

SUMMARY
Identifying molecular circuits that control adipose tissue macrophage (ATM) function is necessary to understand how ATMs contribute to tissue homeostasis and obesity-induced insulin resistance. In this study, we find that mice with a myeloid-specific knockout of the miR-23–27-24 clusters of microRNAs (miRNAs) gain less weight on a high-fat diet but exhibit worsened glucose and insulin tolerance. Analysis of ATMs from these mice shows selectively reduced numbers and proliferation of a recently reported subset of lipid-associated CD9+Trem2+ ATMs (lipid-associated macrophages [LAMs]). Leveraging the role of miRNAs to control networks of genes, we use RNA sequencing (RNA-seq), functional screens, and biochemical assays to identify candidate target transcripts that regulate proliferation-associated signaling. We determine that miR-23 directly targets the mRNA of Eif4ebp2, a gene that restricts protein synthesis and proliferation in macrophages. Altogether, our study demonstrates that control of proliferation of a protective subset of LAMs by noncoding RNAs contributes to protection against diet-induced obesity metabolic dysfunction.
In brief
Trem2+CD9+ lipid-associated macrophage (LAM) accrual has been reported to support white adipose tissue function in obesity. Sprenkle et al. demonstrate that myeloid-specific expression of miR-23–27-24 confers protection against obesity-induced defects in glucose metabolism and promotes LAM proliferation, in part, by suppressing the expression of Eif4ebp2, a negative regulator of translation.
Graphical Abstract
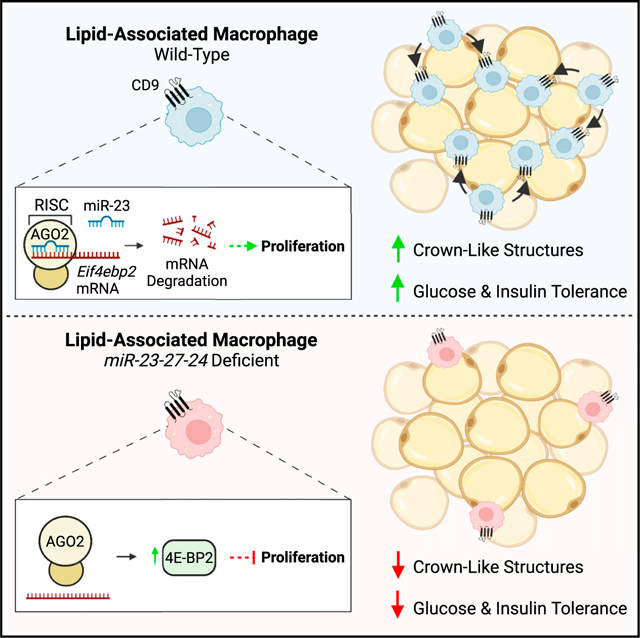
INTRODUCTION
Obesity is a risk factor for metabolic diseases, including type 2 diabetes and cardiovascular disease, by promoting peripheral insulin resistance (IR).1 Visceral white adipose tissue (WAT) IR is a crucial contributor to obesity-induced metabolic impairments due to its roles as a primary lipid storage depot and dynamic endocrine organ.2 Accompanying WAT dysfunction in obesity is dramatic cellular and physiological remodeling within the myeloid cell compartment.3,4 A critical event contributing to WAT IR is macrophage accumulation and sterile activation. In lean WAT, adipose tissue macrophages (ATMs) maintain tissue integrity by fostering an immunosuppressive microenvironment and regulating tissue iron and lipid homeostasis.5–8 In contrast, chronic overnutrition and increased visceral adiposity incite local proliferation of tissue-resident macrophages and recruitment of monocyte-derived macrophages into WAT, where they acquire an activation program that promotes aberrant production of pro-inflammatory factors that disrupt adipocyte function.9–14
While early studies assigned ATMs as key drivers of WAT dysfunction by propagating low-grade tissue inflammation, recent evidence indicates that the lipid-handling and phagocytic capabilities of monocyte-derived CD9+Trem2+ ATMs can confer protection against adipocyte hypertrophy and systemic metabolic dysregulation in obesity.4 Mechanistically, it has been proposed that these lipid-associated macrophages (LAMs) accrue within crown-like structures and phagocytize dying lipid-laden adipocytes from the microenvironment to prevent lipid spillover that results in pathology.4 Importantly, the regulation of LAMs is poorly defined. While most work has focused on Trem2 as a major driver of the transcriptional and functional profile of LAMs, reports on the metabolic consequences of Trem2 ablation are mixed.4,15,16 This suggests that other key regulators could be involved in LAM function and highlights a complex interaction between innate immunity and WAT physiology. Mechanisms driving macrophage accumulation in obese WAT and their physiologic functions remain incompletely understood. Determining the molecular regulation and function of macrophage subpopulations in obesity is essential for understanding the pathophysiology of obesity-associated metabolic diseases.
Small RNAs, such as microRNAs (miRNAs), have emerged as essential regulators of macrophage function.17–19 Unlike messenger RNAs (mRNAs), miRNAs do not encode protein products and instead contribute to post-transcriptional gene silencing of target mRNAs. Therefore, miRNA expression patterns are negatively correlated with the expression of target transcripts. Although each individual miRNA-mRNA interaction typically results in modest reductions in protein levels, miRNAs have evolved to regulate key signaling hubs and target tens to hundreds of mRNAs in a cell.20,21 Together, these properties allow miRNAs to fine-tune complex gene networks that govern specific functional programs to control immune responses. Many miRNAs are located in co-transcribed clusters containing multiple miRNAs that have evolved to co-regulate common molecular pathways by either silencing the same gene or different genes involved in similar biological processes.22–24
Observations from our group and other laboratories indicate that the cooperative regulation of gene expression by the paralogous miR-23a-27a-24–2 (Mirc11) and miR-23b-27b-24–1 (Mirc22) clusters−together referred to as the miR-23–27-24 clusters here−controls effector immune cell responses.23,25–27 In macrophages, expression of miR-23a or miR-27a has been reported to restrain interleukin (IL)-4-induced alternative “M2-like” macrophage activation, a functional state associated with homeostasis, tissue repair, and wound healing.18,27 Nevertheless, how the miR-23–27-24 clusters regulate ATM programming in obesity and the physiologic outcomes of these interactions remains unknown due to the heterogeneous nature of ATMs in vivo and the lack of published studies utilizing macrophage-specific methods to investigate the biology of the clusters in models of obesity. In this study, we used a myeloid-specific deletion of the miR-23–27-24 clusters to test the function of these miRNAs in ATMs, identify genes regulated by these clusters, and define a role for these miRNAs in obesity-associated metabolic function.
RESULTS
Expression of the miR-23–27-24 clusters in myeloid cells protects against obesity-induced glucose and insulin intolerance
Given the important role of macrophages on obesity-induced metabolic outcomes,4,9–12 and studies demonstrating the ability of the miR-23–27-24 clusters to regulate macrophage function,18,23,27 we sought to study how loss of expression of these miRNAs in macrophages regulates metabolic function in an in vivo murine model of obesity. We generated mice from embryonic stem cells with a targeted allele for Mirc11 (the miR-23a-27a-24–2 cluster)28 and crossed them to mice with a targeted allele for Mirc22 (the miR-23b-27b-24–1 cluster)25 and mice with Lyz2Cre to generate Mirc11fl/flMirc22fl/fl (fl/fl) control and Mirc11fl/fl Mirc22fl/flLyz2Cre (MyelΔ) mice. In bone marrow-derived macrophages (BMDMs) (Figure S1A), peritoneal macrophages (Figure S1B), and bead-selected F4/80+ ATMs from mice fed a high-fat diet (HFD; 60% kcal of fat) (Figure S1C), miR-23, miR-24, and miR-27 expression was reduced by ≥70%–95% in MyelΔ compared to fl/fl controls. These results indicate that the clusters were effectively deleted in bone marrow-derived and tissue-resident macrophage populations.
To determine how expression of the miR-23–27-24 clusters in myeloid cells regulates systemic metabolic function, we first examined metabolic parameters in young lean mice. Neither male nor female MyelΔ mice exhibited altered body weights (Figure S1D), fasting glucose levels (Figure S1E), or glucose tolerance (Figure S1F). Additionally, myeloid cell numbers in visceral-like epididymal WAT (eWAT) were not different between genotypes (Figure S1G). These findings demonstrate that lean fl/fl and MyelΔ mice display similar immunometabolic characteristics.
To test how expression of these clusters in myeloid cells regulates systemic metabolic function in obesity, male fl/fl and MyelΔ mice were fed a HFD (60% kcal of fat) for 20 weeks to induce obesity (Figure 1A). Male MyelΔ mice gained less body weight over 20 weeks of HFD feeding (Figures 1B and 1C). Fat mass was ~3 g lower in obese MyelΔ mice, while lean mass remained unchanged (Figure 1D). Slopes for cumulative energy intake curves differed significantly between fl/fl and MyelΔ mice (98.58 vs. 92.93 kcal/mouse/week, respectively) over the dietary intervention (Figure 1E), suggesting reduced energy consumption may have contributed to reduced fat accumulation. The rate of weight gain was also lower in MyelΔ versus fl/fl female mice on HFD for 20 weeks (Figures S1H and S1I). Like males, MyelΔ female mice accrued less fat mass (~5 g) following dietary intervention (Figure S1J).
Figure 1. Myeloid-specific expression of the miR-23–27-24 clusters protects against obesity-induced glucose and insulin intolerance.

(A) Schematic of dietary interventions. Mirc11fl/flMirc22fl/fl (fl/fl) and Mirc11fl/flMirc22fl/flLyz2Cre (MyelΔ) mice were fed a high-fat diet (HFD) for 20 weeks.
(B–E) Body weight curves (B) (two-way ANOVA with Šídák’s multiple comparison test), final body weights and weight gain (C) (two-tailed t test), final body composition measurements by NMR (D) (two-tailed t test), or cumulative energy intake (E) (simple linear regression).
(F) Fasting glucose and insulin levels were recorded following HFD feeding (two-tailed t test).
(G–H) Intraperitoneal glucose (1 g/kg lean mass) (G) or insulin tolerance tests (0.5 U/kg lean mass) (H) were performed on fl/fl and MyelΔ mice after dietary intervention (two-way ANOVA with Šídák’s multiple comparison test for curves; two-tailed t test for area under curve [AUC] comparisons). Male mice 8–10 weeks old at beginning of dietary intervention. Each dot in bar graphs represents one mouse after HFD feeding. B&E, 18–21 mice per genotype. ±SEM. #p = 0.07, *p ≤ 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
We then assessed glucose tolerance between both genotypes following HFD feeding. While no differences in fasting blood glucose or insulin levels were observed (Figure 1F), obese male MyelΔ mice exhibited a significant delay in glucose clearance during an intraperitoneal (IP) glucose tolerance test (GTT; Figure 1G). To determine if insulin responsiveness was impaired in MyelΔ mice, an IP-insulin tolerance test was performed. Obese male MyelΔ mice exhibited blunted insulin-induced glucose clearance compared to obese control mice (Figure 1H). Myeloid-specific deletion of the miR-23–27-24 clusters did not impair glucose tolerance in HFD-fed female mice (Figures S1K and S1L). Studies testing sex-specific responses toward HFD feeding show that female mice are protected against obesity-driven impairments in glucose metabolism compared to their male counterparts.29–31 This protective response has been proposed to be mediated, in part, by sex-dependent suppression of low-grade tissue inflammation, reduction in ATM accrual, and increased WAT vascularization, leading to improved WAT function. These findings indicate that the myeloid cell-specific expression of the miR-23–27-24 clusters in male mice protects against obesity-induced impairments in glucose regulation.
miR-23–27-24 cluster expression promotes LAM accumulation in obese adipose tissue
Because obese male MyelΔ mice are more glucose and insulin intolerant, we hypothesized that deleting the clusters in myeloid cells would exacerbate obesity-induced inflammatory ATM accumulation. To examine if myeloid-specific expression of the miR-23–27-24 clusters remodels the myeloid cell compartment of obese eWAT, we immunophenotyped the stromal vascular fraction (SVF) of eWAT from male obese fl/fl and MyelΔ mice by flow cytometry. We defined ATMs based on their expression of the surface antigens F4/80 and CD64 and lack of expression of the neutrophil marker Ly6G and the eosinophil marker Siglec F (Figure 2A).32 Contrary to our initial hypothesis, we observed a selective decrease in ATMs in MyelΔ compared to control mice by flow cytometry (Figure 2B).
Figure 2. Expression of the miR-23–27-24 clusters promotes LAM accumulation in obese eWAT.

(A) Representative flow panels identifying ATMs from obese fl/fl and MyelΔ mice.
(B) Myeloid cell numbers were compared between obese fl/fl and MyelΔ mice (two-way ANOVA with Šídák’s multiple comparison test). ATE, adipose tissue eosinophil; ATM, adipose tissue macrophage; ATN, adipose tissue neutrophil.
(C and D) mRNA levels in whole eWAT (C) or serum levels (D) of IL-6 and TNF-α were compared between obese fl/fl and MyelΔ mice (two-tailed t test).
(E–G) Quantification of M1-like and M2-like markers on ATMs via flow cytometry; two-tailed t test for comparison of (E) and (F), two-way ANOVA with Šídák’s multiple comparison test for (G).
(H and I) Expression of CD9 on ATMs (H) and number of CD9+ ATMs (I) by flow cytometry (two-tailed t test).
(J) Trem2 mRNA levels from whole fl/fl and MyelΔ eWAT compared via RT-qPCR (one-sample t test).
(K) Representative image of eWAT sections stained with anti-F4/80 to visualize crown-like structures (orange star) with quantitation (one-tailed t test). Each dot represents one male mouse after HFD feeding. ±SEM. *p ≤ 0.05, ***p < 0.001, ****p < 0.0001. See also Figures S2 and 3.
Next, we sought to determine whether deletion of the miR-23–27-24 clusters in myeloid cells alters the function or polarization of ATMs to enhance tissue inflammation, despite reducing total ATM numbers. However, no differences in levels of mRNA encoding the pro-inflammatory cytokines IL-6 and tumor necrosis factor alpha (TNF-α) in whole eWAT (Figure 2C) or serum levels of IL-6 and TNF-α (Figure 2D) were observed. In addition, flow cytometric analysis showed a significant decrease in surface levels of CD11c on macrophages in the obese adipose tissue of MyelΔ mice, an integrin molecule often expressed by inflammatory ATMs (Figure 2E).33–35 We also examined the expression of markers associated with an insulin-sensitizing M2-like macrophage phenotype on ATMs. However, we did not detect differences in the numbers of CD163+ ATMs or surface levels of the M2-like markers CD71 and CD206 between genotypes (Figures 2F and 2G). Levels of the M2-like macrophage-associated genes Cd38, Chil3, Egr2, Irf4, Mrc1, Retnla, and Lyve1 were also unchanged in eWAT from obese MyelΔ compared to control fl/fl mice (Figure S2A). Together, these findings demonstrate that overall adipose tissue inflammation is not increased after miR-23, miR-24, and miR-27 deletion in myeloid cells.
To test for tissue inflammation in another insulin-sensitive site, we examined the liver in male mice. While we observed significantly elevated Il6 mRNA levels in whole MyelΔ hepatic tissue, we did not detect differences in mRNA expression levels of other pro-inflammatory factors, M2-like macrophage markers, rate-limiting gluconeogenic enzymes (i.e., G6pc and Pck1), or enzymes involved in lipid biosynthesis or storage (Figures S2B–S2E). Furthermore, quantification of neutral lipids in the livers of obese fl/fl and MyelΔ mice showed no differences in pathologic lipid deposition (Figure S2F). These findings suggest that the systemic impairment in glucose metabolism found in obese MyelΔ mice is not correlated with changes in metabolism and inflammation in other insulin-sensitive tissues.
Because we found no evidence to suggest that MyelΔ ATMs exacerbated pathologic inflammation, we sought to determine if deleting the clusters in myeloid cells alters LAM accumulation. CD9+Trem2+ LAMs are a lipid-laden subset of ATMs that accumulate in crown-like structures within obese WAT and confer protection against obesity-induced pathology in some,4,36 but not all,15,16 studies. Flow cytometric analysis for the LAM marker CD9 revealed a significant reduction in surface levels of CD9 on obese MyelΔ ATMs in male mice (Figure 2H). This was accompanied by a selective decrease in the absolute counts of CD9+ ATMs from obese MyelΔ eWAT (Figure 2I) and Trem2 mRNA levels in whole MyelΔ eWAT (Figure 2J). As in males, total ATM and LAM numbers were significantly reduced in female MyelΔ perigonadal WAT (Figures S3A and S3B). We also observed a decrease in crown-like structures in the obese adipose tissue of MyelΔ mice (Figures 2K and S2G). Overall, our findings identify a role for miR-23–27-24 cluster expression in LAMs and link a reduction in the number of protective LAMs in eWAT with worsened systemic glucose regulation in obese MyelΔ mice.
The miR-23–27-24 clusters support LAM proliferation in obesity
Since LAMs may provide a protective adaptation in expanding adipose tissue, we investigated the cellular mechanisms by which loss of expression of the miR-23–27-24 clusters in myeloid cells decreased LAM accumulation. LAMs are derived from monocytes,4 thus we began by comparing numbers of circulating and eWAT-resident monocytes between fl/fl and MyelΔ mice after HFD feeding. We did not detect any differences in blood monocytes by complete blood count (Figure 3A), arguing against a defect in the production or survival of monocytes in circulation. Total SSC-AloLy6C+ monocytes within eWAT were also equivalent in fl/fl and MyelΔ mice (Figure 3B). We then characterized monocytes and ATMs based on the receptor for CCL2, a chemotactic factor important for monocyte infiltration into obese eWAT.9 Although we did not find differences in CCR2+ monocytes in obese mice among both genotypes (Figure 3B), we found significant reductions in both CCR2− and CCR2+ ATMs in MyelΔ eWAT compared to control (Figure 3C). These findings indicate that loss of the miR-23–27-24 clusters in myeloid cells diminishes absolute ATM numbers in this visceral fat depot through a mechanism downstream of monocyte recruitment.
Figure 3. The miR-23–27-24 clusters promote LAM proliferation.

(A–C) Circulating monocyte numbers (A), total monocytes and CCR2+ monocytes in eWAT (B), or CCR2− or CCR2+ ATMs (C) were compared between obese fl/fl and MyelΔ mice (two-tailed t test).
(D–F) Representative flow panel of Ki67 proliferation index of CD9+ ATMs from obese fl/fl and MyelΔ mice (D) with quantitation of number of Ki67+ ATMs (E) (two-way ANOVA with Šídák’s multiple comparison test) and percentage of proliferating cells (F) (one-sample t test).
(G and H) Ki67 (G) and bromodeoxyuridine (BrdU) (H) staining of BMDM from fl/fl and MyelΔ mice cultured under metabolically activating conditions (MMe) (one-sample t test).
(I) Relative changes in Ki67+ WT MMe BMDMs treated with the indicated miRNA locked nucleic acid (LNA) inhibitor (one-sample t test).
(J and K) Representative Annexin V and 7-AAD cell death staining gated on CD9+ ATMs in the SVF from obese fl/fl and MyelΔ mice (J) with quantitation (K) (two-tailed t test).
(L) Caspase 3 activity was quantified in fl/fl and MyelΔ MMe BMDMs (two-way ANOVA with Šídák’s multiple comparisons test). Each dot represents (A–F) one male mouse after HFD feeding or (G–L) one biological replicate. ±SEM. *p ≤ 0.05, **p < 0.01, ***p < 0.001.
Because ATM proliferation represents a key event contributing to ATM accumulation in obese eWAT,37 we next investigated whether loss of the clusters in myeloid cells impaired LAM proliferation by staining for Ki67, a nuclear antigen expressed by cells undergoing proliferation. Consistent with a proliferation defect, we observed a significant reduction in Ki67+ CD9+ LAMs, but not Ki67+ CD9− ATMs, in MyelΔ eWAT (Figures 3D–3F). This included both an ~50% reduction in the total number of Ki67+ cells per gram of adipose tissue (Figure 3E), as well as a relative reduction in the percentage of Ki67+ CD9+ LAMs among CD9+ LAMs in MyelΔ compared to fl/fl controls (Figure 3F). Furthermore, MyelΔ BMDMs cultured under metabolically activating conditions (MMe BMDMs) to promote an obese-like ATM phenotype12 exhibited a significant defect in proliferation compared to fl/fl MMe BMDMs (Figures 3G and 3H). To determine which miRNAs of the cluster were capable of regulating proliferation, we treated wild-type (WT) MMe BMDMs with locked nucleic acids (LNAs) to selectively deplete miR-23, miR-24, or miR-27. We found that acute inhibition of miR-23 or miR-24, but not miR-27, expression in WT BMDMs was sufficient to reduce the number of Ki67+ cells in MMe conditions (Figure 3I). These findings indicate that expression of the miR-23–27-24 clusters supports LAM proliferation in obese eWAT and in MMe macrophages.
Finally, we performed assays to determine whether augmented cell death contributed to reduced LAM accumulation in obese MyelΔ eWAT. We detected no differences in apoptotic (AnnexinV+7-AAD−) or late apoptotic/necrotic (AnnexinV+7-AAD+) LAMs between genotypes (Figures 3J and 3K). Furthermore, Casp ase-3 activity was similar between fl/fl and MyelΔ BMDMs cultured under MMe conditions (Figure 3L). These findings argue against elevated cell death being responsible for the observed defect in LAM numbers. Overall, our data demonstrate that expression of the miR-23–27-24 clusters supports LAM accrual in obese eWAT by promoting proliferation.
The miR-23–27-24 clusters enhance the expression of multiple genes involved in the negative regulation of cell proliferation
Because miRNAs attenuate the expression of networks of genes, we set out to identify candidate target mRNA transcripts regulated by miR-23, miR-24, and miR-27 in ATMs. We bead selected F4/80+ ATMs from male fl/fl and MyelΔ mice after 20 weeks of HFD and performed bulk RNA sequencing (RNA-seq). Differential expression analysis identified multiple genes that were selectively regulated in ATMs with deletion of the miR-23–27-24 clusters (Figure S4A; Tables S1 and S2). Consistent with the proposed function of CD9+Trem2+ macrophages in phagocytosis4 and within the central nervous system,38 pathway analysis identified lysosome, phagosome, and microglial gene signatures as downregulated in MyelΔ ATMs (Figure S4B). To further test whether myeloid-specific deletion of these miRNA clusters leads to reduced LAMs, we performed gene set enrichment analysis of our data with a list of genes generated from published single-cell RNA-seq data comparing LAMs to lean ATMs.4 We found that ATMs from obese fl/fl mice were significantly enriched in this gene signature compared to ATMs from MyelΔ mice (Figure S4C). Also consistent with our previous data, we did not detect global changes in the expression of M2-like macrophage genes, although Mrc1 and Retnla were slightly increased in MyelΔ ATMs (Figure S4D). Finally, we did not detect differences in gene signatures for inflammation or apoptosis between groups (Figure S4E); however, we did detect an increased a leukocyte proliferation signature in fl/fl ATMs (Figure S4E).
Given that we observed a proliferation defect in miR-23–27-24 cluster-deficient BMDMs cultured in MMe conditions, we also performed RNA-seq in these cells (Figure S5A; Tables S3 and S4). In these in vitro conditions, we found similar differentially regulated pathways in MyelΔ macrophages when compared with fl/fl controls (Figure S5B). There was an overall strong concordance between differentially expressed genes within both datasets that highlighted lysosome, phagosome, and microglia-like pathways (Figure S5C and S5D). Gene set enrichment analysis also showed an enrichment in a LAM-like transcriptional profile in control fl/fl BMDMs in MMe conditions and no changes in inflammatory or apoptotic gene signatures (Figure S5E). Together, these RNA-seq data support our previous findings that loss of miR-23, miR-24, and miR-27 expression leads to selective reduction of LAMs in obese adipose tissue and that miR-23–27-24 cluster-deficient BMDMs cultured in MMe conditions share similar transcriptional changes.
Because mRNA decay is the predominant mechanism for miRNA-mediated gene silencing, we first tested whether loss of the clusters resulted in elevated expression of predicted mRNA targets in our RNA-seq data. Cumulative distribution frequency plots showed a statistically significant global upregulation (right shift) of predicted high-confidence 8mer target mRNAs in MyelΔ versus fl/fl ATMs for miR-23, miR-24, and miR-27 (Figure 4A) as well as MyelΔ versus fl/fl MMe BMDMs (Figure S5F). We observed no change in global expression of predicted target mRNAs of miR-21 and miR-223, two unrelated miRNAs that have previously demonstrated important roles in macrophage biology39 (Figures 4A and S5F). Together, these data demonstrate that loss of miR-23–27-24 specifically upregulates numerous predicted target genes regulated by this cluster but not predicted targets of other miRNAs in MMe BMDMs and obese ATMs.
Figure 4. The miR-23–27-24 clusters negatively regulate multiple genes involved in suppressing cell proliferation.

(A) Cumulative distribution frequency plots depicting global mRNA expression by RNA sequencing as log2(MyelΔ/fl/fl) plotted against the cumulative fraction of all genes (black); miR-23, miR-24, or miR-27 8mer targets (red); miR-21 targets (blue); or miR-223 targets (green) (Kolmogorov-Smirnov test). Target predictions were made using TargetScan.
(B) Venn diagram depicting number of shared or unique candidate target genes whose 3′ untranslated region (3′ UTR) is predicted to be regulated by miR-23, miR-24, and/or miR-27.
(C) Table and heatmap representing predicted interactions and expression of candidate target genes involved in negatively regulating cell proliferation or proliferative signaling.
(D and E) mRNA levels of candidate target genes were compared between fl/fl and MyelΔ unstimulated (BSA) or MMe BMDMs (D) (two-way ANOVA with Šídák’s multiple comparisons test) or MyelΔ MMe BMDMs transfected with miR-23, miR-24, or miR-27 mimics via RT-qPCR (E) (one-way ANOVA with Dunnett’s multiple comparison test). Three to five biological replicates per group. ±SEM. *p ≤ 0.05, **p < 0.01, ****p < 0.0001. See also Figures S4, S5, and Tables S1–S5.
Next, to generate a list of candidate target mRNAs regulated by the three miRNAs encoded in the miR-23–27-24 clusters, we leveraged our RNA-seq dataset comparing fl/fl and MyelΔ obese ATMs. Candidate target genes were selected based on the following criteria: (1) the 3′ UTR of the mRNA contained complementary sequences(s) to the 5′ seed sequence of miR-23, miR-24, and/or miR-27; and (2) the transcript was differentially upregulated (adjusted p value ≤0.05) in obese MyelΔ ATMs. Using these criteria, we identified 78, 42, and 76 candidate target genes for miR-23, miR-24, and miR-27, respectively (Figure 4B; Table S5). Finally, since loss of the miR-23–27-24 clusters selectively reduced LAM proliferation (Figures 3D–3F), we hypothesized that these miRNAs would be specifically important for suppressing the expression of negative regulators of cell proliferation. Therefore, we compared the list of candidate targets with gene sets annotated to negatively regulate cell proliferation and biosynthetic processes or signaling pathways involved in cell proliferation. We identified the following candidate genes through this approach: Dusp5, Eif4ebp2, Sesn2, Socs6, Spry1, and Tmem127 (Figure 4C).
In MMe conditions, we also identified numerous predicted targets of miR-23, miR-24, and miR-27 that were significantly upregulated (Figure S5G). More than 60% of significantly differentially expressed predicted target transcripts of all three miRNAs were upregulated in both MMe BMDMs and ATMs, with relatively few targets that were upregulated in one cell population but not the other (Figure S5H). Although gene set enrichment analysis did not show differences in enrichment of leukocyte proliferation genes in MMe conditions (Figure S5E), most predicted targets significantly upregulated in MyelΔ ATMs showed concordant upregulation in MyelΔ MMe BMDMs when compared to fl/fl controls, including candidate targets identified as potential regulators of cell proliferation in ATMs (Figure S5I). qPCR studies confirmed that MyelΔ MMe BMDMs expressed two to four times greater levels of Dusp5, Eif4ebp2, Sesn2, Socs6, Spry1, and Tmem127 mRNAs relative to control MMe macrophages (Figure 4D). Interestingly, there were no significant differences in levels of any of the candidate target transcripts between control and MyelΔ BMDMs under non-stimulating (BSA) conditions (Figure 4D), indicating that the differential expression of these transcripts was selectively observed during metabolic activation.
Next, we reconstituted individual miRNA expression in MyelΔ MMe BMDMs using miRNA mimics to determine which miRNAs were essential to suppress the expression of each candidate target gene. Transfection of miR-23 in MyelΔ MMe BMDMs significantly reduced Dusp5, Eif4ebp2, Socs6, and Tmem127 mRNA levels (Figure 4E). In contrast, reconstituting miR-24 only attenuated the expression of Eif4ebp2 mRNA, even though it was not predicted to be a target by the TargetScan predication algorithm40 (Figures 4C–4E). Finally, despite being predicted to target several of these candidate regulators of cell proliferation, miR-27 overexpression had no notable impact on mRNA levels of any of the genes of interest (Figures 4C–4E). Altogether, we identified candidate target transcripts differentially regulated by the miR-23–27-24 clusters in obese ATMs and obese-like MMe BMDMs that are known regulators of cellular proliferation.
The miR-23–27-24 clusters suppress Eif4ebp2 expression to promote metabolically activated macrophage proliferation
To test which of the candidate target genes might regulate proliferation in macrophages exposed to obese-like conditions, we performed a small interfering RNA (siRNA) screen for the six candidate target genes identified by RNA-seq analysis in MyelΔ ATMs from obese eWAT using our MMe in vitro model. We hypothesized that, if negative regulation of candidate target genes by the miR-23–27-24 clusters promotes proliferation, then siRNA-mediated knockdown of these genes would also enhance macrophage proliferation. Among the candidates, only siRNA-mediated knockdown of Eif4ebp2 increased MMe BMDM proliferation (Figure 5A). Interestingly, this was also the only candidate target suppressed by miR-23 transfection and by miR-24 transfection (Figure 4E), the two miRNAs encoded by this cluster that supported proliferation in these macrophages (Figure 3I). To verify the results of this screen, we tested whether knockdown of Eif4ebp2 could enhance proliferation of primary ATMs from obese adipose tissue. Consistent with our cell culture results, F4/80-selected ATMs from MyelΔ obese eWAT transfected with siEif4ebp2 proliferated more ex vivo compared to obese ATMs transfected with control siRNA (Figure 5B). Together, these findings indicate that the negative regulation of Eif4ebp2 contributes to MMe BMDM and ATM proliferation.
Figure 5. Eif4ebp2 is a direct target of miR-23 and suppresses macrophage proliferation.

(A) Relative changes in Ki67+ MyelΔ MMe BMDMs following treatment with the indicated siRNA (one-way ANOVA with Dunnett’s multiple comparisons test).
(B) Relative changes in Ki67+ MyelΔ obese ATMs following treatment with the indicated siRNA (one-sample t test).
(C) Western blot for AGO2 following pull-down with immunoglobulin (Ig) G-conjugated or anti-AGO2-conjugated Dynabeads. Each lane represents a technical replicate. CL, cell lysate. Relative enrichment of Eif4ebp2 mRNA pulled down from WT MMe BMDM cell lysate using anti-AGO2 antibodies compared to anti-IgG negative control (one-sample t test).
(D) Relative enrichment of Eif4ebp2 mRNA pulled down from cell lysate of fl/fl and MyelΔ MMe BMDMs following AGO2-IP (one-sample t test).
(E) Relative enrichment of Eif4ebp2 mRNA pulled down from cell lysate of immortalized BMDMs cultured under metabolically activating conditions and treated with either control, miR-23, or miR-24 LNA inhibitor following AGO2-IP (one-sample t test for miR-23 LNA data).
(F) Representative blot and quantification (two-tailed t test) of 4E-BP2 expression in fl/fl and MyelΔ MMe BMDMs (each lane in blot represents technical replicate; four biological replicates represented in quantification).
(G) Puromycin incorporation assay to measure nascent protein synthesis in fl/fl or MyelΔ MMe BMDMs. Quantified are values from three independent experiments (two-tailed t test).
(H and I) Western blot for DEC-1 was examined between MMe BMDMs transfected with the indicated siRNA (H) or fl/fl and MyelΔ MMe BMDMs (I). UT, untransfected; NC, negative control siRNA. Each dot represents sample from one biological replicate. ±SEM. *p ≤ 0.05, **p < 0.01.
Since miRNAs can affect the expression of target mRNAs directly or indirectly, we next sought to test whether members of the miR-23–27-24 clusters directly regulate Eif4ebp2 expression. To do this, we performed immunoprecipitation assays of Argonaute 2 (AGO2), an RNA-binding protein that facilitates miRNA-mRNA engagement to facilitate post-transcriptional silencing of target transcripts as part of the RNA-induced silencing complex (RISC).41 We detected a significant enrichment of Eif4ebp2 mRNA after immunoprecipitation of AGO2 (AGO2-IP) in control MMe BMDMs, indicating that AGO2 interacts with Eif4ebp2 mRNA during metabolic activation (Figure 5C). Next, we compared the enrichment of Eif4ebp2 mRNA isolated from primary fl/fl and MyelΔ MMe BMDM lysates following AGO2-IP. Consistent with Eif4ebp2 mRNA being directly regulated by the clusters, we detected a ~35% reduction in enrichment of Eif4ebp2 mRNA associated with AGO2 in MyelΔ MMe BMDMs relative to fl/fl MMe BMDMs (Figure 5D).
Because Eif4ebp2 mRNA is predicted to be directly regulated by miR-23 (Figure 4C), but its expression can be reduced by transfection of MMe macrophages with either miR-23 or miR-24 (Figure 4E), we sought to determine which of these miRNAs directly target Eif4ebp2 mRNA in macrophages. Depletion of miR-23 by LNAs in immortalized WT BMDMs cultured in MMe conditions reduced Eif4ebp2 mRNA enrichment in AGO2-IPs by ~20% (Figure 5E). Contrary to miR-23 inhibition, loss of miR-24 did not reduce enrichment of Eif4ebp2 mRNA in AGO2-containing complexes (Figure 5E). These findings indicate that Eif4ebp2 mRNA is a direct target of miR-23 in macrophages during metabolic activation.
Eif4ebp2 encodes a member of the eukaryotic translation initiation factor 4E binding protein (4E-BP) family. Canonically, 4E-BP2 binds to and sequesters the translation initiation factor eIF4E, repressing translation of eIF4E-sensitive transcripts.42,43 Upon mTORC1 activation, 4E-BP2 releases eIF4E to initiate cap-dependent translation. Therefore, 4E-BP2 normally restricts anabolism and proliferation within cells. In MMe BMDMs lacking miR-23–27-24 cluster expression, we observed increased levels of the protein product of Eif4ebp2 (4E-BP2) (Figure 5F). To test whether this increased protein expression of 4E-BP2 correlated with an inhibition of protein translation within miR-23–27-24 cluster-deficient cells, we performed a puromycin incorporation assay to measure changes in active protein synthesis. Supporting our hypothesis, we observed a reduction in the generation of nascent polypeptides in MyelΔ MMe BMDMs relative to control MMe BMDMs (Figure 5G). To test whether the clusters control Eif4ebp2-regulated protein synthesis, we quantified protein levels of Bhlhe40 (DEC-1), which has previously been reported to depend on loss of 4E-BP1/2 translational repression to promote macrophage proliferation.44,45 siRNA-mediated knockdown of Eif4ebp2 increased levels of DEC-1 in WT MMe BMDMs, supporting that DEC-1 expression is negatively regulated by 4E-BP2 in these cells (Figure 5H). Furthermore, DEC-1 levels were markedly reduced in MyelΔ MMe BMDMs relative to control MMe BMDMs (Figure 5I). Overall, these findings support that the miR-23–27-24 clusters promote protein synthesis during metabolic activation, in part, by relieving inhibition of eIF4E-dependent translation through suppressing Eif4ebp2 mRNA.
DISCUSSION
Accumulation of macrophages in adipose tissue is a hallmark of obesity, a condition with growing worldwide prevalence that can significantly increase the risk of type 2 diabetes, cardiovascular disease, and cancer.46–48 Cells of the innate immune system play a complex role in the regulation of obesity-induced IR. While the accumulation of inflammatory ATMs has been linked to metabolic outcomes associated with chronic obesity,9–14 recent evidence demonstrates that ATMs can also protect against the metabolic complications of obesity through proposed lipid-handling and phagocytic capabilities.4 Because the key molecular mechanisms driving ATM adaptations in obesity and their influence on systemic IR remain incompletely understood, we hypothesized that miRNAs might both serve as important molecular regulators of this critical cell population and provide insight into the gene networks and pathways that control ATMs.
miRNAs control macrophage-driven inflammation in obesity and obesity-associated metabolic diseases. miR-155 promotes macrophage-driven inflammation in atherosclerotic plaques, while miR-146a and miR-223 restrain aberrant inflammatory activation in obese adipose tissue.49–51 In addition, the regulation of lipid metabolism by miR-33 promotes development of atherogenic macrophages and progression of artherosclerosis.52,53 Although published data demonstrate that miRNAs of the miR-23–27-24 clusters can limit M2-like macrophage differentiation and function,18,27 we did not observe an increase in M2-like macrophages in obese adipose tissue. Furthermore, male mice with a myeloid-specific deletion of these clusters showed worsened glucose and insulin tolerance rather than protection from obesity-associated metabolic dysfunction. This finding is consistent with a recent study showing that global deletion of the miR-23b-27b-24–1 (Mirc22) cluster aggravated glucose intolerance in mice fed an HFD.54 Of note, we did not observe differences in systemic glucose handling in female mice with a myeloid-specific deletion of the miR-23–27-24 clusters, which adds to literature showing discordant regulation of obesity and metabolism in females and males.29–31 However, we identified that the miR-23–27-24 clusters support LAM accrual in WAT during chronic obesity in both males and females, a subset of macrophages that are Trem2+CD9+, accumulate late in obesity, and may play a role in improving systemic glucose tolerance.4 Importantly, this impairment of systemic metabolic function occurred in the absence of increased weight gain, uncoupling the degree of adiposity from metabolic function through the selective regulation of these noncoding RNAs in myeloid cells.
Next, we identified the cellular and molecular mechanisms whereby miR-23, miR-24, and miR-27 were required for LAM accumulation in obese adipose tissue. We determined a selective defect in the proliferation of CD9+ LAMs in vivo and metabolically activated BMDMs in vitro in miR-23–27-24-deficient cells. Combining ex vivo RNA-seq analysis with miRNA-mRNA target interaction prediction algorithms allowed us to identify relevant candidate target genes of the clusters in obese ATMs. Importantly, miR-23–27-24 deficiency in metabolically activated BMDMs drove similar transcriptomic changes in LAM-associated gene signatures and pathways observed in obese ATMs, with a congruent expression in many candidate target genes identified in vivo. Subsequent functional siRNA screening in primary cells and studies of a subset of those targets identified Eif4ebp2 as a direct target of the clusters whose suppression supports macrophage proliferation. Interestingly, both miR-23 and miR-24 independently suppress proliferation as well as attenuate the expression of this transcript in macrophages. Therefore, two members of these clusters may be acting collaboratively to control a key regulatory node in the gene networks of this cell, highlighting one mechanism of how miRNAs of a cluster may evolve together. Of note, assaying AGO2-containing complexes identified miR-23 as a direct regulator of this transcript but showed that miR-24 is likely to act indirectly through other undiscovered targets to regulate this gene.
This work also demonstrates how miRNA-directed pathway discovery can be used to identify genes important for immune cell function in physiology and disease. First, the identification of a requirement for the miR-23–27-24 clusters selectively in CD9+ ATMs highlights subset-specific requirements for regulatory function that can be used to query deep mechanisms of immune cell function and could be leveraged for selective therapeutic intervention. They add to the theory that LAMs contribute to maintaining metabolic health in obese adipose and suggest that the miR-23–27-24 clusters may exert broader regulation than even single-gene changes in this cell population, given that loss of Trem2 alone does not worsen diet-induced obesity in all studies. Identification of the translational repressor Eif4ebp2 as a critical target of these miRNA clusters also points to the importance of eIF4E-dependent transcripts and their upstream regulator mTORC142,43 in the function of LAMs. Indeed, studies in the central nervous system of a related population of CD9+Trem2+ microglia in mouse models of Alzheimer’s disease have identified mTORC1 as an important downstream mediator of Trem2 signaling, controlling autophagy and metabolism in these cells.55
In summary, our study indicates that myeloid-specific expression of the miR-23–27-24 clusters supports the proliferation of LAMs through the regulation of Eif4ebp2, with protection against obesity-induced metabolic dysfunction. It highlights opportunities for investigations of the layered complexity of post-transcriptional regulation at the intersection of immunometabolism, cellular immunology, and RNA biology. Findings from this study provide insights into the identification of functionally relevant miRNAs and corresponding downstream signaling pathways in immune cells that may be exploited therapeutically to modify the immune system to improve obesity-induced metabolic outcomes.
Limitations of the study
Although we identified Eif4ebp2 as a target gene important for ATM proliferation, there are likely other critical miRNA-mRNA interactions among the 163 significantly regulated predicted targets in our RNA-seq dataset. miRNAs almost always act to inhibit multiple critical target genes to exert a biological effect. Indeed, it remains possible that the additional candidate targets implicated in negatively regulating pro-proliferative mTORC1 and MAPK signaling pathways, Dusp5,56 Sesn2,57 Socs6,58 Spry1,59 and Tmem127,60 may be critical for the in vivo function of LAMs despite not affecting in vitro proliferation in metabolically activating conditions. Moreover, due to the nature of miRNAs, we anticipate that, in addition to regulating cellular proliferation, target genes inhibited by the miR-23–27-24 clusters may coordinate additional cell-autonomous changes in aspects of ATM biology that dictate macrophage function in systemic metabolic homeostasis and obesity. Therefore, testing additional functions for miR-23, miR-24, and miR-27 within ATMs may identify critical characteristics of cell subsets within this tissue. Finally, we cannot exclude that loss of the miR-23–27-24 clusters in macrophages outside eWAT contributes to the observed metabolic phenotype. For example, loss of this cluster’s expression in liver macrophages may also be important, as liver macrophages have been intimately linked to obesity-induced defects in systemic glucose regulation, and LAM signatures have been reported in hepatic tissue in murine obesity.4,61,62 It will be important in the future to determine whether the miR-23–27-24 clusters contribute to the function of other macrophage populations found in metabolic and other diseases, including in atherosclerosis,63,64 liver dysfunction during obesity,4 within the tumor microenvironment,65,66 and in Alzheimer’s disease.67
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Heather Pua (heather.pua@vumc.org).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
All transcriptomic data have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers for deposited RNA sequencing data are GSE222351 and GSE235186 and are referenced in the key resources table.
All original code used in this study is freely and fully available (DOI listed in the key resources table).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| APC anti-mouse CD9 (MZ3) | BioLegend | Cat# 124812, RRID:AB_2783071 |
| eFluor450 anti-mouse CD9 (KMC8) | Thermo Fisher Scientific | Cat# 48-0091-82, RRID:AB_2574008 |
| violetFluor450 anti-human/mouse CD11b (M1/70) | Tonbo Bioscience | Cat# 75-0112, RRID:AB_2621936 |
| FITC anti-mouse CD11c (N418) | Thermo Fisher Scientific | Cat# 11-0114-85, RRID:AB_464941 |
| eFluro506 anti-CD45 (30-F11) | Thermo Fisher Scientific | Cat# 69-0451-82, RRID:AB_2637147 |
| APC anti-mouse CD64 (X54-5/7.1) | Thermo Fisher Scientific | Cat# 17-0641-82, RRID:AB_2735010 |
| FITC anti-mouse CD64 (X54-5/7.1) | BioLegend | Cat# 139316, RRID:AB_2566556 |
| PerCP/Cy5.5 anti-mouse CD64 (X54-5/7.1) | BioLegend | Cat# 139308, RRID:AB_2561963 |
| PerCP/Cy5.5 anti-mouse CD71 (R17217) | Leinco Technologies | Cat# C1933, RRID:AB_2829014 |
| PE anti-mouse CD163 (TNKUPJ) | Thermo Fisher Scientific | Cat# 12-1631-82, RRID:AB_2716924 |
| PE anti-mouse CD170/Siglec F (1RNM44N) | Thermo Fisher Scientific | Cat# 12-1702-82, RRID:AB_2637129 |
| BV421 anti-mouse CD192/CCR2 (SA203G11) | BioLegend | Cat# 150605, RRID:AB_2571913 |
| PE-Cy7 anti-mouse CD206 (MR6F3) | Thermo Fisher Scientific | Cat# 25-2061-82, RRID:AB_2637424 |
| PE-Cy7 anti-mouse F4/80 (BM8.1) | Tonbo Bioscience | Cat# 60-4801-U100 |
| FITC anti-mouse Ly6G (1A8-Ly6g) | Thermo Fisher Scientific | Cat# 11-9668-82, RRID:AB_2572532 |
| PE-Cy7 anti-mouse TCRβ (H57-597) | Thermo Fisher Scientific | Cat# 25-5961-82, RRID:AB_2573507 |
| Anti-4E-BP2 rabbit Ab | Cell Signaling Technology | Cat# 2845, RRID:AB_10699019 |
| Anti-β-Actin (D6A8) rabbit Ab | Cell Signaling Technology | Cat# 8457, RRID:AB_10950489 |
| Anti-BHLHE40/DEC-1 rabbit Ab | Proteintech | Cat# 17895-1-AP, RRID:AB_2065351 |
| Anti-GAPDH (D16H11) XP® rabbit Ab | Cell Signaling Technology | Cat# 5174, RRID:AB_10622025 |
| Anti-Puromycin (12D10) mouse Ab | Millipore Sigma | Cat# MABE343, RRID:AB_2566826 |
| Goat Anti-mouse IgG (H + L) secondary Ab, HRP | Thermo Fisher Scientific | Cat# A16078, RRID:AB_2534751 |
| Goat Anti-mabbit IgG (H + L) secondary Ab, HRP | Thermo Fisher Scientific | Cat# 31460, RRID:AB_228341 |
| Anti-mouse Ago2 (2D4) | FUJIFILM Wako Shibayagi | Cat# 018-22021, RRID:AB_1106838 |
| Mouse IgG1 kappa Isotype Control (P3.6.2.8.1) | Thermo Fisher Scientific | Cat# 14-4714-82, RRID:AB_470111 |
|
| ||
| Critical commercial assays | ||
|
| ||
| eBioscience™ BrdU Staining Kit for Flow Cytometry APC |
Thermo Fisher Scientific | Cat# 8817-6600-42, RRID:AB_2575274 |
| EnzChek™ Caspase-3 Activity Assay Kits | Thermo Fisher Scientific | Cat# E13183 |
| FITC Annexin V Apoptosis Detection Kit with 7-AAD | BioLegend | Cat# 640922 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| PE Mouse Anti-Ki-67 Set | BD Bioscience | Cat# 556027, RRID:AB_2266296 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed RNA sequencing data - adipose tissue macrophages | This paper | GEO: GSE222351 |
| Raw and analyzed RNA sequencing data - cultured metabolically activated macrophages | This paper | GEO: GSE235186 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Cas9+ immortalized murine bone marrow-derived macrophages | Laboratory of Jeffery Cox. Roberts et al. (2019, eLife) | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| B6.129P2-Lyz2tm1(cre)Ifo/J | The Jackson Laboratory | JAX: 004781 |
| C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Mirc11 fl/fl Mirc22 fl/fl | Laboratory of Mark Ansel. Pua et al. (2016, Cell Rep.). And Laboratory of Heather Pua. This paper. | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S6 for oligonucleotides used in this study | IDT, Qiagen, Dharmacon | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Original code of RNA-seq pipeline | This paper | https://doi.org/10.5281/zenodo.8143940 |
| GSEA | Subramanian, Tamayo, et al. (2005, PNAS) and Mootha, Lindgren, et al. (2003, Nature Genetics). | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Schneider et al.(2012, Nature Methods) | https://imagej.nih.gov/ij/ |
| FlowJo™ v10.6 Software | BD Life Sciences | Flojo.com |
| Graphpad Prism 8 | Dotmatics | https://www.graphpad.com/ |
| R studio | Integrated Development for R | www.r-project.org |
| FastQC v0.11.8 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt v1.18 | Martin et al. (2011, EMBnet) | https://cutadapt.readthedocs.io/en/v1.18/ |
| Salmon | Patro et al. (2017, Nat. Methods) | https://github.com/COMBINE-lab/Salmon |
| DESeq2 | Love et al. (2014, Genome Biol.) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
|
| ||
| Other | ||
|
| ||
| Albumin, Bovine Fraction V [BSA] | RPI Products International | CAS Number: 9048-46-8 |
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| DynaMag™-2 Magnet | Thermo Fisher Scientific | Cat# 12321D |
| Palmitic acid, ≥99% | MP Biomedicals | SKU: 02100905-CF |
| Puromycin | InvivoGen | Cat# Ant-pr-1 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals and diets
All animal studies were performed after obtaining approval from the Vanderbilt Institutional Animal Care and Use Committee. C57BL/6 ES cells were targeted using constructs generated as a resource for the conditional deletion of miRNA clusters as described28 to produce chimeric mice with a conditionally mutant allele of the miRNA cluster containing miR-23a, miR-27a, and miR-24–2 (the Mirc11 cluster). These chimeras were crossed to Rosa26-Flp mice (Gt(ROSA)26Sortm1(FLP1)Dym; JAX: 009086) to delete the selection cassette. Mice previously generated with a conditionally mutant allele containing miR-23b, miR-27b and miR-24–1 (the Mirc22 cluster)25 were backcrossed for 10 generations to C57BL/6J mice (JAX: 000664). These two lines were intercrossed, and the progeny crossed to Lyz2Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J; JAX: 004781) to generate mice with myeloid cells lacking expression of Mirc11 and Mirc22 clusters in myeloid cells (MyelΔ). For the diet-induced obesity model, 7–9-week-old male or female Mirc11fl/flMirc22 fl/fl and Mirc11fl/flMirc22fl/flLyz2Cre mice were fed a high-fat diet (HFD, 60% kcal fat; Research Diets) for 20+ weeks. Body weight and food intake were recorded weekly. Body composition measurements, collection of fasting blood samples, and intraperitoneal glucose and insulin tolerance tests were performed before or during HFD feeding. All other analyses were performed after the mice were euthanized.
Primary and immortalized bone marrow-derived macrophage cultures
Primary bone marrow-derived macrophages (BMDMs) were prepared by culturing bone marrow of female and male mice in RPMI-1640 media supplemented with 20% L929-conditioned medium, 10% FBS, gentamicin, glutamine, HEPES, non-essential amino acids, and penicillin-streptomycin for 7+ days at 37°C. Media was changed on Day 1, then every three days of culture. To plate for experiments, BMDMs were detached from the flask using cold PBS containing 0.5–5 mM EDTA and plated into tissue culture plates at a concentration of 1×106 cells/mL.
Origin and details of the Cas9+ immortalized bone marrow-derived macrophage cell line used in this study are summarized in this paper’s key resources table. Conditionally immortalized macrophages were derived from male Cas9+ mice. These cells are not commercially available, and therefore do not have reference STRs for authentication.
All cell lines were maintained at 37°C in RPMI-1640 medium supplemented with 10% FBS, gentamicin, glutamine, HEPES, non-essential amino acids, and penicillin-streptomycin. To metabolically activate primary and immortalized BMDMs, BMDMs were cultured in supplemented DMEM media containing 25 mM glucose, 10 nM insulin (Novolin), and 150–250 mM palmitic acid (MP Biomedicals) conjugated to BSA (Research Products International) for 24 h–48 h. BMDMs in the control group were cultured in supplemented RPMI-1640 medium (11 mM glucose) containing BSA alone.
METHOD DETAILS
Body composition
Fat and lean mass were quantified at the Vanderbilt University Mouse Metabolic Phenotyping Center via nuclear magnetic resonance (Bruker Minispec, Woodlands, TX).
Bulk RNA sequencing
F4/80-selected ATMs from perigonadal fat pads were homogenized with TRIzol Reagent (Life Technologies). Large RNA (>200 nt) was isolated using the RNeasy Mini Kit (Qiagen) and submitted to the Vanderbilt Technologies for Advanced Genomics Core for sequencing on the NovaSeq 6000 (Illumina). Data were processed on the Vanderbilt computing cluster (ACCRE) using the lab’s RNA-seq pipeline (https://github.com/parkdj1/RNASeq-Pipeline). Briefly, FastQC was used to evaluate data quality, adaptors were trimmed using Cutadapt, and alignment with quantification was performed using Salmon. Primary data was deposited in GEO (accession numbers in the key resources table). Downstream analysis included differential expression (DESeq2) and gene set enrichment (www.broadinstitute.org/gsea). IDs of the following GSEA gene sets were used to identify candidate target transcripts: GO: 0070373, GO: 0008285, GO: 0043409, GO: 1904262, GO: 0009890, GO: 0097696, Reactome: R-HSA-5675221.
Caspase 3 activity assay
BMDMs were plated into tissue culture plates (1 × 106 cells/mL) and allowed to rest overnight. Media was replaced the next day, and cells were subjected to metabolic activation for ~24h. Afterward, cleaved caspase 3 assay was carried out using EnzChek Caspase 3 Assay Kit (Molecular Probes) according to the manufacturer’s instructions.
Electroporation
One to two million BMDMs from the indicated genotype resuspended in 100 μL Buffer R were mixed with 500 nM miRNA mimic (miRIDIAN) or 100 nM siRNA (siGENOME), then electroporated using the Neon Transfection System (Thermo-Fisher) at pulse code (10 ms x 3 pulses) using 100 μL Neon tips (Thermo-Fisher) at 1500 V. Following electroporation, cells were slowly pipetted into wells of tissue culture plates containing prewarmed supplemented RPMI-1640 medium containing 20% L929 conditioned media. Cells incubated in media for 24–48h, then were subjected to metabolic activation.
Fasting glucose, insulin, and glucose/insulin tolerance tests
Fasting blood samples were collected from the tail vein following a 5–6 h fast. After collecting baseline blood samples, mice underwent an intraperitoneal glucose or insulin tolerance test. In brief, blood samples were collected by massaging the tail at the indicated time points after receiving an intraperitoneal injection of glucose (1 g/kg lean body mass) or insulin (0.5 U/kg lean body mass), respectively. Blood glucose levels were measured using a CONTOURNEXT glucometer. Fasting plasma insulin concentrations were determined using a radioimmunoassay; insulin measurements were carried out by the Hormone Assay & Analytical Services Core at Vanderbilt University.
Locked nucleic acid treatments
BMDMs were plated into tissue culture plates (1 × 106 cells/mL) and allowed to rest overnight. Media was replaced the next day, and cells were treated with 50 nM of the indicated locked nucleic acid inhibitor (miRCURY LNA miRNA Power Inhibitors). Cells incubated with inhibitors for 24h, then subjected to metabolic activation.
Luminex analysis
Serum samples from mice fed an HFD were sent to Vanderbilt Hormone Assay & Analytical Services Core for Luminex analysis. Analytes examined in this study were chosen from their “Mouse Metabolic Hormone Panel”.
miRNA and mRNA quantification via RT-qPCR
TRIzol Reagent (Life Technologies) was used to extract and purify RNA from tissues and cultured cells. Small RNA (<200 nt) of F4/80-selected adipose tissue macrophages from perigonadal fat pads was isolated using the RNeasy Mini Kit (Qiagen). For miRNA quantitation, the Mir-X miRNA First-Strand Synthesis Kit (Takara) was used for cDNA synthesis. For mRNA quantitation, the SuperScript IV First-Strand Synthesis System Kit (Invitrogen) was used for cDNA synthesis. Real-time PCR of cDNA was performed on a CFX96 Real-Time PCR Detection System (BioRad) located at the Vanderbilt Cell & Developmental Biology Equipment Resource Core using FastStart Universal SYBR Green master mix (Roche). Expression of the indicated genes was normalized to β-actin (Actb) using the 2−ΔΔCt method.
Palmitic acid-BSA preparation
Palmitic acid (MP Biomedical) was dissolved in Molecular Biology Grade Ethanol (Fisher BioReagents) by alternating between heating at 70°C and vortexing to produce a 1M palmitic acid solution. Immediately afterward, the dissolved palmitic acid was conjugated to BSA by gently mixing and heating (70°C) 40 μL of the 1 M palmitic acid solution with 960 μL of a 10% BSA in HEPES (Gibco) solution for 20–30 min, creating a 40 mM palmitic acid-BSA solution. Before cell culture treatment, the 40 mM palmitic acid-BSA solution was shaken at 50°C for 20 min prior to dispensing into a tube containing supplemented DMEM media which was then incubated with cells. 10% BSA in HEPES solution was used as the vehicle control.
Puromycin incorporation assay
Puromycin (10 μg/mL; InvivoGen) was added to BMDMs 15 min before harvest. Cells were then washed with ice-cold PBS twice, and protein was collected and processed for Western blotting. A “no puromycin” treatment group served as a negative control.
RNA-Argonaute 2 immunoprecipitation
Primary or immortalized BMDMs68 were cultured in 150 mm tissue culture plates until they reached 100% confluence. Following indicated treatments, ribonucleoprotein complexes were crosslinked with 300 mJ/cm2 UV (wavelength 254 nm). To isolate RNAs complexed with Argonaute 2 (AGO2), we first conjugated 5 μg of anti-AGO2 (Wako; clone 2D4) to 50 μL of protein G Dynabeads (Invitrogen; 10004D) by incubating both at room temperature for 1 h while rotating. Beads were then collected using the DynaMag-2 magnet (Invitrogen; 12321D) and washed 3x with lysis buffer. After which, cell lysate from crosslinked cells incubated with anti-AGO2 conjugated beads for 2 h at 4°C while rotating. Beads were washed 3x with high-salt buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate), then 3x with wash buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl, 0.2% Tween). Proteins were then digested with proteinase K to release AGO2-associated RNA. TRIzol LS Reagent (Life Technologies) was used to isolate RNA, then treated with DNase to remove contaminating genomic DNA. cDNA synthesis and qPCR of cDNA were carried out as described above. Prior to proteinase K digestion, a small fraction of beads was collected and used to confirm successful pull-down of AGO2. Pull-down with mouse IgG1κ isotype control (Invitrogen) served as a negative control.
Stromal vascular fraction isolation and flow cytometric analysis
The stromal vascular fraction (SVF) from perigonadal fat pads were isolated following collagenase digestion and differential centrifugation.69 Adipose tissue (AT) myeloid cells were characterized via flow cytometry using fluorescent anti-mouse antibodies (described in the key resources table) against the following antigens: F4/80, Ly6G, Siglec F, CD9, CD64, CD11c, CD163, CD206, CCR2, Ly6C, TCRβ. Dead cells were identified with e780 viability dye (ThermoFisher).
For Ki67 staining, cells were first stained with surface antigen fluorescent antibodies, then were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (ThermoFisher). Afterward, intracellular staining was performed using either PE anti-Ki67 or PE IgG1, κ isotype control from the PE Mouse Anti-Ki67 Set (BD Biosciences) according to the manufacturer’s instructions. % Ki67+ cells were calculated by subtracting % PE+ cells stained with isotype control from % PE+ cells stained with anti-Ki67 [% PE+ (anti-Ki67) - % PE+ (isotype)]. For Annexin V and 7-AAD staining, SVF cells were stained using reagents from the FITC Annexin V Apoptosis Detection Kit with 7-AAD (TONBO) as described by manufacturers. For BrdU staining, BMDMs were metabolically activated for 24 h then labeled with 10 μM BrdU overnight with the eBioscience BrdU Staining Kit (Invitrogen). Afterward, cells were harvested, stained with e780 viability dye, fixed, subjected to DNase I digestion, then intracellularly stained with APC anti-BrdU as described by manufacturers. All flow cytometry was performed on an 8-color FACSCANTO II flow cytometer (BD Biosciences) provided by the Division of Molecular Pathogenesis. FlowJo was used to process data.
Western blotting
Cells were lysed in Pierce Ripa buffer (ThermoFischer) supplemented with a Halt protease and phosphatase inhibitor cocktail (ThermoFischer) on ice. Lysed cells were scraped off tissue culture plates, and lysates were cleared by centrifugation at 4°C at 12,000 x g for 15 min. Protein concentrations were quantified with the Pierce BCA Protein Assay Kit (ThermoFischer), and equal amounts of protein (20–30 μg) were loaded on BoltTM 4–12% Bis-Tris Plus precast gels for SDS-PAGE. Proteins were transferred onto nitrocellulose membranes with a tank blotting system (Invitrogen), blocked with 5% milk in TBS-T for 30min – 1h after transfer, then incubated with antibodies specific for the following proteins of interest overnight: 4E-BP2 (2845), β-Actin (8457), GAPDH (5174), puromycin (MABE343), and BHLHE40/DEC-1 (17895). Anti-4E-BP2, anti-β-Actin, and anti-GAPDH antibodies were from Cell Signaling Technology. Anti-puromycin antibody (clone 12D10) was from Millipore. Anti-BHLHE40/DEC-1 was from Proteintech. Following overnight incubation with primary antibodies, the membrane was washed with TBS-T, then incubated with HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies (ThermoFisher) for 1h. Blots were imaged using an AI600 Imager (GE Healthcare) housed at the Vanderbilt Cell & Developmental Biology Equipment Resource Core. Images and quantifications were obtained using ImageJ software.
QUANTIFICATIONS AND STATISTICAL ANALYSIS
Results are shown as mean ± SEM. Statistics were done on experiments with 3+ biological replicates or independent experiments as outlined in figure legends. Analyses were performed using R (www.r-project.org) and GraphPad Prism8 (GraphPad Software, Version 8.02). p values of less than 0.05 were considered significant.
Supplementary Material
Highlights.
miR-23-27-24 expression in myeloid cells supports glucose metabolism in obesity
Genetic deletion of miR-23-27-24 impairs Trem2+ CD9+ macrophage proliferation
miR-23-27-24 regulates a network of genes in obese adipose tissue macrophages
Suppression of Eif4ebp2 by miR-23 promotes macrophage proliferation
ACKNOWLEDGMENTS
We would like to thank Mr. Grant Nation, Mr. Zachary Bressman, Ms. Cherie Saffold, and Dr. Nathan Klopfenstien for technical assistance. We also thank the Cell & Developmental Biology Equipment Core, Hormone Assay & Analytical Services Core (NIH DK059637 and DK020593), Mouse Metabolic Phenotyping Center (NIH DK059637), Vanderbilt Technologies for Advanced Genomics, and VUMC Flow Cytometry Shared Resource Core at Vanderbilt University for their services. Pilot work for this project was supported by a voucher from the Vanderbilt Institute for Clinical and Translational Research (VICTR, UL1TR002243). This work was supported by a grant from the NIAID (R21 AI156292) to H.H.P.; start-up funds from the Department of Pathology, Microbiology and Immunology at VUMC to H.H.P.; the Cellular, Biochemical, and Molecular Sciences training grant (T32 GM008554) from Vanderbilt University to N.T.S.; and a pre-doctoral fellowship from the AHA (836819) to N.T.S. A.H.H. is supported by a Career Scientist award from Veterans Affairs (IK6 BX005649). N.C.W. is supported by an AHA postdoctoral fellowship (21POST834990).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112928.
REFERENCES
- 1.Haffner SM, Stern MP, Hazuda HP, Mitchell BD, and Patterson JK (1990). Cardiovascular Risk Factors in Confirmed Prediabetic Individuals. JAMA 263, 2893–2898. 10.1001/jama.1990.03440210043030. [DOI] [PubMed] [Google Scholar]
- 2.Rosen ED, and Spiegelman BM (2006). Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444, 847–853. 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surmi BK, and Hasty AH (2008). Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 3, 545–556. 10.2217/17460875.3.5.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698.e14. 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubler MJ, Peterson KR, and Hasty AH (2015). Iron homeostasis: A new job for macrophages in adipose tissue? Trends Endocrinol. Metabol 26, 101–109. 10.1016/j.tem.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flaherty SE, Grijalva A, Xu X, Ables E, Nomani A, and Ferrante AW (2019). A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 363, 989–993. 10.1126/science.aaw2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubler MJ, Erikson KM, Kennedy AJ, and Hasty AH (2018). MFe hi adipose tissue macrophages compensate for tissue iron perturbations in mice. Am. J. Physiol. Physiol 315, 319–329. 10.1152/ajpcell.00103.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lumeng CN, Delproposto JB, Westcott DJ, and Saltiel AR (2008). Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246. 10.2337/db08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, and Ferrante AW (2006). CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest 116, 115–124. 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa KI, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, and Kasuga M. (2006). MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest 116, 1494–1505. 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng C, Yang Q, Cao J, Xie N, Liu K, Shou P, Qian F, Wang Y, and Shi Y. (2016). Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 7, e2167. 10.1038/cddis.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metabol. 20, 614–625. 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christ A, Lauterbach M, and Latz E. (2019). Western Diet and the Immune System: An Inflammatory Connection. Immunity 51, 794–811. 10.1016/j.immuni.2019.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Hotamisligil GS (2017). Inflammation, inflammation and immunometabolic disorders. Nature 542, 177–185. 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- 15.Winn NC, Wolf EM, Garcia JN, and Hasty AH (2022). Exon 2-mediated deletion of Trem2 does not worsen metabolic function in diet-induced obese mice. J. Physiol 600, 4485–4501. 10.1113/JP283684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharif O, Brunner JS, Korosec A, Martins R, Jais A, Snijder B, Vogel A, Caldera M, Hladik A, Lakovits K, et al. (2021). Beneficial Metabolic Effects of TREM2 in Obesity Are Uncoupled From Its Expression on Macrophages. Diabetes 70, 2042–2057. 10.2337/DB20-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu XQ, Dai Y, Yang Y, Huang C, Meng XM, Wu BM, and Li J. (2016). Emerging role of microRNAs in regulating macrophage activation and polarization in immune response and inflammation. Immunology 148, 237–248. 10.1111/imm.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boucher A, Klopfenstein N, Hallas WM, Skibbe J, Appert A, Jang SH, Pulakanti K, Rao S, Cowden Dahl KD, and Dahl R. (2021). The miR-23a~27a~24–2 microRNA Cluster Promotes Inflammatory Polarization of Macrophages. J. Immunol 206, 540–553. 10.4049/jimmunol.1901277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN, and Tsatsanis C. (2009). The Kinase Akt1 Controls Macrophage Response to Lipopolysaccharide by Regulating MicroRNAs. Immunity 31, 220–231. 10.1016/j.immuni.2009.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebert MS, and Sharp PA (2012). Roles for MicroRNAs in conferring robustness to biological processes. Cell 149, 515–524. 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farh KKH, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, and Bartel DP (2005). Biochemistry: The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310, 1817–1821. 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 22.Kabekkodu SP, Shukla V, Varghese VK, D’Souza J, Chakrabarty S, and Satyamoorthy K. (2018). Clustered miRNAs and their role in biological functions and diseases. Biol. Rev. Camb. Phil. Soc 93, 1955–1986. 10.1111/brv.12428. [DOI] [PubMed] [Google Scholar]
- 23.Cho S, Wu C-J, Yasuda T, Cruz LO, Khan AA, Lin L-L, Nguyen DT, Miller M, Lee H-M, Kuo M-L, et al. (2016). miR-23~27~24 clusters control effector T cell differentiation and function. J. Exp. Med 213, 235–249. 10.1084/jem.20150990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Luo J, Zhang H, and Lu J. (2016). MicroRNAs in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol 33, 2232–2247. 10.1093/molbev/msw089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pua HH, Steiner DF, Patel S, Gonzalez JR, Ortiz-Carpena JF, Kageyama R, Chiou NT, Gallman A, de Kouchkovsky D, Jeker LT, et al. (2016). MicroRNAs 24 and 27 Suppress Allergic Inflammation and Target a Network of Regulators of T Helper 2 Cell-Associated Cytokine Production. Immunity 44, 821–832. 10.1016/j.immuni.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho S, Wu C-J, Nguyen DT, Lin L-L, Chen M-C, Khan AA, Yang B-H, Fu W, and Lu L-F (2017). A Novel miR-24–TCF1 Axis in Modulating Effector T Cell Responses. J. Immunol 198, 3919–3926. 10.4049/jimmunol.1601404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma S, Liu M, Xu Z, Li Y, Guo H, Ge Y, Liu Y, Zheng D, and Shi J. (2016). A double feedback loop mediated by microRNA-23a/27a/24–2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget 7, 13502–13519. 10.18632/oncotarget.6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park CY, Jeker LT, Carver-Moore K, Oh A, Liu HJ, Cameron R, Richards H, Li Z, Adler D, Yoshinaga Y, et al. (2012). A Resource for the Conditional Ablation of microRNAs in the Mouse. Cell Rep. 1, 385–391. 10.1016/j.celrep.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettersson US, Waldén TB, Carlsson PO, Jansson L, and Phillipson M. (2012). Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue. PLoS One 7, e46057. 10.1371/journal.pone.0046057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ingvorsen C, Karp NA, and Lelliott CJ (2017). The role of sex and body weight on the metabolic effects of high-fat diet in C57BL/6N mice. Nutr. Diabetes 7, e261. 10.1038/nutd.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudnicki M, Abdifarkosh G, Rezvan O, Nwadozi E, Roudier E, and Haas TL (2018). Female mice have higher angiogenesis in perigonadal adipose tissue than males in response to high-fat diet. Front. Physiol 9, 1452. 10.3389/fphys.2018.01452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, Geletka L, Meyer KA, O’Rourke RW, and Lumeng CN (2016). Adipose Tissue Dendritic Cells Are Independent Contributors to Obesity-Induced Inflammation and Insulin Resistance. J. Immunol 197, 3650–3661. 10.4049/jimmunol.1600820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Wu H, An J, Ballantyne CM, and Cyster JG (2018). Critical role of integrin CD11c in splenic dendritic cell capture of missing-self CD47 cells to induce adaptive immunity. Proc. Natl. Acad. Sci. USA 115, 6786–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukácsi S, Nagy-Baló Z, Erdei A, Sá ndor N, and Bajtay Z. (2017). The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett 189, 64–72. 10.1016/j.imlet.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Lumeng CN, Bodzin JL, and Saltiel AR (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest 117, 175–184. 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A, et al. (2018). Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 115, 5096–5105. 10.1073/pnas.1802611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, and Aouadi M. (2014). Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metabol. 19, 162–171. 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, and Weiner HL (2011). MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-a-PU.1 pathway. Nat. Med 17, 64–70. 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sprenkle NT, Serezani CH, and Pua HH (2023). MicroRNAs in Macrophages: Regulators of Activation and Function. J. Immunol 210, 359–368. 10.4049/jimmunol.2200467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis BP, Burge CB, and Bartel DP (2005). Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets We predict regulatory targets of vertebrate micro-RNAs. Cell 120, 15–20. 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 41.Gebert LFR, and MacRae IJ (2019). Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol 20, 21–37. 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter JD, and Sonenberg N. (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 43.Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras AC, Sonenberg N, and Wagner G. (1997). Structure of translation factor elF4E bound to m7GDP and interaction with 4E-binding protein. Nat. Struct. Biol 4, 717–724. 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- 44.Jarjour NN, Schwarzkopf EA, Bradstreet TR, Shchukina I, Lin CC, Huang SCC, Lai CW, Cook ME, Taneja R, Stappenbeck TS, et al. (2019). Bhlhe40 mediates tissue-specific control of macrophage proliferation in homeostasis and type 2 immunity. Nat. Immunol 20, 687–700. 10.1038/s41590-019-0382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colina R, Costa-Mattioli M, Dowling RJO, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, et al. (2008). Translational control of the innate immune response through IRF-7. Nature 452, 323–328. 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 46.Sanders JW, Fuhrer GS, Johnson MD, and Riddle MS (2008). The epidemiological transition: The current status of infectious diseases in the developed world versus the developing world. Sci. Prog 91, 1–37. 10.3184/003685008X284628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao Z, Zheng X, Yang H, Li S, Xu F, Yang X, and Wang Y. (2020). Association of obesity status and metabolic syndrome with site-specific cancers: a population-based cohort study. Br. J. Cancer 123, 1336–1344. 10.1038/s41416-020-1012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skinner AC, Perrin EM, Moss LA, and Skelton JA (2015). Cardiometabolic risks and severity of obesity in children and young adults. N. Engl. J. Med 373, 1307–1317. 10.1056/NEJMoa1502821. [DOI] [PubMed] [Google Scholar]
- 49.Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J, et al. (2012). MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Invest 122, 4190–4202. 10.1172/JCI61716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ying W, Tseng A, Chang RCA, Morin A, Brehm T, Triff K, Nair V, Zhuang G, Song H, Kanameni S, et al. (2015). MicroRNA-223 is a crucial mediator of PPARg-regulated alternative macrophage activation. J. Clin. Invest 125, 4149–4159. 10.1172/JCI81656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Runtsch MC, Nelson MC, Lee SH, Voth W, Alexander M, Hu R, Wallace J, Petersen C, Panic V, Villanueva CJ, et al. (2019). Anti-inflammatory microRNA-146a protects mice from diet-induced metabolic disease. PLoS Genet. 15, e1007970. 10.1371/journal.pgen.1007970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, Van Solingen C, Fullerton MD, Cecchini K, et al. (2015). MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Invest 125, 4334–4348. 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rayner KJ, Suá rez Y, Dá valos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, and Fernández-Hernando C. (2010). MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573. 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang YH, Man YY, Liu Y, Yin CJ, Li JL, Shi HC, Zhao H, and Zhao SG (2021). Loss of mir-23b/27b/24–1 cluster impairs glucose tolerance via glycolysis pathway in mice. Int. J. Mol. Sci 22, 550. 10.3390/ijms22020550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlyle WC, Mcclain JB, Tzafriri AR, Bailey L, Zani BG, Markham PM, Stanley JR, Edelman ER, Sciences CB, Technologies E, et al. (2012). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 162, 561–567. 10.1016/j.cell.2017.07.023. [DOI] [Google Scholar]
- 56.Grasset M-F, Gobert-Gosse S, Mouchiroud G, and Bourette RP (2010). Macrophage differentiation of myeloid progenitor cells in response to M-CSF is regulated by the dual-specificity phosphatase DUSP5. J. Leukoc. Biol 87, 127–135. 10.1189/jlb.0309151. [DOI] [PubMed] [Google Scholar]
- 57.Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al. (2012). Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metabol. 16, 311–321. 10.1016/j.cmet.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayle J, Letard S, Frank R, Dubreuil P, and De Sepulveda P. (2004). Suppressor of Cytokine Signaling 6 Associates with KIT and Regulates KIT Receptor Signaling. J. Biol. Chem 279, 12249–12259. 10.1074/jbc.M313381200. [DOI] [PubMed] [Google Scholar]
- 59.Hanafusa H, Torii S, Yasunaga T, and Nishida E. (2002). Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol 4, 850–858. 10.1038/ncb867. [DOI] [PubMed] [Google Scholar]
- 60.Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, et al. (2010). Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat. Genet 42, 229–233. 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgantini C, Jager J, Li X, Levi L, Azzimato V, Sulen A, Barreby E, Xu C, Tencerova M, Näslund E, et al. (2021). Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab 3, 287. 10.1038/s42255-021-00343-5. [DOI] [PubMed] [Google Scholar]
- 62.Tencerova M, Aouadi M, Vangala P, Nicoloro SM, Yawe JC, Cohen JL, Shen Y, Garcia-Menendez L, Pedersen DJ, Gallagher-Dorval K, et al. (2015). Activated Kupffer cells inhibit insulin sensitivity in obese mice. Faseb. J 29, 2959–2969. 10.1096/fj.15-270496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, and Zernecke A. (2018). Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res 122, 1661–1674. 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 64.Willemsen L, and de Winther MP (2020). Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol 250, 705–714. 10.1002/path.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song Q, Hawkins GA, Wudel L, Chou PC, Forbes E, Pullikuth AK, Liu L, Jin G, Craddock L, Topaloglu U, et al. (2019). Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 8, 3072–3085. 10.1002/cam4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, Brioschi S, Bugatti M, Omodei AS, Ricci B, et al. (2020). TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182, 886–900.e17. 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Z. BH, Gilfillan S, Krishnan GM, Sudhakar S, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kiritsy MC, Ankley LM, Trombley J, Huizinga GP, Lord AE, Orning P, Elling R, Fitzgerald KA, and Olive AJ (2021). A genetic screen in macrophages identifies new regulators of ifng-inducible mhcii that contribute to T cell activation. Elife 10, e65110. 10.7554/eLife.65110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orr JS, Kennedy AJ, and Hasty AH (2013). Isolation of Adipose Tissue Immune Cells. J. Vis. Exp, e50707. 10.3791/50707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All transcriptomic data have been deposited at the Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers for deposited RNA sequencing data are GSE222351 and GSE235186 and are referenced in the key resources table.
All original code used in this study is freely and fully available (DOI listed in the key resources table).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| APC anti-mouse CD9 (MZ3) | BioLegend | Cat# 124812, RRID:AB_2783071 |
| eFluor450 anti-mouse CD9 (KMC8) | Thermo Fisher Scientific | Cat# 48-0091-82, RRID:AB_2574008 |
| violetFluor450 anti-human/mouse CD11b (M1/70) | Tonbo Bioscience | Cat# 75-0112, RRID:AB_2621936 |
| FITC anti-mouse CD11c (N418) | Thermo Fisher Scientific | Cat# 11-0114-85, RRID:AB_464941 |
| eFluro506 anti-CD45 (30-F11) | Thermo Fisher Scientific | Cat# 69-0451-82, RRID:AB_2637147 |
| APC anti-mouse CD64 (X54-5/7.1) | Thermo Fisher Scientific | Cat# 17-0641-82, RRID:AB_2735010 |
| FITC anti-mouse CD64 (X54-5/7.1) | BioLegend | Cat# 139316, RRID:AB_2566556 |
| PerCP/Cy5.5 anti-mouse CD64 (X54-5/7.1) | BioLegend | Cat# 139308, RRID:AB_2561963 |
| PerCP/Cy5.5 anti-mouse CD71 (R17217) | Leinco Technologies | Cat# C1933, RRID:AB_2829014 |
| PE anti-mouse CD163 (TNKUPJ) | Thermo Fisher Scientific | Cat# 12-1631-82, RRID:AB_2716924 |
| PE anti-mouse CD170/Siglec F (1RNM44N) | Thermo Fisher Scientific | Cat# 12-1702-82, RRID:AB_2637129 |
| BV421 anti-mouse CD192/CCR2 (SA203G11) | BioLegend | Cat# 150605, RRID:AB_2571913 |
| PE-Cy7 anti-mouse CD206 (MR6F3) | Thermo Fisher Scientific | Cat# 25-2061-82, RRID:AB_2637424 |
| PE-Cy7 anti-mouse F4/80 (BM8.1) | Tonbo Bioscience | Cat# 60-4801-U100 |
| FITC anti-mouse Ly6G (1A8-Ly6g) | Thermo Fisher Scientific | Cat# 11-9668-82, RRID:AB_2572532 |
| PE-Cy7 anti-mouse TCRβ (H57-597) | Thermo Fisher Scientific | Cat# 25-5961-82, RRID:AB_2573507 |
| Anti-4E-BP2 rabbit Ab | Cell Signaling Technology | Cat# 2845, RRID:AB_10699019 |
| Anti-β-Actin (D6A8) rabbit Ab | Cell Signaling Technology | Cat# 8457, RRID:AB_10950489 |
| Anti-BHLHE40/DEC-1 rabbit Ab | Proteintech | Cat# 17895-1-AP, RRID:AB_2065351 |
| Anti-GAPDH (D16H11) XP® rabbit Ab | Cell Signaling Technology | Cat# 5174, RRID:AB_10622025 |
| Anti-Puromycin (12D10) mouse Ab | Millipore Sigma | Cat# MABE343, RRID:AB_2566826 |
| Goat Anti-mouse IgG (H + L) secondary Ab, HRP | Thermo Fisher Scientific | Cat# A16078, RRID:AB_2534751 |
| Goat Anti-mabbit IgG (H + L) secondary Ab, HRP | Thermo Fisher Scientific | Cat# 31460, RRID:AB_228341 |
| Anti-mouse Ago2 (2D4) | FUJIFILM Wako Shibayagi | Cat# 018-22021, RRID:AB_1106838 |
| Mouse IgG1 kappa Isotype Control (P3.6.2.8.1) | Thermo Fisher Scientific | Cat# 14-4714-82, RRID:AB_470111 |
|
| ||
| Critical commercial assays | ||
|
| ||
| eBioscience™ BrdU Staining Kit for Flow Cytometry APC |
Thermo Fisher Scientific | Cat# 8817-6600-42, RRID:AB_2575274 |
| EnzChek™ Caspase-3 Activity Assay Kits | Thermo Fisher Scientific | Cat# E13183 |
| FITC Annexin V Apoptosis Detection Kit with 7-AAD | BioLegend | Cat# 640922 |
| Foxp3/Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| PE Mouse Anti-Ki-67 Set | BD Bioscience | Cat# 556027, RRID:AB_2266296 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed RNA sequencing data - adipose tissue macrophages | This paper | GEO: GSE222351 |
| Raw and analyzed RNA sequencing data - cultured metabolically activated macrophages | This paper | GEO: GSE235186 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Cas9+ immortalized murine bone marrow-derived macrophages | Laboratory of Jeffery Cox. Roberts et al. (2019, eLife) | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| B6.129P2-Lyz2tm1(cre)Ifo/J | The Jackson Laboratory | JAX: 004781 |
| C57BL/6J | The Jackson Laboratory | JAX:000664 |
| Mirc11 fl/fl Mirc22 fl/fl | Laboratory of Mark Ansel. Pua et al. (2016, Cell Rep.). And Laboratory of Heather Pua. This paper. | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S6 for oligonucleotides used in this study | IDT, Qiagen, Dharmacon | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Original code of RNA-seq pipeline | This paper | https://doi.org/10.5281/zenodo.8143940 |
| GSEA | Subramanian, Tamayo, et al. (2005, PNAS) and Mootha, Lindgren, et al. (2003, Nature Genetics). | https://www.gsea-msigdb.org/gsea/index.jsp |
| ImageJ | Schneider et al.(2012, Nature Methods) | https://imagej.nih.gov/ij/ |
| FlowJo™ v10.6 Software | BD Life Sciences | Flojo.com |
| Graphpad Prism 8 | Dotmatics | https://www.graphpad.com/ |
| R studio | Integrated Development for R | www.r-project.org |
| FastQC v0.11.8 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt v1.18 | Martin et al. (2011, EMBnet) | https://cutadapt.readthedocs.io/en/v1.18/ |
| Salmon | Patro et al. (2017, Nat. Methods) | https://github.com/COMBINE-lab/Salmon |
| DESeq2 | Love et al. (2014, Genome Biol.) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
|
| ||
| Other | ||
|
| ||
| Albumin, Bovine Fraction V [BSA] | RPI Products International | CAS Number: 9048-46-8 |
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| DynaMag™-2 Magnet | Thermo Fisher Scientific | Cat# 12321D |
| Palmitic acid, ≥99% | MP Biomedicals | SKU: 02100905-CF |
| Puromycin | InvivoGen | Cat# Ant-pr-1 |