

Abstract
In addition to housing the major energy producing pathways in cells, mitochondria are active players in innate immune responses. One critical way mitochondria fulfill this role is by releasing damage associated molecular patterns (mtDAMPs) that are recognized by innate sensors to activate pathways including, but not limited to, cytokine expression, selective autophagy, and cell death. Mitochondrial reactive oxygen species (mtROS) is a multifunctional mtDAMP linked to pro- and anti-microbial immune outcomes. Formed as a byproduct of energy generation, mtROS links mitochondrial metabolism with downstream innate immune responses. As a result, altered cellular metabolism can change mtROS levels and impact downstream antimicrobial responses in a variety of ways. MtROS has emerged as a particularly important mediator of pathogenesis during infection with Mycobacterium tuberculosis, an intracellular bacterial pathogen that continues to pose a significant threat to global public health. Here, we will summarize how Mtb modulates mtROS levels in infected macrophages and how mtROS dictates Mtb infection outcomes by controlling inflammation, lipid peroxidation, and cell death. We propose that mtROS may serve as a biomarker to predict tuberculosis patient outcomes and/or a target for host-directed therapeutics.
MtROS in cellular metabolism and homeostasis
Generation of mtROS is a natural consequence of the electron transport chain (ETC). The ETC operates as a series of complexes spanning the inner mitochondrial membrane that shuttle high energy electrons sourced from NADH or FADH2, produced by glycolysis or the Krebs cycle, to generate an electrochemical proton gradient. As this gradient is generated, some electrons escape the normal ETC. The cell typically manages these reactive electrons, most of which are in the form of ROS, through a series of enzymatic reactions and antioxidant pathways. For example, superoxide species can be dismutated to hydrogen peroxide by superoxide dismutase 2 (SOD2), then converted into water with catalase. Lipid peroxides, created when ROS interacts with membrane phospholipids, can be reduced at the expense of glutathione using enzymes such as glutathione peroxidase 4 (GPX4), which is active in the mitochondria (Amaral et al., 2022). However, when there is a dramatic shift in metabolism, such as during infection, the balance between ETC usage and the ability to deal with high energy electrons can be disrupted and high levels of mtROS can accumulate.
MtROS in infection and immunity
Generation of cellular ROS is traditionally thought of as an antimicrobial strategy. Work from the O’Riordan lab and others show that endoplasmic reticulum (ER) stress stimulates mitochondria via IRE1α to produce vesicles containing SOD2. SOD2-containing vesicles are delivered to bacteria-containing phagosomes where SOD2 catalyzes peroxide production, which can control bacterial burdens in the case of methicillin-resistant Staphylococcus aureus infections (Abuaita et al., 2018). In Mycobacterium avium infection, pyruvate-driven reverse electron transport (RET, a process whereby high energy electrons from ubiquinol are transferred back to Complex I, reducing NAD+ to NADH (Scialò et al., 2017)), generates mtROS to control intracellular bacterial burdens (Røst et al., 2022). Several antimicrobials, including moxifloxacin (Shee et al., 2022) have been shown to act by increasing levels of mtROS (Leferman et al., 2023, Zhao et al., 2022) and current antimycobacterials can promote ROS formation, which may contribute to their antibacterial activity (Howell Wescott et al., 2017, Yano et al., 2011). Indeed, the antidiabetic drug metformin, which can act as an adjunctive against Mtb, works by increasing mtROS (Singhal et al., 2014).
Because mtROS can be antimicrobial, many pathogens, including Mtb, have evolved specific measures to mitigate its effects. For example, the major Mtb antigen and ESX-1 type VII secretion system substrate ESAT-6 can decrease mtROS levels in cells (Yabaji et al, 2020). ESAT-6 plays a well-known role in Mtb virulence by destabilizing the Mtb phagosomal membrane to promote release of Mtb secreted proteins into the cytosol and allow cytosolic sensors access to the endosomal lumen (Bell et al., 2021, Conrad et al., 2017, Osman et al., 2022). When ESAT-6 is expressed in Mycobacterium bovis BCG (BCG), it can upregulate macrophage Sod2 to catalyze the dismutation of superoxide into hydrogen peroxide (Yabaji et al., 2020). The authors of this study link loss of SOD2 with increased acidification of the BCG phagosome and decreased BCG intracellular survival, although the dysregulated nature of ESAT-6 secretion in this system (which is directed via addition of the FbpB sec-dependent signal peptide and not ESX-1, as BCG lacks a type VII secretion system) presents a caveat for interpretation. Mtb may also encode its own ROS detoxification molecules, such as MTS1338, a small RNA induced by oxidative stress that is associated with increased catalase activity (Singh et al., 2021). Thus, while ROS may limit survival of avirulent mycobacterial species in in vitro models, virulent Mtb has evolved strategies to survive and replicate in a high ROS environment and/or directly modulate cellular ROS levels.
There is also evidence that Mtb controls mtROS generation by manipulating cellular energy production. Mtb infection of macrophages depresses the rate of mitochondrial respiration, which limits production of mtROS at ETC complexes I and II (Cumming et al., 2018) (Fig. 1A). Whether this decrease is accompanied by a compensatory increase in glycolysis (commonly known as the Warburg shift) remains a topic of debate. There is some transcriptional evidence for the Warburg shift in animal models of tuberculosis (Shi et al., 2015), but metabolic profiling of infected human macrophages ex vivo argues instead for Mtb promoting a quiescent state characterized by a low glycolytic proton efflux rate (Cumming et al., 2018). It is likely that Mtb dynamically reprograms macrophages in different ways, depending on factors like multiplicity of infection, macrophage cell type, and time after infection. Whether this reprogramming primarily serves to metabolically switch macrophages from oxidative phosphorylation to fatty acid metabolism (Huang et al., 2018), manipulate mtROS levels, or both remains an important outstanding question.
Figure 1: Altered mitochondrial function and mtROS are intimately linked with mycobacterial infection outcomes.
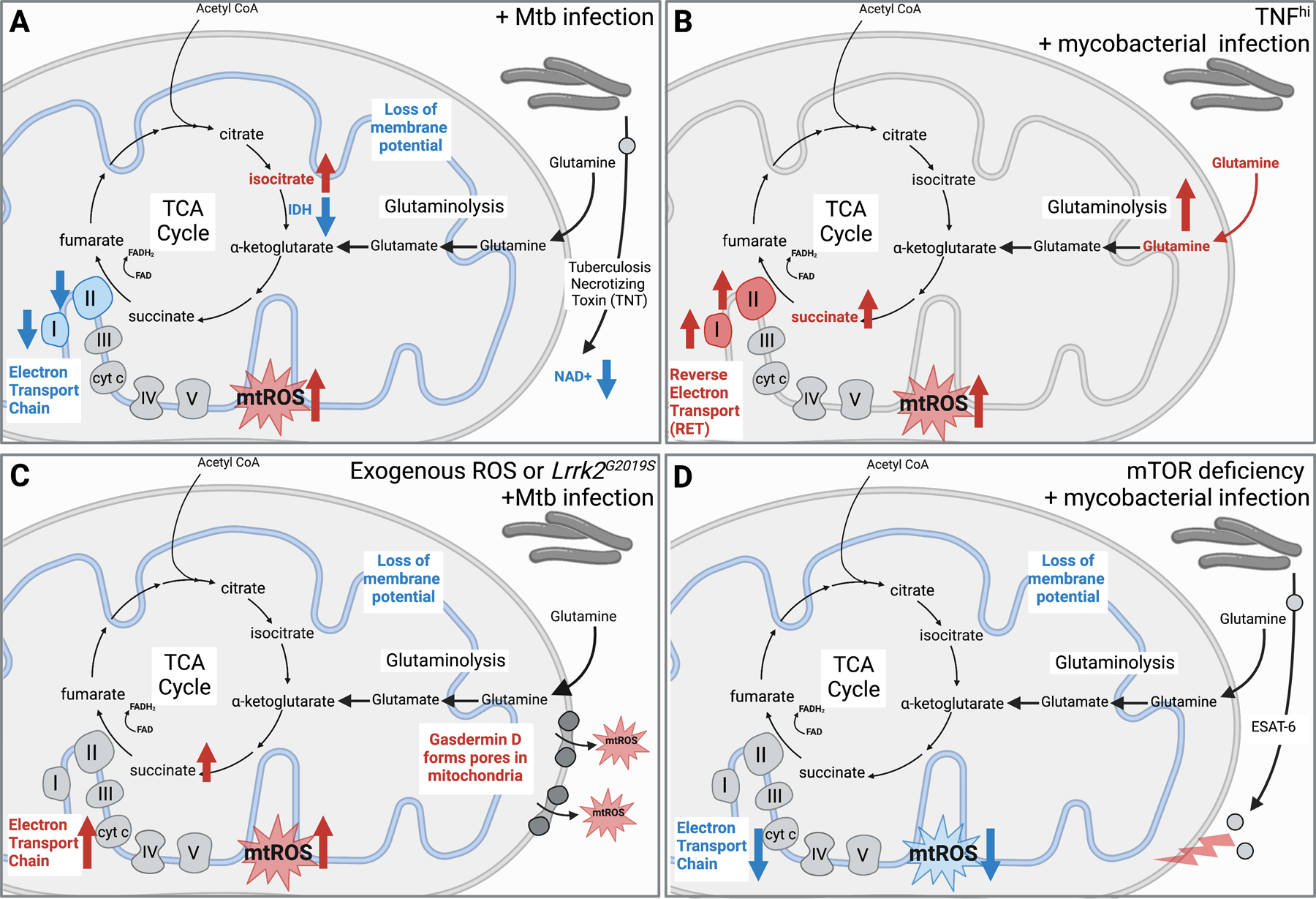
A. Smulan et al. show that Mtb infection increases isocitrate, reduces complex I and II activity in the electron transport chain, and results in loss of the mitochondrial membrane potential. Pajuelo et al. demonstrate NAD+ hydrolysis mediated by the Mtb secreted protein Tuberculosis necrotizing toxin (TNT) promotes mtROS generation and necrotic cell death.
B. Roca et al. show that mycobacterial infection in the presence of high TNF levels leads to increased glutamine uptake, glutaminolysis, and accumulation of succinate, which promotes succinate oxidation and reverse electron transport to produce large amount of mtROS. They link this mtROS to mycobacterial replication/cording and necroptotic cell death.
C. Weindel et al. report that in the presence of the Lrrk2G2019S mutation or in the presence of excess ROS, canonical inflammasome activation promotes GSDMD pore formation in mitochondrial membranes, which is associated with higher mtROS, necroptotic cell death, and hyperinflammation during Mtb infection in vivo.
D. Pagan et al. demonstrate that mycobacterial infection in mTOR deficient cells leads to reduced electron transport and mtROS, which they link to increased sensitivity to ESAT-6-mediated mitochondrial damage.
Mtb also dampens mitochondrial respiration by downregulating the TCA cycle via IDH2 and succinate dehydrogenase (Smulan et al., 2021) (Fig. 1A). Because IDH2 is responsible for the regeneration of the antioxidant glutathione, its downregulation limits the pool of glutathione available for antioxidant function (Smulan et al., 2021). Regulation of glutathione by IDH2 is dependent on Sirtuin 3 (SIRT3), a NAD+ dependent mitochondrial protein, that is downregulated in host macrophages during infection with Mtb. Consequently, murine BMDMs deficient in SIRT3 have increased necrosis and increased mtROS compared to wild type BMDMs and Sirtuin 3-deficient mice suffer higher bacterial burdens (Smulan et al., 2021). These data begin to suggest that mtROS is associated with increased Mtb pathogenesis, despite its antibacterial capacity.
MtROS in cell death
One way that mtROS promotes mycobacterial pathogenesis is by acting as a DAMP to activate cell death pathways that help Mtb spread to neighboring cells and/or create a pro-bacterial inflammatory milieu. Early studies linked mtROS to a form of inflammatory cell death known as pyroptosis (Zhou et al., 2010). During pyroptosis, assembly of the inflammasome activates caspase-1, which cleaves IL-1ꞵ and gasdermin D (GSDMD), the latter of which forms pores in the plasma membrane and promotes membrane rupture with the help of the protein NINJ1 (Bjanes et al., 2021). Subsequent studies have shown that mtROS is not required for NLRP3 activation (Billingham et al., 2022). Instead, as several steps in pyroptosis can be pushed forward by oxidation (e.g., oxidation of GSDMD promotes its oligomerization and pore formation (Devant et al., 2023, Kang et al., 2018)), mtROS likely contributes to pyroptotic cell death downstream of inflammasome activation. Accordingly, during Mtb infection, mitochondrial damage and depolarization is observed with inflammasome activation—not prior to it (Beckwith et al, 2020). Studies of Mtb strains that hyperactivate the inflammasome (i.e., ΔpknF) also argue against a role for mtROS in activating NLRP3, suggesting instead that modulation of xanthine oxidase activity is important in the context of Mtb infection (Rastogi et al., 2021). Consistent with Mtb infection introducing many PAMPs/DAMPs capable of priming/stimulating pyroptosis (Bronner et al., 2015), it seems unlikely that mtROS is a major driver of inflammasome activation during Mtb infection.
Despite debate surrounding mtROS’s ability to directly activate the inflammasome, there is strong evidence linking mtROS to activation of RIPK1 and necroptosis (Zhang et al., 2017). Necroptosis is a regulated form of cell death characterized by cell swelling and rupture induced by MLKL plasma membrane pores. Necroptosis has been repeatedly associated with poor Mtb disease outcomes in mouse models of disease and in human patients (Roca & Ramakrishnan, 2013, Srinivasan et al., 2016, Weindel et al., 2022). Curiously, the only toxin characterized to date in Mtb has been linked to mtROS and necroptosis. Tuberculosis necrotizing toxin (TNT), the C-terminal domain of Mtb outer membrane channel protein CpnT, has NAD+ glycohydrolase activity (Pajuelo et al., 2018 and 2020) (Fig. 1A). Deleting the CpnT gene from Mtb decreases ROS levels and limits cell death during infection, although some ROS made during Mtb infection is TNT-independent (Pajuelo et al., 2020). Treatment with cyclosporin A, a selective inhibitor of the mitochondrial permeability transition pore (MPTP) reduces TNT-dependent ROS production, strongly linking NAD+ depletion and formation of the MPTP to ROS production and cell death. In reductionist experiments, NAD+ depletion was sufficient to trigger macrophage necroptosis in a RIPK1/RIPK3-dependent fashion, but ROS generated by addition of the redox cycling agent menadione was not (Pajuelo et al, 2020), suggesting that additional NAD+ functions (e.g., regulating mitochondrial metabolism or fatty acid oxidation) are important for TNT-mediated necroptosis. Additional Mtb virulence factors have also been associated with mitochondrial damage and necrosis. For example, loss of the Mtb protein Rv3167c, a transcriptional repressor of the cell wall lipid PDIM (Quigley et al., 2017) has been linked to mitochondrial ROS-dependent cell death (Srinivasan et al., 2016). Furthermore, infection of human monocytes with the H37Rv strain of Mtb induces loss of mitochondrial inner membrane potential leading to necrosis but infection with the avirulent strain H37Ra (which carries mutations in a variety of virulence-associated factors including PE/PPE genes and PhoP (Zheng et al., 2008)) does not (Chen et al., 2006). These data suggest that Mtb has evolved multiple strategies to manipulate the mitochondrial network and push cells towards necrotic forms of cell death.
Host genetics also play a role in regulating the mtROS-necroptosis axis during Mtb infection. Recent work out of our labs showed that macrophages harboring a common human SNP in the leucine rich repeat kinase 2 (LRRK2) gene are prone to undergo inflammatory cell death in response to canonical inflammasome activation and Mtb infection (Weindel et al, 2022). Curiously, despite being triggered by inflammasome stimuli via either NLRP3 or AIM2, this death has hallmarks of necroptosis as opposed to pyroptosis (i.e. IL-1β release is limited and death is RIPK1/RIPK3/MLKL-dependent). In dissecting this pathway, we found that mitochondrial dysfunction associated with the Lrrk2G2019S mutation, including overreliance on the electron transport chain following innate stimuli, generates high levels of mtROS. This mtROS renders the mitochondria susceptible to permeabilization by GSDMD (cleaved/activated downstream of inflammasome activation), which promotes mtROS release, and pushes cells to die from necroptosis downstream of inflammasome activation in a GSDMD-dependent fashion (dubbed GSDMD-mediated necroptosis) (Fig. 1C). This form of cell death is novel and whether it occurs in contexts outside of Lrrk2G2019S mutants, and what those contexts might be, remains to be seen. Because GSDMD-mediated necroptosis is amplified via the addition of menadione, which generates mtROS through redox cycling, and rescued by the redox scavenger Necrox-5, our data position mtROS as the executioner of hyperinflammatory, necroptotic cell death during Mtb infection. Notably, Lrrk2G2019S mice are extremely susceptible to Mtb infection: they experience hyperinflammation in the lung and a dramatic influx of neutrophils. In this way, enhanced activation of the necroptosome, here, via GSDMD-mediated mitochondrial damage and mtROS release, seems to promote Mtb pathogenesis (Weindel et al. 2022). These data, together with the TNT literature above strongly argue that disruption of mitochondrial membranes (through either GSDMD pore formation or MPTP opening) is a major contributor to inflammatory cell death during Mtb infection and a possible point for therapeutic intervention.
Consistent with mtROS playing a preeminent role in dictating cell death and inflammatory outcomes during Mtb infection, work out of the Ramakrishnan lab has repeatedly implicated these same pathways in the zebrafish model of mycobacterial pathogenesis. Their work shows that high levels of TNF render zebrafish susceptible to Mycobacterium marinum infection via mtROS-enhanced necrosis (Roca and Ramakrishnan 2013; Roca et al., 2022). Using zebrafish, but validating several key results in human macrophages, the lab has worked out a model whereby high TNF stimulates mtROS by inducing reverse electron transport at Complex I downstream of TNF-induced glutamine uptake and increased glutaminolysis (Fig. 1B). This leads to an increased concentration of succinate, which when oxidized by Complex II, drives reverse electron transport and mtROS superoxide at Complex I. This TNF-glutamine-succinate pathway is pushed forward by RIPK3 and PGAM5 in a manner that is seemingly functionally analogous to RIPK3-dependent mtROS formation described in mammalian cells, which occurs via phosphorylation of pyruvate dehydrogenase by RIPK3 (Yang et al. 2019). In the zebrafish, the consequences of this high TNF pathway are dramatic, with enhanced necrosis and extracellular bacilli replication (cording) that is not observed in wild-type fish (Roca et al., 2022), reminiscent of the hyperinflammatory phenotype observed in Lrrk2G2019S mice during Mtb infection (Weindel et al., 2022). Together, these data suggest that pre-existing conditions or co-infection, where Mtb-infected macrophages could face high TNF and/or oxidative stress, may steer cells towards necroptosis even when other cell death pathways (pyroptosis, apoptosis, etc.) are engaged.
Further supporting the idea that mitochondrial dysfunction renders cells more sensitive to cell death during Mtb infection, mTOR-deficient zebrafish and macrophages are susceptible to cytotoxicity associated with mycobacterial ESAT-6 (Pagán et al., 2022). While cell death in mTOR-deficient animals/macrophages was not associated with high mtROS, it was proceeded by loss of mitochondrial membrane potential and ATP production (Fig. 1D). Because mTOR is a master regulator of many critical cellular functions, it will be important to continue to investigate the mechanisms through which mTOR protects against Mtb cytotoxicity. For example, translation of GPX4, the enzyme that catalyzes the reduction of toxic lipid peroxides in the mitochondria, is mediated by mTORC1 in response to cystine availability (Zhang et al., 2021), highlighting the complex roles mTOR may play in regulating cellular redox during Mtb infection.
Concluding remarks
As studies continue to link mitochondrial dysfunction and oxidative stress with poor tuberculosis disease outcomes, the mycobacterium field is experiencing a paradigm shift regarding the role of antimicrobial mediators like mtROS in Mtb pathogenesis. Outstanding questions of key importance include how mtROS impacts the biology of both host and bacterial lipids. We know that fatty acids, rather than carbohydrates, are the major energy source for Mtb inside macrophages (Pandey & Sassetti, 2008; Ehrt et al., 2018). Are these lipids modified in the face of high mtROS? Does this change their availability as an energy source for Mtb? MtROS might also impact the structural integrity of the Mtb phagosome phospholipid bilayer, thus impacting Mtb cytosolic access. Likewise, mtROS could modify Mtb-derived PAMPs/DAMPs such that they have altered affinity for pattern recognition receptors (e.g., oxidation of dsDNA from Mtb or the mitochondria could alter its ability to engage cytosolic DNA sensors). Careful mechanistic experiments will be needed to assess the degree to which each of these potential roles for mtROS impacts macrophage biology and innate immune outcomes in response to Mtb.
The role of redox reactions that might mitigate the effects of mtROS in the context of Mtb infection also demands further investigation. We know that GPX4 limits Mtb pathogenesis in large part by preventing cell death via the inflammatory cell death modality ferroptosis (Amaral et al., 2019, Amaral et al., 2022). However, we do not fully understand how GPX4 might regulate cellular redox pathways through mechanisms independent of iron/ferroptosis (Amaral et al., 2019, Amaral et al., 2022). Mtb produces effectors known to traffic to the nucleus and downregulate Gpx4 expression during infection of host macrophages, suggesting a role for GPX4-dependent pathways in controlling infection (Qiang et al., 2023). In situations where GPX4 is not able to keep up with oxidative stress (e.g., in cells harboring mitochondrial mutations or subject to mitochondrial stress), oxidized phospholipids may accumulate, which could alter cell death modalities that rely on pore formation in phospholipid membranes via MLKL and GSDMD/NINJ1. Coalescing these studies highlights the myriad host- and pathogen-dependent variables that dictate whether mtROS promotes Mtb clearance or aids in its survival and dissemination. Unravelling how signals generated by mtROS are integrated into a balanced immune response to control Mtb will be key to identifying regulatory tipping points that we can target for therapeutic intervention.
Acknowledgements
Our apologies to the authors of reports that could not be highlighted due to space limitations. This work was supported by NIH/NIAID R01AI155621 to ROW and KP.
Footnotes
Declaration of Interests: The authors declare no competing interests.
References
- Abuaita BH, Schultz TL, O’Riordan MX: Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host & Microbe 2018, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, Sher A: A major role for ferroptosis in mycobacterium tuberculosis–induced cell death and tissue necrosis. Journal of Experimental Medicine 2019, 216:556–570. * This critical report demonstrates that ferroptosis, a newly described iron-dependent cell death pathway associated with oxidative stress, contributes to Mtb pathogenesis in vivo and can be rescued with ferroptosis inhibitors.
- Amaral EP, Foreman TW, Namasivayam S, Hilligan KL, Kauffman KD, Barbosa Bomfim CC, Costa DL, Barreto-Duarte B, Gurgel-Rocha C, Santana MF, et al. : GPX4 regulates cellular necrosis and host resistance in Mycobacterium tuberculosis infection. Journal of Experimental Medicine 2022, 219. * A follow-up to their 2019 study, here, Amaral et al., implicate GPX4 in controlling oxidative stress and ferroptosis during Mtb infection and suggest modulation of GPX4 levels/activity as potential tuberculosis host-direct therapy.
- Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, Åsberg SE, Strand TA, Haug M, Niederweis M, et al. : Plasma membrane damage causes NLRP3 activation and pyroptosis during mycobacterium tuberculosis infection. Nature Communications 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SL, Lopez KL, Cox JS, Patrick KL, Watson RO: Galectin-8 senses phagosomal damage and recruits selective autophagy adapter TAX1BP1 to control Mycobacterium tuberculosis infection in macrophages. mBio 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, Jacobs HT, Reczek CR, Rashidi A, Zhang P, et al. : Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nature Immunology 2022, 23:692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjanes E, Sillas RG, Matsuda R, Demarco B, Fettrelet T, DeLaney AA, Kornfeld OS, Lee BL, Rodríguez López EM, Grubaugh D, et al. : Genetic targeting of CARD19 is linked to disrupted NINJ1 expression, impaired cell lysis, and increased susceptibility to yersinia infection. PLOS Pathogens 2021, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, Yin X-M, O’Riordan MXD: Endoplasmic Reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 2015, 43:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Gan H, Remold HG: A mechanism of virulence: Virulent Mycobacterium tuberculosis strain H37RV, but not attenuated H37RA, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. The Journal of Immunology 2006, 176:3707–3716. [DOI] [PubMed] [Google Scholar]
- Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, Hopkinson-Woolley D, Brosch R, Ramakrishnan L: Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proceedings of the National Academy of Sciences 2017, 114:1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devant P, Boršić E, Ngwa EM, Xiao H, Chouchani ET, Thiagarajah JR, Hafner-Bratkovič I, Evavold CL, Kagan JC: Gasdermin D pore-forming activity is redox-sensitive. Cell Reports 2023, 42:112008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S, Schnappinger D, Rhee KY: Metabolic principles of persistence and pathogenicity in mycobacterium tuberculosis. Nature Reviews Microbiology 2018, 16:496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell Wescott HA, Roberts DM, Allebach CL, Kokoczka R, Parish T: Imidazoles induce reactive oxygen species in Mycobacterium tuberculosis which is not associated with cell death. ACS Omega 2017, 2:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, Russell DG: Growth of mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. Journal of Experimental Medicine 2018, 215:1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, et al. : Lipid peroxidation drives Gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host & Microbe 2018, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leferman C-E, Stoica L, Stoica BA, Ciubotaru AD, Badescu AC, Bogdanici C-M, Neagu TP, Ghiciuc C-M: Mitochondria-targeted curcumin: A potent antibacterial agent against methicillin-resistant Staphylococcus aureus with a possible intracellular ROS accumulation as the mechanism of action. Antibiotics 2023, 12:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienard J, Nobs E, Lovins V, Movert E, Valfridsson C, Carlsson F: The Mycobacterium marinum ESX-1 system mediates phagosomal permeabilization and type I interferon production via separable mechanisms. Proceedings of the National Academy of Sciences 2019, 117:1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Tao J, Duan F, Li H, Tan H: HHCY induces pyroptosis and atherosclerosis via the lipid raft-mediated NOx-Ros-NLRP3 inflammasome pathway in apoe−/− mice. Cells 2022, 11:2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li M, Chen Z, Yu Y, Shi H, Yu Y, Wang Y, Chen R, Ge J: Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Research in Cardiology 2022, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman MM, Shanahan JK, Chu F, Takaki KK, Pinckert ML, Pagán AJ, Brosch R, Conrad WH, Ramakrishnan L: The C terminus of the mycobacterium ESX-1 secretion system substrate ESAT-6 is required for phagosomal membrane damage and virulence 2022, doi: 10.1101/2022.01.14.476355. [DOI] [PMC free article] [PubMed]
- Pagán AJ, Lee LJ, Edwards-Hicks J, Moens CB, Tobin DM, Busch-Nentwich EM, Pearce EL, Ramakrishnan L: MTOR-regulated mitochondrial metabolism limits mycobacterium-induced cytotoxicity. Cell 2022, 185. * Through a powerful forward genetic screen and follow-up experiments in mTOR-deficient zebrafish, the authors identify mTOR as a host resistance factor to mycobacterial infection. Specifically, they report that mTOR supports oxidative phosphorylation and resistance to mitochondrial dysfunction/cytotoxicity elicited by the secreted virulence factor ESAT-6.
- Pajuelo D, Gonzalez-Juarbe N, Tak U, Sun J, Orihuela CJ, Niederweis M: NAD+ depletion triggers macrophage necroptosis, a cell death pathway exploited by mycobacterium tuberculosis. Cell Reports 2018, 24:429–440. * Suggesting that Mtb evolved strategies to specifically induce mitochondrial stress and necroptosis, the authors link TNT-dependent NAD+ hydrolysis and opening of the mitochondrial permeability transition pore to loss of mitochondrial inner membrane potential and mtROS accumulation.
- Pajuelo D, Gonzalez‐Juarbe N, Niederweis M: NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages. Cellular Microbiology 2019, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM: Mycobacterial persistence requires the utilization of host cholesterol. Proceedings of the National Academy of Sciences 2008, 105:4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri G, Naura AS: Implication of mitochondrial Ros-NLRP3 inflammasome axis during two-hit mediated acute lung injury in mice. Free Radical Research 2022, 56:1–16. [DOI] [PubMed] [Google Scholar]
- Qiang L, Zhang Y, Lei Z, Lu Z, Tan S, Ge P, Chai Q, Zhao M, Zhang X, Li B, et al. : A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nature Communications 2023, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley J, Hughitt VK, Velikovsky CA, Mariuzza RA, El-Sayed NM, Briken V. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. mBio 2017. Mar 7;8(2):e00148–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi S, Ellinwood S, Augenstreich J, Mayer-Barber KD, Briken V: Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase pknf. PLOS Pathogens 2021, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca FJ, Ramakrishnan L: TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153:521–534. ** This seminal paper demonstrates that TNF can promote necroptosis and exuberant extracellular mycobacterial growth through a mechanism that depends on mitochondrial ROS.
- Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L: Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376. * A follow-up to their 2013 paper, here, Roca et al. implicate reverse electron transport at mitochondrial Complex I in generating mtROS and promoting necroptosis in TNFhi zebrafish/macrophages during mycobacterial infection.
- Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L: TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell 2019, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Røst LM, Louet C, Bruheim P, Flo TH, Gidon A: Pyruvate supports RET-dependent mitochondrial ROS production to control mycobacterium avium infection in human primary macrophages. Frontiers in Immunology 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialò F, Fernández-Ayala DJ, Sanz A: Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Frontiers in Physiology 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shee S, Singh S, Tripathi A, Thakur C, Kumar TA, Das M, Yadav V, Kohli S, Rajmani RS, Chandra N, et al. : Moxifloxacin-mediated killing of mycobacterium tuberculosis involves respiratory downshift, reductive stress, and accumulation of reactive oxygen species. Antimicrobial Agents and Chemotherapy 2022, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML: Infection with mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Scientific Reports 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Nirban R, Dutta T: MTS1338 in mycobacterium tuberculosis promotes detoxification of reactive oxygen species under oxidative stress. Tuberculosis 2021, 131:102142. [DOI] [PubMed] [Google Scholar]
- Singhal A, Jie L, Kumar P, Hong GS, Leow MK-S, Paleja B, Tsenova L, Kurepina N, Chen J, Zolezzi F, et al. : Metformin as adjunct antituberculosis therapy. Science Translational Medicine 2014, 6. [DOI] [PubMed] [Google Scholar]
- Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao L-Y: Evidence for pore formation in host cell membranes by ESX-1-secreted Esat-6 and its role in Mycobacterium marinum escape from the vacuole. Infection and Immunity 2008, 76:5478–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulan LJ, Martinez N, Kiritsy MC, Kativhu C, Cavallo K, Sassetti CM, Singhal A, Remold HG, Kornfeld H: Sirtuin 3 downregulation in mycobacterium tuberculosis-infected macrophages reprograms mitochondrial metabolism and promotes cell death. mBio 2021, 12. * This report links Sirtuin 3 to modulation of the TCA cycle and increased reactive oxygen species during Mtb infection of macrophages. Importantly, they show that mice harboring Sirt3−/− myeloid cells experience higher bacterial burdens and greater immunopathology, directly linking SIRT3 activity in macrophages to tuberculosis disease outcomes.
- Srinivasan L, Gurses SA, Hurley BE, Miller JL, Karakousis PC, Briken V: Identification of a transcription factor that regulates host cell exit and virulence of mycobacterium tuberculosis. PLOS Pathogens 2016, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, Coleman AK, VanPortfliet JJ, Zhao B, Wagner AR, et al. : Mitochondrial ROS promotes susceptibility to infection via GASDERMIN D-mediated necroptosis. Cell 2022, 185. ** This study demonstrates that mitochondrial dysfunction can direct immune outcomes via cell death modality switching. Specifically, the authors describe a new type of cell death whereby inflammasome activation promotes necroptosis in cells with high mitochondrial ROS and link this cell death to poor disease outcomes in a mouse model of tuberculosis.
- Xie H, Peng J, Zhang X, Deng L, Ding Y, Zuo X, Wang F, Wu Y, Zhang J, Zhu Q: Effects of mitochondrial reactive oxygen species-induced NLRP3 inflammasome activation on trichloroethylene-mediated kidney immune injury. Ecotoxicology and Environmental Safety 2022, 244:114067. [DOI] [PubMed] [Google Scholar]
- Yabaji SM, Dhamija E, Mishra AK, Srivastava KK: Esat-6 regulates autophagous response through SOD-2 and as a result induces intracellular survival of mycobacterium bovis BCG. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2020, 1868:140470. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang Y, Zhang Y, He X, Zhong C-Q, Ni H, Chen X, Liang Y, Wu J, Zhao S, et al. : RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nature Cell Biology 2018, 20:186–197. [DOI] [PubMed] [Google Scholar]
- Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H: Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase. Journal of Biological Chemistry 2011, 286:10276–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Pan C, Feng C, Yan C, Yu Y, Chen Z, Guo C, Wang X: Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Report 2022, 27:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Su SS, Zhao S, Yang Z, Zhong C-Q, Chen X, Cai Q, Yang Z-H, Huang D, Wu R, et al. : RIP1 autophosphorylation is promoted by mitochondrial Ros and is essential for rip3 recruitment into necrosome. Nature Communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, Lei G, Mao C, Koppula P, Cheng W, et al. : MTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nature Communications 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Du B, Xu J, Xie Q, Lu Z, Kang Y: Baicalin promotes antibacterial defenses by modulating mitochondrial function. Biochemical and Biophysical Research Communications 2022, 621:130–136. [DOI] [PubMed] [Google Scholar]
- Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, Shi W, Zhang L, Wang H, Wang S, Zhao G, Zhang Y. Genetic Basis of Virulence Attenuation Revealed by Comparative Genomic Analysis of Mycobacterium tuberculosis Strain H37Ra versus H37Rv. 2008. PLoS One; 3(6):e2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J: A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469:221–225. * A seminal paper describing links between mitochondrial ROS and NLRP3 inflammasome activation. This was one of the first studies to link mitochondria with inflammatory cell death pathways that are distinct from apoptosis.