventually inhibiting ferroptosis in NAFLD. All the findings suggest a regulatory mechanism of metformin in NAFLD and demonstrate that targeting FPN have the therapeutic potential for treating NAFLD and related disorders.
Iron causes harmful effects, that is, excessive iron promotes the Fenton response, produces irrepressible free radicals that lead to severe cell and tissue damage.24 Regarding dysregulation of hepatic iron in NAFLD, there have been some conflicting results. Some demonstrated HIO in patients with NAFLD/NASH, and HIO is associated with increased likelihood of higher fibrosis stages in NAFLD patients.25 Additionally, iron reduction therapies, such as phlebotomy and iron chelation, improved hepatic function and insulin resistance in NAFLD patients.26 However, other report failed to find an increase in HIO in NAFLD and indicated that studies of phlebotomy in patients with NAFLD were generally negative.27 Differences in ethnicity, frequency of steatohepatitis, genetic background underlying iron accumulation, and beta-globin mutations may explain the discrepancy.28,29,30 In the present study, we found that serum ferritin and hepatic iron concentration were increased in HFD-fed rats, a commonly used NAFLD model. And in WRL68 cells, to which FAC was supplied as an iron source, PA treatment induced increases in total iron, Fe2+, and Fe3+ in the pellets of cells. These data support the notion of dysregulation of hepatic iron in NAFLD. Lipid accumulation has been suggested to be the “first hit” for NAFLD.31 Our in vitro experiments found that FAC alone could lead to lipid accumulation, which has also been reported by Fisher et al., indicating that dysregulation of hepatic iron may initiate the pathogenesis of NAFLD.32
Notably, we found that metformin treatment improved HIO in NAFLD. There are few studies about the effects of metformin on iron homeostasis. Here, metformin reduced HIO in HFD-fed rats, and in PA+FAC-treated WRL68 cells, metformin reduced the intracellular iron levels. The essential nature of ferroptosis is regarded as intracellular iron (especially Fe2+) accumulation.33 Our data indicated that metformin might inhibit ferroptosis through improvement of HIO and iron dysregulation in NAFLD.
Further, metformin has gained attention in recent years due to its pleiotropic effects, such as anti-cancer, and anti-aging effects.34 Evidences suggest that iron dysregulation is involved in the pathophysiology of obesity, diabetes, cancer, and aging diseases,35 a link between metformin and iron homeostasis in the present study may open new avenues on mechanisms of metformin in these diseases.
Metformin is well-established to activate AMPK.36 Accumulating evidence indicates a link between AMPK and ferroptosis.37,38 However, the results remain largely inconclusive. Some studies showed that AMPK activation promotes ferroptosis,39,40 while some proposed that AMPK activation inhibits ferroptosis.41 Interestingly, a recent study revealed that either AMPK activation or inhibition can induce ferroptosis in breast cancer cell lines.42 Different cell lines (e.g., non-cancer cell lines and cancer cell lines), culture conditions may account for the inconsistency. Alternatively, these findings may suggest that AMPK imbalance is critical for ferroptosis. Our study demonstrated inhibitory effects of AMPK activation on ferroptosis, which agree with Lee H et al. observations.43 Mechanistically, they have linked AMPK regulation of ferroptosis to AMPK-mediated phosphorylation of acetyl-CoA carboxylase and polyunsaturated fatty acid biosynthesis, but not to iron metabolism, while our data suggest that iron metabolism is involved in the effects of AMPK on ferroptosis. Based on these findings, we speculated that the exact role of AMPK in ferroptosis or iron metabolism is context-dependent and varies with the disease types, different cell lines (e.g., human WRL68 cells and cancer cell lines) et al. As a matter of fact, Lee et al. found a marginally significant differences in cellular iron between AMPK double knockout cells and the wild type cells (p = 0.07–0.09).43 More studies are needed to dissect the role of AMPK or metformin on ferroptosis or cellular iron metabolism.
FPN, the only known cellular iron exporter in mammals, has been reported to be downregulated in NAFLD patients and animal models and closely related to HIO in NAFLD.44 Here, we also observed a decrease in FPN protein expression in HFD-C rats and PA-treated WRL68 cells, indicating that HIO in NAFLD maybe be partially the result of decreased iron output. A decrease in FPN is expected to increase the labile iron pool and promotes ferroptosis.45 Indeed, evidence has shown that FPN acts as a negative regulator of ferroptosis.46 In HFD rats or PA-treated WRL68 cells, we found that metformin increased expression of FPN, and siRNA-FPN reversed the amelioration of iron overload in NAFLD by metformin, indicating that FPN acts as a substrate for metformin in regulating iron homeostasis, metformin inhibits HIO and ferroptosis in NAFLD through upregulating FPN expression, and FPN may represent a suitable therapeutic intervention target for NAFLD.
The regulation of FPN expression is complex, with important layers of control at transcriptional, translational, and posttranslational levels.21 Our data showed that metformin significantly increased FPN expression at both mRNA and protein levels in liver tissues of HFD-C rats or PA-treated WRL68 cells, suggesting multiple levels of regulation were involved. As the functional activity of FPN is predominantly regulated at the posttranslational level,47 we then explored the posttranslational mechanisms. At posttranslational levels, FPN may be degraded via lysosome and proteasome.48 Here we found that lysosome inhibitor (CQ), but not proteasome inhibitor MG132, prevented the reduction in FPN in PA-treated WRL68 cells, and metformin inhibited the lysosomal ubiquitination degradation of FPN, upregulated FPN in PA-treated WRL68 cells. Our data indicate that lysosomal pathways are vital for the posttranslational regulation of metformin on FPN. In WRL68 cells, neither mRNA nor protein levels of FPN were altered by metformin treatment, implying that metformin may have no direct effect on FPN. Nevertheless, AMPK-mediating FPN degradation by metformin beyond in lysosome should be addressed, and the transcriptional mechanism by which metformin regulates FPN in PA-treated WRL68 cells need further studies. Also, we found an increase in FTH in liver tissue of HFD-C rats and PA-treated WRL68 cells, and metformin reduced its expression. It may account to the changes of FPN, but the direct effects of metformin on FTH cannot be ruled out.
In summary, our study demonstrates that metformin reduces the lysosomal degradation pathway of FPN through activation AMPK, thus upregulates the expression of FPN protein, alleviates HIO and ferroptosis, and prevents progression of NAFLD. These findings discovered a mechanism of metformin, suggesting that targeting FPN may have the therapeutic potential for treating NAFLD and related disorders.
Limitations of the study
There are several limitations. Firstly, the study was performed in male rats. Sex differences seem to be considered in the expression and effect of FPN on NAFLD and HIO.44 It remains to be seen whether there are any sex differences of metformin on NAFLD subjects. Secondly, FPN was regarded as the only identified iron exporter in mammals. However, a recent study reported an FPN-independent iron export pathway, i.e., extracellular vesicles (EV)-dependent iron export mechanism that lead to hepatocyte iron deficiency and hepatic stellate cell iron overload, and which contributes to the development of liver steatosis and fibrosis in HFD-fed mice.14 Although our data showed that hepatic iron concentration was increased in HFD-fed rats, and hepatocyte iron accumulation may initiate the pathogenesis of NAFLD, and metformin alleviated HIO through FPN pathway, effects of metformin on the hepatic iron distribution and the pathophysiological significance of the iron redistribution are needed to clarify in the follow studies. Thirdly, GPX4 plays a master role in blocking ferroptosis by eliminating phospholipid hydroperoxides.18 We found metformin increased hepatic GPX4 protein expression. We cannot rule out the possibility that metformin inhibits ferroptosis by upregulation of GPX4 directly.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-AMPKα | Cell Signaling Technology | Cat#5831; RRID: AB_10622186 |
| Rabbit monoclonal anti-phospho-AMPKα (Thr172) | Cell Signaling Technology | Cat#50081; RRID: AB_2799368 |
| Rabbit monoclonal anti-GPX4 | Abcam | Cat# ab125066; RRID: AB_10973901 |
| Rabbit monoclonal anti-FTH | Abcam | Cat# ab75972; RRID: AB_1310223 |
| Mouse monoclonal anti-FPN | Santa cruz | Cat# sc-518125 |
| Mouse monoclonal anti-β-actin | Abcam | Cat# ab6276; RRID: AB_2223210 |
| Rabbit monoclonal anti-GAPDH | Abcam | Cat# ab181602; RRID: AB_2630358 |
| Secondary antibody goat anti-mouse IgG | Beverly | Cat#7074 |
| Rabbit polyclonal anti-Ubiquitin | Cell Signaling Technology | Cat#3933S; RRID: AB_2180538 |
| Chemicals, peptides, and recombinant proteins | ||
| minimum Eagle’s medium | Thermo Fisher Scientific | Cat#11095098 |
| fetal bovine serum (FBS) | Sigma | Cat#12103C |
| Palmitic acid | Sigma | Cat# P0500 |
| metformin | Sigma | Cat# PHR1084 |
| erastin | Sigma | Cat# E7781 |
| Ferric ammonium citrate | Sigma | Cat# F5879 |
| interleukin-6 | Cusabio | Cat# CSB-E04640r |
| ferritin | Cusabio | Cat# CSB-E08826r |
| protease inhibitors | Thermo Fisher Scientific | Cat# 36978 |
| Bicinchoninic acid | Thermo Fisher Scientific | Cat# 23227 |
| DAPI | Sigma | Cat# D9542 |
| Lipofectamine 2000 | Invitrogen | Cat#11668-019 |
| Ferrostatin-1 (Fer-1) | Sigma | Cat# SML0583 |
| chloroquine | Sigma | Cat# C6628 |
| AICAR | Sigma | Cat# A1393 |
| MG132 | Sigma | Cat# M7449 |
| Critical commercial assays | ||
| UltraSYBR Mixture RT-PCR reagent | CWBIO | Cat# CW2569M |
| Cell Counting Kit-8 | Sigma | Cat# 96992 |
| triglyceride | Nanjing Jiancheng Bioengineering Institute | Cat# A110-1-1 |
| total cholesterol | Nanjing Jiancheng Bioengineering Institute | Cat# F002-1-1 |
| malondialdehyde | Nanjing Jiancheng Bioengineering Institute | Cat# A003-1-2 |
| glutathione | Nanjing Jiancheng Bioengineering Institute | Cat# A006-2-1 |
| total superoxide dismutase | Nanjing Jiancheng Bioengineering Institute | Cat# A001-1-2 |
| iron assay kit | Abcam | Cat# ab83366 |
| Experimental models: Cell lines | ||
| WRL68 | iCell Bioscience Inc | Cat# CL-48 |
| Experimental models: Organisms/strains | ||
| Rats: Sprague-Dawley (SD) | Hunan Slac-Jinda Animal Company | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S1 | This paper | N/A |
| siRNA targeting sequence: FPN #1: GGATGGGTCTCCTACTACA | RiboBio | N/A |
| siRNA targeting sequence: FPN #2: GGACAAGAATGCTAGACTT | RiboBio | N/A |
| siRNA targeting sequence: FPN #3: GCACAGCTTTCCTGTTTGA | RiboBio | N/A |
| shRNA targeting sequence: AMPKα #1: GCGGCTCTTTCAGCAGATTCT | Honor Gene | N/A |
| shRNA targeting sequence: AMPKα #2: GCTGAAGTTTACCGAGCTATG | Honor Gene | N/A |
| shRNA targeting sequence: AMPKα #3: GCTGTGAAAGAAGTGTGTGAA | Honor Gene | N/A |
| Recombinant DNA | ||
| Plasmid: GFP-FPN | OriGene | Cat# RG205219 |
| Software and algorithms | ||
| GraphPad prism 5 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Standard chow diet | Hunan Slac-Jinda Animal Company | N/A |
| High-fat diet | Hunan Slac-Jinda Animal Company | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dongmei Zhang (drdmzhang@csu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
For animal studies
Twenty-four Specified Pathogen Free six-week-old male Sprague-Dawley (SD) rats (body weight 250-280g) were purchased from Hunan Slac-Jinda Animal Company (Changsha, China). All the rats housed two per cage were maintained in a controlled environment (22 ± 2°C, 40–50% humidity) with a standard 12 h light/dark cycle (lights on 9 a.m.-9 p.m.) and free access to water and food.
Ethics statement
All the animal procedures and experimental protocols were approved by the Medical Ethics Committee of Central South University (No.2018sydw184).
Cell line and culture
Normal human hepatocytes cell line WRL68 was purchased from iCell Bioscience company (iCell Bioscience Inc, Shanghai, China). WRL68 cells were cultured in minimum Eagle’s medium (MEM) (Cat. 11095098, Gibco, Thermo Fisher Scientific Inc.), added with 1% Penicillin-Streptomycin, 10% fetal bovine serum (Cat. 12103C, Sigma, USA) at 37°C with 5% CO2. The cell line was examined for mycoplasma contamination, and mycoplasma negative status of cell line was confirmed. Authentication testing of WRL68 cell line have been performed by Shanghai Biowing Applied Biotechnology Co.,Ltd via short tandem repeat (STR) DNA profiling. STR profiles match the standards recommended for WRL68 cell lines authentication.
Animal studies
Animal studies were performed in accordance with the guidelines established by the Animal Ethnics Committee of the Xiangya Medical Collage of Central South University (No.2018sydw184).
Method details
Experimental group and samples collection
After one week’s adaptation, rats were either fed on a normal chow diet (ND) (fat 12%, protein 22%, carbohydrate 66%, 3.48 kcal/g) or a HFD (fat 37%, protein 17.5%, carbohydrate 45.5%, 4.50 kcal/g). At the beginning of 9th week, HFD-fed rats were randomly divided into three groups (n = 6 for each): (1) HFD+Met 150 group: rats were administered with metformin (150 mg/kg/day) intragastrically for 8 weeks (2) HFD+Met 300 group: rats were administered with metformin (300 mg/kg/day) intragastrically for 8 weeks,49,50 and (3) rats were administered intragastrically with sterilized water as HFD control (HFD-C) group. And ND-fed rats were administered intragastrically with sterilized water as NC group (n = 6). Body weights were recorded weekly throughout the whole experiment for 16 weeks. Then, all the rats were sacrificed to collect blood samples and liver tissues. Serum samples were obtained after centrifugation (2000×g, 20 min), and was temporarily stored at 4°C, finally it was frozen at −80°C for subsequent measurements. The collected liver tissues were weighed, fixed with 4% paraformaldehyde solution or stored in liquid nitrogen or at −80°C for subsequent experiments.
Cell treatment
PA (Cat. P0500, Sigma, USA) was added to cell culture medium to reach a final concentration of 0.4 mM at 70%–80% confluency in the presence or absence of different concentrations of metformin (Cat. PHR1084, Sigma, USA) for 24 h. An in vitro model of ferroptosis was established by treatment with 20 μM erastin (Cat. E7781, Sigma, USA). FAC (Cat. F5879, Sigma, USA), as an iron source, was co-incubated with PA at 0.1 mM or 0.2 mM to establish iron overload model.
Serum biochemical analyses
Serum ALT, and aspartate aminotransferase (AST) were measured on an automatic analyzer (Hitachi, Japan) using commercial reagents (Serotec Co., Sapporo, Japan).
Enzyme-linked immunosorbent (ELISA) assay
Serum interleukin-6 (IL-6), and ferritin were measured using ELISA kits (Cat. CSB-E04640r, and Cat. CSB-E08826r respectively, Cusabio, Wuhan, China), according to manufacturer’s instructions.
Histopathology
Liver tissues were harvested, and fixed in 4% paraformaldehyde for 4 h at 4°C. Then, they were washed with PBS for about 30 min and dehydrated in a series of ethanol (50%, 70%, 90%, and 100%) for 2 h. Paraffin-embedded liver tissues were used for histopathology assessments.
Hematoxylin and eosin (H&E) staining
Paraffin-embedded liver tissues were cut into 5-μm-thick slices, and dissolved in xylene. After re-hydration with a serial change of ethanol (100%, 95%, and 70%). Hemotoxylin was used for nuclei staining (Cat. G1120, Solarbio, Beijing biotechnology company), all the slides were incubated for 8 min at room temperature. Then they were differentiated with 1% acid ethanol and liver tissues were stained with eosin for 30s. Images were captured by a light microscope (Zeiss, Germany). NAS was assessed by a modified semiquantitative Brunt score. Specifically, indices of steatosis, lobular inflammation, and hepatocellular ballooning were evaluated semi-quantitatively, steatosis(0–3): 5–33% (1), 34–66% (2), more than 66% (3); lobular inflammation (0–3): none (0), mild (1), moderate (2), and many (3); and hepatocellular ballooning (0–2): none (0), few balloon cells (1), and prominent ballooning (2).51
Masson’s Trichrome staining
Liver tissues were differentiated by Masson's Trichrome staining after dewaxing, rehydrating, and fixing for about 2 h. Subsequently, nuclear were stained with Hematoxylin (Cat. G1120, Solarbio, Beijing biotechnology company) for 5 min and dehydrated in ethanol and xylene. Images were captured by a light microscope (Leica, Germany). Liver fibrosis score was assessed by Scheuer’s classification, ranging from 0 to 4. In brief, no fibrosis (0), expansion of portal area (1), portal fibrosis, formation of fiber septa (2), fiber septa with lobular structural disorder (3), and probable or definite cirrhosis (4).52
Prussian blue staining
Liver iron content was assessed by Perl’s Prussian blue staining kit (Polysciences, Inc, Warrington, USA) according to the manufacturer’s instructions. The images were captured by a light microscope (Leica, Germany) for iron deposition.
RNA extraction, cDNA synthesis, and qRT-PCR
Total RNA was extracted from liver tissues or WRL68 cells, and 1 μg RNA was used for cDNA synthesis in accordance with manufacturer’s instructions. qPCR was carried out using UltraSYBR Mixture RT-PCR reagent (Cat. CW2569M, CWBIO, Beijing, China) on Quantstudio 7 Flex System (Corbett Life Science Pty. Ltd., Mortlake, NSW, Australia) according to manufacturer’s instructions. Briefly, total mixed volume of 20 μL was used for amplification reaction and the amplification conditions were performed as follows: 95°C for 10 min, 40 cycles at 95°C for 15s and 60°C for 60s. The relative expressions of genes were normalized to GAPDH. All the results were analyzed by Optical System Software version 2.0. The primers were obtained from Shanghai Boshang company and sequences were listed in Table S1.
Western blotting
Liver tissues or WRL68 cells were lysed by RIPA buffer with protease inhibitors (Thermo Fisher Scientific, Dartford, UK) on ice for about 15 min. The supernatants were obtained after centrifugation at 4°C (14,000×g, 20 min), and quantified by the Bicinchoninic acid (BCA) kit (Thermo Fisher Scientific, Dartford, UK). In brief, 30 μg of total protein were separated on 10–12% SDS-PAGE (Cat.3450124, Bio-Rad, USA), and transferred to PVDF membrane. The membranes were incubated with primary antibodies overnight at 4°C after blocking by TBST containing 5% skim milk for about 1 h. The primary antibodies used were listed as follows: anti-AMPKα (Cat. 5831, CST, USA), anti-phospho-AMPKα (Cat. 50081, CST, USA), anti-GPX4 (Cat. ab125066, Abcam, UK), anti-FTH (Cat. ab75972, Abcam, UK), anti-FPN (Cat. sc-518125, Santa Cruz, USA), anti-β-actin (Cat. ab6276, Abcam, UK), and anti-GAPDH (Cat. ab181602, Abcam, UK) were used in our study. Then, the membranes were washed by TBST for three times, and incubated with peroxidase-conjugated secondary antibodies (Cat. 7074, Beverly, USA) for about 1 h at room temperature. The western blots were performed on ChemiDoc Touch Imaging System Version 1.2 (Bio-Rad).
Immunofluorescence staining
Paraffin-embedded liver sections (5-μm thick) were incubated with anti-GPX4 for about 30 min at room temperature. All the slides were washed with phosphate buffer solution (PBS) for three times after staining, and incubated with a FITC-conjugated secondary antibody (Thermo Fisher Scientific, Dartford, UK) for 1 h at room temperature. Then the nucleus was stained with 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) (Cat. D9542, Sigma, USA). All the images were captured by fluorescence microscopy (Leica, Germany).
Cell viability
The WRL68 cells (5×103 per well) were seeded in 96-well plates with the cell density of 1000 cells/well, and cultured at 37°C under 5% CO2 for 24 h. The viability of treated cells was determined by Cell Counting Kit-8 (CCK-8) assay kit (Cat. 96992, Sigma, USA) according to manufacturer’s instructions. A complete medium containing CCK-8 (10 μL/well) was added and incubated for another 2 h at 37°C. The optical density (OD) was assessed by a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) (absorbance at OD 450 nm).
Intracellular lipid assay
WRL68 cells were seeded into 6-well plates in 2 mL of medium, and treated with FAC (0.1 mM, 0.2 mM) without or with PA (0.4 mM) in the absence or presence of metformin (2.5 mM) for 24 h with a starvation for 4 h in serum and glucose-free medium. Then, the cells were collected and lysed by PBS buffer. Intracellular triglyceride (TG) and TC levels were determined by commercial reagents (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. Total cellular protein content was quantified by BCA Protein Assay kit. Concentration of intracellular TG and TC were normalized to total cellular protein concentration.
Oil Red O staining
WRL68 cells were washed twice and fixed with 4% paraformaldehyde solution for 1 h at room temperature. Cells were stained with Oil Red O solution for about 30 min after three washes using 60% isopropanol solution. Finally, the cells were washed with distilled water for four times after removing the stain.
Malondialdehyde assay
Hepatic homogenates and WRL68 cells were collected. The MDA levels were determined by lipid peroxidation colorimetric assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to manufacturer’s instructions. In brief, liver tissues or WRL68 cells were homogenized in RIPA buffer on ice. Then, tissue or cell lysates supernatant were collected by centrifugation after the reaction. The OD was determined by a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) (absorbance at OD 532 nm). MDA levels were normalized to protein concentration.
Glutathione and total superoxide dismutase (T-SOD) assay
Liver tissues or WRL68 cells were homogenized in RIPA buffer on ice, then the supernatant was collected by centrifugation. The concentration of GSH and T-SOD were determined by colorimetric assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to manufacturer’s instructions. The absorbance was measured at 550 nm or 420 nm with a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). GSH and T-SOD levels were normalized to protein concentration.
Iron content measurements in tissues and cells
The concentration of nonheme iron content in liver tissues was assessed.53 In brief, liver tissue samples were dry weighted, and digested in mixed acid solution (3 mol/L hydrochloric acid, 10% trichloroacetic acid) for 20 h at 65°C. Then, 100 μL acid extract was mixed with 2 mL bathophenanthroline chromagen reagent, and were incubated at room temperature for 10 min. OD was assessed by a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) (absorbance at OD 535 nm).
Intracellular total iron, ferrous (Fe2+), and ferric iron (Fe3+) in WRL68 cells were assessed by iron assay kit (Cat. ab83366, Abcam, UK) in accordance with manufacturer’s instructions. Briefly, WRL68 cells were harvested, and homogenized in iron assay buffer after washing with PBS. The supernatant was collected by centrifugation, and incubated with iron reducer. Iron probe was added and incubated for about 1 h finally. The results were determined by a colorimetric microplate reader (absorbance at OD 593 nm).
RNA interference and transfection
FPN-targeting siRNAs (siRNA-FPN) or non-targeting control siRNAs (siRNA-NC) were synthesized by RiboBio (Guangzhou, China). Three different siRNAs were designed according to different target sites in the FPN gene, and the sequences were listed as follows: #1 GGATGGGTCTCCTACTACA; #2 GGACAAGAATGCTAGACTT; and #3 GCACAGCTTTCCTGTTTGA. WRL68 cells were seeded in 3.5 cm culture dishes in the logarithmic growth phase and transiently transfected with FPN siRNAs or a control siRNA.
For siRNA transfection, WRL68 cells were cultured in six-well plates with approximately 50%–60% confluence, and transfected with 1 mg of siRNA (siRNA NC, siRNA FPN) by Lipofectamine 2000 (Cat. 11668-019, Invitrogen, USA) in accordance with manufacturer’s instructions. In brief, FPN siRNAs were transfected, and incubated with siRNA complexes at a final concentration of 100 nM for 48 h at 37°C with 5% CO2. Cells were harvested 48 h after transfection, and total cellular protein or mRNA was extracted for western blotting and qRT-PCR respectively, as well as iron content measurement.
Construction of shRNA plasmids and transfection
Short hairpin RNA (shRNA) constructs for transient silencing of AMPKα gene (sh-AMPKα) or non-targeting control shRNA (sh-NC) were synthesized by Honor Gene (Abiowell, Changsha, China) according to manufacturer’s instructions. Three different shRNAs were designed according to different target sites in the AMPKα gene, and the sequences were listed as follows: #1: GCGGCTCTTTCAGCAGATTCT; #2: GCTGAAGTTTACCGAGCTATG; and #3: GCTGTGAAAGAAGTGTGTGAA.
WRL68 cells with cell density of 2.5×105 were seeded in 6-well plate and incubated with MEM without antibiotics for 24 h, obtaining 70–80% confluency. Then, WRL68 cells were transfected with sh-AMPKα vector sh-AMPKα-1, sh-AMPKα-2, or sh-AMPKα-3, in each well, and empty shRNA as negative controls. The silencing efficiency of sh-AMPKα constructs were determined by RT-PCR, and the one with the highest silencing efficiency (sh-AMPKα-2) was used for the following experiments.
Plasmid transfections
WRL68 cells (1×107 per well) were seeded onto a 6-well plate with 10% MEM medium, and were washed completely. Then antibiotic-free medium was added for 6 h. WRL68 cells were transfected with GFP-tagged human overexpression FPN plasmid (Cat. RG205219, OriGene, USA), or empty vector as control, using Lipofectamine 2000 (Cat. 11668-019, Invitrogen, USA), according to the manufacturer’s protocol.
Co-immunoprecipitation
WRL68 cells were transiently transfected with 2 μg GFP/FPN for 48 h, then were treated with 0.4 mM PA for 24 h and/or 100 μM chloroquine (CQ) (Cat. C6628, Sigma, USA) for 2 h. Metformin (2.5 mM), AICAR (1 mM) (Cat. A1393, Sigma, USA), shRNA-AMPKα and shRNA-NC (Abiowell, Changsha, China) were co-incubated for 24 h. Cell proteins were extracted from cell lysates by RIPA lysis buffer, 1% protease inhibitor (Thermo Scientific, Dartford, UK), and cell extracts were incubated with GFP/FPN overnight at 4°C. Then, immunocomplexes were precipitated and incubated with 40 μL of protein A/G agarose beads for 2 h (Beyotime, P2012). The cell lysates and immunoprecipitates were separated on 10% SDS-PAGE, and transferred to PVDF membrane. Immunostaining with anti-FPN (Cat. sc-518125, Santa Cruz, USA), anti-Ubiquitin (Cat. 3933S, CST, USA), and anti-β-actin (Cat. ab6276, Abcam, UK) was performed, and western blotting was performed as described above.
Quantification and statistical analysis
All the results were analyzed by GraphPad Prism version 5.0 (GraphPad Software). The data were expressed as mean ± SEM. The comparisons between two groups were assessed by Student’s t tests, and the statistical differences among groups were determined by one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison. Correlation analysis between hepatic iron levels and IL-6, NAS, liver fibrosis scores was assessed by Pearson’s or Spearman’s test. p < 0.05 was defined as statistical significance, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (NO. 82070884), Natural Science Foundation of Hunan Province (No. 2021JJ31123), and Key laboratory of diabetes immunology, ministry of education (No. DKME202213).
Author contributions
Conceptualization, F.Z.Y. and D.M.Z.; methodology, F.Z.Y., Y.S., S.Y.W., L.X., L.W., A.Q.Q., and D.H.; investigation, F.Z.Y. and L.X.; writing – original draft, F.Z.Y. and D.M.Z.; writing – review & editing, F.Z.Y., D.M.Z., and R.R.; funding acquisition, D.M.Z.; resources, D.M.Z.; supervision, D.M.Z. All authors read and approved the final manuscript.
Declaration of interests
All the authors declare no conflicts of interest.
Published: November 22, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108560.
Supplemental information
References
- 1.Nakatsuka T., Tateishi R., Koike K. Changing clinical management of NAFLD in Asia. Liver Int. 2022;42:1955–1968. doi: 10.1111/liv.15046. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z., Anstee Q.M., Marietti M., Hardy T., Henry L., Eslam M., George J., Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018;15:11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 3.Sodum N., Kumar G., Bojja S.L., Kumar N., Rao C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021;167 doi: 10.1016/j.phrs.2021.105484. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Rangel E., Inzucchi S.E. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60:1586–1593. doi: 10.1007/s00125-017-4336-x. [DOI] [PubMed] [Google Scholar]
- 5.de Oliveira S., Houseright R.A., Graves A.L., Golenberg N., Korte B.G., Miskolci V., Huttenlocher A. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J. Hepatol. 2019;70:710–721. doi: 10.1016/j.jhep.2018.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wabitsch S., McCallen J.D., Kamenyeva O., Ruf B., McVey J.C., Kabat J., Walz J.S., Rotman Y., Bauer K.C., Craig A.J., et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J. Hepatol. 2022;77:748–760. doi: 10.1016/j.jhep.2022.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gawrieh S., Harlow K.E., Pike F., Yates K.P., Wilson L.A., Cummings O.W., Rosenberg W.M., Chalasani N., Molleston J.P. Relationship of Enhanced Liver Fibrosis Score with Pediatric Nonalcoholic Fatty Liver Disease Histology and Response to Vitamin E or Metformin. J. Pediatr. 2021;239:161–167.e5. doi: 10.1016/j.jpeds.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoni S., Tovoli F., Napoli L., Serio I., Ferri S., Bolondi L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018;24:3361–3373. doi: 10.3748/wjg.v24.i30.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsurusaki S., Tsuchiya Y., Koumura T., Nakasone M., Sakamoto T., Matsuoka M., Imai H., Yuet-Yin Kok C., Okochi H., Nakano H., et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10:449. doi: 10.1038/s41419-019-1678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu Y., Cao Y., Cao W., Jia Y., Lu N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020;159 doi: 10.1016/j.phrs.2020.104919. [DOI] [PubMed] [Google Scholar]
- 12.Li X., Wang T.X., Huang X., Li Y., Sun T., Zang S., Guan K.L., Xiong Y., Liu J., Yuan H.X. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020;40:1378–1394. doi: 10.1111/liv.14428. [DOI] [PubMed] [Google Scholar]
- 13.Rochette L., Dogon G., Rigal E., Zeller M., Cottin Y., Vergely C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022;24:449. doi: 10.3390/ijms24010449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao H., Jin Z., Bandyopadhyay G., Wang G., Zhang D., Rocha K.C.E., Liu X., Zhao H., Kisseleva T., Brenner D.A., et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022;34:1201–1213.e5. doi: 10.1016/j.cmet.2022.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corradini E., Buzzetti E., Dongiovanni P., Scarlini S., Caleffi A., Pelusi S., Bernardis I., Ventura P., Rametta R., Tenedini E., et al. Ceruloplasmin gene variants are associated with hyperferritinemia and increased liver iron in patients with NAFLD. J. Hepatol. 2021;75:506–513. doi: 10.1016/j.jhep.2021.03.014. [DOI] [PubMed] [Google Scholar]
- 16.Donovan A., Lima C.A., Pinkus J.L., Pinkus G.S., Zon L.I., Robine S., Andrews N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Knovich M.A., Storey J.A., Coffman L.G., Torti S.V., Torti F.M. Ferritin for the clinician. Blood Rev. 2009;23:95–104. doi: 10.1016/j.blre.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ursini F., Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020;152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 19.Ayala A., Muñoz M.F., Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014;2014 doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogt A.C.S., Arsiwala T., Mohsen M., Vogel M., Manolova V., Bachmann M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021;22:4591. doi: 10.3390/ijms22094591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 22.Shaaban H.H., Alzaim I., El-Mallah A., Aly R.G., El-Yazbi A.F., Wahid A. Metformin, pioglitazone, dapagliflozin and their combinations ameliorate manifestations associated with NAFLD in rats via anti-inflammatory, anti-fibrotic, anti-oxidant and anti-apoptotic mechanisms. Life Sci. 2022;308 doi: 10.1016/j.lfs.2022.120956. [DOI] [PubMed] [Google Scholar]
- 23.Huang S.W., Ou Y.C., Tang K.S., Yu H.R., Huang L.T., Tain Y.L., Lin I.C., Sheen J.M., Hou C.Y., Tsai C.C., Tiao M.M. Metformin ameliorates maternal high-fat diet-induced maternal dysbiosis and fetal liver apoptosis. Lipids Health Dis. 2021;20:100. doi: 10.1186/s12944-021-01521-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbasi U., Abbina S., Gill A., Takuechi L.E., Kizhakkedathu J.N. Role of Iron in the Molecular Pathogenesis of Diseases and Therapeutic Opportunities. ACS Chem. Biol. 2021;16:945–972. doi: 10.1021/acschembio.1c00122. [DOI] [PubMed] [Google Scholar]
- 25.Buzzetti E., Petta S., Manuguerra R., Luong T.V., Cabibi D., Corradini E., Craxì A., Pinzani M., Tsochatzis E., Pietrangelo A. Evaluating the association of serum ferritin and hepatic iron with disease severity in non-alcoholic fatty liver disease. Liver Int. 2019;39:1325–1334. doi: 10.1111/liv.14096. [DOI] [PubMed] [Google Scholar]
- 26.Beaton M.D., Chakrabarti S., Levstik M., Speechley M., Marotta P., Adams P. Phase II clinical trial of phlebotomy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013;37:720–729. doi: 10.1111/apt.12255. [DOI] [PubMed] [Google Scholar]
- 27.Murali A.R., Gupta A., Brown K. Systematic review and meta-analysis to determine the impact of iron depletion in dysmetabolic iron overload syndrome and non-alcoholic fatty liver disease. Hepatol. Res. 2018;48:E30–E41. doi: 10.1111/hepr.12921. [DOI] [PubMed] [Google Scholar]
- 28.Czaja A.J. Review article: iron disturbances in chronic liver diseases other than haemochromatosis - pathogenic, prognostic, and therapeutic implications. Aliment. Pharmacol. Ther. 2019;49:681–701. doi: 10.1111/apt.15173. [DOI] [PubMed] [Google Scholar]
- 29.Kang W., Barad A., Clark A.G., Wang Y., Lin X., Gu Z., O'Brien K.O. Ethnic Differences in Iron Status. Adv. Nutr. 2021;12:1838–1853. doi: 10.1093/advances/nmab035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dongiovanni P., Fracanzani A.L., Fargion S., Valenti L. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J. Hepatol. 2011;55:920–932. doi: 10.1016/j.jhep.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Buzzetti E., Pinzani M., Tsochatzis E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism. 2016;65:1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Fisher A.L., Srole D.N., Palaskas N.J., Meriwether D., Reddy S.T., Ganz T., Nemeth E. Iron loading induces cholesterol synthesis and sensitizes endothelial cells to TNFalpha-mediated apoptosis. J. Biol. Chem. 2021;297 doi: 10.1016/j.jbc.2021.101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang D., Chen X., Kang R., Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J., Massey S., Story D., Li L. Metformin: An Old Drug with New Applications. Int. J. Mol. Sci. 2018;19:2863. doi: 10.3390/ijms19102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nairz M., Weiss G. Iron in health and disease. Mol. Aspects Med. 2020;75 doi: 10.1016/j.mam.2020.100906. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C.S., Li M., Ma T., Zong Y., Cui J., Feng J.W., Wu Y.Q., Lin S.Y., Lin S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016;24:521–522. doi: 10.1016/j.cmet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Zhong S., Chen W., Wang B., Gao C., Liu X., Song Y., Qi H., Liu H., Wu T., Wang R., Chen B. Energy stress modulation of AMPK/FoxO3 signaling inhibits mitochondria-associated ferroptosis. Redox Biol. 2023;63 doi: 10.1016/j.redox.2023.102760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.You Y., Liu C., Liu T., Tian M., Wu N., Yu Z., Zhao F., Qi J., Zhu Q. FNDC3B protects steatosis and ferroptosis via the AMPK pathway in alcoholic fatty liver disease. Free Radic. Biol. Med. 2022;193:808–819. doi: 10.1016/j.freeradbiomed.2022.10.322. [DOI] [PubMed] [Google Scholar]
- 39.Song X., Zhu S., Chen P., Hou W., Wen Q., Liu J., Xie Y., Liu J., Klionsky D.J., Kroemer G., et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(-) Activity. Curr. Biol. 2018;28:2388–2399.e5. doi: 10.1016/j.cub.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y., Li M., Yao X., Fei Y., Lin Z., Li Z., Cai K., Zhao Y., Luo Z. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108487. [DOI] [PubMed] [Google Scholar]
- 41.Li C., Dong X., Du W., Shi X., Chen K., Zhang W., Gao M. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target. Ther. 2020;5:187. doi: 10.1038/s41392-020-00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J., Zhou Y., Xie S., Wang J., Li Z., Chen L., Mao M., Chen C., Huang A., Chen Y., et al. Metformin induces Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res. 2021;40:206. doi: 10.1186/s13046-021-02012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee H., Zandkarimi F., Zhang Y., Meena J.K., Kim J., Zhuang L., Tyagi S., Ma L., Westbrook T.F., Steinberg G.R., et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020;22:225–234. doi: 10.1038/s41556-020-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H.Y., Kwon W.Y., Park J.B., Lee M.H., Oh Y.J., Suh S., Baek Y.H., Jeong J.S., Yoo Y.H. Hepatic STAMP2 mediates recombinant FGF21-induced improvement of hepatic iron overload in nonalcoholic fatty liver disease. FASEB J. 2020;34:12354–12366. doi: 10.1096/fj.202000790R. [DOI] [PubMed] [Google Scholar]
- 45.Dev S., Kumari S., Singh N., Kumar Bal S., Seth P., Mukhopadhyay C.K. Role of extracellular Hydrogen peroxide in regulation of iron homeostasis genes in neuronal cells: Implication in iron accumulation. Free Radic. Biol. Med. 2015;86:78–89. doi: 10.1016/j.freeradbiomed.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 46.Li Y., Zeng X., Lu D., Yin M., Shan M., Gao Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021;36:951–964. doi: 10.1093/humrep/deaa363. [DOI] [PubMed] [Google Scholar]
- 47.Drakesmith H., Nemeth E., Ganz T. Ironing out Ferroportin. Cell Metab. 2015;22:777–787. doi: 10.1016/j.cmet.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang L., Wang J., Wang K., Wang H., Wu Q., Yang C., Yu Y., Ni P., Zhong Y., Song Z., et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood. 2021;138:689–705. doi: 10.1182/blood.2020008986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green C.J., Marjot T., Tomlinson J.W., Hodson L. Of mice and men: Is there a future for metformin in the treatment of hepatic steatosis? Diabetes Obes. Metab. 2019;21:749–760. doi: 10.1111/dom.13592. [DOI] [PubMed] [Google Scholar]
- 50.Zabielski P., Hady H.R., Chacinska M., Roszczyc K., Gorski J., Blachnio-Zabielska A.U. The effect of high fat diet and metformin treatment on liver lipids accumulation and their impact on insulin action. Sci. Rep. 2018;8:7249. doi: 10.1038/s41598-018-25397-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 52.Liu J., Wang J., Yan X., Xue R., Zhan J., Jiang S., Geng Y., Liu Y., Mao M., Xia J., et al. Presence of Liver Inflammation in Asian Patients With Chronic Hepatitis B With Normal ALT and Detectable HBV DNA in Absence of Liver Fibrosis. Hepatol. Commun. 2022;6:855–866. doi: 10.1002/hep4.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garuti C., Tian Y., Montosi G., Sabelli M., Corradini E., Graf R., Ventura P., Vegetti A., Clavien P.A., Pietrangelo A. Hepcidin expression does not rescue the iron-poor phenotype of Kupffer cells in Hfe-null mice after liver transplantation. Gastroenterology. 2010;139:315–322.e1. doi: 10.1053/j.gastro.2010.03.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.