

Abstract
This review provides an update about cardiac sympathetic denervation in Lewy body diseases. The family of Lewy body diseases includes Parkinson’s disease (PD), pure autonomic failure (PAF), and dementia with Lewy bodies (DLB). All three feature intra-neuronal cytoplasmic deposits of the protein, alpha-synuclein. Multiple system atrophy (MSA), the parkinsonian form of which can be difficult to distinguish from PD with orthostatic hypotension, involves glial cytoplasmic inclusions that contain alpha-synuclein. By now there is compelling neuroimaging, neuropathologic, and neurochemical evidence for cardiac sympathetic denervation in Lewy body diseases. In addition to denervation, there is decreased storage of catecholamines in the residual terminals. The degeneration develops in a centripetal, retrograde, “dying back” sequence. Across synucleinopathies the putamen and cardiac catecholaminergic lesions seem to occur independently of each other, whereas non-motor aspects of PD (e.g., anosmia, dementia, REM behavior disorder, OH) are associated with each other and with cardiac sympathetic denervation. Cardiac sympathetic denervation can be caused by synucleinopathy in inherited PD. According to the catecholaldehyde hypothesis, 3,4-dihydroxyphenylacetaldehyde (DOPAL), an intermediary metabolite of dopamine, causes or contributes to the death of catecholamine neurons, especially by interacting with proteins such as alpha-synuclein. DOPAL oxidizes spontaneously to DOPAL-quinone, which probably converts alpha-synuclein to its toxic oligomeric form. Decreasing DOPAL production and oxidation might slow the neurodegenerative process. Tracking cardiac sympathetic innervation over time could be the basis for a proof of principle experimental therapeutics trial targeting DOPAL.
Search terms: Parkinson’s disease, sympathetic nervous system, norepinephrine
Introduction: The family of Lewy body diseases
Lewy body diseases are a family of neurodegenerative disorders characterized by intra-neuronal cytoplasmic inclusions that have a characteristic histopathologic appearance upon hematoxylin/eosin staining. The inclusions were first described by Friedrich Heinrich Lewy in 1912 (Holdorff, 2002) in brainstem regions including the dorsal motor nucleus of the vagus nerve in patients with idiopathic Parkinson’s disease (PD). In 1919 Tretiakoff noted what he called “Lewy bodies” in the substantia nigra in PD (Lees et al., 2008).
In 1997 it was reported that Lewy bodies contain the protein, alpha-synuclein (Spillantini et al., 1997) and that mutation of the gene encoding alpha-synuclein causes familial PD in a kindred in which the disease is transmitted as an autosomal dominant trait (Polymeropoulos et al., 1997). Subsequent genome wide association studies agreed that variants of the alpha-synuclein gene increase the risk of developing PD (Edwards et al., 2010; Satake et al., 2009). The current understanding is that sporadic PD is a Lewy body form of synucleinopathy.
Pure autonomic failure (PAF) is a rare but scientifically important Lewy body disease in which the patients have neurogenic orthostatic hypotension (OH) without signs of central neurodegeneration (Hague et al., 1997; Miura et al., 2001; van Ingelghem et al., 1994). Dementia with Lewy bodies (DLB) is characterized clinically by fluctuating cognitive function, visual hallucinations, and parkinsonism and neuropathologically by Lewy bodies in the brainstem or cortex (McKeith et al., 1996). When dementia follows PD by at least 1 year, then by consensus this is called PD with dementia (PDD) (Gomperts, 2016). Incidental Lewy body disease (ILBD) is defined by Lewy bodies discovered upon post-mortem neuropathology, without PD having been diagnosed during life (Iacono et al., 2015). Lewy bodies have also been reported in cases of neurodegeneration with brain iron accumulation (Jellinger, 2003), a heterogeneous group of rare inherited neurodegenerative diseases (Tello et al., 2017).
Neuroimaging and neuropathologic evidence of cardiac sympathetic denervation in Lewy body diseases
The year 1997, which saw the first reports of an identified genetic cause of PD (Polymeropoulos et al., 1997) and of alpha-synuclein deposition in Lewy bodies (Spillantini et al., 1997), also saw the first report of neuroimaging evidence indicating cardiac sympathetic denervation in Lewy body diseases (Goldstein et al., 1997) (Figure 1).
Figure 1: 13N-Ammonia and 18F-dopamine thoracic positron emission tomographic images from a normal control subject, a patient with pure autonomic failure (PAF), a patient with multiple system atrophy (MSA), and a patient with Parkinson’s disease (PD).
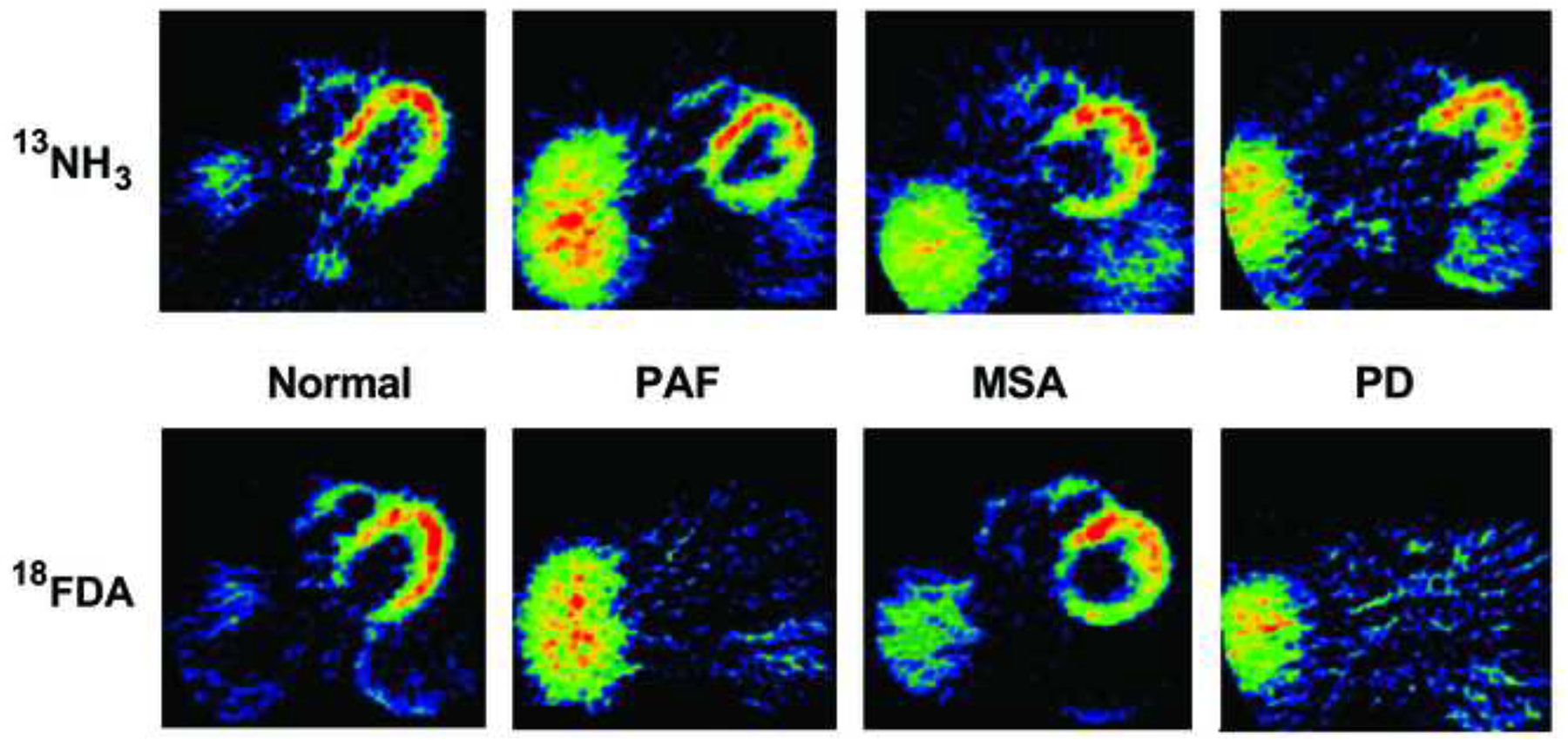
Note markedly decreased myocardial 18F-dopamine-derived radioactivity in PAF and PD.
Numerous subsequent studies, based mainly on 123I-metaiodobenzylguanidine (123I-MIBG) scanning, have agreed universally on the finding of decreased myocardial noradrenergic innervation in PD, PAF, and, more recently DLB (Goldstein, 2003; Jost et al., 2010; Kashihara et al., 2006; Suzuki et al., 2006; Treglia et al., 2012). 123I-MIBG scanning can be used to distinguish DLB from Alzheimer’s disease (Manabe et al., 2017; Miyoshi et al., 2013).
Post-mortem neuropathological studies have agreed on the finding of decreased immunoreactive tyrosine hydroxylase (TH, a marker of sympathetic noradrenergic innervation) in epicardial, myocardial, or sympathetic ganglion tissue from patients with PD, PAF, DLB, and ILBD (Dickson et al., 2008; Fujishiro et al., 2008; Orimo et al., 2005a; Orimo et al., 2006; Orimo et al., 2007b; Orimo et al., 2008b).
In confirmation of the validity of cardiac sympathetic neuroimaging to identify myocardial noradrenergic deficiency, the extent of myocardial sympathetic innervation by 123I-MIBG scanning has been found to correlate positively with myocardial immunoreactive TH (Takahashi et al., 2015). We recently found a strong positive correlation between myocardial 18F-dopamine-derived radioactivity and myocardial norepinephrine content (unpublished observations).
Profound myocardial norepinephrine deficiency in Lewy body diseases
Only recently has the extreme extent of myocardial norepinephrine depletion in PD become evident (Goldstein et al., 2014). In an extension of the published study, we simultaneously assayed apical myocardial concentrations of norepinephrine, dopamine (from which norepinephrine is synthesized in storage vesicles in sympathetic nerves), and 3,4-dihydroxyphenylglycol (DHPG, the main neuronal metabolite of norepinephrine) in patients with autopsy-proven PD and control subjects. Compared to controls, PD patients had a 96% mean decrease in tissue norepinephrine content (Figure 2), accompanied by a 90% mean decrease in dopamine, which in sympathetic nerves is the precursor of norepinephrine, and an 89% decrease in DHPG, the main intra-neuronal metabolite of norepinephrine.
Figure 2: Myocardial tissue contents of (left) norepinephrine and (right) 6 endogenous catechols in PD, PAF, and MSA.
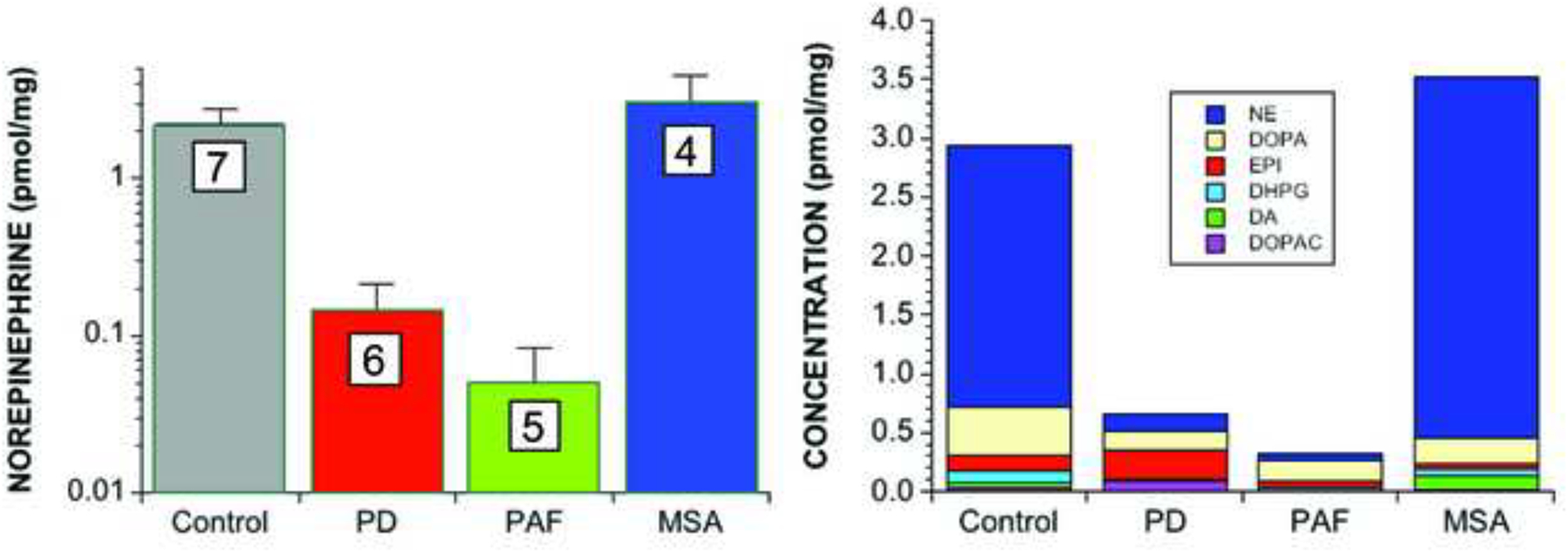
Numbers in white boxes show the numbers of patients. Note large decreases in norepinephrine and the sum of endogenous catechols in PD and PAF.
Limited data so far suggest if anything even more severe myocardial norepinephrine deficiency in PAF than in PD (unpublished observations; Figure 2). In contrast, MSA is associated with generally normal immunoreactive TH in epicardial nervous tissue (Orimo et al., 2008b; Orimo et al., 2012) and myocardial norepinephrine content (Figure 2), although cases of MSA with cardiac sympathetic denervation have been described (Cook et al., 2014; Orimo et al., 2007a).
About 30–40% of PD patients have orthostatic hypotension (OH) (Velseboer et al., 2011). In PD, OH often is an early rather than late finding (Goldstein, 2006; Kim et al., 2012). All patients with PD+OH have neuroimaging evidence of cardiac sympathetic denervation (Goldstein and Orimo, 2009; Jain and Goldstein, 2012).
The results therefore demonstrate drastic cardiac norepinephrine deficiency in Lewy body diseases. The myocardial noradrenergic lesion in PD is about equal in severity to the putamen dopaminergic lesion that underlies the motor signs (Goldstein et al., 2011c; Kish et al., 1988).
Decreased vesicular storage: A common theme in catecholaminergic neurodegeneration
Considering the decrease in immunoreactive TH in epicardial nerve tissue from PD patients, one might presume that myocardial norepinephrine deficiency in PD directly and solely reflects cardiac sympathetic denervation. Quantification of TH axon area/total area in apical myocardial tissue has indicated about an 84% decrease in cardiac sympathetic innervation in PD (Takahashi et al., 2015). This value agrees well with the estimated value of 88% based on the rate of endogenous DOPA production in the heart (Goldstein et al., 2014).
The extent of loss of myocardial norepinephrine, however, is more severe than the loss of innervation. A potential resolution of this discrepancy would be a decreased ability to store catecholamines in the residual terminals. Accumulating evidence supports the concept of decreased vesicular sequestration of cytoplasmic catecholamines in myocardial sympathetic nerves in Lewy body diseases.
The first evidence for decreased vesicular storage in residual sympathetic nerves in Lewy body diseases came from a study of simultaneously measured myocardial 18F-dopamine-derived radioactivity and arterial 18F-dihydroxyphenylacetic acid (18F-DOPAC) levels. As shown in the concept diagram in Figure 3, reduced vesicular uptake would result in higher 18F-DOPAC levels than expected for myocardial 18F-dopamine-derived radioactivity. This pattern was observed in patients with PD or PAF and not in MSA (Goldstein et al., 2011a).
Figure 3: Concept diagram showing effects of denervation, decreased vesicular uptake, and combined denervation and decreased vesicular uptake on myocardial 18F-dopamine-derived radioactivity and arterial plasma 18F-dihydroxyphenylacetic acid (18F-DOPAC).
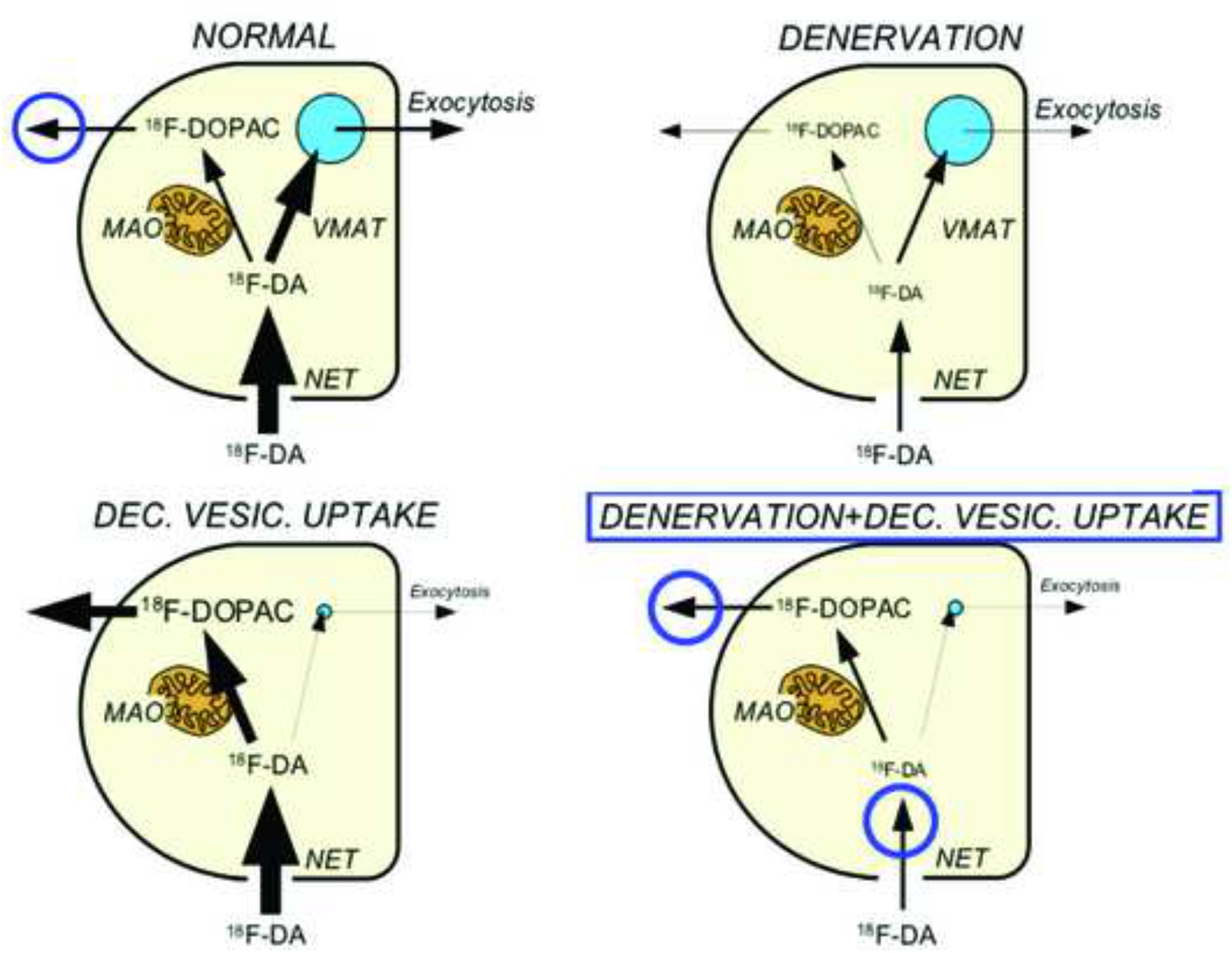
Circles placed to indicate increased arterial 18F-DOPAC/18F-dopamine-derived radioactivity as an inverse index of vesicular uptake.
According to the concept diagram in Figure 4, a vesicular storage defect would also be expected to increase the slope of the mono-exponential decline in 18F-dopamine-derived radioactivity (Goldstein et al., 1991). We observed this phenomenon in patients with PD or PAF—including patients with inherited PD from abnormalities of the alpha-synuclein gene (Goldstein et al., 2011a) (Figure 5).
Figure 4: Concept diagram showing effects of denervation, and combined denervation and decreased vesicular uptake on myocardial 18F-dopamine-derived radioactivity.
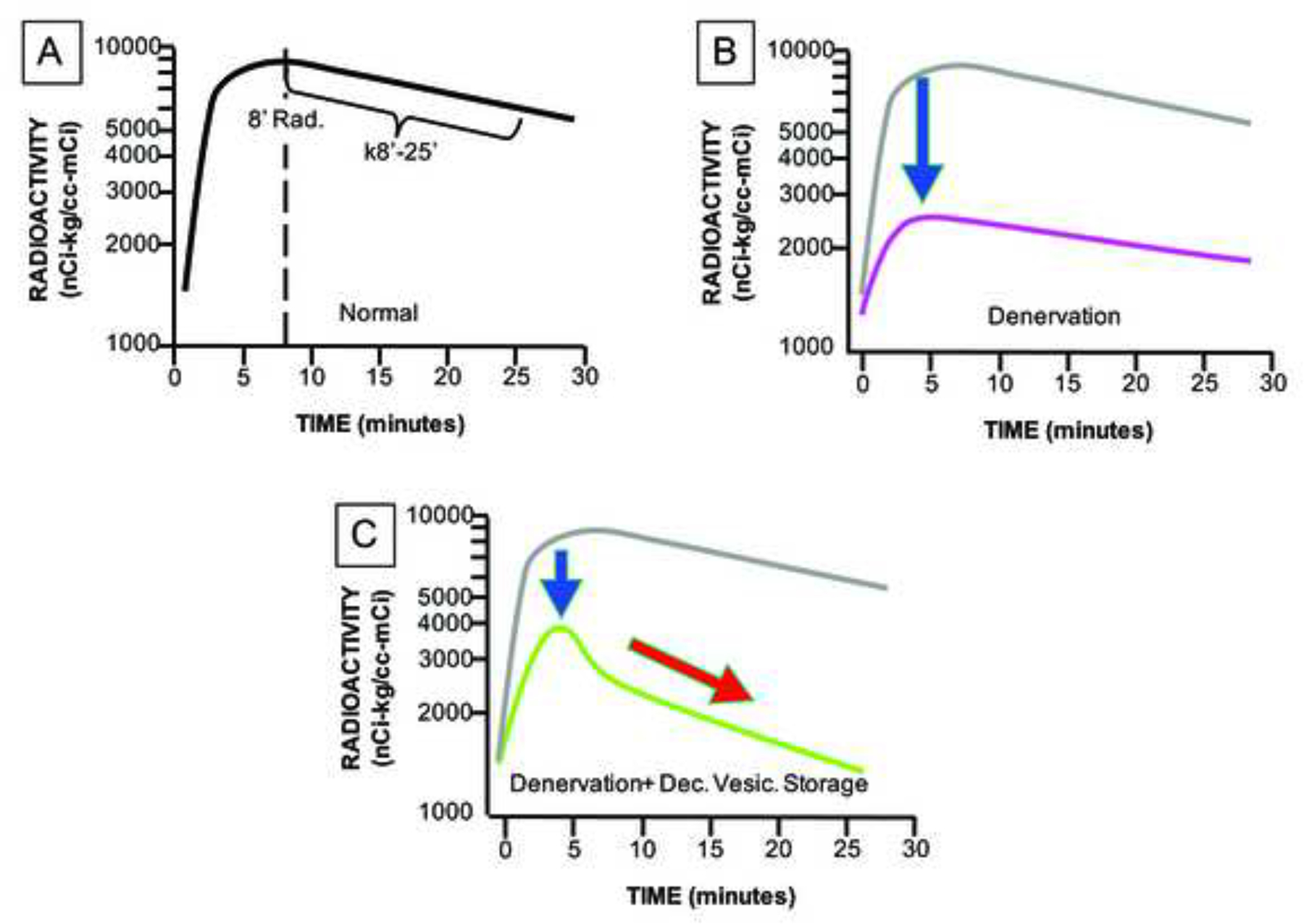
Denervation should decrease peak initial radioactivity (8’ Rad.), and decreased vesicular uptake should increase the slope of decline of radioactivity (k8’-25’).
Figure 5: Mean (± SEM) interventricular septal myocardial concentrations of 18F-dopamine-derived radioactivity in groups with synucleinopathies or desipramine (DMI)-treated control subjects.
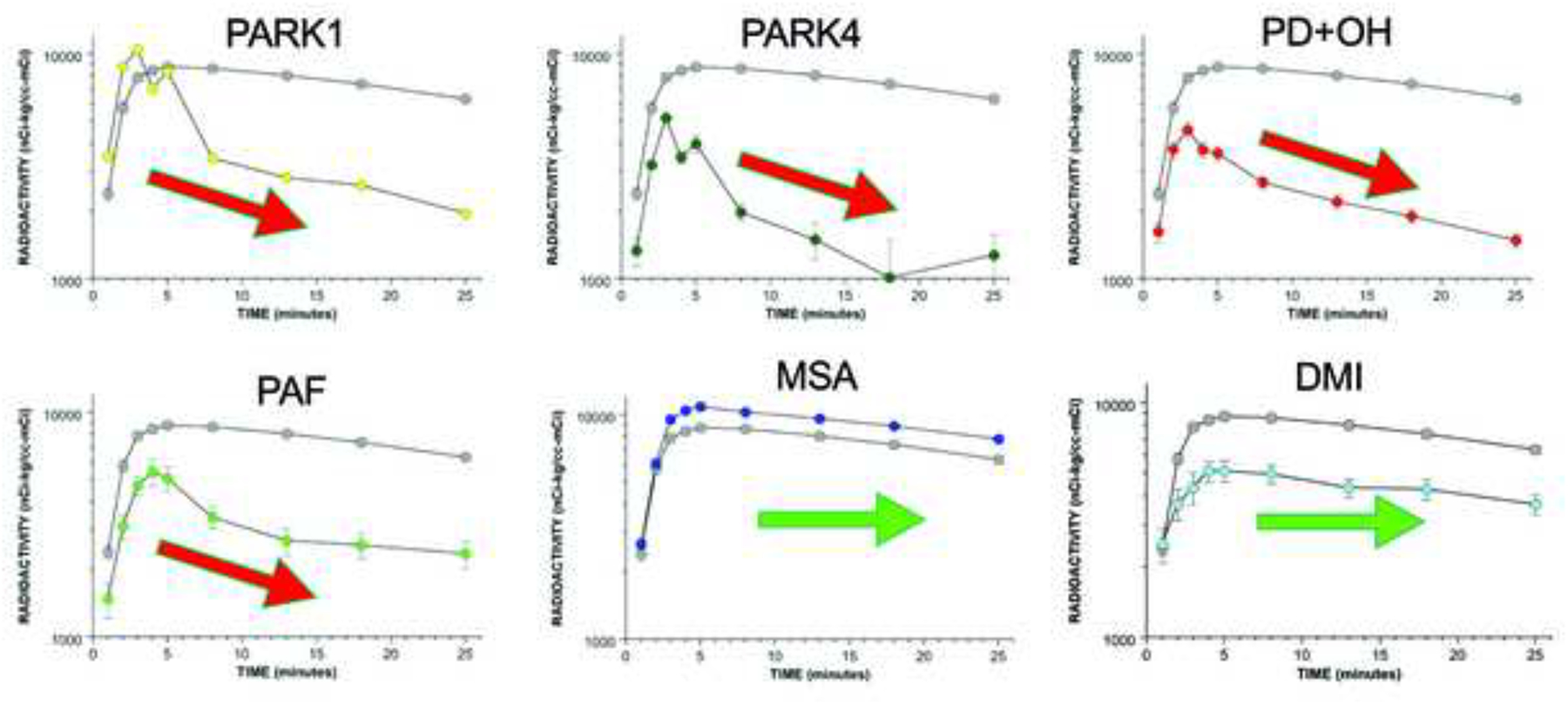
PARK1, PARK4, PD with orthostatic hypotension (PD+OH), and PAF are associated with accelerated declines in radioactivity, whereas MSA and DMI treatment are not.
Further support for decreased vesicular storage in sympathetic noradrenergic nerves in Lewy body diseases has come from a study on arterial plasma levels of 6F-DOPAC and DHPG in PD+OH vs. the parkinsonian form of MSA. PD+OH was associated with elevated ratios of 6F-DOPAC/DHPG, consistent with decreased vesicular sequestration after taking sympathetic denervation into account; MSA-P was not (Goldstein et al., 2015).
A post-mortem study provided evidence for a vesicular storage defect in the residual myocardial noradrenergic terminals in PD (Goldstein et al., 2014). Based on the known intracellular pathways of norepinephrine synthesis and turnover (Figure 6), we validated 5 different indices of vesicular sequestration in mice with inherited deficient activity of the type-2 vesicular monoamine transporter (VMAT), the form of the VMAT that is expressed in sympathetic nerves. By all 5 indices, PD patients who had myocardial norepinephrine depletion also had remarkably decreased vesicular sequestration of cytoplasmic catecholamines (Figure 7). From the observed abnormalities, coupled with previously obtained data about cardiac norepinephrine spillover (Goldstein et al., 2000) and arterial and coronary sinus concentrations of DOPA, DOPAC, and DHPG, we estimated a 67% decrease in norepinephrine synthesis or vesicular storage in the residual myocardial nerves in PD (Figure 8).
Figure 6: Concept diagram about sources and metabolic fates of catecholamines in myocardial sympathetic nerves.
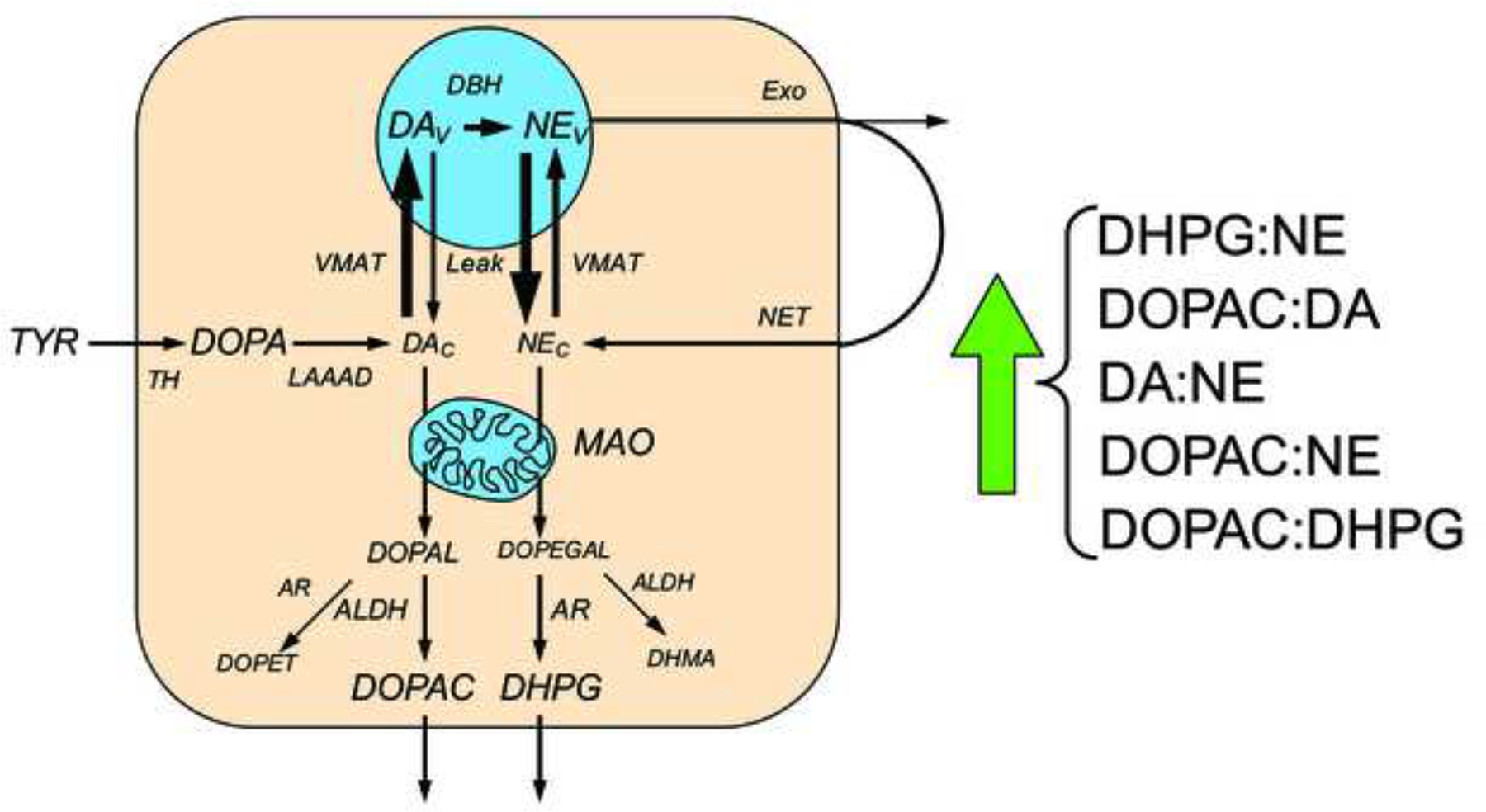
Under resting conditions, loss of norepinephrine (NE) from the neurons is due mainly to passive leakage from the vesicles (NEv) into the cytosol (NEc), followed by enzymatic deamination catalyzed by monoamine oxidase (MAO). Cytosolic NE is taken up into the vesicles via the type 2 vesicular monoamine transporter (VMAT). Release by exocytosis from the vesicles, with escape of reuptake via the cell membrane NE transporter (NET), is a minor determinant of NE turnover. NE loss is balanced by catecholamine biosynthesis from the action of cytosolic L-aromatic-amino-acid decarboxylase (LAAAD) on 3,4-dihydroxyphenylalanine (DOPA) produced from tyrosine (TYR) by tyrosine hydroxylase (TH) and of dopamine-beta-hydroxylase (DBH), which is localized in the vesicles. The action of MAO on cytosolic DA produces 3,4-dihydroxyphenylacetaldehyde (DOPAL) and on NE produces 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL). DOPEGAL is mainly reduced by aldehyde/aldose reductase (AR), to form 3,4-dihydroxyphenylglycol (DHPG), and DOPAL is mainly oxidized by aldehyde dehydrogenase (ALDH) to form 3,4-dihydroxyphenylacetic acid (DOPAC). When vesicular uptake is attenuated, as in VMAT2-Lo mice, myocardial NE depletion reflects decreased NE synthesis, because less DA is taken up into the vesicles and more is deaminated to form DOPAC. Furthermore, decreased reuptake of NE that leaks from vesicles into the cytoplasm, where the NE is deaminated and is converted to DHPG, accelerates the turnover of NE. Decreased vesicular sequestration of cytoplasmic catecholamines would be expected to produce 5 alterations in ratios of catechols in myocardial tissue.
Figure 7: Mean (± SEM) ratios of catechols in myocardial tissue from (left) PD patients with norepinephrine depletion (NE Depl.) and control subjects (CON) and (right) mice with very low VMAT2 activity (VMAT2-Lo) and wild type (WT) mice.
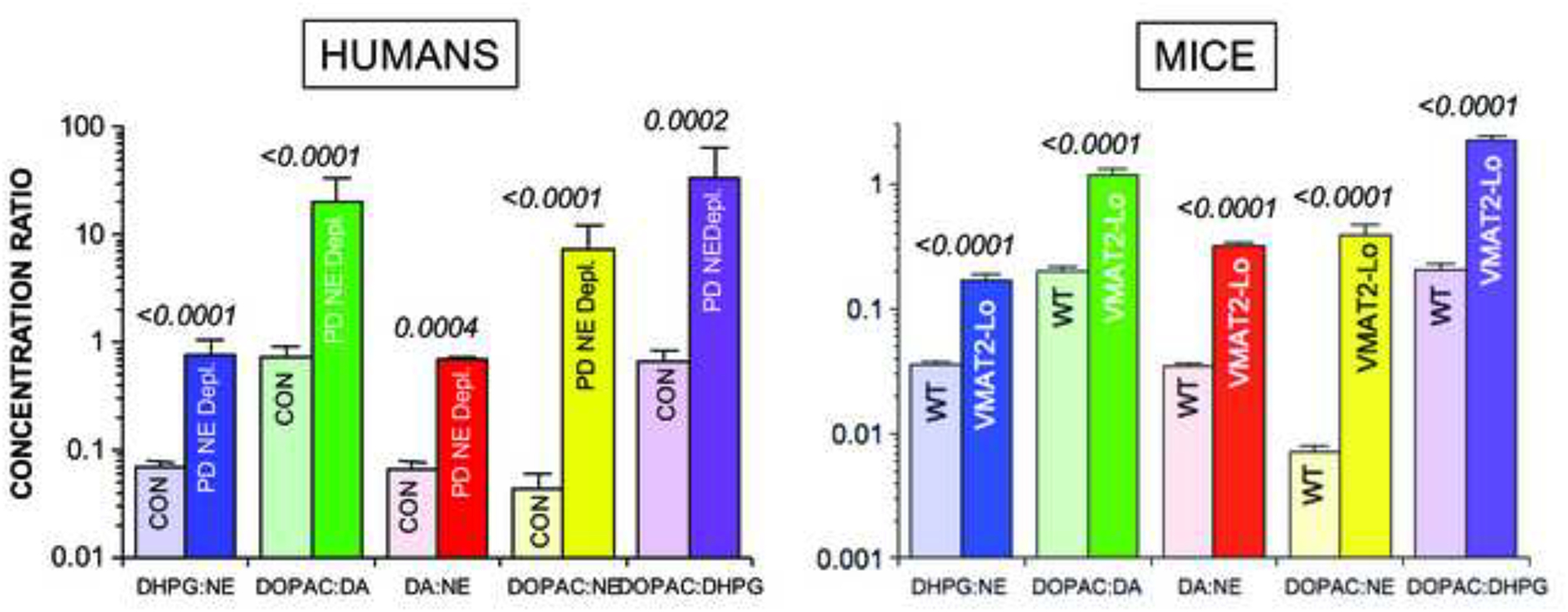
Note similar abnormalities of indices of vesicular storage in PD patients with norepinephrine depletion and in VMAT-2 Lo mice.
Figure 8: Estimated rates of processes in myocardial sympathetic nerves in control subjects and in PD patients.
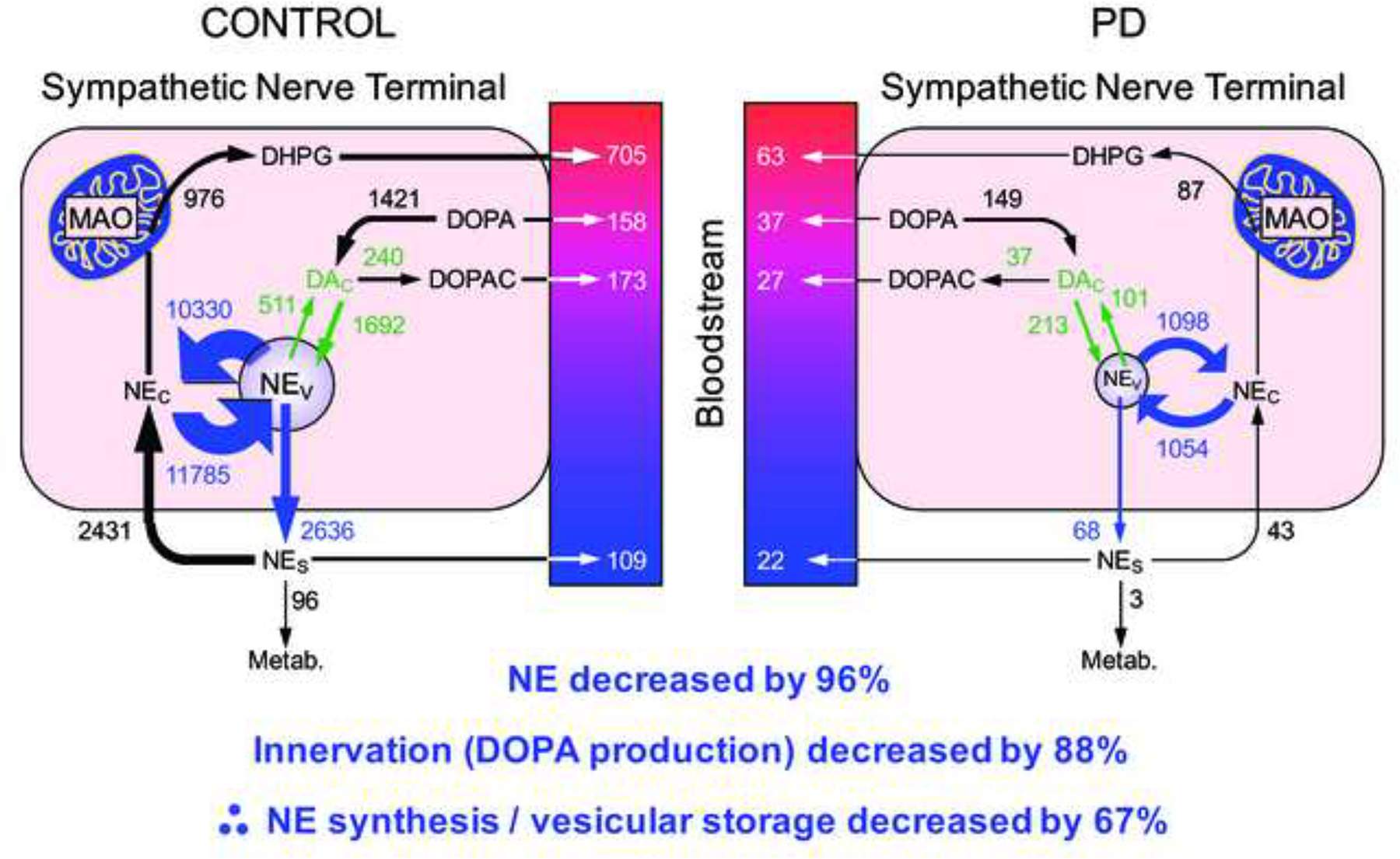
Norepinephrine (NE) depletion reflects both denervation (decreased DOPA production) and decreased vesicular storage in the residual sympathetic nerves. The model cannot distinguish decreased vesicular uptake from increased vesicular leakage and decreased intra-vesicular NE synthesis as determinants of tissue NE depletion.
Vesicular uptake limits the buildup of toxic products of enzymatic and spontaneous oxidation of catecholamines. A vesicular storage defect could therefore play a pathogenic role in the death of catecholaminergic neurons, as explained below in the section on catecholamine autotoxicity.
Analogously decreased vesicular storage in putamen dopaminergic terminals
Analogous to the findings in the heart, there is accumulating evidence for a vesicular storage defect also in the residual nigrostriatal dopaminergic terminals in parkinsonian synucleinopathies. We used putamen tissue levels and ratios of cysteinyl and parent catechols to explore possible denervation-independent abnormalities of dopamine synthesis and fate in PD and MSA (Figure 9). 5-S-Cysteinyldopa (Cys-DOPA) is produced from spontaneous oxidation of DOPA and 5-S-cysteinyldopamine (Cys-DA) from spontaneous oxidation of DA. Post-mortem putamen tissue samples from PD and MSA patients and controls were assayed for endogenous catechols including DA, its cytoplasmic metabolites (Cys-DA, 3,4-dihydroxyphenylacetic acid, 3,4-dihydroxyphenylethanol, and 3,4-dihydroxyphenylacetaldehyde), and tyrosine hydroxylation products proximal to DA (DOPA and Cys-DOPA). The PD and MSA groups did not differ in mean values of parent or cysteinyl catechols, and the data for the two groups were combined. In the patients an index of vesicular storage of DA (the ratio of DA to the sum of its cytoplasmic metabolites) averaged 54% of control; an index of L-aromatic-amino-acid decarboxylase (LAAAD) activity (the ratio of DA and the sum of its cytoplasmic metabolites to the sum of DOPA+Cys-DOPA) averaged 21% of control; and an index of innervation (the sum of DOPA+Cys-DOPA) averaged 63% of control. Based on the patterns of parent and cysteinyl catechols in putamen tissue, PD and MSA therefore both involve decreased vesicular uptake and decreased LAAAD activity in the residual dopaminergic terminals (Goldstein et al., 2017b).
Figure 9: Concept diagram about sources and metabolic fate of dopamine in putamen tissue.
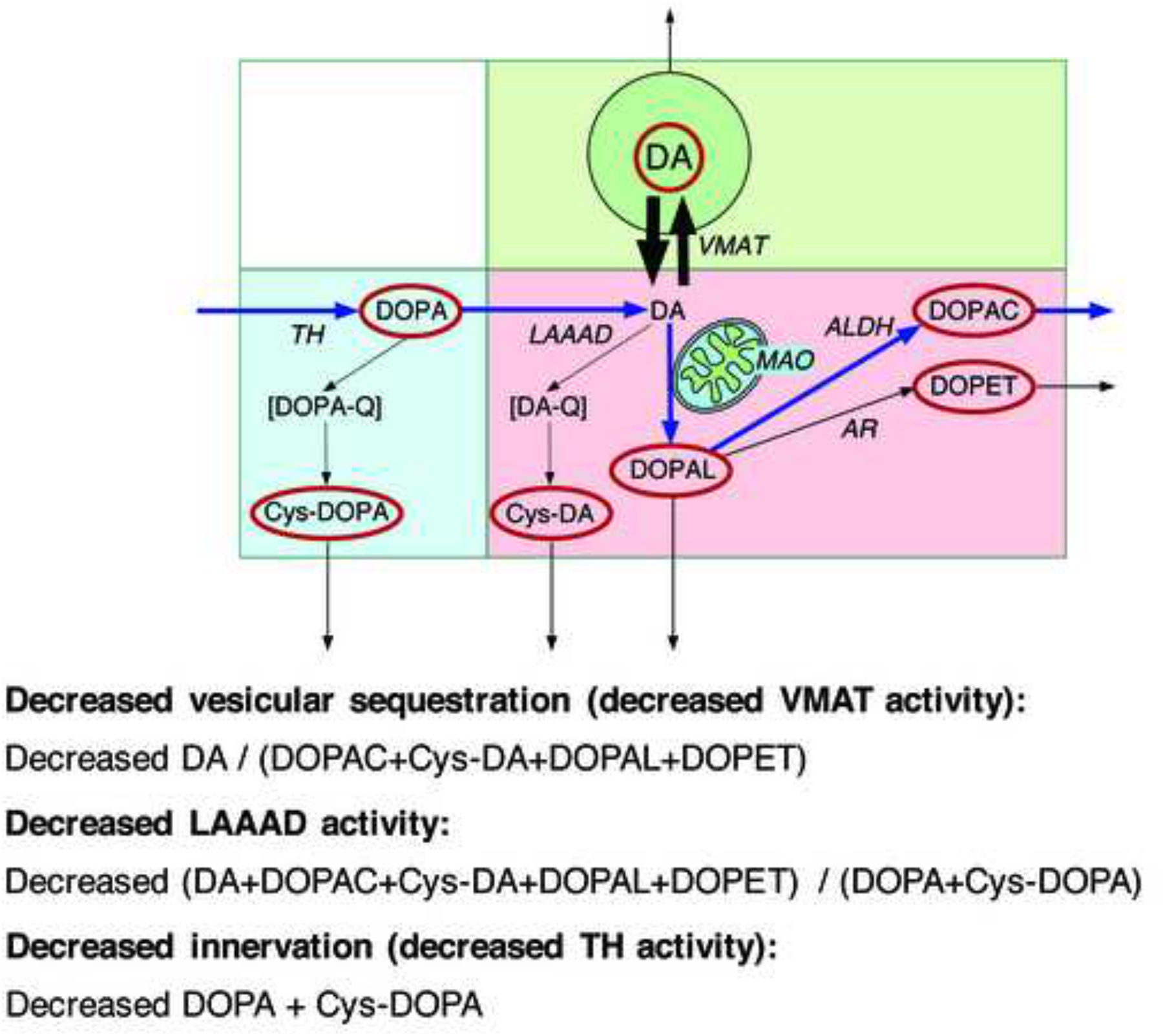
Tyrosine hydroxylase (TH) catalyzes the conversion of tyrosine to DOPA, and L-aromatic-amino-acid decarboxylase (LAAAD) converts DOPA to dopamine (DA). Most of the DA in putamen tissue is in vesicles, due to uptake mediated by the vesicular monoamine transporter (VMAT). Cytoplasmic DA can be metabolized by monoamine oxidase (MAO) in the outer mitochondrial membrane to form 3,4-dihydroxyphenylacetaldehyde (DOPAL), which is metabolize by aldehyde dehydrogenase (ALDH) to form 3,4-dihydroxyphenylacetic acid (DOPAC) or by aldehyde/aldose reductase (AR) to form 3,4-dihydroxyphenylethanol (DOPET). Cytoplasmic DA can oxidize spontaneously to form DA-quinone (DA-Q) and then 5-S-cysteinyl-DA (Cys-DA), and cytoplasmic DOPA can oxidize spontaneously to form DOPA-quinone (DOPA-Q) and then 5-S-cysteinyl-DOPA (Cys-DOPA). The rectangle in aqua corresponds to products of TH proximal to DA; in pink to cytoplasmic DA metabolites; and in green to vesicular DA.
There seem to be multiple determinants of decreased vesicular sequestration of cytoplasmic catecholamines in the striatum in patients with parkinsonian synucleinopathies. Decreased vesicular storage could be due to limited availability of ATP required for the energy-requiring vesicular uptake process, decreased expression or activity of VMAT2 (Taylor et al., 2014), decreased axonal transport of vesicles or vesicle-associated proteins (Chu et al., 2012; Gaugler et al., 2012; Lundblad et al., 2012), or vesicle permeabilization (Plotegher et al., 2017). Pifl et al. (Pifl et al., 2014) isolated dopamine storage vesicles from the striatum of autopsied brains of PD patients and found that (1) vesicular uptake of dopamine was decreased by 87–90% and VMAT2 binding by 71–80%; (2) after correcting for dopaminergic terminal loss, dopamine uptake per VMAT2 transport site was reduced by 55%; and dopamine efflux studies and measurements of acidification in the vesicular preparations suggested that the dopamine storage impairment was localized at the VMAT2 protein itself.
Centripetal, retrograde degeneration of cardiac sympathetic nerves in Lewy body diseases
Previous reports have provided immunohistochemical evidence for a particular sequence of catecholaminergic denervation in PD and other Lewy body diseases. Loss of the myocardial axons occurs before loss of the neuronal cell bodies in sympathetic ganglia, suggesting a centripetal, or “die-back,” sequence (Orimo et al., 2008b). There is earlier loss of the relatively more distal inferolateral free wall than anterobasal septum innervation (Jain and Goldstein, 2012; Li et al., 2002) (Figure 10).
Figure 10: Myocardial 18F-dopamine-derived radioactivity as a function of time in (top) a PD patient with initially normal radioactivity and (bottom) a PD patient with decreased radioactivity in the apex and free wall.
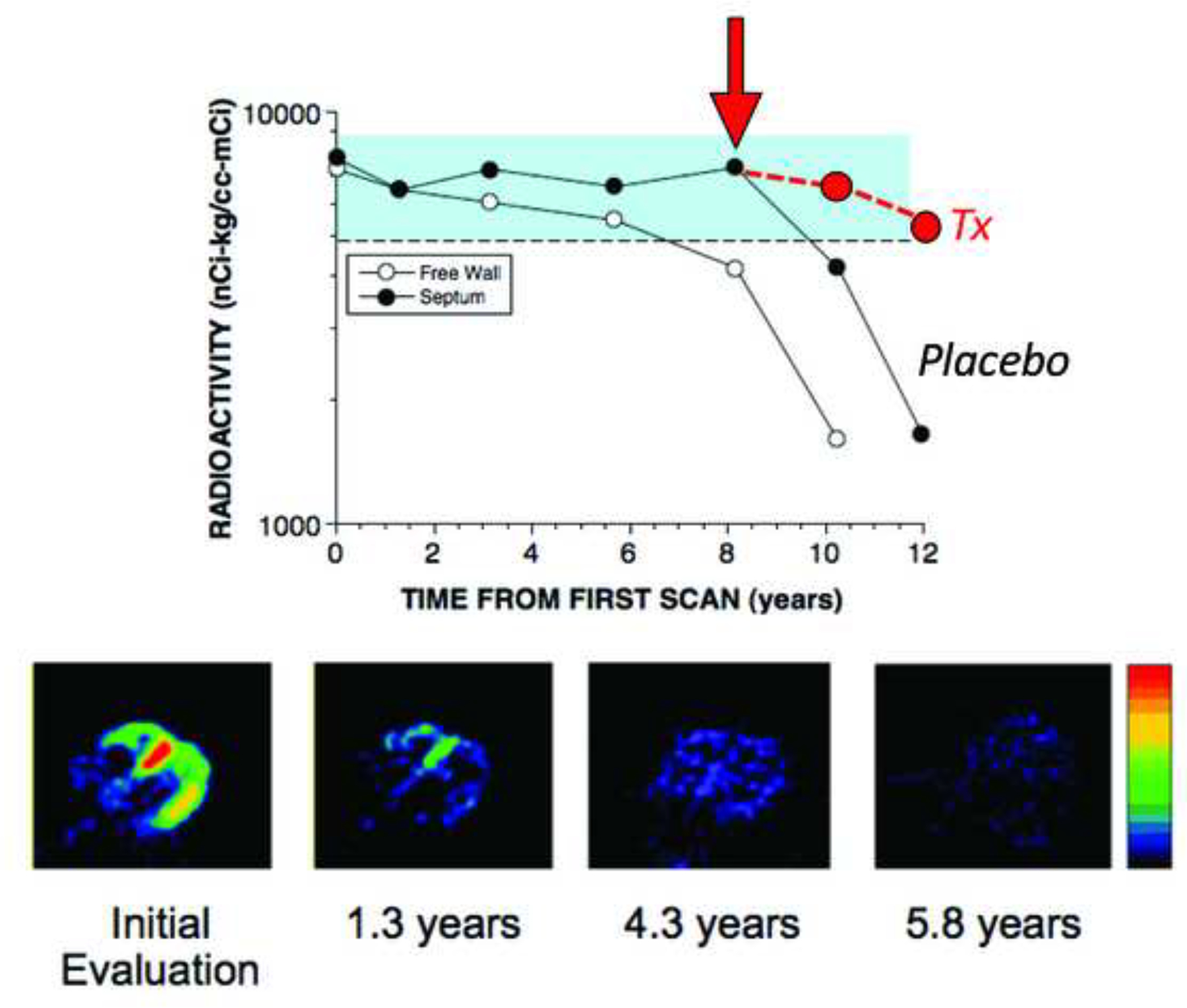
In the PD patient at the top, radioactivity was normal for about 8 years, then free wall radioactivity declined, and then septal radioactivity followed after about 2 years. In the PD patient at the bottom, there was rapid loss of radioactivity over a few years. Red circles placed to indicate hypothesized slowing of cardiac sympathetic denervation in an experimental therapeutics trial.
We recently evaluated this hypothesis by measuring apical myocardial and sympathetic ganglionic concentrations of catechols in patients with PD or PAF and in control subjects. Myocardial norepinephrine in the Lewy body disease group averaged 3.4% that in the control group, whereas sympathetic ganglion norepinephrine in the Lewy body disease group averaged 64% that in the control group—about a 20-fold difference in severity of norepinephrine depletion in the myocardium compared to sympathetic ganglion. The results indicate far greater loss of norepinephrine in the myocardial target tissue than in the ganglia, consistent with the die-back concept (unpublished observations).
Independent putamen and cardiac catecholamine lesions in synucleinopathies
Braak’s neuropathological staging concept imputes early autonomic involvement (Braak et al., 2004; Del Tredici et al., 2010; Del Tredici and Braak, 2012). Consistent with this concept, we reported a case of neuroimaging evidence of cardiac sympathetic denervation preceding by about 4 years the motor onset of PD (Goldstein et al., 2007b).
Across individual PD patients, however, neither the severity of sympathetic denervation as assessed by cardiac 18F-dopamine neuroimaging nor values for measures of other non-motor manifestations are related to the severity of loss of nigrostriatal dopaminergic neurons as assessed by 18F-DOPA neuroimaging (Goldstein et al., 2008; Goldstein et al., 2011b). Similar results have been reported based on cardiac 11C-meta-hydroxyephedrine and striatal 11C-dihydrotetrabenazine binding (Raffel et al., 2006) and cardiac 123I-MIBG and brain 123I-ioflupane scanning (Lamotte et al., 2015). Therefore, autonomic dysfunction, at least as reflected by sympathetic noradrenergic denervation, seems to occur independently of the dopaminergic lesion that produces the movement disorder. Because of this independence, although neuroimaging evidence of cardiac sympathetic denervation invariably progresses over time in PD and can come on years before the movement disorder, the denervation can also be a late finding (Figure 10).
Myocardial sympathetic lesion and non-motor manifestations of Lewy body diseases
In PD, anosmia, REM behavior disorder, cognitive dysfunction, and OH all have been associated with cardiac sympathetic denervation (Goldstein et al., 2002) (Goldstein et al., 2009; Kashihara et al., 2010; Kim et al., 2009).
Almost all patients with PD have at least some olfactory dysfunction, and a substantial minority have absent sense of smell—anosmia. Olfactory function can be quantified by the University of Pennsylvania Smell Identification Test (UPSIT). Among patients with alpha-synucleinopathies, UPSIT scores are correlated positively with myocardial noradrenergic innervation as assessed by cardiac sympathetic neuroimaging (Goldstein et al., 2008).
Several sleep disorders are associated with PD; however, REM behavior disorder stands out. In REM behavior disorder, the patients act out their dreams (“dream enactment behavior”) and thrash about in bed. Polysomnography shows absence of the atonia that normally accompanies REM sleep. REM behavior disorder patients have substantial myocardial noradrenergic denervation as indicated by cardiac sympathetic neuroimaging. Indeed, heart/mediastinum ratios of 123I-MIBG-derived radioactivity have been reported to be even more severely decreased in REM behavior disorder than in PD (Kashihara et al., 2010).
PD patients commonly eventually develop dementia. In some patients, dementia precedes the movement disorder or dominates the clinical picture, or the presence of visual hallucinations and fluctuating cognition leads to a diagnosis of dementia with Lewy bodies (DLB) when the clinical findings are insufficient to diagnose PD. DLB features neuroimaging evidence of substantial cardiac sympathetic denervation. In this regard, DLB differs from Alzheimer’s disease, in which cardiac sympathetic innervation is thought to be generally intact. Because of this distinction, cardiac sympathetic neuroimaging has been proposed as a means to diagnose DLB differentially from Alzheimer’s disease (Kobayashi et al., 2009) (Yoshita et al., 2006).
It has been reported that among PD patients, it is specifically the subgroup with non-motor manifestations that has neuroimaging evidence of cardiac sympathetic denervation (Kim et al., 2017). About ½ of PD patients without OH have diffusely decreased 18F-dopamine-derived radioactivity in the left ventricular myocardium, in contrast with all PD+OH patients (Goldstein et al., 2011a; Jain and Goldstein, 2012).
Mechanisms for the association between non-motor manifestations and neuroimaging evidence for cardiac sympathetic denervation remain unknown. A hypothesis for future research is that they are linked because they involve depletion of norepinephrine.
Cardiac sympathetic denervation linked to synucleinopathy in Lewy body diseases
Cardiac sympathetic denervation seems to be linked etiologically to synucleinopathy. Patients with PD due to inherited alpha-synucleinopathy, either from mutation of the gene encoding alpha-synuclein or from replication of the normal alpha-synuclein gene, have neuroimaging or neuropathologic evidence of cardiac sympathetic denervation (Goldstein et al., 2001; Orimo et al., 2008a; Singleton et al., 2004). Patients carrying the E46K mutation of the alpha-synuclein gene have neuroimaging evidence of cardiac sympathetic denervation (Tijero et al., 2013), even before abnormal striatal 123I-FP-CIT scanning (Tijero et al., 2010). Cardiac sympathetic denervation has also been reported in Gaucher/PD and in PD associated with LRRK2 mutation, both of which can involve Lewy bodies (Goldstein et al., 2007a; Lebouvier et al., 2014). Meanwhile, patients with familial PD due to parkin gene mutation, which is not typically associated with Lewy bodies, have been reported to have normal cardiac sympathetic innervation (Orimo et al., 2005b).
Both PARK1 and PARK4 are associated with physiological evidence of baroreflex-sympathoneural failure and neuroimaging evidence of cardiac noradrenergic denervation (Goldstein et al., 2001; Singleton et al., 2004), establishing that inherited alpha-synucleinopathy can cause catecholaminergic neurodegeneration both in the brain and periphery. A recent study reported alpha-synuclein aggregates in the sympathetic ganglia or myocardium in all patients with PD or DLB (Gelpi et al., 2014).
Functional and symptomatic effects of cardiac sympathetic denervation
The functional effects of cardiac sympathetic denervation in Lewy body diseases are poorly understood. Cardiac sympathetic denervation probably does not affect cardiac structure or function under resting conditions; however, patients with cardiac sympathetic denervation due to PD+OH or PAF have a failure to increase myocardial contractility in response to stimuli that depend on endogenous norepinephrine release (Imrich et al., 2008). Thus, during stresses such as exercise, there might be limited ability to increase cardiac inotropy, and the patient might have dyspnea on exertion or fatigue. Even in this situation, however, a combination of denervation supersensitivity, generally unaffected adrenomedullary release of epinephrine, and baroreflex failure could mask the effect.
The catecholamine autotoxicity theory and catecholaldehyde hypothesis
Alpha-synuclein overexpression increases cytoplasmic dopamine concentrations (Mosharov et al., 2006), inhibits VMAT2 activity (Guo et al., 2008), and down-regulates VMAT2 gene expression (Lotharius et al., 2002). Conditional over-expression of the protein in PC12 cells exerts a cytotoxic effect that is prevented by blocking dopamine synthesis (Ito et al., 2010). These abnormalities promote build-up of the deaminated dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL). The “catecholaldehyde hypothesis” states that increased levels of DOPAL cause or contribute to the death of nigrostriatal dopaminergic neurons in PD (Burke et al., 2003) (Figure 11).
Figure 11: Overview of the catecholamine autotoxicity theory as applied to cardiac sympathetic denervation in Lewy body diseases.
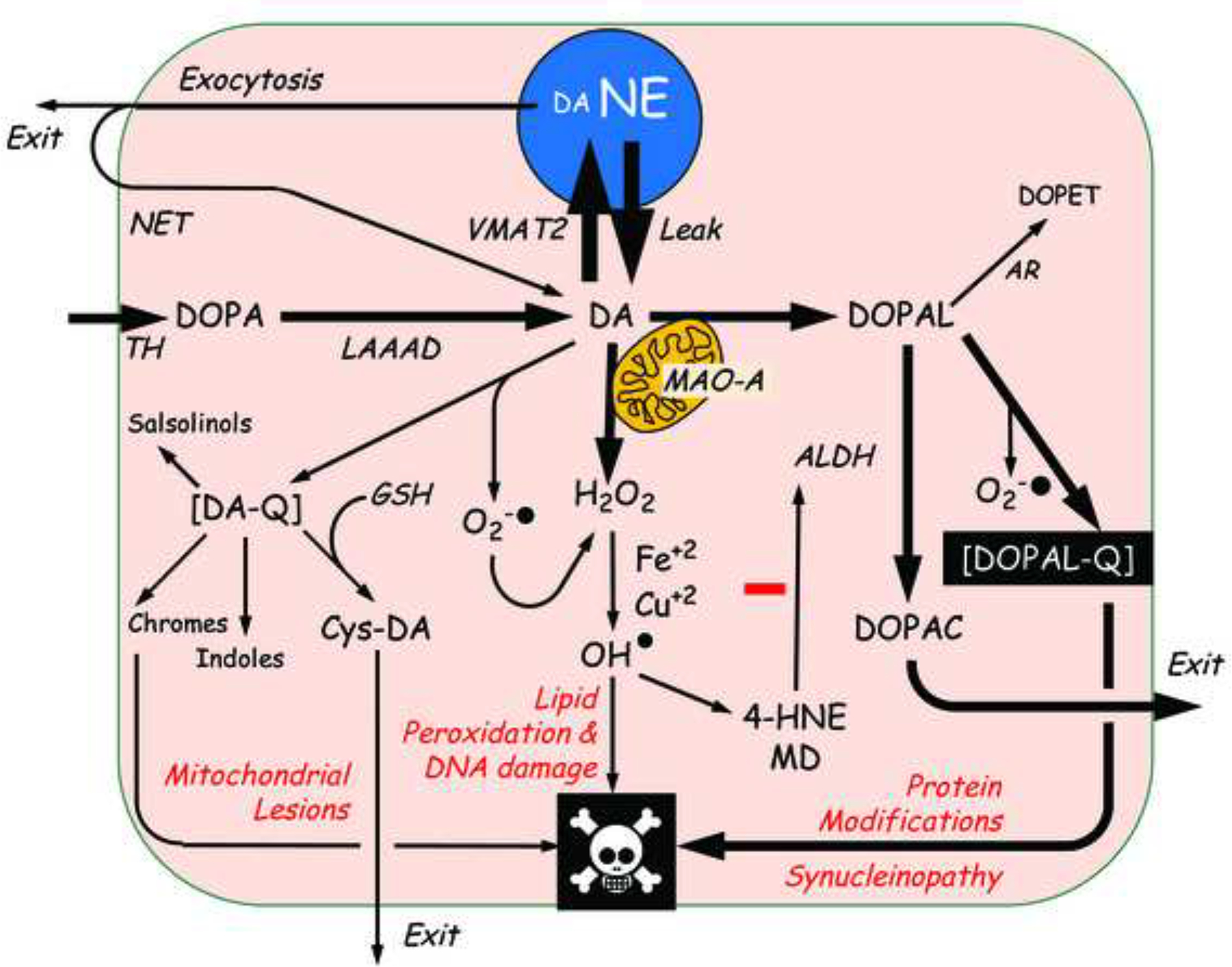
The theory explains PD in terms of toxic effects of products of enzymatic and spontaneous oxidation of cytoplasmic dopamine (DA), via mitochondrial lesions; lipid peroxidation and DNA damage, and protein modifications including alpha-synucleinopathy. The complex inter-relationships set the stage for allostatic load and multiple positive feedback loops that threaten neuronal integrity. Not shown are cytoplasmic norepinephrine (NE), its deamination via monoamine oxidase A (MAO-A) to form the catecholaldehyde 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL), and the reduction of DOPEGAL via aldehyde reductase (AR) to form 3,4-dihydroxyphenylglycol (DHPG). Other abbreviations: ALDH=aldehyde dehydrogenase; Cys-DA=5-S-cysteinyldopamine; DA-Q=dopamine quinone; DOPAC=3,4-dihydroxyphenylacetic acid; DOPAL=3,4-dihydroxyphenylacetaldehyde; DOPAL-Q=DOPAL quinone; GSH=reduced glutathione; 4-HNE=4-hydroxynonenal; LAAAD=L-aromatic-amino-acid decarboxylase; MD=malondialdehyde; NET=cell membrane norepinephrine transporter; TH=tyrosine hydroxylase; VMAT2=type 2 vesicular monoamine transporter.
DOPAL potently oligomerizes alpha-synuclein (Burke et al., 2008; Mazzulli et al., 2006), and the oligomers are thought to be toxic (Winner et al., 2011). DOPAL-induced oligomers of alpha-synuclein impede vesicular sequestration of catecholamines by permeabilizing vesicles (Plotegher et al., 2017). Interactions of DOPAL with alpha-synuclein, via decreased vesicular uptake or augmented vesicular leakage, therefore could set the stage for multiple pathogenic positive feedback loops.
DOPAL spontaneously oxidizes to form DOPAL-quinone. DOPAL-quinone may be responsible for DOPAL-induced oligomerization of alpha-synuclein, since anti-oxidants interfere with or prevent the oligomerization (Follmer et al., 2015). It has been proposed that DOPAL-quinone forms adducts with lysine residues in proteins (Follmer et al., 2015), altering protein function functions or causing them to form toxic oligomers (Anderson et al., 2016; Follmer et al., 2015). The oxidation of DOPAL also generates reactive oxygen species, which can lead to lipid peroxidation via formation of hydroxyl radicals (Li et al., 2001). Lipid peroxidation products inhibit ALDH and therefore build up cytoplasmic DOPAL (Florang et al., 2007)—another positive feedback loop.
There are several other ways that DOPAL may act alone or in concert with other compounds to cause death of dopaminergic neurons. DOPAL can bind covalently to dopamine to form tetrahydropapaveroline (Marchitti et al., 2007; Walsh et al., 1970), it can react with hydrogen peroxide and divalent metal cations to produce extremely harmful hydroxyl radicals (Li et al., 2001), and it can oxidize spontaneously to form reactive oxygen species and DOPAL-quinone.
There is indirect evidence that elevated levels of DOPAL play a pathogenetic role in PD. Exogenous DOPAL is cytotoxic (Panneton et al., 2010), decreased DOPAL detoxification by ALDH is associated with aging-related parkinsonism in genetic animal models (Wey et al., 2012), and exogenous agents that elevate DOPAL levels produce acquired parkinsonism in animals (Lamensdorf et al., 2000a; Lamensdorf et al., 2000b; Sherer et al., 2003). Nevertheless, as yet there is no evidence that manipulations decreasing DOPAL formation or decreasing DOPAL oxidation to DOPAL-quinone slow the neurodegenerative process in PD.
Treatment implications of cardiac sympathetic denervation in Lewy body diseases
Based on the concept diagram in Figure 11, combining an MAO inhibitor with an anti-oxidant that is bioavailable to catecholaminergic neurons should decrease DOPAL formation and toxicity.
Hydroxytyrosol (synonymous with 3,4-dihydroxyphenylethanol, DOPET) is the main anti-oxidant in olives (Raederstorff, 2009). We recently reported that exogenous DOPET (hydroxytyrosol) prevents MAO inhibitor-induced increases in Cys-DA levels in PC12 cells (Goldstein et al., 2016). Potential limitations of DOPET are that as a catechol it feedback-inhibits TH (Laschinski et al., 1986), and its bioavailability is poor (de la Torre, 2008; Goldstein et al., 2017a; Kountouri et al., 2007).
N-Acetylcysteine (NAC) is a precursor of the cellular anti-oxidant, glutathione, and in PD patients NAC increases brain glutathione levels (Holmay et al., 2013). Oral NAC increases cerebrospinal fluid NAC concentrations to about 10 μM (Reyes et al., 2016), and in mice neuronal reduced glutathione levels and anti-oxidant capacity are augmented at NAC doses that produce peak cerebrospinal fluid NAC concentrations of ≥50 nM. Thus, NAC seems to have good bioavailability to the brain.
As noted above, DOPAL-quinone may mediate reactivity of DOPAL with lysine-rich proteins such as alpha-synuclein (Follmer et al., 2015). NAC inhibits the protein reactivity of DOPAL, presumably via decreased DOPAL-quinone production (Anderson et al., 2016). Application of a recently described method to measure ortho-quinones by near-infrared spectroscopy (Mazzulli et al., 2016) has shown that NAC inhibits the oxidation of DOPAL to DOPAL-quinone (unpublished observations). Finally, NAC inhibits the DOPAL-induced oligomerization of alpha-synuclein (Anderson et al., 2016). A case is building for a proof of principle experimental therapeutics trial to test whether combining an MAO inhibitor with NAC slows the loss of myocardial sympathetic neurons in Lewy body diseases.
ACKNOWLEDGEMENTS
The research reported here was supported by the Division of Intramural Research of the National Institute of Neurological Disorders and Stroke.
Financial support:
Division of Intramural Research, NINDS, NIH.
ABBREVIATIONS
- ALDH
aldehyde dehydrogenase
- AR
aldehyde/aldose reductase
- Cys-DA
5-S-cysteinyldopamine
- Cys-DOPA
5-S-Cysteinyldopa
- DA
dopamine
- DA-Q
dopamine quinone
- DHPG
3,4-dihydroxyphenylglycol
- DLB
dementia with Lewy bodies
- DOPAC
3,4-dihydroxyphenylacetic acid
- DOPAL
3,4-dihydroxyphenylacetaldehyde
- DOPAL-Q
DOPAL quinone
- DOPET
3,4-dihydroxyphenylethanol
- GSH
reduced glutathione
- 4-HNE
4-hydroxynonenal
- ILBD
Incidental Lewy body disease
- LAAAD
L-aromatic-amino-acid decarboxylase
- MD
malondialdehyde
- 123I-MIBG
123I-metaiodobenzylguanidine
- MSA
multiple system atrophy
- NE
norepinephrine
- NET
cell membrane norepinephrine transporter
- OH
orthostatic hypotension
- PAF
pure autonomic failure
- PD
Parkinson’s disease
- PDD
Parkinson’s disease with dementia
- TH
tyrosine hydroxylase
- VMAT2
type 2 vesicular monoamine transporter
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
- Anderson DG, Florang VR, Schamp JH, Buettner GR, Doorn JA, 2016. Antioxidant-mediated modulation of protein reactivity for 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Chem. Res. Toxicol 29, 1098–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K, 2004. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS, 2003. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res. 989, 205–13. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE, 2008. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 115, 193–203. [DOI] [PubMed] [Google Scholar]
- Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH, 2012. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain. 135, 2058–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook GA, Sullivan P, Holmes C, Goldstein DS, 2014. Cardiac sympathetic denervation without Lewy bodies in a case of multiple system atrophy. Parkinsonism Relat. Disord 20, 926–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, 2008. Bioavailability of olive oil phenolic compounds in humans. Inflammopharmacology. 16, 245–247. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H, 2010. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 119, 703–13. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, Braak H, 2012. Lewy pathology and neurodegeneration in premotor Parkinson’s disease. Movement Dis. 27, 597–607. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, Ahlskog JE, 2008. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 115, 437–44. [DOI] [PubMed] [Google Scholar]
- Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER, 2010. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet 74, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florang VR, Rees JN, Brogden NK, Anderson DG, Hurley TD, Doorn JA, 2007. Inhibition of the oxidative metabolism of 3,4-dihydroxyphenylacetaldehyde, a reactive intermediate of dopamine metabolism, by 4-hydroxy-2-nonenal. Neurotoxicology. 28, 76–82. [DOI] [PubMed] [Google Scholar]
- Follmer C, Coelho-Cerqueira E, Yatabe-Franco DY, Araujo GD, Pinheiro AS, Domont GB, Eliezer D, 2015. Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of alpha-synuclein with the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL). J Biol Chem. 290, 27660–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishiro H, Frigerio R, Burnett M, Klos KJ, Josephs KA, Delledonne A, Parisi JE, Ahlskog JE, Dickson DW, 2008. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov. Disord 23, 1085–92. [DOI] [PubMed] [Google Scholar]
- Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun JC, Schneggenburger R, Knott GW, Aebischer P, Schneider BL, 2012. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 123, 653–669. [DOI] [PubMed] [Google Scholar]
- Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, Marti MJ, Hernandez I, Valldeoriola F, Rene R, Ribalta T, 2014. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord. 29, 1010–8. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Grossman E, Tamrat M, Chang PC, Eisenhofer G, Bacher J, Kirk KL, Bacharach S, Kopin IJ, 1991. Positron emission imaging of cardiac sympathetic innervation and function using 18F-6-fluorodopamine: effects of chemical sympathectomy by 6-hydroxydopamine. J. Hypertens 9, 417–423. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Cannon RO 3rd, Eisenhofer G, Kopin IJ, 1997. Sympathetic cardioneuropathy in dysautonomias. N. Engl. J. Med 336, 696–702. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO 3rd, 2000. Cardiac sympathetic denervation in Parkinson disease. Ann. Intern. Med 133, 338–47. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Li ST, Kopin IJ, 2001. Sympathetic neurocirculatory failure in Parkinson disease: Evidence for an etiologic role of alpha-synuclein. Ann. Intern. Med 135, 1010–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes CS, Dendi R, Bruce SR, Li ST, 2002. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology. 58, 1247–55. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, 2003. Dysautonomia in Parkinson’s disease: neurocardiological abnormalities. Lancet Neurol. 2, 669–676. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, 2006. Orthostatic hypotension as an early finding in Parkinson disease. Clin. Auton. Res 16, 46–64. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Imrich R, Peckham E, Holmes C, Lopez G, Crews C, Hardy J, Singleton A, Hallett M, 2007a. Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation. Neurology. 69, 1580–4. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sharabi Y, Karp BI, Bentho O, Saleem A, Pacak K, Eisenhofer G, 2007b. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Clin. Auton. Res 17, 118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, Imrich R, Conant S, Eldadah BA, 2008. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat. Disord 14, 600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Orimo S, 2009. Cardiac sympathetic neuroimaging: summary of the First International Symposium. Clin. Auton. Res 19, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sewell L, Holmes C, 2009. Association of anosmia with autonomic failure in Parkinson disease. Neurology. 74, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Kopin IJ, Sharabi Y, 2011a. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J. Clin. Invest 121, 3320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sewell L, Sharabi Y, 2011b. Autonomic dysfunction in PD: A window to early detection? J. Neurol. Sci 310, 118–122. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC, 2011c. Catechols in post-mortem brain of patients with Parkinson disease. Eur. J. Neurol 18, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Miller GW, Sharabi Y, Kopin IJ, 2014. A vesicular sequestration to oxidative deamination shift in myocardial sympathetic nerves in Parkinson disease. J. Neurochem 131, 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Kopin IJ, Sharabi Y, Holmes C, 2015. Plasma biomarkers of decreased vesicular storage distinguish Parkinson disease with orthostatic hypotension from the parkinsonian form of multiple system atrophy. Clin. Auton. Res 25, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Jinsmaa Y, Sullivan P, Kopin IJ, Sharabi Y, 2016. 3,4-Dihydroxyphenylethanol (hydroxytyrosol) mitigates the increase in spontaneous oxidation of dopamine during monoamine oxidase inhibition in PC12 cells. Neurochem. Res 41, 2173–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Holmes C, Cherup J, Sharabi Y, 2017a. Plasma catechols after eating olives. Clin. Trans. Sci (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS, Sullivan P, Holmes C, Mash DC, Kopin IJ, Sharabi Y, 2017b. Determinants of denervation-independent depletion of putamen dopamine in Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord. 35, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts SN, 2016. Lewy Body Dementias: Dementia With Lewy Bodies and Parkinson Disease Dementia. Continuum. 22, 435–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Chen AQ, Kong Q, Zhu H, Ma CM, Qin C, 2008. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell. Mol. Neurobiol 28, 35–47. [DOI] [PubMed] [Google Scholar]
- Hague K, Lento P, Morgello S, Caro S, Kaufmann H, 1997. The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol. 94, 192–6. [DOI] [PubMed] [Google Scholar]
- Holdorff B, 2002. Friedrich Heinrich Lewy (1885–1950) and his work. J. Hist. Neurosci 11, 19–28. [DOI] [PubMed] [Google Scholar]
- Holmay MJ, Terpstra M, Coles LD, Mishra U, Ahlskog M, Oz G, Cloyd JC, Tuite PJ, 2013. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol 36, 103–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono D, Geraci-Erck M, Rabin ML, Adler CH, Serrano G, Beach TG, Kurlan R, 2015. Parkinson disease and incidental Lewy body disease: Just a question of time? Neurology. 85, 1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imrich R, Eldadah BA, Bentho O, Pechnik S, Sharabi Y, Holmes C, Goldstein DS, 2008. Attenuated pre-ejection period response to tyramine in patients with cardiac sympathetic denervation. Ann. N .Y. Acad. Sci 1148, 486–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Nakaso K, Imamura K, Takeshima T, Nakashima K, 2010. Endogenous catecholamine enhances the dysfunction of unfolded protein response and alpha-synuclein oligomerization in PC12 cells overexpressing human alpha-synuclein. Neurosci. Res 66, 124–30. [DOI] [PubMed] [Google Scholar]
- Jain S, Goldstein DS, 2012. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol. Dis 46, 572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, 2003. Neuropathological spectrum of synucleinopathies. Mov Disord. 18 Suppl 6, S2–12. [DOI] [PubMed] [Google Scholar]
- Jost WH, Del Tredici K, Landvogt C, Braune S, 2010. Importance of 123I-metaiodobenzylguanidine scintigraphy/single photon emission computed tomography for diagnosis and differential diagnostics of Parkinson syndromes. Neurodegener Dis. 7, 341–7. [DOI] [PubMed] [Google Scholar]
- Kashihara K, Ohno M, Kawada S, Okumura Y, 2006. Reduced cardiac uptake and enhanced washout of 123I-MIBG in pure autonomic failure occurs conjointly with Parkinson’s disease and dementia with Lewy bodies. J. Nucl. Med 47, 1099–1101. [PubMed] [Google Scholar]
- Kashihara K, Imamura T, Shinya T, 2010. Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat Disord. 16, 252–5. [DOI] [PubMed] [Google Scholar]
- Kim JS, Shim YS, Song IU, Yoo JY, Kim HT, Kim YI, Lee KS, 2009. Cardiac sympathetic denervation and its association with cognitive deficits in Parkinson’s disease. Parkinsonism & related disorders. 15, 706–8. [DOI] [PubMed] [Google Scholar]
- Kim JS, Oh YS, Lee KS, Kim YI, Yang DW, Goldstein DS, 2012. Association of cognitive dysfunction with neurocirculatory abnormalities in early Parkinson disease. Neurology. 79, 1323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Park HE, Park IS, Oh YS, Ryu DW, Song IU, Jung YA, Yoo IR, Choi HS, Lee PH, Lee KS, 2017. Normal ‘heart’ in Parkinson’s disease: is this a distinct clinical phenotype? Eur. J. Neurol 24, 349–356. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O, 1988. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N. Engl. J. Med 318, 876–880. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Tateno M, Morii H, Utsumi K, Saito T, 2009. Decreased cardiac MIBG uptake, its correlation with clinical symptoms in dementia with Lewy bodies. Psychiatry Res. 174, 76–80. [DOI] [PubMed] [Google Scholar]
- Kountouri AM, Mylona A, Kaliora AC, Andrikopoulos NK, 2007. Bioavailability of the phenolic compounds of the fruits (drupes) of Olea europaea (olives): impact on plasma antioxidant status in humans. Phytomedicine. 14, 659–67. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Eisenhofer G, Harvey-White J, Hayakawa Y, Kirk K, Kopin IJ, 2000a. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J. Neurosci. Res 60, 552–8. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Eisenhofer G, Harvey-White J, Nechustan A, Kirk K, Kopin IJ, 2000b. 3,4-Dihydroxyphenylacetaldehyde potentiates the toxic effects of metabolic stress in PC12 cells. Brain Res. 868, 191–201. [DOI] [PubMed] [Google Scholar]
- Lamotte G, Morello R, Lebasnier A, Agostini D, Defer GL, 2015. Accuracy and cutoff values of delayed heart to mediastinum ratio with (123)I-metaiodobenzylguanidine cardiac scintigraphy for Lewy body disease diagnoses. BMC Neurol. 15, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschinski G, Kittner B, Brautigam M, 1986. Direct inhibition of tyrosine hydroxylase from PC-12 cells by catechol derivatives. Naunyn Schmiedebergs Arch. Pharmacol 332, 346–50. [DOI] [PubMed] [Google Scholar]
- Lebouvier T, Clairembault T, Devos D, Pallardy A, Coron E, Neunlist M, Derkinderen P, 2014. Peripheral autonomic nervous system involvement in Gaucher-related parkinsonism. J. Parkinsons Dis 4, 29–32. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Selikhova M, Andrade LA, Duyckaerts C, 2008. The black stuff and Konstantin Nikolaevich Tretiakoff. Mov. Disord 23, 777–83. [DOI] [PubMed] [Google Scholar]
- Li ST, Dendi R, Holmes C, Goldstein DS, 2002. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann. Neurol 52, 220–3. [DOI] [PubMed] [Google Scholar]
- Li SW, Lin TS, Minteer S, Burke WJ, 2001. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: possible role in Parkinson’s disease pathogenesis. Brain Res. Mol. Brain Res 93, 1–7. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P, 2002. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem 277, 38884–94. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Bjorklund A, 2012. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U.S.A 109, 3213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Inui Y, Toyama H, Kosaka K, 2017. 123I-metaiodobenzylguanidine myocardial scintigraphy with early images alone is useful for the differential diagnosis of dementia with Lewy bodies. Psychiatry Res. 261, 75–79. [DOI] [PubMed] [Google Scholar]
- Marchitti SA, Deitrich RA, Vasiliou V, 2007. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol. Rev 59, 125–150. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H, 2006. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 26, 10068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Burbulla LF, Krainc D, Ischiropoulos H, 2016. Detection of free and protein-bound ortho-quinones by near-infrared fluorescence. Anal. Chem 88, 2399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH, 1996. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 47, 1113–24. [DOI] [PubMed] [Google Scholar]
- Miura H, Tsuchiya K, Kubodera T, Shimamura H, Matsuoka T, 2001. An autopsy case of pure autonomic failure with pathological features of Parkinson’s disease. Rinsho Shinkeigaku. 41, 40–44. [PubMed] [Google Scholar]
- Miyoshi F, Ogawa T, Kitao SI, Kitayama M, Shinohara Y, Takasugi M, Fujii S, Kaminou T, 2013. Evaluation of Parkinson disease and Alzheimer disease with the use of neuromelanin MR imaging and (123)I-metaiodobenzylguanidine scintigraphy. AJNR Am J Neuroradiol. 34, 2113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Staal RG, Bove J, Prou D, Hananiya A, Markov D, Poulsen N, Larsen KE, Moore CM, Troyer MD, Edwards RH, Przedborski S, Sulzer D, 2006. Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J. Neurosci 26, 9304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H, 2005a. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol. 109, 583–8. [DOI] [PubMed] [Google Scholar]
- Orimo S, Amino T, Yokochi M, Kojo T, Uchihara T, Takahashi A, Wakabayashi K, Takahashi H, Hattori N, Mizuno Y, 2005b. Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov. Disord 20, 1350–1353. [DOI] [PubMed] [Google Scholar]
- Orimo S, Amino T, Takahashi A, Kojo T, Uchihara T, Mori F, Wakabayashi K, Takahashi H, 2006. Cardiac sympathetic denervation in Lewy body disease. Parkinsonism Relat. Disord 12 Suppl 2, S99–S105. [DOI] [PubMed] [Google Scholar]
- Orimo S, Kanazawa T, Nakamura A, Uchihara T, Mori F, Kakita A, Wakabayashi K, Takahashi H, 2007a. Degeneration of cardiac sympathetic nerve can occur in multiple system atrophy. Acta Neuropathol (Berl). 113, 81–86. [DOI] [PubMed] [Google Scholar]
- Orimo S, Takahashi A, Uchihara T, Mori F, Kakita A, Wakabayashi K, Takahashi H, 2007b. Degeneration of cardiac sympathetic nerve begins in the early disease process of Parkinson’s disease. Brain Pathol. 17, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Ikeuchi T, Onodera O, Nishizawa M, Ishikawa A, Kakita A, Wakabayashi K, Takahashi H, 2008a. Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol. 116, 575–7. [DOI] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H, 2008b. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain. 131, 642–50. [DOI] [PubMed] [Google Scholar]
- Orimo S, Suzuki M, Inaba A, Mizusawa H, 2012. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat. Disord 18, 494–500. [DOI] [PubMed] [Google Scholar]
- Panneton WM, Kumar VB, Gan Q, Burke WJ, Galvin JE, 2010. The neurotoxicity of DOPAL: behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS One. 5, e15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pifl C, Rajput A, Reither H, Blesa J, Cavada C, Obeso JA, Rajput AH, Hornykiewicz O, 2014. Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci 34, 8210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Berti G, Ferrari E, Tessari I, Zanetti M, Lunelli L, Greggio E, Bisaglia M, Veronesi M, Girotto S, Dalla Serra M, Perego C, Casella L, Bubacco L, 2017. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci Rep. 7, 40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Raederstorff D, 2009. Antioxidant activity of olive polyphenols in humans: a review. Int. J. Vitam. Nutr. Res 79, 152–65. [DOI] [PubMed] [Google Scholar]
- Raffel DM, Koeppe RA, Little R, Wang CN, Liu S, Junck L, Heumann M, Gilman S, 2006. PET measurement of cardiac and nigrostriatal denervation in parkinsonian syndromes. J. Nucl. Med 47, 1769–1777. [PubMed] [Google Scholar]
- Reyes RC, Cittolin-Santos GF, Kim JE, Won SJ, Brennan-Minnella AM, Katz M, Glass GA, Swanson RA, 2016. Neuronal glutathione content and antioxidant capacity can be normalized in situ by N-acetyl cysteine concentrations attained in human cerebrospinal fluid. Neurotherapeutics. 13, 217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T, 2009. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT, 2003. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol 179, 9–16. [DOI] [PubMed] [Google Scholar]
- Singleton A, Gwinn-Hardy K, Sharabi Y, Li ST, Holmes C, Dendi R, Hardy J, Crawley A, Goldstein DS, 2004. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain. 127, 768–72. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, 1997. Alpha-synuclein in Lewy bodies. Nature. 388, 839–40. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kurita A, Hashimoto M, Fukumitsu N, Abo M, Ito Y, Urashima M, Inoue K, 2006. Impaired myocardial 123I-metaiodobenzylguanidine uptake in Lewy body disease: comparison between dementia with Lewy bodies and Parkinson’s disease. J. Neurol. Sci 240, 15–9. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Ikemura M, Oka T, Uchihara T, Wakabayashi K, Kakita A, Takahashi H, Yoshida M, Toru S, Kobayashi T, Orimo S, 2015. Quantitative correlation between cardiac MIBG uptake and remaining axons in the cardiac sympathetic nerve in Lewy body disease. J Neurol Neurosurg Psychiatry. 86, 939–44. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Alter SP, Wang M, Goldstein DS, Miller GW, 2014. Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology. 76 Pt A, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello C, Darling A, Lupo V, Perez-Duenas B, Espinos C, 2017. On the complexity of clinical and molecular bases of neurodegeneration with brain iron accumulation. Clin Genet. [DOI] [PubMed] [Google Scholar]
- Tijero B, Gomez-Esteban JC, Llorens V, Lezcano E, Gonzalez-Fernandez MC, de Pancorbo MM, Ruiz-Martinez J, Cembellin JC, Zarranz JJ, 2010. Cardiac sympathetic denervation precedes nigrostriatal loss in the E46K mutation of the alpha-synuclein gene (SNCA). Clin. Auton. Res 20, 267–9. [DOI] [PubMed] [Google Scholar]
- Tijero B, Gomez-Esteban JC, Lezcano E, Fernandez-Gonzalez C, Somme J, Llorens V, Martinez A, Ruiz-Martinez J, Foncea N, Escalza I, Berganzo K, Aniel-Quiroga MA, Ruiz V, Teran N, Kaufmann H, Zarranz JJ, 2013. Cardiac sympathetic denervation in symptomatic and asymptomatic carriers of the E46K mutation in the alpha synuclein gene. Parkinsonism Relat Disord. 19, 95–100. [DOI] [PubMed] [Google Scholar]
- Treglia G, Cason E, Stefanelli A, Cocciolillo F, Di Giuda D, Fagioli G, Giordano A, 2012. MIBG scintigraphy in differential diagnosis of Parkinsonism: a meta-analysis. Clin Auton Res. 22, 43–55. [DOI] [PubMed] [Google Scholar]
- van Ingelghem E, van Zandijcke M, Lammens M, 1994. Pure autonomic failure: a new case with clinical, biochemical, and necropsy data. J. Neurol. Neurosurg. Psychiatry 57, 745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM, 2011. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat. Disord 17, 724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MJ, Davis VE, Yamanaka Y, 1970. Tetrahydropapaveroline: an alkaloid metabolite of dopamine in vitro. J. Pharmacol. Exp. Ther 174, 388–400. [PubMed] [Google Scholar]
- Wey M, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R, 2012. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: Implications for Parkinson’s disease. PLoS ONE. 7, e31522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R, 2011. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A 108, 4194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshita M, Taki J, Yokoyama K, Noguchi-Shinohara M, Matsumoto Y, Nakajima K, Yamada M, 2006. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 66, 1850–4. [DOI] [PubMed] [Google Scholar]