

Abstract
Hepatic inflammation is a common trigger of chronic liver disease. Macrophage activation is a predictive parameter for survival in patients with cirrhosis. Ring finger protein 41 (RNF41) negatively regulates proinflammatory cytokines and receptors; however, the precise involvement of macrophage RNF41 in liver cirrhosis remains unknown. Here, we sought to understand how RNF41 dictates macrophage fate in hepatic fibrosis and repair within the inflammatory milieu. We found that RNF41 expression is down-regulated in CD11b+ macrophages recruited to mouse fibrotic liver and to patient cirrhotic liver regardless of cirrhosis etiology. Prolonged inflammation with TNF-α progressively reduced macrophage RNF41 expression. We designed a macrophage-selective gene therapy with dendrimer-graphite nanoparticles (DGNPs) to explore the influence of macrophage RNF41 restoration and depletion in liver fibrosis and regeneration. RNF41 expression induced in CD11b+ macrophages by DGNP-conjugated plasmids ameliorated liver fibrosis, reduced liver injury, and stimulated hepatic regeneration in fibrotic mice with or without hepatectomy. This therapeutic effect was mainly mediated by the induction of insulin-like growth factor 1. Conversely, depletion of macrophage RNF41 worsened inflammation, fibrosis, hepatic damage, and survival. Our data reveal implications of macrophage RNF41 in the control of hepatic inflammation, fibrosis, and regeneration and provide a rationale for therapeutic strategies in chronic liver disease and potentially other diseases characterized by inflammation and fibrosis.
INTRODUCTION
Chronic liver disease accounts for nearly 2 million deaths per year worldwide. Cirrhosis is within the top 20 causes of disability-adjusted life years and years of life lost (1). No curative solutions exist for cirrhosis except for organ transplantation, which requires substantial surgery and lifelong immunosuppression. However, only 50% of eligible patients receive a liver transplant, which translates into a shortage of about 13,000 donors per year (2). Alternative strategies to treat cirrhosis and stimulate hepatic regeneration are thus being investigated, including nanotherapeutics and cell therapies (3–5).
Macrophages are cellular regulators involved in all stages of liver disease, from initial tissue injury to chronic inflammation, fibrosis, and repair (6). Resident hepatic macrophages release signals that promote local immune response and limit initial injury through the classic path of inflammatory cell recruitment and subsequent activation of hepatic stellate cells (HSCs) with production of a supporting extracellular matrix (ECM) (7–9). When injury abates, macrophages remodel fibrosis primarily by releasing matrix metalloproteinases (MMPs). MMPs then promote fibrotic ECM degradation and repair through elaboration of factors that reduce the inflammatory response and boost liver regeneration (10, 11).
Ring finger protein 41 (RNF41), also known as neuregulin receptor degradation protein 1 (Nrdp1) or fetal liver ring finger, is an E3 ubiquitin protein ligase that plays an essential role in the degradation of various proinflammatory cytokine receptors, adaptors, and kinases (12). This ligase inhibits the production of proinflammatory cytokines in Toll-like receptor–triggered macrophages via suppression of MyD88 and nuclear factor κB (NF-κB) activation and confers resistance to lipopolysaccharide-induced endotoxin shock (13). RNF41 also promotes anti-inflammatory macrophage polarization by ubiquitination and activation of the transcription factor CCAAT/enhancer-binding protein β (C/EBPβ) (14), which has been associated with muscle injury repair (15). Collectively, these data induced our investigation of the roles of RNF41 on the control of macrophage behavior in the context of chronic liver injury and regeneration. To our knowledge, nothing is known about the regulation of macrophage RNF41 expression in a prolonged tissue inflammatory environment or its pathophysiological roles in liver fibrosis and regeneration. Considering the crucial influence that macrophages exert on the modulation of the hepatic cellular response to injury, we further explored the use of a nanoscale gene therapy delivery system designed to modulate inflammatory macrophages for the harmonization of fibrosis resolution and hepatic regeneration. We recently reported that graphene-derived nanoparticles linked to polyamidoamine (PAMAM) dendrimers preferentially accumulate in inflammatory macrophages within the fibrotic liver, where they function as a precision gene therapy system (16). A modified version of this system was used in this investigation to explore the role of macrophage RNF41 in chronic liver disease.
Here, we investigated whether RNF41 is regulated by the sustained inflammatory milieu of the cirrhotic liver and how altered RNF41 expression in macrophages from cirrhotic livers affects hepatic inflammation, damage, and survival. We used macrophage-selective nanoparticles linked to plasmids to study the effects that the modulation of macrophage RNF41 expression exert on liver fibrosis and regeneration. We further explored the relationship between macrophage RNF41 and the synthesis of inflammatory and profibrogenic cytokines in different models of liver fibrosis and hepatic regeneration and the downstream molecular signals associated with these effects.
RESULTS
Macrophage RNF41 decays in human cirrhotic and mouse fibrotic liver
To determine the RNF41 gene expression in macrophages recruited to cirrhotic liver (CD11bhigh), we isolated CD11b+ macrophages from liver biopsy specimens of patients with liver cirrhosis and healthy participants. This cell surface marker is a selective macrophage marker in liver injury and regeneration (2). Twelve patients (n = 3 female and n = 9 males, 58.7 ± 6.1 years) with decompensated liver cirrhosis and MELD (Model for End-Stage Liver Disease) scores between 13 and 30 were selected from a single center (Hospital Clinic of Barcelona, Spain). Demographic and baseline characteristics of study participants are shown in table S1. Participants in the diseased group displayed a mean duration of cirrhosis of 4.3 ± 5.8 years. RNF41 macrophage mRNA expression was notably lower in macrophages from cirrhotic than healthy liver (Fig. 1A) regardless of cirrhosis etiology (alcoholic, nonalcoholic, hepatitis C, or autoimmune). This also occurred with ubiquitin-specific peptidase 8 (USP8) expression, a known stabilizer of RNF41 activity (Fig. 1B) (17). We also evaluated macrophage RNF41 expression in an animal model of CCl4-induced chronic liver injury and fibrosis in the BALB/c mouse strain because these mice are most sensitive to induction of liver fibrosis (18). CCl4 is a hepatotoxic molecule classically used to promote chronic liver injury, fibrosis, and infiltration of proinflammatory macrophages (18, 19). Quantification of RNF41 in CD11b+ macrophages from liver specimens obtained from healthy and fibrotic mice mirrored what we saw in human specimens with down-regulated RNF41 (Fig. 1C) and USP8 (Fig. 1D). Although human and mouse CD11b+ macrophages were isolated after removing other CD11b+ cells such as neutrophils and dendritic cells, we cannot exclude that RNF41 and USP8 down-regulation is shared by different specific macrophage subpopulations with diverse abundances of CD11b, suggesting that these findings should be considered for all liver CD11b+ macrophages. No changes in RNF41 or USP8 expression were found in hepatocytes isolated from hepatic specimens obtained from healthy participants, cirrhotic patients, or mice with liver fibrosis (fig. S1). Neither RNF41 nor USP8 expression was down-regulated in HSCs and up-regulated in liver sinusoidal endothelial cells (LSECs) from fibrotic mice (fig. S1).
Fig. 1. Macrophage RNF41 and its stabilizer USP8 are down-regulated in cirrhotic liver in part due to chronic inflammation.
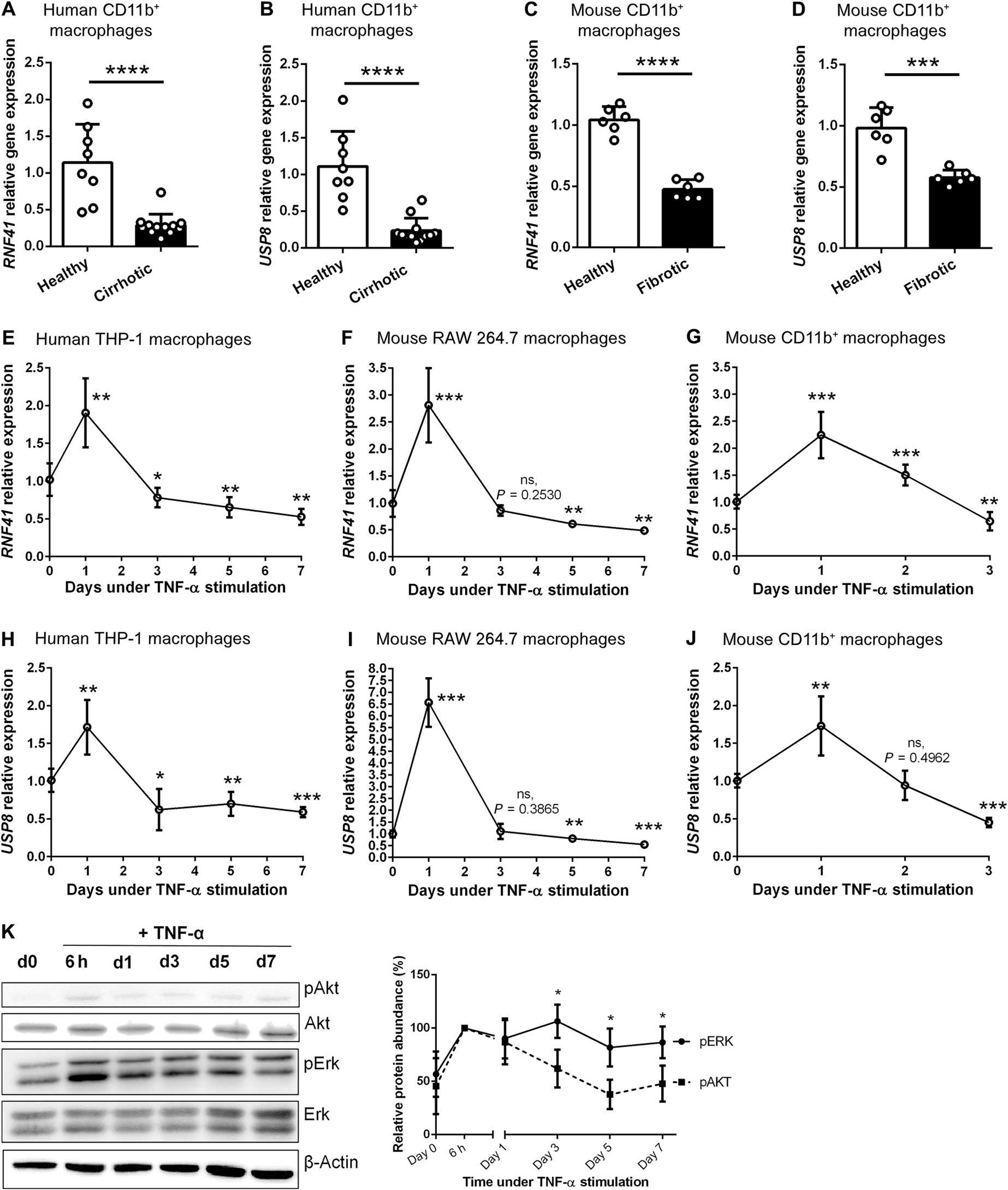
(A) RNF41 expression in CD11b+ macrophages isolated from the livers of patients with liver cirrhosis (n = 12) and healthy participants (n = 8). (B) USP8 expression in CD11b+ macrophages isolated from the livers of patients with liver cirrhosis (n = 12) and healthy participants (n = 8). (C) RNF41 expression in CD11b+-macrophages isolated from the livers of healthy and fibrotic mice (n = 6 per group). (D) USP8 expression in CD11b+-macrophages isolated from the livers of healthy and fibrotic mice (n = 6 per group). (E) RNF41 expression in THP-1 macrophages stimulated with TNF-α for 7 days. (F) RNF41 expression in RAW 264.7 macrophages stimulated with TNF-α for 7 days. (G) RNF41 expression in freshly isolated primary hepatic CD11b+ macrophages stimulated with TNF-α for 3 days. (H) USP8 expression in THP-1 macrophages stimulated with TNF-α for 7 days. (I) USP8 expression in RAW 264.7 macrophages stimulated with TNF-α for 7 days. (J) USP8 expression in freshly isolated primary hepatic CD11b+ macrophages stimulated with TNF-α for 3 days. Experiments in (E) and (F) to (J) were performed in triplicates in two independent experiments. (K) Western blot analysis of phospho-Akt, total Akt, phospho-Erk, total Erk, and β-actin in RAW 264.7 macrophages stimulated with TNF-α for 7 days and relative protein abundance (%) of phospho-Akt and phospho-Erk relative to β-actin (n = 3). For (A) to (D), Student’s t test. For (E) to (J), versus day 0 using Student’s t test with Benjamini-Hochberg correction for multiple comparisons. For (K), comparison between pAKT protein abundance and pERK protein abundance in each time point using Student’s t test. Data are shown as means ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. ns, not significant.
We hypothesized that hepatic chronic inflammation could be behind the down-regulation of macrophage RNF41 due to the known cross-talk between RNF41 and cytokine receptors (12) and some indications on the human and mouse gene expression atlas and bulk data (table S2). We used human THP-1 and mouse RAW 264.7 macrophages to design an in vitro model of prolonged inflammation (independent of infection and lipopolysaccharide) using tumor necrosis factor–α (TNF-α), a prominent cytokine driving inflammation in chronic liver disease (20). RNF41 expression was up-regulated in human THP-1 and mouse RAW 264.7 macrophages during the first 24 hours of induction with TNF-α and then decreased from day 1 to day 5, becoming lower than that in untreated macrophages (Fig. 1, E and F). The same pattern of initial up-regulation and subsequent drop in expression of RNF41 after day 1 was observed in primary mouse hepatic macrophage cultures (Fig. 1G). This pattern was also found with USP8, the RNF41 stabilizer (Fig. 1, H, I, and J). It is known that phospho-Akt (pAkt) phosphorylates USP8 and that the latter stabilizes RNF41 (21). To understand the connection between the inflammatory activity of TNF-α and RNF41, we analyzed downstream transduction pathways engaged by TNF-α, including Akt (22) and mitogen-activated protein kinases (MAPKs) [such as extracellular signal–regulated kinases (ERKs) (23)]. Phosphorylation of Erk1/2 and Akt increased during the first 6 hours after TNF-α stimulation, but only pAkt substantially dropped afterward (Fig. 1K), coinciding with the observed down-regulation pattern of RNF41 and its stabilizer USP8.
Plasmid-dendrimer-graphite nanoparticles selectively induce RNF41 in inflammatory macrophages
An expression plasmid for RNF41 was designed with a CD11b promoter (to assure that only recruited inflammatory macrophages express this protein) and an enhanced green fluorescent protein (EGFP) gene reporter under the control of a cytomegalovirus (CMV) promoter (fig. S2). To synthesize the gene therapy nanosystem, we first oxidized graphite nanoparticles (GNPs) to obtain GNPs decorated with a carboxylated surface. Then, we chemically attached PAMAM generation 5 dendrimers, which are established to bind nucleic acids (24) (such as plasmids) by electrostatic forces (Fig. 2A). Transmission electron microscopy (TEM) images revealed GNPs with a diameter of 29.9 ± 2.9 nm (Fig. 2B) that rose to 36.8 ± 4.2 nm when PAMAM dendrimers were covalently incorporated (Fig. 2C). GNP diameters visualized by TEM were more than eight times smaller than the size of nanoparticles dispersed in phosphate-buffered saline (PBS) and measured by dynamic light scattering. The hydrodynamic diameter of GNPs resulted in a mean particle diameter (Z average) of 255.6 nm, denoting a highly hydrated corona and a high aggregation of GNPs in PBS with rather narrow particle size distributions [polydispersity index (PDI) < 0.20] (Fig. 2D). The Z-average of dendrimer-GNPs (DGNPs) increased to 280.3 nm, preserving a narrow particle size distribution (Fig. 2E). As expected, GNPs showed a negative zeta potential (−43.2 mV) because of their carboxylic groups and isotonic properties (Fig. 2F). The chemical binding of dendrimers to GNPs promoted a switch to a positive zeta potential (49.01 mV), resulting in hypertonic nanoparticle dispersions (Fig. 2F). The addition of a RNF41 plasmid to DGNPs (pRNF41-DGNPs) switched the zeta potential back to negative (−31.52 mV), returning the composition to physiological osmolality (Fig. 2F).
Fig. 2. Dendrimer-graphite nanoparticles are macrophage-selective plasmid-delivery vectors for effective gene therapy.
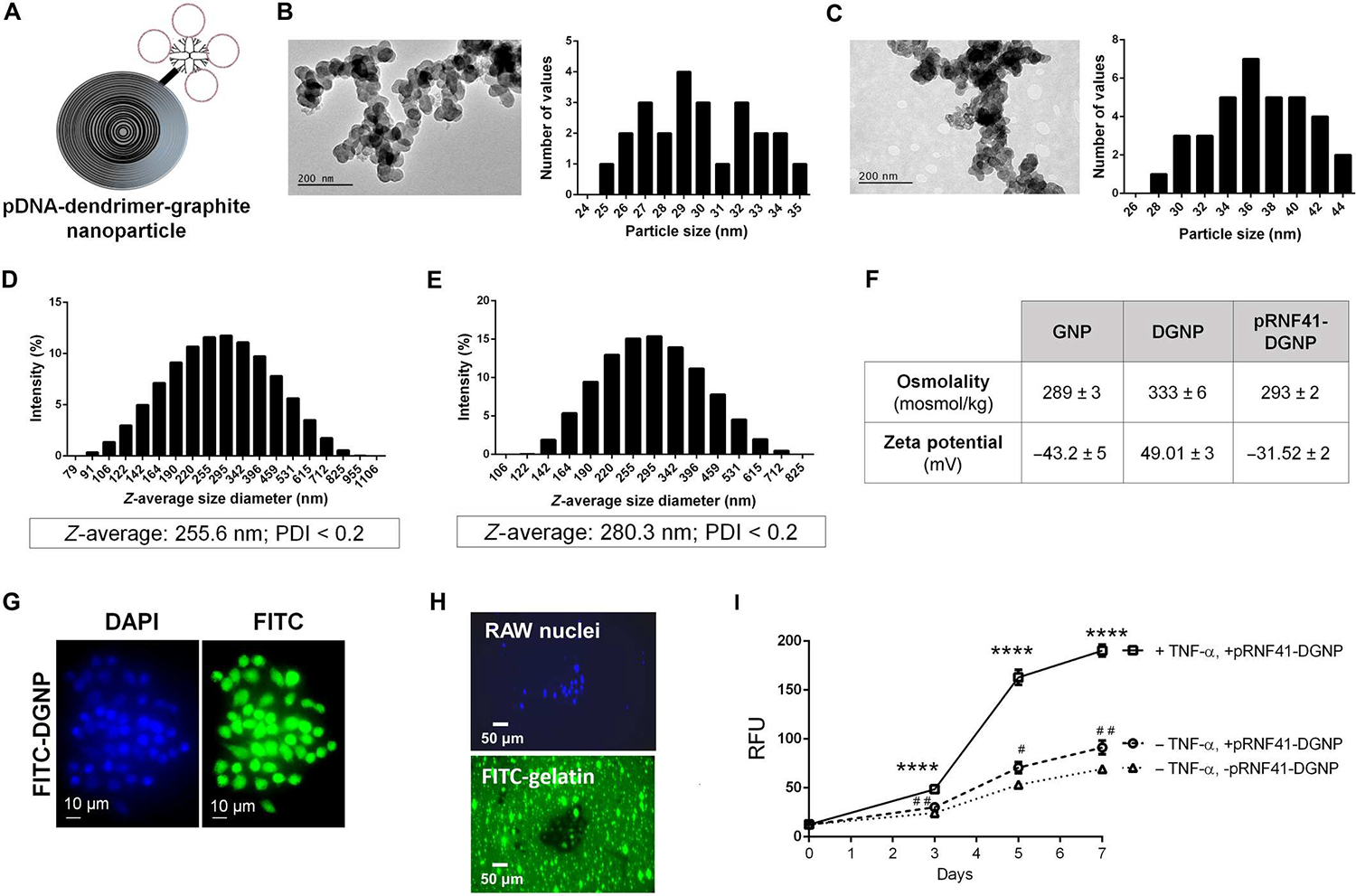
(A) Structure of graphite nanoparticles linked to dendrimer and plasmid DNA. (B) Particle size (in nanometers) of graphite nanoparticles measured using TEM images. (C) Particle size (in nanometers) of dendrimer-graphite nanoparticles measured using TEM images. (D) Z-average size and polydispersity index (PDI) of graphite nanoparticles measured using dynamic light scattering. (E) Z-average size and PDI of dendrimer-graphite nanoparticles measured using dynamic light scattering. (F) Osmolality and zeta potential of every graphite nanoparticle composite. (G) RAW 264.7 macrophage intracellular distribution of FITC-dendrimer-graphite nanoparticles. (H) Fluorescence images of RAW 264.7 macrophages seeded on FITC-gelatin–coated plates and treated with dendrimer-graphite nanoparticles linked to pRNF41 (pRNF41-DGNP) for 5 days displaying a black halo indicating collagen digestion and green nuclear staining indicating EGFP expression. (I) Time-course quantitative analysis of FITC released to the medium in the gelatinase activity assay in the presence or absence of TNF-α or pRNF41-DGNP for 7 days (n = 3 to 5). For (I), ****P ≤ 0.0001 versus macrophages without TNF-α and with or without pRNF41-DGNP, #P ≤ 0.05 versus macrophages without TNF-α and pRNF41-DGNP, and ##P ≤ 0.01 versus macrophages without TNF-α and pRNF41-DGNP at the same time point using a one-way analysis of variance (ANOVA) with posthoc Newman-Keuls test. RFU, relative fluorescence units. Data are shown as means ± SD.
Isotonic dispersions of pRNF41-DGNPs were then tested for biocompatibility with human endothelial cells, the standard primary cell barrier in blood vessels. No harmful effects of the nanoparticles were found on human umbilical vein endothelial cells (fig. S3A). The uptake of nanoparticles over 200 nm is conceptually assumed to involve mainly macrophages, especially proinflammatory macrophages at diseased sites (25). The incorporation of pRNF41-DGNPs in RAW 264.7 macrophages activated by TNF-α increased over time up to 45 min and then only rose in the presence of TNF-α, reaching most of the cells after 180 min (fig. S3B) and lasting for at least 24 hours (fig. S3C). These results indicate that the GNP core is involved in selective macrophage uptake. Fluorescein isothiocyanate (FITC)–decorated DGNPs confirmed the intracellular fate of dendrimers after internalization in inflamed macrophages. FITC-DGNPs were internalized and degraded by macrophages, distributing the dendrimer-FITC molecules throughout the cell, including the cell nucleus (Fig. 2G). These results suggested that these nanoparticles could be useful for selective gene therapy to inflammatory macrophages in chronically inflamed livers. Certainly, dendrimers are known to escape from lysosomes by the proton sponge effect, opening pores in the nuclear membrane for pDNA or small interfering RNA gene therapy (26).
We next sought to confirm the effectiveness of plasmid-DGNPs for gene therapy in vitro. In line with the previous uptake outcomes, pRNF41-DGNPs were mainly phagocytized by macrophages stimulated with TNF-α, and plasmid expression efficiency was functionally highlighted by the high abundance of intracellular EGFP in most cells after 3 days of incubation (fig. S3D). Moreover, macrophages incubated with pRNF41-DGNP and TNF-α (a CD11b promoter activator) displayed a switch in macrophage morphology (fig. S4A) and phenotype, exemplified by elevated expression of CD206 (mannose receptor, an anti-inflammatory macrophage marker) (fig. S4B). Induced RNF41 expression has previously been associated with anti-inflammatory macrophage polarization (14). Anti-inflammatory macrophages produce high amounts of MMPs to degrade ECM proteins such as collagen (27). Because this collagenase activity of anti-inflammatory macrophages might be beneficial for the treatment of liver fibrosis, we tested the capacity of macrophages treated with pRNF41-DGNPs to digest collagen using FITC-gelatin. RAW 264.7 macrophages seeded on FITC-gelatin–coated plates and treated with pRNF41-DGNPs for 5 days displayed a black halo and green nuclear staining due to collagen digestion and EGFP expression, respectively (Fig. 2H). FITC released during the gelatinase assay revealed that collagen degradation rapidly increased because of pRNF41-DGNPs 3 days after incubation only when TNF-α was present, because collagen degradation was much lower in the absence of inflammatory stimulus (Fig. 2I). This functional experiment was the rationale for our administration schedule of 3 days for in vivo experiments in animals with liver fibrosis using pRNF41-DGNPs.
RNF41 restoration in macrophages orchestrates fibrosis regression and hepatocyte proliferation
We intravenously administered pRNF41-DGNPs or DGNPs with the same plasmid but with an RNF41 scrambled sequence (pSCR-DGNP) every 3 days for a total of 10 days to mice with CCl4-induced liver fibrosis (Fig. 3A). To determine whether pDNA-DGNPs were incorporated into hepatic inflammatory macrophages, we isolated all hepatic cells from fibrotic mice 24 hours after receiving pSCR-DGNPs. We mainly found an intense fluorescence signal in CD11b+ macrophages corresponding to the plasmid EGFP reporter and a negligible signal in hepatocytes, HSCs, and LSECs (Fig. 3B). This was confirmed via visualization of hepatic EGFP+ cells in fibrotic mice treated with pSCR-DGNPs. We found high intracellular EGFP abundance specifically in Ly6c-stained inflammatory macrophages (an inflammatory surface marker of macrophages in transition from monocytes) (Fig. 3, C and D). These EGFP+ inflammatory macrophages lacked CD206 all along the fibrotic tracts in fibrotic livers (Fig. 3, E and F). In contrast, we mainly observed macrophages expressing both EGFP and CD206 in liver fibrotic tracts from animals treated with pRNF41-DGNPs (Fig. 3, G and H). The presence of functional pSCR-DGNP was negligible in other organs such as the kidney (Fig. 3I) and low in the spleen (Fig. 3J) and the lung (Fig. 3K) from fibrotic animals, denoting the selectivity of these nanoparticles for inflammatory and scar-associated macrophages present in injured livers. Negative controls for these immunofluorescence stainings can be found in fig. S5.
Fig. 3. Dendrimer-graphite nanoparticles efficiently and selectively transfect a RNF41-encoding plasmid into macrophages recruited to mouse fibrotic liver.
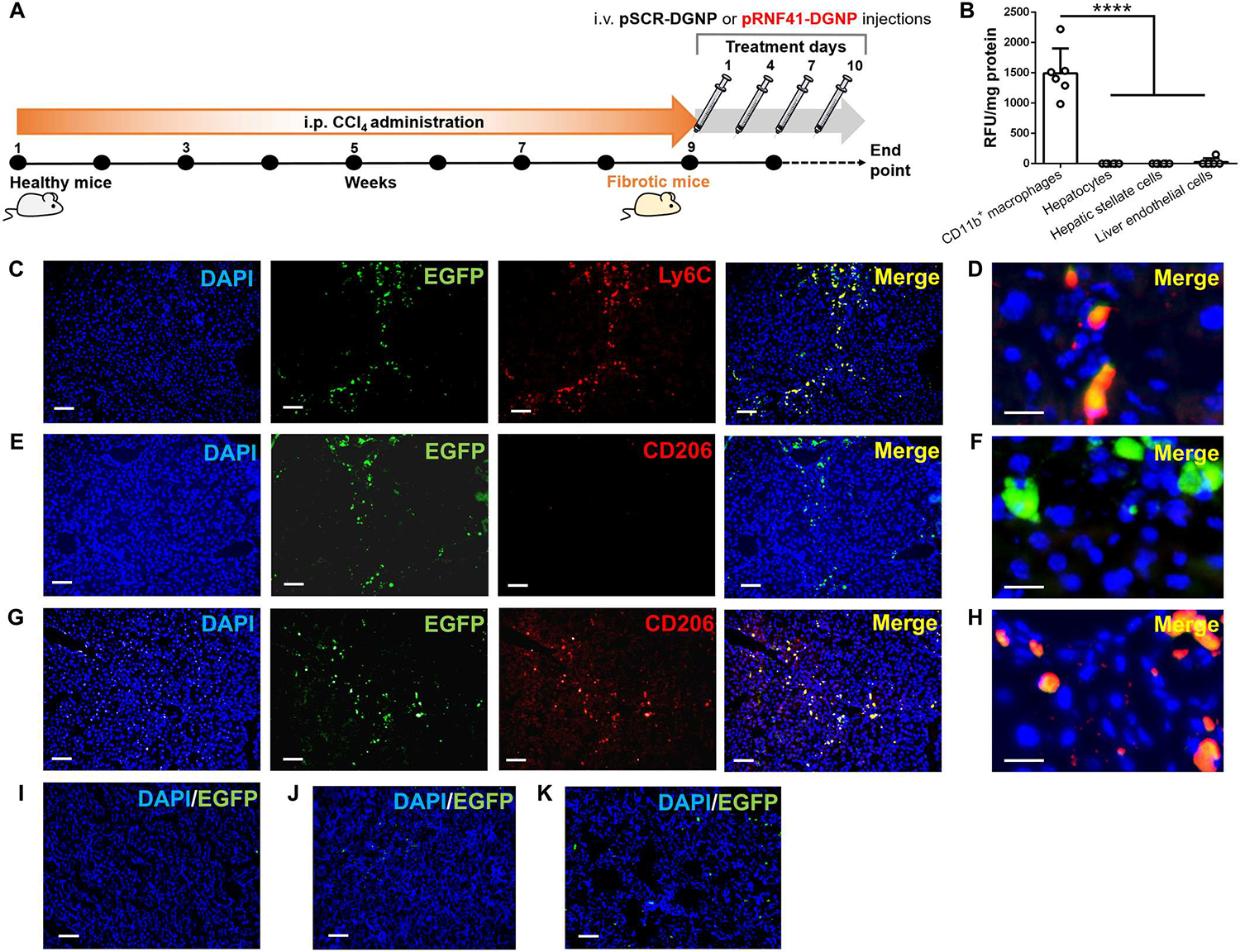
(A) Schematic figure illustrating the time points of fibrosis induction with CCl4 and the administration schedule of dendrimer-graphite nanoparticles linked to plasmid pRNF41 (pRNF41-DGNPs) or scrambled pSCR (pSCR-DGNPs). (B) Relative fluorescence units (RFU) of EGFP per milligram of protein of liver isolated CD11b+ macrophages, hepatocytes, hepatic stellate cells, and liver endothelial cells from mice treated with pSCR-DGNPs (n = 6). (C) Immunofluorescence (IF) staining for Ly6C and simultaneous detection of EGFP fluorescence in the livers of fibrotic mice treated with pSCR-DGNPs. (D) High-power image for Ly6C and simultaneous detection of EGFP fluorescence in the livers of fibrotic mice treated with pSCR-DGNPs. (E) IF staining for CD206 and simultaneous detection of EGFP fluorescence in the livers of fibrotic mice treated with pSCR-DGNPs. (F) High-power image for CD206 and simultaneous detection of EGFP fluorescence in the liver of fibrotic mice treated with pSCR-DGNPs. (G) IF staining for CD206 and simultaneous detection of EGFP fluorescence in the livers of fibrotic mice treated with pRNF41-DGNPs. (H) High-power image for CD206 and simultaneous detection of EGFP fluorescence in the livers of fibrotic mice treated with pRNF41-DGNPs. (I) EGFP fluorescence in the kidneys of fibrotic animals treated with pSCR-DGNPs. (J) EGFP fluorescence in the spleens of fibrotic animals treated with pSCR-DGNPs. (K) EGFP fluorescence in the lungs of fibrotic mice treated with pSCR-DGNPs. All plasmids constitutively expressed EGFP under the control of a CMV promoter. Scale bars, 200 μm (C, E, G, I, J, and K) and 20 μm (D, F, and H). For (B), ****P ≤ 0.0001 versus CD11b+ macrophages using a one-way analysis of variance (ANOVA) with the posthoc Newman-Keuls test. i.p., intraperitoneal; i.v., intravenous.
Macrophage expression of RNF41 was substantially reduced in animals with fibrosis treated with pSCR-DGNPs compared with control mice receiving corn oil and restored to physiological amounts after pRNF41-DGNP exposure (Fig. 4A). The first hepatic effect of macrophage RNF41 recovery after plasmid administration was visually appreciated as a change in the macroscopic aspect of fibrotic liver from micronodular pathology to a nonfibrotic liver appearance (Fig. 4B). Rescue of RNF41 expression in macrophages of fibrotic liver promoted an 86% reduction in the hepatic fibrosis area and a recovery of physiological parenchymal structure (Fig. 4C), along with a diminished abundance of hydroxyproline (Fig. 4D). This decrease in the collagen fibers in the fibrotic liver was associated with decreased collagen-I expression (fig. S6A) and mitigated HSC activity, as illustrated by reduced expression and presence of α–smooth muscle actin (α-SMA) (fig. S6B) and tissue inhibitor of metalloproteinases-1 (TIMP-1) (fig. S6C). These beneficial antifibrotic effects translated into substantially reduced liver injury (Fig. 4E and fig. S6D).
Fig. 4. RNF41 restoration in macrophages located into the fibrotic liver orchestrates fibrosis and inflammation regression and reduction of hepatic injury in mice.
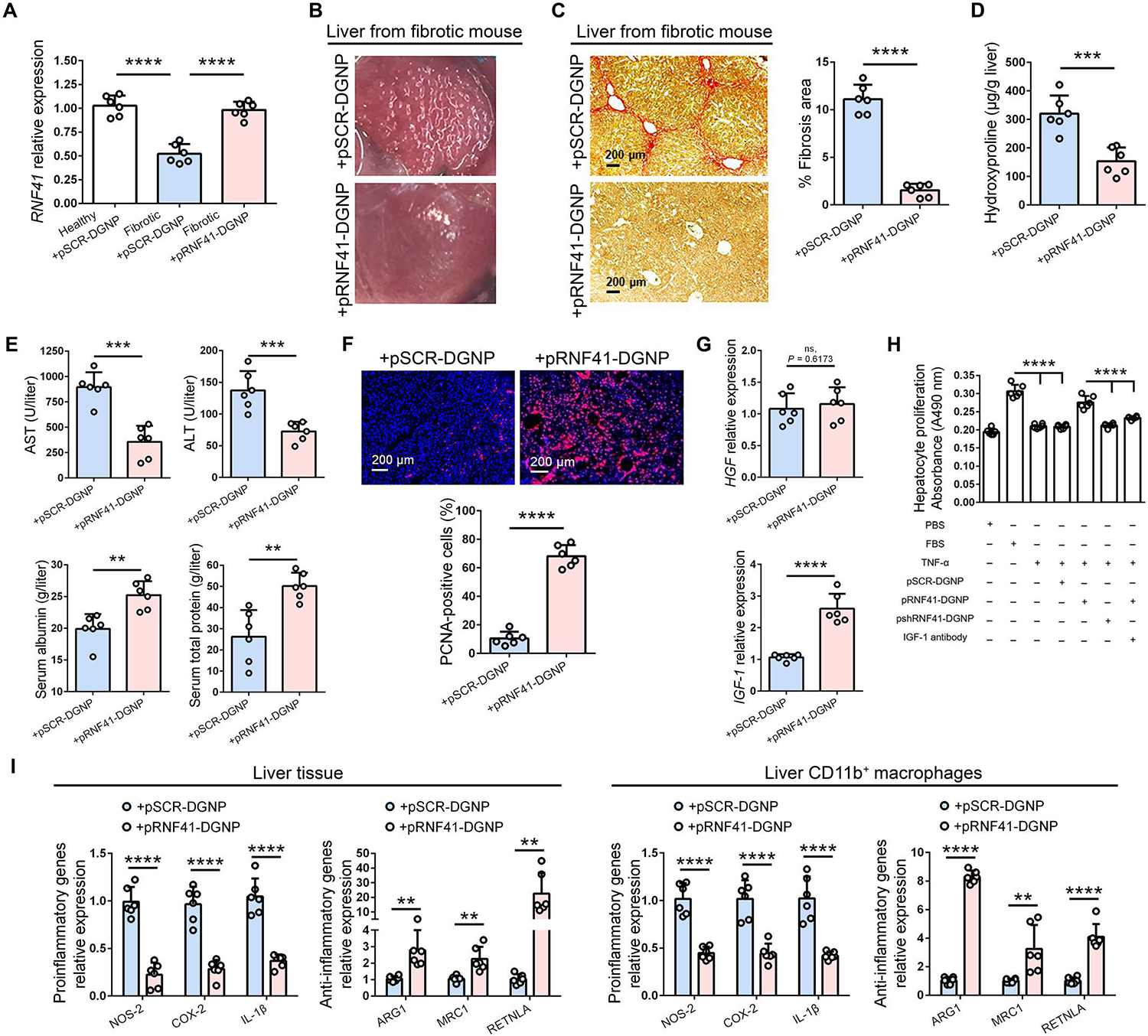
(A) RNF41 abundance in healthy and fibrotic mice treated with dendrimer-graphite nanoparticles linked to plasmid pRNF41 (pRNF41-DGNPs) or scrambled pSCR (pSCR-DGNPs). (B) Macroscopic aspect of fibrotic liver after treatment with pRNF41-DGNPs. (C to G) Sirius Red staining and quantification of liver fibrosis area (C), hydroxyproline measurements (D), serum liver injury parameters [ALT (alanine aminotransferase), AST (aspartate aminotransferase), serum albumin, and serum total protein] (E), hepatic PCNA immunofluorescence staining (F), and hepatic expression of HGFand IGF-1 (G) in fibrotic mice treated with pSCR-DGNPs or pRNF41-DGNPs. (H) Cell proliferation in isolated mouse hepatocytes treated for 24 hours with conditioned medium from RAW 264.7 cultures treated with FBS, TNF-α, pSCR-DGNPs, pRNF41-DGNPs, or IGF-1 antibody for 3 days. Experiments were performed in triplicates in two independent experiments. (I) Expression of proinflammatory and anti-inflammatory genes in liver tissue and in CD11b+ macrophages isolated from the livers of fibrotic mice treated with pSCR-DGNPs or pRNF41-DGNPs. n = 6 animals per group. Student’s t test was used for (A), (C) to (G), and (I), and a one-way ANOVA with posthoc Newman-Keuls test was used for (H). Data are shown as means ± SD. **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
Inflammatory macrophages stimulate HSC activation and subsequent fiber production during liver fibrosis through the synthesis and release of agents such as oncostatin M (OSM), platelet-derived growth factor-BB (PDGF-BB), and transforming growth factor–β (TGF-β) (28, 29). Accordingly, it is consistent that pRNF41-DGNPs promoted a substantial reduction in hepatic OSM (fig. S7A), PDGF-BB (fig. S7B), and TGF-β expression (fig. S7C), indicating that macrophage RNF41 hinders ECM excessive production in fibrosis though the down-regulation of major macrophage-derived signals involved in HSC activation. RNF41 not only promoted the synthesis of HSC-inhibitory factors in macrophages but also macrophage overproduction of the collagenase MMP-9 to boost collagenous fiber digestion (fig. S8A). Fibrotic tracts spatially limit hepatocyte expansion. The reduction in these collagenous chains in the livers of fibrotic mice treated with pRNF41-DGNPs was associated with an increase in proliferating cell nuclear antigen+ (PCNA+) cells (Fig. 4F), most of which corresponded to proliferating hepatocytes (fig. S8B). This promoted the liver mass repair observed in scarred fibrotic livers from mice treated with pRNF41-DGNPs (fig. S8C).
We then wondered whether major trophic factors might be also directly involved in the hepatocyte proliferation induced by macrophage RNF41 recovery. Hepatocyte growth factor (HGF; the main hepatocyte proliferative factor) was not affected by pRNF41-DGNPs in fibrotic mice, but insulin-growth factor 1 (IGF-1) expression was up-regulated in the livers of these animals (Fig. 4G). IGF-1 is related to hepatocyte proliferation and HSC inactivation (30, 31). For this reason, we hypothesized that IGF-1 synthesized by macrophages treated with pRNF41-DGNPs might be directly associated with the effects observed in hepatocyte proliferation and HSC activation. To test this hypothesis, we incubated hepatocytes isolated from mouse livers with conditioned medium from macrophages stimulated with pRNF41-DGNPs, pSCR-DGNPs, or DGNPs containing a plasmid with an inhibitory shRNF41 (shRNF41-DGNPs) in the presence or absence of a specific antibody against IGF-1. Only conditioned medium from macrophages treated with pRNF41-DGNPs stimulated hepatocyte proliferation similar to fetal bovine serum (FBS) (10%), and this proliferative induction was reduced with the addition of an antibody blocking IGF-1 effects (Fig. 4H). We also tested the possible effects of macrophage RNF41-induced IGF-1 production on LX-2 human HSC activation using the same conditioned medium and experimental conditions. Conditioned medium from macrophages treated with TNF-α and pSCR-DGNPs up-regulated HSC expression of collagen I (fig. S9A), α-SMA (fig. S9B), and TIMP-1 (fig. S9C), which was abolished by the treatment with pRNF41-DGNPs and then recovered when IGF-1 was blocked with a specific antibody.
Anti-inflammatory macrophages may influence the response of HSCs, endothelial cells, and other immune cells to injury (32). To investigate whether RNF41 restoration was related to the hepatic macrophage phenotype, we quantified gene expression of proinflammatory and anti-inflammatory macrophage markers in fibrotic livers from animals treated with either pSCR-DGNPs or pRNF41-DGNPs. We found a quantitative increase in anti-inflammatory markers ARG1, MRC1, and RETNLA and a decrease in proinflammatory markers NOS2, COX-2, and IL-1β in liver tissue and isolated CD11b+ macrophages, which denoted that this RNF41-restorative gene therapy switched proinflammatory to anti-inflammatory macrophages in fibrotic livers (Fig. 4I and fig. S10D). It is known that peroxisome proliferator–activated receptor γ (PPAR-γ) activation triggers macrophages into an alternative anti-inflammatory phenotype (33). We quantified the expression of both downstream PPAR-γ target genes IL-10 and CD36 in fibrotic livers from animals treated with either pSCR-DGNPs or pRNF41-DGNPs. Induced macrophage RNF41 expression stimulated the expression of IL-10 (fig. S11A) and CD36 (fig. S11B) in fibrotic liver, denoting increased PPAR-γ activation. Last, we validated the therapeutic effects of pRNF41-DGNPs in a second model of liver fibrosis based on intraperitoneal injections of thioacetamide (TAA) (fig. S12A). Livers from fibrotic animals treated with pRNF41-DGNPs displayed a nonfibrotic liver appearance (fig. S12B), a substantial reduction in the fibrosis area (fig. S12C), HSC-related gene expression (fig. S12D), hepatic damage (fig. S12E), macrophage profibrotic genes (fig. S12F), and proinflammatory markers (fig. S12G), along with a substantial increase in anti-inflammatory markers (fig. S12H), liver mass repair (fig. S12I) and IGF-1 expression (fig. S12J).
Macrophage RNF41 depletion aggravates inflammation and hepatic damage and reduces survival
Either pshRNF41-DGNPs or pshSCR-DGNPs were intravenously administered to mice with liver fibrosis every 3 days for a total of 10 days (Fig. 5A). RNF41 expression was considerably reduced in macrophages isolated from fibrotic mice treated with pshRNF41-DGNPs (Fig. 5B). The first observable effect of RNF41 depletion was a considerable decrease in survival (Fig. 5C). Collagen fiber staining revealed an increase in fibrosis area in mice receiving pshRNF41-DGNPs (Fig. 5D). This was associated with HSC hyperactivation, as indicated by the increase in collagen I, α-SMA, and TIMP-1 (Fig. 5E). Treatment with pshRNF41-DGNPs promoted an increase in hepatic abundance of the macrophage-derived HSC activators OSM, PDGF-BB, and TGF-β (Fig. 5F). Liver injury was also enhanced in fibrotic animals treated with pshRNF41-DGNPs in comparison with fibrotic mice receiving pshSCR-DGNPs (Fig. 5G). These detrimental effects of pshRNF41-DGNP treatment were associated with a further increase in proinflammatory macrophage-derived inflammatory cytokines without affecting anti-inflammatory macrophage genes (Fig. 5H). This exacerbated inflammation resulted in lower hepatocyte proliferation and liver mass repair (Fig. 5I), effects associated with a decrease in IGF-1 abundance (Fig. 5J). No changes in HGF were observed (Fig. 5J).
Fig. 5. Depletion of macrophage RNF41 worsens fibrosis, inflammation, and hepatic damage in mice with liver fibrosis.
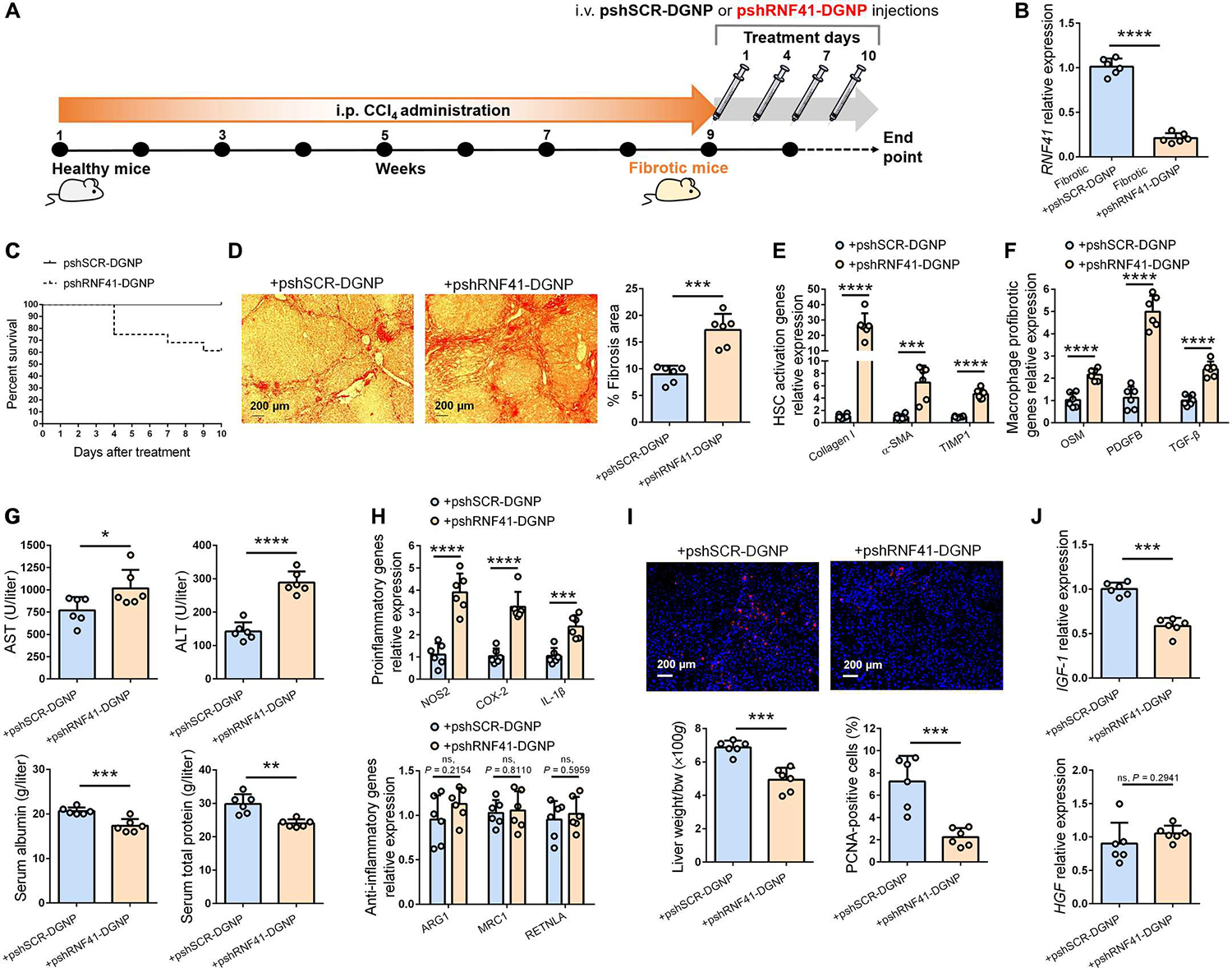
(A) Schematic figure illustrating time points of fibrosis induction with CCl4 and administration schedule of dendrimer-graphite nanoparticles linked to pshSCR (pshSCR-DGNPs) or pshRNF41 (pshRNF41-DGNPs). (B and C) Hepatic gene expression of RNF41 (B) and survival rate (C) of fibrotic mice treated with pshSCR-DGNPs or pshRNF41-DGNPs. (D to J) Sirius red staining (D), gene expression related to hepatic stellate cell (HSC) activation (E) or profibrogenic agents produced by liver-resident macrophages (F), serum parameters of liver injury (ALT, AST, serum albumin, and serum total protein) (G), expression of inflammatory and anti-inflammatory macrophage genes (H), PCNA immunofluorescence staining (I), and HGF and IGF-1 expression (J) in liver of fibrotic mice treated with pshSCR-DGNPs or pshRNF41-DGNPs. n = 6 per group of animals. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 using Student’s t test. Data are shown as means ± SD.
RNF41 induction promotes hepatic regeneration after hepatectomy
We wondered whether induction of macrophage RNF41 could also be beneficial in the context of liver resection. Administration of pRNF41-DGNPs to healthy mice undergoing 70% hepatectomy (Fig. 6A) showed greater hepatic restoration than animals receiving pSCR-DGNPs (Fig. 6B). This effect was associated with a higher hepatic proliferative signal, highlighted by the increase in PCNA+ cells (Fig. 6C). However, treatment with pRNF41-DGNPs did not reduce hepatocyte damage caused by liver resection, because no changes in serum transaminases, albumin, or proteins were observed compared with animals receiving pSCR-DGNPs (Fig. 6D). In line with the outcomes in fibrotic mice, hepatectomized mice receiving pRNF41-DGNPs did not show up-regulation of HGF but did show more abundant IGF-1 (Fig. 6E) concomitant with increased expression of hepatic PPAR-γ target genes IL-10 and CD36 (fig. S11, C and D).
Fig. 6. Macrophage RNF41 induces liver regeneration after hepatectomy.
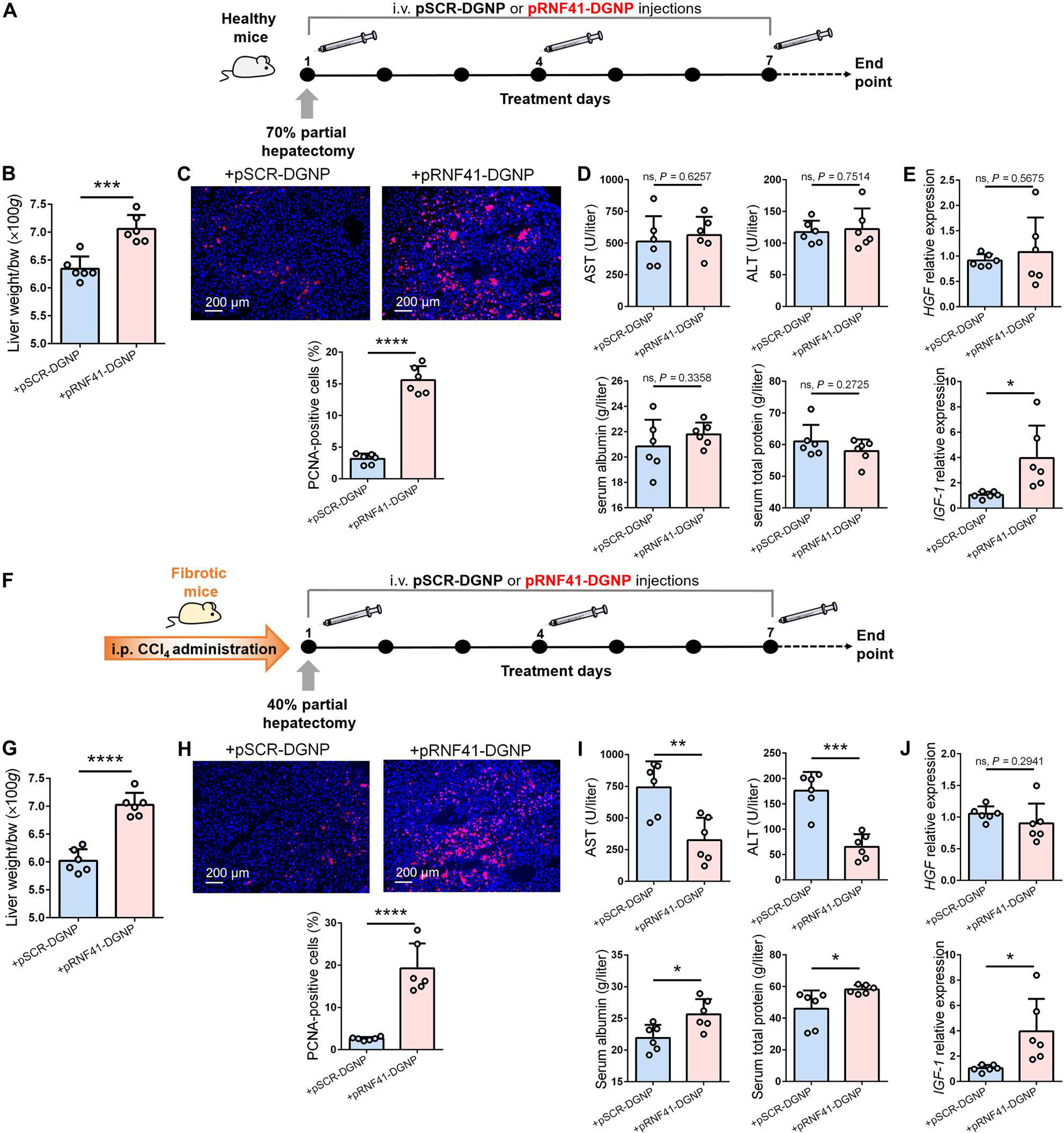
(A) Schematic figure illustrating 70% hepatectomy in healthy mice and administration schedule of dendrimer-graphite nanoparticles linked to pRNF41 (pRNF41-DGNPs) or pSCR (pSCR-DGNPs). (B to E) Liver restoration rate (B), PCNA immunofluorescence staining (C), serum liver injury parameters [ALT, AST, serum albumin, and serum total protein] (D), and HGF and IGF-1 expression (E) in healthy mice undergoing 70% hepatectomy and treated with pSCR-DGNPs or pRNF41-DGNPs. (F) Schematic figure illustrating the time points of fibrosis induction with CCl4, 40% hepatectomy in fibrotic mice, and the administration schedule of pSCR-DGNPs or pRNF41-DGNPs. (G to J) Liver restoration rate (G), PCNA immunofluorescence staining (H), serum liver injury parameters (ALT, AST, serum albumin, and serum total protein) (I), and expression of HGF and IGF-1 in the livers of fibrotic mice undergoing 40% hepatectomy and treated with pSCR-DGNPs or pRNF41-DGNPs. n = 6 animals per group. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 using Student’s t test. Data are shown as means ± SD.
Fibrotic mice operated for 40% hepatectomy and administered pRNF41-DGNPs (Fig. 6F) also displayed a higher liver restoration rate than animals receiving pSCR-DGNPs (Fig. 6G). This effect was proportional with an elevated hepatic proliferative signal demonstrated by the rise in PCNA+ cells (Fig. 6H). In this case, treatment with pRNF41-DGNPs did reduce liver injury (Fig. 6I). In agreement with outcomes in fibrotic mice, hepatectomized mice treated with pRNF41-DGNPs did not display up-regulation of HGF but did show higher IGF-1 (Fig. 6J). Treatment with pRNF41-DGNPs also promoted an increased expression of hepatic PPAR-γ target genes IL-10 and CD36 in hepatectomized fibrotic animals (fig. S11, E and F).
DISCUSSION
We sought to determine whether emerging concepts on macrophage RNF41 function are applicable to chronic liver disease and translatable to humans. By examining isolated hepatic macrophages in specimens obtained from cirrhotic patients and mice with liver fibrosis, we tested the hypothesis that macrophage RNF41 expression is altered in chronic liver disease. We demonstrated that RNF41 expression is negatively regulated in macrophages isolated from the liver specimens of both states of liver fibrosis: cirrhosis in patients and fibrosis in mice. Our data reveal that sustained inflammatory signals from TNF-α promote down-regulation of macrophage RNF41 and its stabilizer USP8. In contrast, short-term TNF-α exposition induces RNF41 and USP8 expression in macrophages, as also described using lipopolysaccharide (13). This rapid induction is likely driven by Akt phosphorylation, because TNF-α activates Akt independently of NF-κB (22), and Akt regulates the function of RNF41 and its stabilizer USP8 (21). Down-regulation of RNF41 and USP8 expression under long-term TNF-α stimulation is likely due to the continuous and progressive increase in stress-activated MAPK (23), the subsequent Erk-induced Akt down-regulation (34), and the reciprocal inhibitory effects between RNF41 and Erk (35).
This investigation describes the importance of RNF41 in chronic liver injury. However, it is known that mice deficient for the RNF41-stabilizer USP8 are embryonic lethal, and USP8 inactivation in adulthood causes fatal liver failure (36). We speculate that part of the harmful hepatic effects caused by USP8 knockdown could be mediated by RNF41 deficiency in macrophages, but there are no conditional and macrophage-specific knockout mice to study this possibility yet. Here, our investigations substantiate that selective macrophage recovery of RNF41 in the livers of fibrotic animals using pRNF41-DGNPs promotes the switch of hepatic macrophages from a proinflammatory to anti-inflammatory phenotype. This macrophage switch by induced RNF41 has also been seen in thioglycollate-elicited peritoneal macrophages (14). However, the extraordinary complexity of the hepatic niche defines different macrophage subpopulations from health to disease (37). Whereas resident Kupffer cells are the major source in the hepatic macrophage pool in homeostasis, monocyte-derived macrophages predominate in acute and chronic liver injury (38). Namely, scar-associated macrophages display a profibrogenic profile and expand similarly in cirrhotic patients and in fibrotic mice (37). Notably, pRNF41-DGNPs selectively accumulated into these macrophages within fibrotic septae in mouse fibrotic liver and reduced their synthesis of profibrogenic factors such as OSM, PDGF-BB, or TGF-β1 [the most potent profibrogenic cytokine (39)]. In contrast, a further RNF41 reduction with pshRNF41-DGNPs exerted the opposite effect, an increase in HSC-activating factors. This suggests that RNF41 is a central negative modulator of profibrogenic and proinflammatory signals in scar-associated macrophages during chronic liver disease. RNF41 is behaving as a node connecting the mechanisms driving fibrogenesis with those regulating inflammation (fig. S13). Signals coming from macrophages with low RNF41 expression seem to perpetuate inflammation and fibrosis that ultimately hampers hepatic function recovery. Hepatocyte damage was diminished by macrophage RNF41 restoration only in mice with chronic liver injury. This shows that macrophage RNF41 activation is not directly involved in hepatocyte protection. Instead, it leads to inflammation and fibrosis resolution, and these homeostatic effects likely allow for the hepatic function recovery.
A previous study described the predominant effect of macrophage RNF41 on the activation of adenosine 3′,5′-monophosphate response element–binding protein-C/EBP-β cascade (14), and because the activation of this cascade has been associated with muscle injury repair (15), this prompted us to explore the possible role of RNF41 in hepatic repair after liver resection. In this scenario, we demonstrated that RNF41 induction in macrophages results in faster hepatic regeneration after liver resection in healthy and fibrotic mice. IGF-1 rather than HGF participates in the proregenerative effects derived from macrophage RNF41 induction. IGF-1 is not only a trophic factor mainly produced by the liver but also delivered by monocytes/macrophages to stimulate tissue growth (30). In chronic liver disease, IGF-1 deficiency is a common condition independent of chronic liver damage etiology (40). However, IGF-1 has effects beyond proliferation, including inhibition of HSC activation and activity (31). We cannot exclude other participants in the therapeutic effects of macrophage RNF41 induction in liver injury. The PPAR-γ signaling pathway might, for example, link macrophage biology, inflammation, and insulin-related factor biochemistry. This pathway is involved in the nuclear regulation of inflammation, glucose metabolism, and macrophage phenotype (33). Because RNF41 activates C/EBP-β (14) and the latter is a PPAR-γ inducer (41), this may explain the macrophage phenotype switch observed in fibrotic animals treated with pRNF41-DGNPs. Our findings then combine macrophage RNF41 activation and its downstream phenotypic effects on inflammation, fibrosis, and tissue regeneration, expanding our understanding of the relationship between RNF41, PPAR-γ, and IGF-1 with respect to chronic liver injury and hepatic regeneration. It has long been thought that inflammation is necessary to promote tissue repair and that the mechanisms driving fibrogenesis are distinct from those regulating inflammation; however, we suggest a slightly different paradigm where RNF41 may behave as a central regulator of macrophage behavior, balancing both inflammatory and fibrogenic signals toward tissue repair.
Several limitations and open questions remain. First, human THP-1 cells were used as an alternative to primary human macrophages because of the difficulty of obtaining enough viable primary cells from small control biopsies for experiments involving prolonged incubation with TNF-α. Therefore, other advanced approaches such as precision-cut liver slices or perfusable three-dimensional liver-on-a-chip models are required to better understand the effects of inflammatory factors such as TNF-α on the regulation of RNF41 in human macrophages. Second, the precise role of IGF-1 on the effects driven by macrophage RNF41 induction needs further investigation to depict the mechanistic link between these two proteins. Third, specific deletion of RNF41 in macrophages would solidify its role in fibrosis resolution and hepatic regeneration. Macrophage-specific conditional gene knockout mice for RNF41 are still needed to study all the potential downstream effects modulated by macrophage RNF41.
In conclusion, our findings provide evidence that RNF41 may be an immune regulatory node that regulates the response of macrophages to restore homeostasis after tissue injury—down-regulated in chronic liver injury in mice and humans. Therapeutics targeting macrophage RNF41 may represent a therapeutic target for patients with chronic liver disease characterized by inflammation and fibrosis.
MATERIALS AND METHODS
Study design
This study aimed to evaluate RNF41 expression in macrophages from livers of cirrhotic patients and fibrotic mice and to analyze the influence of macrophage RNF41 modulation in the balance of hepatic fibrosis and repair in response to the inflammatory milieu. To achieve these objectives, we first quantified RNF41 gene expression in CD11b+ macrophages isolated from the livers of patients with cirrhosis and BALB/c fibrotic mice using a multistep protocol of magnetic-activated cell sorting. We also designed macrophage-selective gene therapy with DGNPs to explore the influence of macrophage RNF41 in liver fibrosis and regeneration. In addition, we designed in vitro studies to explore how macrophage RNF41 influences other hepatic cells to regulate fibrosis and regeneration. Patients (n = 12) with decompensated liver cirrhosis and MELD scores between 13 and 30 from the hospital Clinic of Barcelona were enrolled in this study. Healthy participant samples (n = 8) with normal liver histology were used as controls. For animal studies, we used littermates as much as possible and randomized them in diseased or healthy groups. The study was performed according to the criteria of the Investigation and Ethics Committees of the Hospital Clínic and University of Barcelona and according to Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Two models of mouse liver fibrosis were performed with intraperitoneal injections of CCl4 diluted 1:8 (v/v) in corn oil or TAA (200 mg/kg) twice a week for 9 weeks. Mouse models of partial hepatectomy (70% in healthy and 40% in fibrotic mice induced with CCl4) were also performed. Dispersions of macrophage-selective plasmid-DGNPs were intravenously injected to regulate RNF41 gene expression. We determined sample size for each experimental group on the basis of our experience with similar studies. The sample size (n) of each experimental group (n = 6) is indicated in the figure legends. For cell culture studies, a minimum of three experimental replicates were performed (exact number of replicates is presented in figure legends).
Human specimens
All protocols conformed to the ethical guidelines of the 1975 Declaration of Helsinki and were approved by the Ethics Committee of the Hospital Clinic of Barcelona. All the patients included in this study provided written and signed informed consent. Human normal liver samples were obtained from small biopsies from donor liver lobules during transplantation (n = 8). All participants had normal hepatic histology and no declared acute or chronic diseases. Human cirrhotic liver samples were obtained from liver explants of patients with end-stage cirrhosis caused by liver disease (n = 12) undergoing liver transplantation.
Animals and in vivo procedures
Male BALB/c mice (7 weeks old) were purchased from Charles River Laboratories (Charles River). All animals were maintained in a temperature-controlled room (22°C) on a 12-hour light-dark cycle. After arrival, mice were continuously fed ad libitum until euthanasia. To induce liver fibrosis, mice were injected intraperitoneally twice a week with CCl4 diluted 1:8 (v/v) in corn oil for 9 weeks. A second fibrosis mouse model was generated with intraperitoneal injections of TAA (200 mg/kg) twice a week for 9 weeks. Dispersions of plasmid-DGNPs were then intravenously injected (50 μg/kg in a ratio plasmid/DGNP, 1:10) every 3 days for 9 days (four injections in total). Animals were euthanized on day 10 after the start of treatment. Liver samples and serum were collected and frozen for further analysis. Serum parameters were measured using a BS-200E Chemistry Analyzer (Mindray Medical international Ltd.).
Partial hepatectomy (70% in healthy and 40% in fibrotic mice) was performed as previously described (42). Hepatectomy was performed at 40% in fibrotic mice to avoid unnecessary animal losses of these already diseased animals according to the criteria of the Investigation and Ethics Committees of the Hospital Clínic and University of Barcelona. Dispersions of plasmid-DGNP were intravenously injected to hepatectomized animals, which were euthanized 7 days after hepatectomy to obtain and analyze tissue and serum samples as described above.
Isolation of hepatic CD11b+-macrophages, stellate cells, and hepatocytes
Freshly isolated primary hepatic CD11b+ macrophages were obtained from the livers of control and cirrhotic patients and from the livers of healthy mice (control) receiving corn oil and fibrotic mouse. Briefly, hepatocytes, macrophages and HSCs were purified after collagenase A (Roche Diagnostics) administration via retrograde perfusion in mice or via an intravenous catheter in human liver samples and subsequent Histodenz gradient (Sigma-Aldrich). Purification was optimized using magnetic beads (magnetic-activated cell sorting system, Miltenyi Biotec) with a modified protocol previously reported (43, 44). First, the gradient fraction corresponding to polymorphonuclear cells was purified using Ly6G (R&D Systems, reference: 25872-1-AP) and PDCA-1 (Invitrogen, reference: PA5-23505) antibodies in mouse and CD15 (R&D Systems, reference: MAB7368) and BDCA-1 (R&D Systems, reference: AF5910) antibodies in human with MACS to remove neutrophils and dendritic cells, respectively. Then, the remaining eluted fraction was incubated with CD11b magnetic beads (Miltenyi Biotec, reference: 130-049-601), and MACS-purified CD11b+ macrophages were resuspended in TRIzol (Gibco-Invitrogen) for total RNA extraction or in Dulbecco’s modified Eagle’s medium supplemented with FBS (1%) for prolonged inflammation assays. A 0.5-μg aliquot of total RNA was reverse-transcribed using a complementary DNA synthesis kit (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems) for further analysis of gene expression using real-time polymerase chain reaction.
Synthesis and functionalization of DGNPs
Carbon GNPs were supplied by Graphene Supermarket. The Generation 5 PAMAM dendrimer was purchased from Dendritech Inc. Scrambled RNF41 or shRNF41 expression plasmids (fig. S2) were obtained from Cyagen Biosciences. One Shot Top 10 Chemically Competent Escherichia coli and the QIAGEN Endofree Plasmid Maxi Kit, used for transformation, amplification, and purification of ultrapure, transfection-grade plasmid DNA, were purchased from Thermo Fisher Scientific Inc. and QIAGEN Inc., respectively. Luria broth (LB broth) and LB agar ampicillin-100 plates for bacterial selection were obtained from Sigma-Aldrich. Deionized water was obtained from a Milli-Q water purification system (Millipore). GNPs were dispersed in deionized water (500 μg/ml) and oxidized using a modified Hofmann method (68% HNO3/96% H2SO4 at a 3:1 ratio in the presence of 70 μM KClO3) in continuous magnetic stirring for 96 hours. Dispersion was then neutralized with NaOH until reaching pH 7 and centrifuged at 21,000g for 30 min. Supernatant with small graphene oxide sheets was discarded, and graphite oxide nanoparticles were washed four times with distilled water and centrifuged at 21,000g for 30 min. Oxidized GNPs were separated by incubating the dispersion in an ultrasound bath (Selecta) at a frequency of 50 kHz and a potency of 360 W for 15 min. Afterward, 100 μl of GNPs were mixed with 900 μl of EDC/N-hydroxysuccinimide (1 mg/ml; 1:1) containing 30 μl of PAMAM dendrimer 25% (v/v) and incubated for 2 hours in the ultrasound bath at a constant temperature (25° ± 2°C). Then, dispersions were centrifuged at 21,000g for 10 min and washed three times with PBS for subsequent in vitro and in vivo experiments. Plasmids were incubated with dispersions of DGNP in a ratio of 1:10 for 2 hours in a rotatory shaker, centrifuged, and washed three times with PBS before use for transfection and functional assays. The ratio of plasmid/nanoparticles was established using the variations in zeta potential from positive (DGNP) to negative charge, when coating with plasmid, and evaluated with a Zetasizer Nano ZS (Malvern Instruments Ltd.).
Collagen degradation assay
Gelatin and FITC were obtained from Sigma-Aldrich. The preparation of FITC-conjugated gelatin and the quantitative analysis of collagen degradation assay were performed as previously described (16). Briefly, gelatin was dissolved (1 mg/ml) in a buffer containing 61 mM NaCl and 50 mM Na2B4O7 (pH 9.3) and then incubated at 37°C for 1 hour. After this incubation period, FITC was added (2 mg/ml) and mixed for 2 hours in complete darkness. This mixture was then dialyzed at room temperature in PBS in complete darkness for 4 days with two or three PBS changes per day. After a quick spin to remove insoluble material, small aliquots were stored in the dark at 4°C. FITC-conjugated gelatin-coated plates were prepared covering the surface of each well with FITC-gelatin and fixed with 1 drop of 0.5% ice-cold formaldehyde in PBS at 4°C for 15 min. Wells were then gently washed three times with PBS and lastly quenched in complete medium for 1 hour at 37°C. Cells were cultured for variable lengths of time up to 7 days, and supernatants were collected. Cells were fixed, washed, and stained with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) and visualized with an epifluorescence microscope. Supernatants were centrifuged and fluorescence was quantified with a Hitachi F-2500 Fluorescence Spectrophotometer (Hitachi High Technologies Corp.).
Statistical analysis
All data were expressed as means ± SD. Statistical analysis of the results was performed by one-way analysis of variance (ANOVA) with the post hoc Newman-Keuls test or by Student’s t tests, where appropriate (GraphPad Prism v6.0a). Data were tested for assumptions before the use of these statistical tests. Differences were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
Funding:
This work was supported by grants to P.M-L. and W.J. from Ministerio de Ciencia, Innovación y Universidades (grants RTI2018-094734-B-C21 and PID2021-123426OB-I00/funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”). P.M-L. was additionally supported by a fellowship from the Ramon y Cajal Program (RYC2018-0Z23971-I) funded by the Spanish Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033 and FSE invierte en tu futuro. P.M-L. was additionally granted a fellowship from the AGAUR Beatriu de Pinós Program 2016 (BP-00236) of Generalitat de Catalunya and a Research Fellowship Sheila Sherlock from the European Association for the Study of Liver (EASL). A.M.-L. had a Formación de Personal Investigador (FPI) grant from Ministerio de Ciencia, Innovación y Universidades and FSE invierte en tu futuro (reference: PRE2019-088097). M.M-B. had a Formación de Profesorado Universitario (FPU) grant from Ministerio de Ciencia, Innovación y Universidades and FSE invierte en tu futuro (reference: FPU19/03323). E.R.E. was supported by a grant (R01 GM 49039) from the National Institutes of Health. The Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBERehd) is funded by the Instituto de Salud Carlos III. This work was also supported by RedFibro (RED2022-134485-T) of the 2022 call for aid to «RESEARCH NETWORKS», within the framework of the Programa Estatal del Plan Estatal de Investigación Científica, Técnica y de Innovación 2021–2023, and the Consolidated Research Group of the Generalitat de Catalunya AGAUR (2021 SGR 00881).
Footnotes
Competing interests: The following patent is associated with the study: “Methods and compositions to target and treat macrophages”, PCT international application number 18/163.253, by co-inventors P.M.-L. and E.R.E. The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are in the paper or the Supplementary Materials. Raw data from figures are available in data file S1.
REFERENCES AND NOTES
- 1.Asrani SK, Devarbhavi H, Eaton J, Kamath PS, Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Melgar-Lesmes P, Edelman ER, Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 63, 917–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melgar-Lesmes P, Balcells M, Edelman ER, Implantation of healthy matrix-embedded endothelial cells rescues dysfunctional endothelium and ischaemic tissue in liver engraftment. Gut 66, 1297–1305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oro D, Yudina T, Fernandez-Varo G, Casals E, Reichenbach V, Casals G, González de la Presa B, Sandalinas S, Carvajal S, Puntes V, Jiménez W, Cerium oxide nanoparticles reduce steatosis, portal hypertension and display anti-inflammatory properties in rats with liver fibrosis. J. Hepatol. 64, 691–698 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Cordoba-Jover B, Arce-Cerezo A, Ribera J, Pauta M, Oró D, Casals G, Fernández-Varo G, Casals E, Puntes V, Jiménez W, Morales-Ruiz M, Cerium oxide nanoparticles improve liver regeneration after acetaminophen-induced liver injury and partial hepatectomy in rats. J. Nanobiotechnol. 17, 112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forbes SJ, Rosenthal N, Preparing the ground for tissue regeneration: From mechanism to therapy. Nat. Med. 20, 857–869 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Tsuchida T, Friedman SL, Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 14, 397–411 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Trautwein C, Friedman SL, Schuppan D, Pinzani M, Hepatic fibrosis: Concept to treatment. J. Hepatol. 62, S15–S24 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Sole C, Sola E, Morales-Ruiz M, Fernàndez G, Huelin P, Graupera I, Moreira R, de Prada G, Ariza X, Pose E, Fabrellas N, Kalko SG, Jiménez W, Ginès P, Characterization of inflammatory response in acute-on-chronic liver failure and relationship with prognosis. Sci. Rep. 6, 32341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP, Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. U.S.A. 109, E3186–E3195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, Mitchell D, Glover A, Laurie A, Doig S, Hargreaves E, Fraser AR, Turner ML, Campbell JDM, McGowan NWA, Barry J, Moore JK, Hayes PC, Leeming DJ, Nielsen MJ, Musa K, Fallowfield JA, Forbes SJ, Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 25, 1560–1565 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Wauman J, De Ceuninck L, Vanderroost N, Lievens S, Tavernier J, RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J. Cell Sci. 124, 921–932 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, Cao X, The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Ye S, Xu H, Jin J, Yang M, Wang C, Yu Y, Cao X, The E3 ubiquitin ligase neuregulin receptor degradation protein 1 (Nrdp1) promotes M2 macrophage polarization by ubiquitinating and activating transcription factor CCAAT/enhancer-binding protein β (C/EBPβ). J. Biol. Chem. 287, 26740–26748 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C, A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. U.S.A. 106, 17475–17480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melgar-Lesmes P, Luquero A, Parra-Robert M, Mora A, Ribera J, Edelman ER, Jiménez W, Graphene-dendrimer nanostars for targeted macrophage overexpression of metalloproteinase 9 and hepatic fibrosis precision therapy. Nano Lett. 18, 5839–5845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Yen L, Irwin L, Sweeney C, Carraway III KL, Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol. Cell. Biol. 24, 7748–7757 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, Tolba R, Trautwein C, Trebicka J, Weiskirchen R, Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 6, 19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muñoz-Luque J, Ros J, Fernandez-Varo G, Tugues S, Morales-Ruiz M, Alvarez CE, Friedman SL, Arroyo V, Jiménez W, Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J. Pharmacol. Exp. Ther. 324, 475–483 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, Pachter HL, Bar-Sagi D, Frey AB, Miller G, In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-α. J. Clin. Invest. 119, 3213–3225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Z, Wu X, Yen L, Sweeney C, Carraway III KL, Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol. Cell. Biol. 27, 2180–2188 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madge LA, Pober JS, A phosphatidylinositol 3-kinase/Akt pathway, activated by tumor necrosis factor or interleukin-1, inhibits apoptosis but does not activate NFκB in human endothelial cells. J. Biol. Chem. 275, 15458–15465 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Sabio G, Davis RJ, TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conde J, Oliva N, Atilano M, Song HS, Artzi N, Self-assembled RNA-triple-helix hydrogel scaffold for microRNA modulation in the tumour microenvironment. Nat. Mater. 15, 353–363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H, Nanoparticle uptake: The phagocyte problem. Nano Today 10, 487–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos JL, Pandita D, Rodrigues J, Pêgo AP, Granja PL, Balian G, Tomás H, Receptor-mediated gene delivery using PAMAM dendrimers conjugated with peptides recognized by mesenchymal stem cells. Mol. Pharm. 7, 763–774 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Carroll MJ, Kapur A, Felder M, Patankar MS, Kreeger PK, M2 macrophages induce ovarian cancer cell proliferation via a heparin binding epidermal growth factor/matrix metalloproteinase 9 intercellular feedback loop. Oncotarget 7, 86608–86620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, Dragomir AC, Aloman C, Schwabe RF, Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reichenbach V, Fernandez-Varo G, Casals G, Oró D, Ros J, Melgar-Lesmes P, Weiskirchen R, Morales-Ruiz M, Jiménez W, Adenoviral dominant-negative soluble PDGFRβ improves hepatic collagen, systemic hemodynamics, and portal pressure in fibrotic rats. J. Hepatol. 57, 967–973 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, Schneider MD, Musarò A, Rosenthal N, Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther. 23, 1189–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishizawa H, Iguchi G, Fukuoka H, Takahashi M, Suda K, Bando H, Matsumoto R, Yoshida K, Odake Y, Ogawa W, Takahashi Y, IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 6, 34605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, Roy S, Monocyte and macrophage plasticity in tissue repair and regeneration. Am. J. Pathol. 185, 2596–2606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G, PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Arkun Y, Dynamic modeling and analysis of the cross-talk between insulin/AKT and MAPK/ERK signaling pathways. PLOS ONE 11, e0149684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turowec JP, Lau EWT, Wang X, Brown KR, Fellouse FA, Jawanda KK, Pan J, Moffat J, Sidhu SS, Functional genomic characterization of a synthetic anti-HER3 antibody reveals a role for ubiquitination by RNF41 in the anti-proliferative response. J. Biol. Chem. 294, 1396–1409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Ceuninck L, Wauman J, Masschaele D, Peelman F, Tavernier J, Reciprocal cross-regulation between RNF41 and USP8 controls cytokine receptor sorting and processing. J. Cell Sci. 126, 3770–3781 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC, Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krenkel O, Tacke F, Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 17, 306–321 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Fabregat I, Moreno-Caceres J, Sanchez A, Dooley S, Dewidar B, Giannelli G, Dijke PT; IT-LIVER Consortium, TGF-β signalling and liver disease. FEBS J. 283, 2219–2232 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Bonefeld K, Moller S, Insulin-like growth factor-I and the liver. Liver Int. 31, 911–919 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ Jr., Liu XS, Lazar MA, PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22, 2941–2952 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitchell C, Willenbring H, A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 3, 1167–1170 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF, High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 10, 305–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ribera J, Pauta M, Melgar-Lesmes P, Córdoba B, Bosch A, Calvo M, Rodrigo-Torres D, Sancho-Bru P, Mira A, Jiménez W, Morales-Ruiz M, A small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 313, G492–G504 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or the Supplementary Materials. Raw data from figures are available in data file S1.