

Abstract
INTRODUCTION:
Millions of people have received genome and exome sequencing to date, a collective effort that has illuminated for the first time the vast catalog of small genetic differences that distinguish us as individuals within our species. However, the effects of most of these genetic variants remain unknown, limiting their clinical utility and actionability. New approaches that can accurately discern disease-causing from benign mutations and interpret genetic variants on a genome-wide scale would constitute a meaningful initial step towards realizing the potential of personalized genomic medicine.
RATIONALE:
As a result of the short evolutionary distance between humans and nonhuman primates, our proteins share near-perfect amino acid sequence identity. Hence, the effects of a protein-altering mutation found in one species are likely to be concordant in the other species. By systematically cataloging common variants of nonhuman primates, we aimed to annotate these variants as being unlikely to cause human disease as they are tolerated by natural selection in a closely related species. Once collected, the resulting resource may be applied to infer the effects of unobserved variants across the genome using machine learning.
RESULTS:
Following the strategy outlined above we obtained whole-genome sequencing data for 809 individuals from 233 primate species and cataloged 4.3 million common missense variants. We confirmed that human missense variants seen in at least one nonhuman primate species were annotated as benign in the ClinVar clinical variant database in 99% of cases. By contrast, common variants from mammals and vertebrates outside the primate lineage were substantially less likely to be benign in the ClinVar database (71 to 87% benign), restricting this strategy to nonhuman primates. Overall, we reclassified more than 4 million human missense variants of previously unknown consequence as likely benign, resulting in a greater than 50-fold increase in the number of annotated missense variants compared to existing clinical databases.
To infer the pathogenicity of the remaining missense variants in the human genome, we constructed PrimateAI-3D, a semisupervised 3D-convolutional neural network that operates on voxelized protein structures. We trained PrimateAI-3D to separate common primate variants from matched control variants in 3D space as a semisupervised learning task. We evaluated the trained PrimateAI-3D model alongside 15 other published machine learning methods on their ability to distinguish between benign and pathogenic variants in six different clinical benchmarks and demonstrated that PrimateAI-3D outperformed all other classifiers in each of the tasks.
CONCLUSION:
Our study addresses one of the key challenges in the variant interpretation field, namely, the lack of sufficient labeled data to effectively train large machine learning models. By generating the most comprehensive primate sequencing dataset to date and pairing this resource with a deep learning architecture that leverages 3D protein structures, we were able to achieve meaningful improvements in variant effect prediction across multiple clinical benchmarks.
Personalized genome sequencing has revealed millions of genetic differences between individuals, but our understanding of their clinical relevance remains largely incomplete. To systematically decipher the effects of human genetic variants, we obtained whole-genome sequencing data for 809 individuals from 233 primate species and identified 4.3 million common protein-altering variants with orthologs in humans. We show that these variants can be inferred to have nondeleterious effects in humans based on their presence at high allele frequencies in other primate populations. We use this resource to classify 6% of all possible human protein-altering variants as likely benign and impute the pathogenicity of the remaining 94% of variants with deep learning, achieving state-of-the-art accuracy for diagnosing pathogenic variants in patients with genetic diseases.
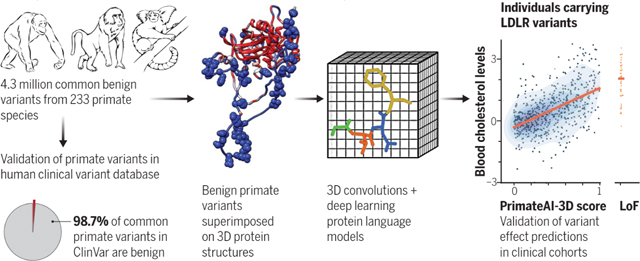
Graphical Abstract
PrimateAI-3D, a deep learning model trained on millions of benign primate variants. Common primate variants generated from 233 primate species (left) were validated as benign (98.7%) in the human ClinVar database. Voxelized protein structures (middle) with benign primate variants (spheres) were used to train a 3D convolution neural network to predict variant pathogenicity based on regional enrichment or depletion of primate variants. The resulting model was validated in independent clinical cohorts, as illustrated by the correlation of PrimateAI-3D scores and blood cholesterol levels for UK Biobank individuals (right).
A scalable approach for interpreting the effects of human genetic variants and their impact on disease risk is urgently needed to realize the promise of personalized genomic medicine (1–3). Out of more than 70 million possible protein-altering variants in the human genome, only ~0.1% are annotated in clinical variant databases such as ClinVar (4), with the remainder being variants of uncertain clinical significance (5,6). Despite collaborative efforts by the scientific community, the rarity of most human genetic variants has meant that progress toward deciphering personal genomes has been incremental (7,8). Consequently, clinical sequencing tests frequently return without definitive diagnoses, a frustrating outcome for both patients and clinicians (9, 10). In certain cases patients must be recontacted and diagnoses reversed when the presumed pathogenic variant was later found to be a common variant in previously understudied human populations (11–13). Common variants can often be ruled out as the cause of penetrant genetic disease, because their high frequency in the population indicates that they are tolerated by natural selection, aside from rare exceptions due to founder effects and balancing selection (14–16).
An emerging strategy for solving clinical variant interpretation on a genome-wide scale is the use of information from closely related primate species to infer the pathogenicity of orthologous human variants (17). Because chimpanzees and humans share 99.4% protein sequence identity (18), a protein-altering variant present in one species can be expected to produce similar effects on the protein in the other species. By conducting population sequencing studies in closely related nonhuman primate species, it is feasible to systematically catalog common variants and rule these out as pathogenic in humans, analogous to how sequencing more diverse human populations has helped to advance clinical variant interpretation (8,17). Nonetheless, earlier work (17) was limited by the very small primate population sequencing datasets available, which bounded the number of common variants discovered and the scale of machine learning classifiers that could be trained.
Results
A database of 4.3 million benign missense variants across the primate lineage
To expand upon this strategy, we sequenced 703 individuals from 211 primate species and aggregated these with data from previous studies (19–26), yielding a total of 809 individuals from 233 species. We identified 4.3 million unique missense (protein-altering) variants and 6.7 million unique synonymous (nonprotein altering) variants (Fig. 1A), after excluding variants at positions that lacked unambiguous 1:1 mapping with humans, or that resulted in nonconcordant amino acid translation outcomes because of changes at neighboring nucleotides (fig. SI). The species selected for sequencing represent close to half of the 521 extant primate species on Earth (27) and cover all major primate families, from Old World monkeys and New World monkeys to lemurs and tarsiers. We targeted a small number of individuals per species (3.5 on average) to ensure that we primarily sampled common variants that have been filtered by natural selection rather than rare mutations (fig. S2).
Fig. 1. Common primate variants are largely benign in humans.

(A) Counts of missense (solid green) and synonymous (shaded gray) variants from primates compared with the gnomAD database. Missense:synonymous counts and ratios are displayed above each bar. (B) Fractions of all possible human synonymous (gray) and missense variants (green) observed in primates. (C) Counts of benign (gray) and pathogenic (red) missense variants with two-star review status or above in the overall ClinVar database (left pie chart), compared with ClinVar variants observed in gnomAD (middle), and compared with ClinVar variants observed in primates (right). Conflicting benign and pathogenic annotations and variants interpreted only with uncertain significance were excluded. (D) Observed gnomAD (green) or primate (blue) missense variants in each amino acid position in the CACNA1A gene. Red circles represent the positions of annotated ClinVar pathogenic missense variants. Bottom scatterplot shows PrimateAI-3D predicted pathogenicity scores for all possible missense substitutions along the gene. (E) Multiple sequence alignment showing the ClinVar pathogenic variant chrll:77181548 G>A (red arrow) creating a cryptic splice site in human sequence (extended splice motif, blue). This variant is tolerated in Cebus Albifrons and other species with a G>C synonymous change in the adjacent nucleotide that stops the splice motif from forming. (F) Pie charts showing the fraction of benign (gray) and pathogenic (red) missense variants with ClinVar two-star review status or above in great apes, Old World monkeys, New World monkeys, lemurs/tarsiers, mammals, chicken, and zebrafish. (G) Missense:synonymous ratios (MSR) across the human allele frequency spectrum, with MSR of human variants seen in primates shown for comparison. The blue dashed line represents the expected missense:synonymous ratio of de novo variants. Colors and legend are the same as (A).
Compared with the genome Aggregation Database (gnomAD) cohort of 141,456 human individuals from diverse populations (28, 29), the primate sequencing cohort contained ~20% more exome variants despite sequencing 1/175th the number of individuals (Fig. 1A and fig. S3), attesting to the notable genetic diversity present in nonhuman primate species (19, 30), many of which are critically endangered (31). The overlap of primate variants with gnomAD was low, consistent with independent mutational origins in each species (fig. S3). Out of the 22 million possible synonymous variants in the human genome, 30% were observed in the primate cohort, compared with just 6% of possible missense mutations (Fig. 1B). Because de novo mutations would have laid down unbiased proportions of missense and synonymous variants, the observed depletion of missense mutations in the primate cohort is consistent with most of the newly-arising human missense mutations being removed by natural selection as a result of their deleteriousness (8,32–34). The surviving missense variants are seen at high frequencies in primate populations and represent a subset of missense variants that have tolerated filtering by natural selection and are unlikely to be pathogenic (35).
Missense variants from the primate cohort are strongly enriched for benign consequence in the ClinVar clinical variant database (Fig. 1C). Among ClinVar variants with higher review levels (two stars or above, indicating consensus by multiple submitters) (4), missense variants found in at least one nonhuman primate species were benign or likely benign ~99% of the time, compared with 63% for ClinVar missense variants in general and 80% for missense variants seen in gnomAD (Fig. 1C). The high fraction of pathogenic variants in gnomAD is consistent with most of these variants having arisen recently. Indeed, recent exponential human population growth introduced large numbers of rare variants through random de novo mutation (95% of variants in the gnomAD cohort are at <0.01% population allele frequency), without sufficient time for selection to purge deleterious variants from the population (36–40). Consequently, the gnomAD cohort provides a comparatively unfiltered look at variation caused by random mutations, whereas primate common variants represent the subset of random mutations that have survived.
The regions of human disease genes that were most densely populated by ClinVar pathogenic variants were also strongly depleted for primate common variants, with examples shown for CACNA1A (Fig. ID) and CREBBP (fig. S4), genes responsible for familial epilepsy (41, 42) and Rubinstein-Taybi syndrome (43,44). Missense variants in the gnomAD cohort were partially depleted within these same critical regions (Fig. ID and fig. S4), indicating that humans and primates experience similar selective pressures. However, deleterious variants were incompletely removed in humans, consistent with the shorter amount of time they were exposed to natural selection.
Prior to using primate data as an indicator of benign consequence in a diagnostic setting, it is vital to understand why a handful of human pathogenic ClinVar variants appear as tolerated common variants in primates. Our clinical laboratory independently reviewed evidence for each of the 36 ClinVar pathogenic variants that appeared in the primate cohort, according to ACMG guidelines (14). Among these 36 variants, 8 were reclassified as variants of uncertain significance based on insufficient evidence of pathogenicity in the literature and an additional 9 were hypomorphic or mild clinical variants (table SI). The remaining 19 variants appear to be truly pathogenic in humans and are presumably tolerated in primates because of primate-human differences, such as interactions with changes in the neighboring sequence context (45, 46). In one such example, a compensatory synonymous sequence change at an adjacent nucleotide explains why the variant is benign in primates but creates a pathogenic splice defect in humans (Fig. IE). We also expect that some of the variants identified among primates are rare pathogenic variants by chance, despite the small number of individuals sequenced within each species. By expanding our cohort to sequence a large number of individuals per species, we would definitively exclude rare variation from our catalog of primate variation, as well as grow the database of benign variants to improve clinical variant interpretation.
As evolutionary distance from humans increases, cases in which the surrounding sequence context has changed sufficiently to alter the effect of the variant should also increase until common variants in more-distant species could no longer be reliably counted on as benign in humans. We examined variation in each major branch of the primate tree as well as variation from mammals (mouse, rat, cow, dog), chicken, and zebrafish and evaluated their pathogenicity in ClinVar (Fig. 1F). Common variants from species throughout the primate lineage, including more-distant branches such as lemurs and tarsiers, varied from 98.6 to 99% benign in the human ClinVar database, but this dropped to 87% for placental mammals and 71% for chicken. The high fraction of variants that are pathogenic in humans yet tolerated as common variants in more distant vertebrates indicates that selection on orthologous variants diverges substantially in distantly related species as a consequence of changes in the surrounding sequence context and other differences in species’ biology (fig. S5).
We have made the primate population variant database, which contains more than 4.3 million likely benign missense variants, publicly available at https://primad.basespace.illumina.com as a reference for the genomics community. Overall, this resource is over 50 times larger than ClinVar in terms of number of annotated missense variants and consists almost entirely of variants of previously unknown significance. Most primate variants are rare or absent in the human population, with 98% of these variants at allele frequency <0.01% (fig. S6). This makes it challenging to establish their pathogenicity through other means, because even the largest sequencing laboratories would be unlikely to observe any given variant in more than one unrelated patient. Despite their rarity, the subset of human variants that appear in primates have a low missense:synonymous ratio consistent with being depleted of deleterious missense variants (Fig. 1G). This contrasts with the high missense:synonymous ratio for rare human variants in the overall gnomAD cohort, which approaches the 2.2:1 ratio expected for random de novo mutations in the absence of selective constraint (47). At higher allele frequencies, natural selection has had more time to purge deleterious missense variants, allowing the human missense:synonymous ratio to start to converge toward the ratio observed for the subset of human variants that are present in other primates.
Gene-level selective constraint in humans versus nonhuman primates
The primate variant resource makes it possible to compare natural selection acting on individual genes across the primate lineage and identify human-specific evolutionary differences. Because the current primate cohort only contains an average of 3 to 4 individuals per species, we focused on comparing selective constraint in human genes versus primates as a whole. We found that the missense:synonymous ratios of individual genes were well-correlated between humans and primates (Spearman r = 0.637) (Fig. 2A), indicating that genes that were depleted for deleterious missense mutations in humans were also consistently depleted throughout the primate lineage. Moreover, the missense:synonymous ratios of both human and primate genes correlated similarly well with the probability of genes being loss of function intolerant (pLI) (Spearman correlation −0.534 and −0.489, respectively) (28). Had there been substantial divergence between humans and primates, pLI, an independent metric derived from human protein-truncating variation, would have been expected to show much clearer agreement with human missense: synonymous ratios than primate.
Fig. 2. Selective constraint of primate genes compared with humans.

(A) Scatter plot of missense: synonymous ratios between primate and human genes. Each gene is colored by its pLI score, with darker points showing haploinsufficient genes. (B) Observed and expected counts of synonymous (top) and missense (bottom) variants per gene in gnomAD (left) and primates (right). Genes are colored by their pLI scores. (C) Distributions of observed and expected ratios of synonymous (dashed lines) and missense (solid lines) variants for all genes. Results for primate genes (orange) and gnomAD genes (blue) are shown. (D) Scatter plot of missense:synonymous ratios between primate and human genes. Highlighted points are genes that are under significantly stronger (blue) or weaker (red) constraint in humans compared with nonhuman primates under both methods (Benjamini-Hochberg FDR < 0.05) and gray points show nonsignificant genes. The top 10 genes with the largest effect sizes in either direction are labeled.
To measure the selective constraint on each gene, we calculated the observed versus expected number of variants per gene, using trinucleotide mutation rates to model the expected probability of observing each variant (fig. S7) (28, 29). We modeled each primate species separately to account for differences in genetic diversity and the number of individuals sampled per species. The expected and observed counts of synonymous variants were highly correlated in both the gnomAD and primate cohorts, indicating that our model accurately captured the background distribution of neutral mutations (Fig. 2B; Spearman correlation 0.933 and 0.949, respectively). By contrast, for missense variants the expected and observed counts per gene diverged substantially (Spearman correlation 0.896 and 0.561 for humans and primates, respectively), due to depletion of deleterious missense variants by natural selection in highly constrained genes (for example, high pLI genes). The most highly constrained genes were almost completely scrubbed of common missense variants in the primate cohort, whereas rare missense variants in the gnomAD cohort were depleted to a more modest extent because of the large sample size of gnomAD (Fig. 2C).
We next aimed to identify genes whose selective constraint was different in humans compared with the rest of the primate lineage, a task made difficult by differences in diversity, allele frequency, and sample size between the human and primate cohorts (34, 48, 49). To this end, we developed two orthogonal strategies and took the intersection of genes identified under both approaches. First, we used population genetic modeling (34, 50, 51) to estimate the average selection coefficient, s, ranging from 0 (benign) to 1 (severely pathogenic) of missense mutations in each gene, using a model of recent human population growth (figs. S7 and S8). We fit a single value of s per gene across nonhuman primate species and identified genes that differed between Sprimate and Shuman using a likelihood ratio test, which we validated using population simulations (fig. S9). In a second approach, we fit a curve approximating the relationship between human and primate missense:synonymous ratios using a Poisson generalized linear mixed model (52) and identified genes in which the observed human missense:synonymous ratio deviated from what would have been expected given the gene’s missense:synonymous ratio in primates (fig. S10). We also adjusted for gene length to account for shorter genes having more variability in their missense:synonymous ratio measurements than longer genes. The two methods were broadly concordant, with a Spearman correlation of 0.80 between the genes’ effect sizes in the two tests. Estimates of selection coefficients and observed and expected counts for each gene in humans and primate are provided in table S2.
In total, we found 39 genes in which selective constraint differed significantly between humans and other primates under both methods [Benjamini-Hochberg FDR < 0.05 (53); Fig. 2D]. The top three genes in which shuman decreased the most relative to sprimate were CFTR, GJB2, and CD36, autosomal recessive disease genes for cystic fibrosis (54), hereditary deafness (55), and platelet glycoprotein deficiency (56), respectively. All three genes are known for deleterious mutations that are unusually common in local geographic human populations (57–60), suggesting that they may be experiencing reduced selection due to heterozygote advantage that protects against specific environmental pathogens (60–64). On the other end of the spectrum, TERT, known for its role in maintaining telomere length (65,66), was among the top genes in which shuman increased the most relative to sprimate Humans have adapted to a much longer life span compared with other primate species, which have a median life span of 20 to 30 years, suggesting that increased selection on TERT may have occurred as part of human adaption toward extended longevity. We note that with the current size of the primate cohort, it is not possible to distinguish whether the increased selection on TERT occurred only in humans, or if it is part of a gradual trend toward extended longevity that began earlier in the great ape lineage, which also have longer life spans relative to other primates (~40 years). Expanding the primate cohort by sequencing more individuals per species would improve detection of additional species-specific and lineage-specific evolutionary adaptations and shed light on the evolutionary path that led to the present human condition.
PrimateAI-3D, a deep learning network for classifying protein-altering variants
We constructed PrimateAI-3D, a semisupervised 3D convolutional neural network for variant pathogenicity prediction, which we trained using 4.5 million common missense variants with likely benign consequence (Fig. 3A). In a departure from prior deep learning architectures that operated on linear sequences (17, 67), we voxelized the 3D structure of the protein at 2 Å resolution (figs. Sll and S12) and used 3D convolutions to enable the network to recognize key structural regions that may not be apparent from sequence alone (Fig. 3A). As an example, we show PrimateAI-3D predictions for STK11 (Fig. 3B), the tumor suppressor gene responsible for Peutz-Jeghers hereditary polyposis syndrome (68–71), with each amino acid position colored by the average PrimateAI-3D score at that position. Common primate variants used for training and annotated ClinVar pathogenic variants from separate parts of the linear sequence form distinct clusters in 3D space. Although ClinVar variants are shown for illustration, it should be noted that the network was not trained on either human-engineered features or annotated variants from clinical variant databases, thereby avoiding potential human biases in variant annotation. Rather, it learns to infer pathogenicity based on the local enrichment or depletion of common primate variants, taking only the protein’s multiple sequence alignment and 3D structure as inputs.
Fig. 3. PrimateAI-3D architecture and variant classification performance.

(A) PrimateAI-3D workflow. Human protein structures and multiple sequence alignments are voxelized (left) as input to a 3D convolutional neural network that predicts pathogenicity of all possible point mutations of a target residue (middle). The network is trained using a loss function with three components (right): common human and primate variants; fill-in-the-blank of a protein structure; score ranks from language models. (B) Protein structure of the STK11 gene, colored by PrimateAI-3D pathogenicity prediction scores (blue, benign; red, pathogenic). Spheres indicate residues with common human and primate variants (left) or residues with pathogenic mutations from ClinVar (right). For spheres, the color corresponds to the pathogenicity score of only the variant. For other residues, pathogenicity scores are averaged over all variants at that site. (C) Scatterplot shows performance of methods that predict missense variant pathogenicity in two clinical benchmarks (DDD and UKBB). Datasets are a subset of variants for which all methods have predictions. (D) Six barplots show method performance for six testing datasets (DMS assays, UKBB, ClinVar, DDD, ASD, and CHD).
PrimateAI-3D can use protein structures from either experimental sources or computational prediction (72–76); we used AlphaFold DB (72, 73) and HHpred (74) predicted structures for the broadest coverage across human genes. For training data, we incorporated all common missense variants from the 233 nonhuman primate species (17) and common human missense variants (allele frequency > 0.1% across populations) in gnomAD (28,29), TOPMed (77, 78), and UK Biobank (UKBB) (79,80), resulting in a total of 4.5 million unique missense variants of likely benign consequence. This dataset covers 6.34% of all possible human missense variants and is over 50 times larger than the current ClinVar database (79,381 missense variants after excluding variants of uncertain significance and those with conflicting annotations), greatly enlarging the training dataset available for machine learning approaches. Because the training dataset consists only of variants labeled as benign, we created a control set of randomly selected variants that were matched to the common variants by trinucleotide mutation rate and trained PrimateAI-3D to separate common variants from matched controls as a semisupervised learning task.
In parallel with the variant classification task, we generated amino acid substitution probabilities for each position in the protein by masking the residue and using the sequence context to predict the missing amino acid, borrowing from language model architectures that are trained to predict missing words in sentences (81, 82). We trained both a 3D convolutional “fill-in-the-blank” model, which tasked the network with predicting the missing amino acid in a gap in the voxelized 3D protein structure, and separately, a language model using the transformer architecture to predict the missing amino acid using the surrounding multiple sequence alignment as context (83). We implemented these models as additional loss functions to farther refine the PrimateAI-3D predictions (fig. S13). We also trained a variational autoencoder (67) on multiple sequence alignments and found that it performed comparably to our transformer architecture (fig. S14). Hence, we incorporated the average of their predictions in the loss function, which performed better than either alone.
We evaluated PrimateAI-3D and 15 other published machine learning methods (67, 84) on their ability to distinguish between benign and pathogenic variants along six different axes (Fig. 3, C and D, and fig. S15): predicting the effects of rare missense variants on quantitative clinical phenotypes in a cohort of 200,643 individuals from the UKBB; distinguishing missense de novo mutations (DNM) seen in 31,058 patients with neurodevelopmental disorders (DDD) (85–87) from de novo missense mutations in 2555 healthy controls (88–93); distinguishing de novo missense mutations seen in 4295 patients with autism spectrum disorders (ASD) (88–94) from de novo missense mutations in the shared set of 2555 healthy controls; distinguishing de novo missense mutations seen in 2871 patients with congenital heart disease (CHD) (95) from de novo missense mutations in the shared set of 2555 healthy controls; separating annotated ClinVar benign and pathogenic variants (ClinVar) (4); and average correlation with in vitro deep mutational scan (DMS) experimental assays across nine genes (96–105). Our set of clinical benchmarks is the most comprehensive to date and has a particular focus on rigorously testing the performance of classifiers on large patient cohorts across a diverse range of real-world clinical settings (table S3).
For the UKBB benchmark, we analyzed 200,643 individuals with both exome sequencing data and broad clinical phenotyping and identified 42 genes in which the presence of rare missense variants was associated with changes in a quantitative clinical phenotype controlling for confounders such as population stratification, age, sex, and medications (table S4). These gene-phenotype associations included diverse clinical lab measurements such as low-density lipoprotein (LDL) cholesterol (increased by rare missense variants in LDLR, decreased by variants in PCSK9), blood glucose (increased by variants in GCK), and platelet count (increased by variants in JAK2, decreased by variants in GP1BB), as well as other quantitative phenotypes such as standing height (increased by variants in ZFAT) (table S4). To test each classifier’s ability to distinguish between pathogenic and benign missense variants, we measured the correlation between pathogenicity prediction score and quantitative phenotype for patients carrying rare missense variants in each of these genes. We report the average correlation across all gene-phenotype pairs for each classifier, taking the absolute value of the correlation because these genes may be associated with either increase or decrease in the quantitative clinical phenotype.
The DDD, ASD, and CHD cohorts are among the largest published trio-sequencing studies to date and consist of thousands of families with a child with rare genetic disease and their unaffected parents. In each cohort, we cataloged de novo missense mutations that appeared in affected probands but were absent in their parents, as well as de novo missense mutations that appeared in a set of shared healthy controls. We evaluated the ability of each classifier to separate the de novo missense mutations that appear in cases versus controls on the basis of their prediction scores, using the Mann-Whitney U test to measure performance.
PrimateAI-3D outperformed all other classifiers at distinguishing pathogenic from benign variants in the four patient cohorts we tested (UKBB, DDD, ASD, CHD); it was also the top performer at separating pathogenic from benign variants in the ClinVar annotation database and had the highest average correlation with the deep mutational scan assays (Fig. 3D and fig. SI5). After PrimateAI-3D there was no clear runner-up, with second place occupied by six different classifiers in the six different benchmarks. We observed a moderate correlation between the performance of different classifiers in UKBB and DDD (Spearman r = 0.556; Fig. 3C), which are the two largest clinical cohorts and therefore likely the most robust for benchmarking (with 200,643 and 33,613 patients, respectively), but outside of PrimateAI-3D, strong performance of a classifier on one task had limited generalizability to other tasks. Our results underscore the importance of validating machine learning classifiers along multiple dimensions, particularly in large real-world cohorts, to avoid overgeneralizing a classifier’s performance based upon a notable showing along a single axis.
PrimateAI-3D’s top-ranked performance at separating benign and pathogenic missense variants in ClinVar was unexpected, as the other machine learning classifiers (with the exception of EVE) were trained either directly on ClinVar or on other variant annotation databases with a high degree of content overlap. Because they are primarily based on variants described in the literature, clinical variant databases are subject to ascertainment bias (12,106,107), which may have contributed to supervised classifiers picking up on tendencies of human variant annotation that are unrelated to the task of separating benign from pathogenic variants (figs. S16, S17, and S18). Given the challenges with human annotation, we also investigated whether PrimateAI-3D could assist in revising incorrectly labeled ClinVar variants, by comparing annotations in the current ClinVar database and those from a September 2017 snapshot. Disagreement between PrimateAI-3D and the 2017 version of ClinVar was highly predictive of future revision and the odds of revision increased with PrimateAI-3D confidence (fig. S19). Among variants with the 10% most confident PrimateAI-3D predictions, the odds of revision were elevated by a factor of 10 if PrimateAI-3D was in disagreement with the ClinVar label (P < 10−14).
The performance of PrimateAI-3D on clinical variant benchmarks scaled directly with training dataset size, indicating that additional primate sequencing data will be the key to unlocking farther gains (Fig. 4 and fig. S20). The current primate cohort already covers 30% of all possible synonymous variants in the human genome, despite containing only 809 individuals from 233 species (Fig. 4B). By increasing the number of species and the number of individuals sequenced per species, we expect to saturate most of the remaining tolerated substitutions in the human genome (fig. S21), including both coding and non-coding variation, leaving the remaining deleterious variants to be deduced by a process of elimination.
Fig. 4. Impact of training data-set size on classification accuracy.

(A) Improved performance of PrimateAI-3D with increasing number of common human and primate variants in the training dataset (x-axis). Performance of each dataset (y-axis) was divided by the maximum performance observed ; across all training dataset sizes. (B) Cumulative fractions of all possible human synonymous (gray) and missense (green) variants observed as common variants in 234 primate species, including humans (allele frequency > 0.1%). Each point shows the average of 10 permutations, calculated with a different random ordering of the list of primate species each time.
Discovery of candidate disease genes for neurodevelopmental disorders
We applied PrimateAI-3D to improve statistical power for discovering candidate disease genes that are enriched for pathogenic de novo mutations in the neurodevelopmental disorders cohort (fig. S22). De novo missense mutations from affected individuals in the DDD cohort (87) were enriched 1.36-fold above expectation, based on estimates of background mutation rate using trinucleotide context (47). We selected a PrimateAI-3D classification threshold of 0.821, which called an equal number of pathogenic missense mutations (n = 7,238) as the excess of de novo missense mutations in the cohort (Fig. 5A). Stratifying missense mutations by this threshold increased enrichment of pathogenic de novo missense mutations to 2.0-fold, substantially increasing statistical power for disease gene discovery in the cohort (Fig. 5B).
Fig. 5. Enrichment of de novo mutations in the neurodevelopmental disorder cohort over expectation.

(A) Enrichment of DNMs from Kaplanis et al. (87) across all genes. Enrichment ratios are given for synonymous, all missense, and protein-truncating variants (PTV), along with missense split by PrimateAI-3D score into benign (<0.821) and pathogenic (>0.821). (B) Enrichment of benign and pathogenic missense above expectation at varying PrimateAI-3D thresholds for classifying pathogenic missense.
By applying PrimateAI-3D to prioritize pathogenic missense variants, we identified 290 genes associated with intellectual disability at genome-wide significance (P < 6.4 × 10−7) (Table 1), of which 272 were previously discovered genes that either appeared in the Genomics England intellectual disability gene panel (108) or were already identified in the prior study (109) without stratifying missense variants (table S5). We excluded two genes, BMPR2 and RYR1, as borderline significant genes that already had well-annotated non-neurological phenotypes. Further clinical studies are needed to independently validate this list of candidate genes and understand their range of phenotypic effects.
Table 1. Additional genes discovered in intellectual disability.
Genes achieving the genome-wide significance (P < 6.4 × 10−7) are shown when considering only missense de novo mutations with PrimateAI-3D scores ≥0.821. Counts of protein-truncating and missense DNMs are provided. P values for gene enrichment are shown when the statistical test was run only with missense mutations with PrimateAI-3D score ≥0.821 and when it was repeated for all missense mutations.
| Missense | P value | ||||
|---|---|---|---|---|---|
|
| |||||
| HGNC symbol | Protein-truncating variants | PrimateAI-3D score ≥0.821 | All missense | PrimateAI-3D score ≥0.821 | All missense |
|
| |||||
| AP1G1 | 2 | 4 | 5 | 4.1×10−7 | 5.9×l0−5 |
| ATP2B2 | 1 | 9 | 11 | 2 1×10−7 | 1.4×10−3 |
| CELF2 | 2 | 4 | 4 | i.2×10−7 | 6.7×10−5 |
| MAP4K4 | 2 | 6 | 7 | 3.9×10−7 | 5.0×10−4 |
| MED13 | 3 | 6 | 9 | 6.6×10−8 | 3.5×l0−5 |
| MFN2 | 0 | 6 | 8 | 3.4×10−7 | 1.0×10−5 |
| NR4A2 | 2 | 4 | 5 | 3.7×10−7 | 3.3×10−5 |
| PIP5K1C | 0 | 8 | 9 | 2.8×10−8 | 4.9×l0−4 |
| RAB5C | 2 | 4 | 5 | 8.6×10−8 | 1.5×10−5 |
| SPOP | 1 | 4 | 6 | 4.1×10−7 | 1.7×10−6 |
| SPTBN2 | 1 | 10 | 16 | 3.9×10−7 | 4.5×10−3 |
| XP01 | 1 | 7 | 7 | 5.0×10−7 | 7.2×10−4 |
| EIF4A2 | 2 | 4 | 4 | 1.7×10−7 | 2.1×10−4 |
| LMBRD2 | 0 | 3 | 4 | 6.0×10−7 | 1.3×10−4 |
| MARK2 | 4 | 3 | 5 | 2.3×10−7 | 3.8×10−5 |
| N0TCH1 | 4 | 6 | 17 | 4.1×10−7 | 1.3×10−6 |
Discussion
Our results demonstrate the successful pairing of primate population sequencing with state-of-the-art deep learning models to make meaningful progress toward solving variants of uncertain significance. Primate population sequencing and large-scale human sequencing are likely to fill complementary roles in advancing clinical understanding of human genetic variants. From the perspective of acquiring additional benign variants to train PrimateAI-3D, humans are not suitable, as the discovery of common human variants (>0.1% allele frequency) plateaus at ~100,000 missense variants after only a few hundred individuals (17), and further population sequencing into the millions mainly contributes rare variants that cannot be ruled out for deleterious consequence. By contrast, because these rare human variants have not been thoroughly filtered by natural selection, they preserve the potential to exert highly penetrant phenotypic effects, making them indispensable for discovering new gene-phenotype relationships in large population sequencing and biobank studies. Fittingly, classifiers trained on common primate variants may accelerate these target discovery efforts by helping to differentiate between benign and pathogenic rare variation.
The genetic diversity found in the 520 known nonhuman primate species is the result of ongoing natural experiments on genetic variation that have been running uninterrupted for millions of years. Today, more than 60% of primate species on Earth are threatened with extinction in the next decade as a result of man-made factors (31). We must decide whether to act now to preserve these irreplaceable species, which act as a mirror for understanding our genomes and ourselves, and are each valuable in their own right, or bear witness to the conclusion of many of these experiments.
Materials and methods
Primate polymorphism data
We aggregated high-coverage whole genomes of 809 primate individuals across 233 primate species, including 703 newly sequenced samples and 106 previously sequenced samples from the Great Ape Genome project (19). Samples that passed quality evaluation were then aligned to 32 high-quality primate reference genomes (110) and mapped to the GRCh38 human genome build.
We developed a random forest (RF) classifier to identify false positive variant calls and errors resulting from ambiguity in the species mapping. In addition, we removed variants that fell in primate codons that did not match the human codon at that position, as well as those residing in primate transcripts with likely annotation errors. We also devised quality metrics based on the distribution of RF scores and Hardy-Weinberg equilibrium, and developed a unique mapping filter to exclude variants in regions of nonunique mapping between primate species.
Identifying differential selection between humans and primates through population modeling
We first established a neutral background distribution of mutation rates per gene for each primate species by fitting the Poisson Random Field model to the segregating synonymous variants in each species. The observed number of segregating synonymous sites is a Poisson random variable, with the mean determined by mutation rate, demography, and sample size (34). For simplicity, we assumed an equilibrium (i.e., constant) demography for all species besides humans; for humans, we used Moments (51) to find a best-fitting demographic history based on the folded site frequency spectrum of synonymous sites. We adopted a Gamma distributed prior on mutation rates, which also accounts for the impact of GC content on mutation rate. We optimized the prior parameters through maximum likelihood and computed the posterior distribution of the mutation rate per gene.
The number of segregating nonsynonymous sites is modeled as a Poisson random variable similar to synonymous sites with additional selection parameters. We assumed that every nonsynonymous mutation in a gene shares the same population-scaled selection coefficient γig. To explicitly estimate the selection coefficient of each gene per species, we devised a two-step procedure analogous to an expectation-maximization algorithm to control for differences in population size across species.
To identify genes in which human constraint is different from nonhuman primate selection, we developed a likelihood ratio test to test whether population-scaled selection coefficients are significantly different between humans and other primates. We then assessed whether our population genetic modeling improved the correlation of selection estimates of our primate data with previous gene-constraint metrics in humans, including pLI (28) and s_het (111). To validate the performance of our model, we performed population genetic simulations.
Poisson generalized linear mixed modeling of selection between humans and primates
In addition to the population genetics model described above, we also applied an orthogonal approach to detect differences in selection between humans and primates based on missense: synonymous ratios. We fit a Poisson generalized linear mixed model (GLMM) to the pooled polymorphic synonymous and missense mutations across all primates to estimate the depletion of missense variants in each gene. Then, we fit a second Poisson GLMM to the human data, controlling for the primate depletion estimates, and compared the pooled primate MSR with the human MSR for each gene.
PrimateAI-3D model
PrimateAI-3D is a 3D convolutional neural network that uses protein structures and multiple sequence alignments (MSA) to predict the pathogenicity of human missense variants. To generate the input for a 3D convolutional neural network, we voxelized the protein structure and evolutionary conservation in the region surrounding the missense variant. The network was trained to optimize three objectives: distinction between benign and unknown human variants; prediction of a masked amino acid at the variant site; per-gene variant ranks based on protein language models.
Protein structures and multiple sequence alignments
For 341 species, we used vertebrate and mammal MSAs from UCSC MultizlOO (112,113) and Zoonomia (23). Another 251 species appeared in Uniprot for at least 75% of all human proteins (114). For each protein, alignments from all 341+251=592 species were merged. Human protein structures were taken from AlphaFold DB (June 2021) (73). Proteins that did not sequence-match exactly to our hg38 proteins (2590; 13.5%) were homology modeled using HHpred (74) and Modeller (115).
Protein voxelization and voxel features
A regular sized 3D grid of 7×7×7 voxels, each spanning 2Å×2Å×2Å, was centered at the Cα atom of the residue containing the target variant (fig. Sll). For each voxel, we provided a vector of distances between its center and the nearest Cα and Cβ atoms of each amino acid type (fig. Sll; details in Supplementary Text section 1). We also provided additional voxel features including the pLDDT confidence metric from AlphaFold DB (fig. S12), and the evolutionary profile, consisting of each amino acid’s frequency at the corresponding position in the 592 species alignment.
Model architecture
The first layers of the PrimateAI-3D model reduce the voxel tensor to a 64-vector through repeated valid-padded 3D convolutions with a kernel size of 3×3×3. A final hidden dense layer transforms this 64-length vector into a 20-length vector, corresponding to one output unit per amino acid at that position. The model was trained simultaneously using multiple loss functions to optimize the following complementary aspects of pathogenicity:
Benign primate variants
Using 4.5 million benign missense variants from primates, we sampled the same number of unknown variants from the set of all possible human missense variants, with the distribution of mutational probabilities matching the benign set, based on a trinucleotide mutation rate model. Variants for the same protein position were combined in a 20-length vector (benign: 0, unknown: 1) which was the target label for the network. We used mean squared error (MSE) as the loss function for non-missing labels and ignored missing labels.
3D fill-in-the-blank
We removed all atoms of a target residue before voxelization, discarding any information about the residue from the input tensor to the network. The network was then trained to predict a 20-length vector, labeled 0 (benign) for amino acids that occur at the target site in any of the 592 species and 1 (pathogenic) otherwise. All human protein positions with at least one possible missense variant were included in this dataset.
Variant ranks from language models
For each gene, we took the average pathogenicity ranking from two protein language models, PrimateAI language model (PrimateAI LM, described below) and our reimplementation of the EVE variational autoencoder algorithm which we extended to all human proteins (EVE*) (67). We calculated the pairwise logistic rank loss as described in Pasumarthi et al. (116).
PrimateAI language model
The PrimateAI language model (PrimateAI LM) is a MSA transformer (83) for fill-in-the-blank residue classification, which was trained end-to-end on MSAs of UniRef-50 proteins (115,117) to minimize an unsupervised masked language modelling (MLM) objective (81). Our model requires ~50× less computation for training than previous MSA transformers as a result of several improvements in architecture and training (fig. S9).
Model training procedure
Each batch had the same number of samples from each of the three variant datasets (~33 with a batch size of 100). For the language model ranks dataset, all 33 samples had to come from the same protein. The number of times a protein was chosen for a batch was proportional to the length of the protein. In order to make our model robust against protein orientations, we randomly rotated the protein atomic coordinates in 3D before voxelizing a variant.
Model evaluation
We compared performance of our model and other models (84) on variants for which all models had scores. Deep mutational scanning assays were available for 9 human genes: Amyloid-beta (102), YAP1 (96),MSH2 (98), SYUA (101), VKOR1 (97), PTEN (99,100), BRCA1 (104), TP53 (103), and ADRB2 (103). For each assay and prediction model, we calculated the absolute Spearman rank correlation between prediction and assay scores. The UKBB dataset (79, 80) contains 42 gene-phenotype pairs which were significantly associated by rare variant burden testing using all rare missense variants, without applying missense pathogenicity prioritization. The evaluation was the same as with DMS assays, except that correlations were calculated from the quantitative phenotypes of individuals carrying the variant, instead of the assay score for the variant. For ClinVar (4), we filtered to high-quality 2-star variants and evaluated model performance by calculating per-gene area under the receiver operating characteristic curve (AUC). For the rare disease cohorts, we collected de novo missense mutations from patients with developmental disorders (85–87), autism spectrum disorders (88–94) or congenital heart disorders (93). For all three datasets, we compared against DNMs from healthy controls (88–93). We applied the Mann-Whitney U test to measure how well each model’s prediction scores could distinguish patient variants from control variants.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. MacArthur, Y. Song, and M. Daly for helpful discussions and the gnomAD team at the Broad Institute for their assistance with the website.
Funding:
L.F.K.K. was supported by an EMBO STF 8286 (to L.F.K.K.). R.R. was supported by an NIH training grant NIH T32 GM007748. M.K. was supported by "la Caixa” Foundation (ID 100010434 to M.K.), fellowship code LCF/BQ/PR19/11700002 (to M.K.), and the Vienna Science and Technólogy Fund (WWTF) [10.47379/VRG20001] (to M.K.). J.D.O. was supported by "la Caixa” Foundation (ID 100010434) and the European Union’s Horizon 2020 research and innovation programme under the Marie Sktodowska-Curie grant agreement 847648. The fellowship code is LCF/BQ/PI20/11760004. F.E.S. has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skodowska- Curie grant agreement No 801505. F.E.S. also received funds from the Conselho Nacional de Desenvolvimento Cientffico e Tecnologico (CNPq) (Process nos.: 303286/2014-8, 303579/2014-5, 200502/2015-8, 302140/2020-4, 300365/2021-7, 301407/2021-5, 301925/2021-6,; the International Primatological Society (Conservation grant), The Rufford Foundation (14861-1, 23117-2, 38786-B), the Margot Marsh Biodiversity Foundation (SMA-CCO-G0023, SMA-CCOG0037), and Primate Conservation Inc. (#1713 and #1689). The Mamirauá Institute for Sustainable Development received funds from the Gordon and Betty Moore Foundation (grant 5344 to J.V.A. and F.E.S.) Fieldwork for samples collected in the Brazilian Amazon was funded by grants from Conselho Nacional de Desenvolvimento Cientffico e Tecnológico (CNPq/SISBIOTA Program 563348/2010-0 to I.P.F.), Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM/SISBIOTA 2317/2011 to I.P.F.), and Coordenaçào de Aperfeigoamento de Pessoal de Nivel Superior (CAPES AUX 3261/2013) to I.P.F. Sampling of nonhuman primates in Tanzania was funded by the German Research Foundation (KN1097/3-1 to S.K. and R03055/2-1 to C.R.) and by the US National Science Foundation (BNS83-03506 to J.P.C.) No animals in Tanzania were sampled purposely for this study. Details of the original study on Treponema pallidum infection can be requested from S.K. Sampling of baboons in Zambia was funded by US NSF grant BCS-1029451 to J.P.C., C.J.J., and J.R. The research reported in this manuscript was also funded by the Vietnamese Ministry of Science and Technology's Program 562 (grant ƉTƉL.CN-64/19). A.N.C. is supported by l+D+i project PID2021-127792NB-I00 funded by MCIN/AEI/10.13039/501100011033 (FEDER Una manera de hacer Europa)" and by "Unidad de Excelencia María de Maeztu", funded by the AEI (CEX2018-000792-M) and Departament de Recerca i Universitats de la Generalitat de Catalunya (GRC 2021 SGR 0467). A.D.M. was supported by the National Sciences and Engineering Research Council of Canada and Canada Research Chairs program. The authors thank the Veterinary and Zoology staff at Wildlife Reserves Singapore for their help in obtaining the tissue samples, as well as the Lee Kong Chian Natural History Museum for storage and provision of the tissue samples. We thank H. Doddapaneni, D. M. Muzny, and M. C. Gingras for their support of sequencing at the Baylor College of Medicine Human Genome Sequencing Center. We greatly appreciate the support of R. Gibbs, director of HGSC, for this project and thank the Baylor College of Medicine for internal funding. T.M.B. is supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 864203 to T.M.B.), PID2021-126004NB-100 (MICIIN/FEDER, UE) and Secretaria d'Universitats i Recerca and CERCA Programme del Departament d'Economia i Coneixement de la Generalitat de Catalunya (GRC 2021 SGR 00177). H.L.R. receives funding from lllumina, Inc to support rare disease gene discovery and diagnosis. M.CJ, D.d.V. I.G., R.M.D.B., and J.P.B. were supported by a UKRI NERC standard grant (NE/T000341/1). We thank P. Karanth (IISc) and H. N. Kumara (SACON) for collecting and providing us with some of the samples from India. S.M.A. was supported by a BINC fellowship from the Department of Biotechnology (DBT), India. We acknowledge the support provided by the Council of Scientific and Industrial Research (CSIR), India, to G.U. for the sequencing at the Centre for Cellular and Molecular Biology (CCMB), India. We acknowledge the Duke Lemur Center for collecting primate samples. This is Duke Lemur Center publication #1560. Samples from Amazônia, Brazil, were accessed under SisGen no. A8F3D55. Aotus azarae samples from Argentina were obtained with grant support to E.F.D. from the Zoological Society of San Diego, the Wenner-Gren Foundation, the L.S.B. Leakey Foundation, the National Geographic Society, the US National Science Foundation (NSF-BCS-0621020,1232349, 1503753, 1848954; NSF-RAPID-1219368, NSF-FAIN-1952072; NSF-DDIG-1540255; NSF-REU 0837921, 0924352, 1026991) and the US National Institute on Aging (NIA- P30 AG012836-19, NICHD R24 HD-044964-11). E.F.D. thanks the Ministry of Production and the Environment of Formosa Province in Argentina for the research presented here. J.H.S. was supported in part by the NIH under award number P400D024628 - SPF Baboon Research Resource. This research is supported by the National Research Foundation Singapore under its National Precision Medicine Programme (NPM) Phase II Funding (MOH-000588 to P.T. and W.K.L.) and administered by the Singapore Ministry of Health's National Medical Research Council. J.R. is also a Core Scientist at the Wisconsin National Primate Research Center, Univ. of Wisconsin, Madison. K.G. was supported by the Swedish Research Council VR (2020-03398). We acknowledge the institutional support of the Spanish Ministry of Science and Innovation through the Instituto de Salud Carlos III and the 2014-2020 Smart Growth Operating Program, to the EMBL partnership and institutional cofinancing with the European Regional Development Fund (MINECO/FEDER, BI02015-71792-P). We also acknowledge the support of the Centro de Excelencia Severo Ochoa, and the Generalitat de Catalunya through the Departament de Salut, Departament d’Empresa i Coneixement and the CERCA Programme to the institute. The research reported in this manuscript was also funded by the Vietnamese Ministry of Science and Technology’s Program 562 (grant no. DTDL.CN-64/19) to M.D.L..
Competing interests:
Employees of lllumina, Inc. are indicated in the list of author affiliations. Serafim Batzoglou is currently affiliated with Seer, Inc. Heidi L. Rehm receives funding to support rare disease research and tool development from lllumina, Inc. and Microsoft, Inc. Patents related to this work are (1) title: Deep convolutional neural networks to predict variant pathogenicity using three-dimensional (3D) protein structures, filing number US 17/232,056, authors: Tobias Hamp, Kai-How Farh, Hong Gao; (2) title: Transfer learning-based use of protein contact maps for variant pathogenicity prediction, filing No.: US 17/876,481, authors: Chen Chen, Hong Gao, Laksshman Sundaram, Kai-How Farh; (3) title: Multichannel protein voxelization to predict variant pathogenicity using deep convolutional neural networks, filing number US 17/703,935, authors: Tobias Hamp, Kai-How Farh, Hong Gao;(4) title: Transformer language model for variant pathogenicity, filing number US 17/975,536 and US 17/975,547, authors: Jeffrey Ede, Tobias Hamp, Anastasia Dietrich, Yibing Wu, Kai-How Farh. (5) title: Identifying genes with differential selective constraint between humans and nonhuman primates, filing number US 63/294,820, authors: H. G., J. G. Schraiber, K.-H. Farh.
Data and materials availability:
All sequencing data have been deposited at the European Nucleotide Archive under the accession number PRJEB49549. Primate variants and PrimateAI-3D prediction scores are available with a noncommercial license upon request and are displayed on https://primad.basespace.illumina.com. The source code of PrimateAI-3D is accessible via https://github.com/lllumina/PrimateAI-3D and is also archived at https://doi.org/10.5281/zenodo.7738731. To reduce problems with circularity that have become a concern for the field, the authors explicitly request that the prediction scores from the method not be incorporated as a component of other classifiers and instead ask that interested parties employ the provided source code and data to directly train and improve upon their own deep learning models.
REFERENCES AND NOTES
- 1.MacArthur DG et al. , Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 (2014). doi: 10.1038/naturel3127; pmid: 24759409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nussbaum RL, Rehm HL; ClinGen, ClinGen and Genetic Testing. N. Engl. J. Med 373,1379 (2015). pmid: 26430707 [DOI] [PubMed] [Google Scholar]
- 3.Rehm HL et al. , ClinGen—The Clinical Genome Resource.N. Engl. J. Med 372, 2235–2242 (2015). doi: 10.1056/NEJMsrl406261; pmid: 26014595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landrum MJ et al. , ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016). doi: 10.1093/nar/gkvl222; pmid: 26582918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Wu C, Li C, Boerwinkle E, dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat 37, 235–241 (2016). doi: 10.1002/humu.22932; pmid: 26555599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenson PD et al. , The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet 133,1–9 (2014). doi: 10.1007/s00439-013-1358-4; pmid: 24077912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehm HL, Evolving health care through personal genomics. Nat. Rev. Genet 18, 259–267 (2017). doi: 10.1038/nrg.2016.162: pmid: 28138143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiffin N et al. , Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med 19, 1151–1158 (2017). doi: 10.1038/gim.2017.26; pmid: 28518168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caspar SM et al. , Clinical sequencing: From raw data to diagnosis with lifetime value. Clin. Genet 93, 508–519 (2018). doi: 10.1111/cge.l3190; pmid: 29206278 [DOI] [PubMed] [Google Scholar]
- 10.Yang Y et al. , Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312,1870–1879 (2014). doi: 10.1001/jama.2014.14601; pmid: 25326635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.SoRelle JA, Thodeson DM, Arnold S, Gotway G, Park JY, Clinical Utility of Reinterpreting Previously Reported Genomic Epilepsy Test Results for Pediatric Patients. JAMA Pediatr. 173, el82302 (2019). doi: 10.1001/jamapediatrics.2018.2302; pmid: 30398534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah N et al. , Identification of Misclassified ClinVar Variants via Disease Population Prevalence. Am. J. Hum. Genet 102, 609–619 (2018). doi: 10.1016/j.ajhg.2018.02.019; pmid: 29625023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campuzano O et al. , Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 54, 102732 (2020). doi: 10.1016/j.ebiom.2020.102732; pmid: 32268277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards S et al. , Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 17, 405–424 (2015). doi: 10.1038/gim.2015.30; pmid: 25741868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YE, Ki CS, Jang MA, Challenges and Considerations in Sequence Variant Interpretation for Mendelian Disorders. Ann. Lab. Med 39, 421–429 (2019). doi: 10.3343/alm.2019.39.5.421; pmid: 31037860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slatkin M, A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am. J. Hum. Genet 75, 282–293 (2004). doi: 10.1086/423146:pmid: 15208782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundaram L et al. , Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet 50, 1161–1170 (2018). doi: 10.1038/s41588-018-0167-z; pmid: 30038395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sequencing Chimpanzee and Consortium Analysis, Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005).doi: 10.1038/nature04072; pmid: 16136131 [DOI] [PubMed] [Google Scholar]
- 19.Prado-Martinez J et al. , Great ape genetic diversity and population history. Nature 499, 471–475 (2013).doi: 10.1038/naturel2228; pmid: 23823723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan Z et al. , Ancient hybridization and admixture in macaques (genus Macaca) inferred from whole genome sequences. Mol. Phylogenet. Evol 127, 376–386 (2018). doi: 10.1016/j.ympev.2018.03.038; pmid: [DOI] [PubMed] [Google Scholar]
- 21.Liu Z et al. , Genomic Mechanisms of Physiological and Morphological Adaptations of Limestone Langurs to Karst Habitats. Mol. Biol. Evol 37, 952–968 (2020). doi: 10.1093/molbev/msz301; pmid: 31846031 [DOI] [PubMed] [Google Scholar]
- 22.Wang L et al. , A high-quality genome assembly for the endangered golden snub-nosed monkey (Rhinopithecus roxellana). Gigascience 8, giz098 (2019). doi: 10.1093/gigascience/giz098; pmid: 31437279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245 (2020). doi: 10.1038/s41586-020-2876-6; pmid: 33177664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans BJ et al. , Speciation over the edge: Gene flow among non-human primate species across a formidable biogeographic barrier. R. Soc. Open Sci. 4,170351 (2017). doi: 10.1098/rsos.l70351; pmid: 29134059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L et al. , Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat. Genet 48, 947–952 (2016). doi: 10.1038/ng.3615; pmid: 27399969 [DOI] [PubMed] [Google Scholar]
- 26.Osada N, Matsudaira K, Hamada Y, Malaivijitnond S, Testing sex-biased admixture origin of macaque species using autosomal and X-chromosomal genomic sequences. Genome Biol. Evol 13, evaa209 (2021). doi: 10.1093/gbe/evaa209; pmid: 33045051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rylands AB, Mittermeier RA, Primate Behavioral Ecology. (Routledge, 2021), ed. 6, pp. 407–428. [Google Scholar]
- 28.Lek M et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). doi: 10.1038/naturel9057; pmid: 27535533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). doi: 10.1038/s41586-020-2308-7; pmid: 32461654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leffler EM et al. , Revisiting an old riddle: What determines genetic diversity levels within species? PLOS Biol. 10, el001388 (2012). doi: 10.1371/journal.pbio.l001388; pmid: 22984349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estrada A et al. , Impending extinction crisis of the world’s primates: Why primates matter. Sci. Adv 3, el600946 (2017). doi: 10.1126/sciadv.l600946; pmid: 28116351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohta T, Slightly deleterious mutant substitutions in evolution. Nature 246, 96–98 (1973). doi: 10.1038/246096a0; pmid: 4585855 [DOI] [PubMed] [Google Scholar]
- 33.Reich DE, Lander ES, On the allelic spectrum of human disease. Trends Genet 17, 502–510 (2001). doi: 10.1016/S0168-9525(01)02410-6; pmid: 11525833 [DOI] [PubMed] [Google Scholar]
- 34.Sawyer SA, Hartl DL, Population genetics of polymorphism and divergence. Genetics 132,1161–1176 (1992). doi: 10.1093/genetics/132.4.1161; pmid: 1459433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eyre-Walker A, Keightley PD, The distribution of fitness effects of new mutations. Nat. Rev. Genet 8, 610–618 (2007). doi: 10.1038/nrg2146; pmid: 17637733 [DOI] [PubMed] [Google Scholar]
- 36.Fu W et al. , Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2013). doi: 10.1038/naturell690; pmid: 23201682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simons YB, Turchin MC, Pritchard JK, Sella G, The deleterious mutation load is insensitive to recent population history. Nat. Genet 46, 220–224 (2014). doi: 10.1038/ng.2896; pmid: 24509481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Do R et al. , No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet 47,126–131 (2015). doi: 10.1038/ng.3186; pmid: 25581429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albers PK, McVean G, Dating genomic variants and shared ancestry in population-scale sequencing data. PLOS Biol. 18, e3000586 (2020). doi: 10.1371/journal.pbio.3000586; pmid: 31951611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathieson I, McVean G, Demography and the age of rare variants. PLOS Genet. 10, el004528 (2014). doi: 10.1371/journal.pgen.1004528; pmid: 25101869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Damaj L et al. , CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet 23,1505–1512 (2015). doi: 10.1038/ejhg.2015.21; pmid: 25735478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reinson K et al. , Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am. J. Med. Genet. A 170, 2173–2176 (2016). doi: 10.1002/ajmg.a.37678; pmid: 27250579 [DOI] [PubMed] [Google Scholar]
- 43.Bentivegna A et al. , Rubinstein-Taybi Syndrome: Spectrum of CREBBP mutations in Italian patients. BMC Med. Genet 7, 77 (2006). doi: 10.1186/1471-2350-7-77; pmid: 17052327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stef M et al. , Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur. J. Hum. Genet 15, 843–847 (2007). doi: 10.1038/sj.ejhg.5201847;pmid: 17473832 [DOI] [PubMed] [Google Scholar]
- 45.Kondrashov AS, Sunyaev S, Kondrashov FA, Dobzhansky-Muller incompatibilities in protein evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 14878–14883 (2002). doi: 10.1073/pnas.232565499; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jordan DM et al. , Identification of cis-suppression of human disease mutations by comparative genomics. Nature 524, 225–229 (2015). doi: 10.1038/naturel4497; pmid: 26123021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samocha KE et al. , A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950 (2014). doi: 10.1038/ng.3050; pmid: 25086666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bustamante CD, Wakeley J, Sawyer S, Hartl DL, Directional selection and the site-frequency spectrum. Genetics 159,1779–1788 (2001). doi: 10.1093/genetics/159.4.1779; pmid: 11779814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang X et al. , Inferring genome-wide correlations of mutation fitness effects between populations. Molecular Biology and Evolution. 38, 4588–4602 (2021). doi: 10.1093/genetics/159.4.1779: pmid: 11779814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD, Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLOS Genet. 5, el000695 (2009). doi: 10.1371/journal,pgen.1000695; pmid: 19851460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jouganous J, Long W, Ragsdale AP, Gravel S, Inferring the Joint Demographic History of Multiple Populations: Beyond the Diffusion Approximation. Genetics 206,1549–1567 (2017). doi: 10.1534/genetics.ll7.200493; pmid: 28495960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bates D, Machler M, Bolker B, Walker S, Fitting Linear Mixed-Effects Models Using Ime4. J. Stat. Softw 67,1–48 (2015). doi: 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- 53.Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289–300 (1995). doi: 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 54.Rowntree RK, Harris A, The phenotypic consequences of CFTR mutations. Ann. Hum. Genet 67, 471–485 (2003). doi: 10.1046/j.l469-1809.2003.00028.x; pmid: 12940920 [DOI] [PubMed] [Google Scholar]
- 55.Wilcox SA et al. , High frequency hearing loss correlated with mutations in the GJB2 gene. Hum. Genet 106, 399–405 (2000). doi: 10.1007/s004390000273; pmid: 10830906 [DOI] [PubMed] [Google Scholar]
- 56.Shu H et al. , The role of CD36 in cardiovascular disease. Cardiovasc. Res (2020). doi: 10.1002/humu.l0041; pmid: 33210138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bobadilla JL, Macek M Jr., Fine JP, Farrell PM, Cystic fibrosis: A worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum. Mutat 19, 575–606 (2002). doi: 10.1002/humu.l0041; pmid: 12007216 [DOI] [PubMed] [Google Scholar]
- 58.Chaleshtori MH et al. , High carrier frequency of the GJB2 mutation (35delG) in the north of Iran. Int. J. Pediatr. Otorhinolaryngol 71, 863–867 (2007). doi: 10.1016/j.ijporl.2007.02.005; pmid: 17428550 [DOI] [PubMed] [Google Scholar]
- 59.Liu J et al. , Distribution of CD36 deficiency in different Chinese ethnic groups. Hum. Immunol 81, 366–371 (2020). doi: 10.1016/j.humimm.2020.05.004; pmid: 32487483 [DOI] [PubMed] [Google Scholar]
- 60.Aitman TJ et al. , Malaria susceptibility and CD36 mutation. Nature 405, 1015–1016 (2000). doi: 10.1038/35016636;pmid: 10890433 [DOI] [PubMed] [Google Scholar]
- 61.Common JE, Di W-L, Davies D, Kelsell DP, Further evidence for heterozygote advantage of GJB2 deafness mutations: A link with cell survival. J. Med. Genet 41, 573–575 (2004). doi: 10.1136/jmg.2003.017632; pmid: 15235031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D'Adamo P et al. , Does epidermal thickening explain GJB2 high carrier frequency and heterozygote advantage? Eur. J. Hum. Genet 17, 284–286 (2009). doi: 10.1038/ejhg.2008.225; pmid: 19050724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schroeder SA, Gaughan DM, Swift M, Protection against bronchial asthma by CFTR delta F508 mutation: A heterozygote advantage in cystic fibrosis. Nat. Med 1, 703–705 (1995). doi: 10.1038/nm0795-703; pmid: 7585155 [DOI] [PubMed] [Google Scholar]
- 64.Pier GB et al. , Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature 393, 79–82 (1998). doi: 10.1038/30006; pmid: 9590693 [DOI] [PubMed] [Google Scholar]
- 65.Bojesen SE et al. , Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet 45, 371–384, 384el-2 (2013). doi: 10.1038/ng.2566; pmid: 23535731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heidenreich B, Kumar R, TERT promoter mutations in telomere biology. Mutat. Res. Rev. Mutat. Res 771,15–31 (2017). doi: 10.1016/j.mrrev.2016.11.002; pmid: 28342451 [DOI] [PubMed] [Google Scholar]
- 67.Frazer J et al. , Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021). doi: 10.1038/s41586-021-04043-8; pmid: 34707284 [DOI] [PubMed] [Google Scholar]
- 68.Chae HD, Jeon CH, Peutz-Jeghers syndrome with germline mutation of STK11. Ann. Surg. Treat. Res 86, 325–330 (2014). doi: 10.4174/astr.2014.86.6.325; pmid: 24949325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hernan I et al. , De novo germline mutation in the serine-threonine kinase STK11/LKB1 gene associated with Peutz-Jeghers syndrome. Clin. Genet 66, 58–62 (2004). doi: 10.1111/j.0009-9163.2004.00266.x; pmid: 15200509 [DOI] [PubMed] [Google Scholar]
- 70.Nakanishi C et al. , Germline mutation of the LKB1/STK11 gene with loss of the normal allele in an aggressive breast cancer of Peutz-Jeghers syndrome. Oncology 67, 476–479 (2004). doi: 10.1159/000082933; pmid: 15714005 [DOI] [PubMed] [Google Scholar]
- 71.Yang HR, Ko JS, Seo JK, Germline mutation analysis of STK11 gene using direct sequencing and multiplex ligation-dependent probe amplification assay in Korean children with Peutz-Jeghers syndrome. Dig. Dis. Sci 55, 3458–3465 (2010). doi: 10.1007/sl0620-010-1194-5; pmid: 20393878 [DOI] [PubMed] [Google Scholar]
- 72.Jumper J et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). doi: 10.1038/S41586-021-03819-2; pmid: 34265844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Varadi M et al. , AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. (2021). doi: 10.1093/nar/gkabl061; pmid: 34791371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Söding J, Biegert A, Lupas AN, The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–8 (2005). doi: 10.1093/nar/gki408; pmid: 15980461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Källberg M et al. , Template-based protein structure modeling using the RaptorX web server. Nat. Protoc 7,1511–1522 (2012). doi: 10.1038/nprot.2012.085; pmid: 22814390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang S, Li W, Liu S, Xu J, RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 44, W430–5 (2016). doi: 10.1093/nar/gkw306; pmid: 27112573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burgess DJ, The TOPMed genomic resource for human health. Nat. Rev. Genet 22, 200 (2021). doi: 10.1038/s41576-021-00343-x; pmid: 33654294 [DOI] [PubMed] [Google Scholar]
- 78.Taliun D et al. , Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). doi: 10.1038/s41586-021-03205-y; pmid: 33568819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bycroft C et al. , The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). doi: 10.1038/s41586-018-0579-z; pmid: 30305743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sudlow C et al. , UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 12, el001779 (2015). doi: 10.1371/journal.pmed.1001779; pmid: 25826379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Devlin J, Chang M-W, Lee K, Toutanova K, BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding in Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers) (Association for Computational Linguistics, 2019), pp. 4171–4186. [Google Scholar]
- 82.You Y et al. , in International Conference on Learning Representations. (2020). [Google Scholar]
- 83.Rao RM, Liu J, Verkuil R, Meier J, Canny J, Abbeel P, Sercu T, Rives A, MSA Transformer in Proceedings of the 38th International Conference on Machine Learning, pp. 8844–8856 (2021). [Google Scholar]
- 84.Liu X, Li C, Mou C, Dong Y, Tu Y, dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12,103 (2020). doi: 10.1186/S13073-020-00803-9; pmid: 33261662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deciphering Developmental Disorders Study, Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015). doi: 10.1038/naturel4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deciphering Developmental Disorders Study,, Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaplanis J et al. , Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762 (2020). doi: 10.1038/s41586-020-2832-5; pmid: 33057194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.An JY et al. , Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576 (2018). doi: 10.1126/science.aat6576;pmid: 30545852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Rubeis S et al. , Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). doi: 10.1038/naturel3772; pmid: 25363760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.lossifov I et al. , The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). doi: 10.1038/naturel3908; pmid: 25363768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.lossifov I et al. , De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012). doi: 10.1016/j.neuron.2012.04.009; pmid: 22542183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanders SJ et al. , Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). doi: 10.1016/j.neuron.2015.09.016; pmid: 26402605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sanders SJ et al. , De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012). doi: 10.1038/naturel0945; pmid: 22495306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O'Roak BJ et al. , Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). doi: 10.1038/naturel0989; pmid: 22495309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin SC et al. , Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49,1593–1601 (2017). doi: 10.1038/ng.3970; pmid: 28991257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Araya CL et al. , A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. U.S.A. 109,16858–16863 (2012). doi: 10.1073/pnas.l209751109; pmid: 23035249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chiasson MA et al. , Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact. eLife 9, e58026 (2020). doi: 10.7554/eLife.58026; pmid: 32870157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jia X et al. , Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am. J. Hum. Genet 108, 163–175 (2021). doi: 10.1016/j.ajhg.2020.12.003; pmid: 33357406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matreyek KA et al. , Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet 50, 874–882 (2018). doi: 10.1038/s41588-018-0122-z; pmid: 29785012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mighell TL, Evans-Dutson S, O'Roak BJ, A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am. J. Hum. Genet 102, 943–955 (2018). doi: 10.1016/j.ajhg.2018.03.018; pmid: 29706350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Newberry RW, Leong JT, Chow ED, Kampmann M, DeGrado WF, Deep mutational scanning reveals the structural basis for a-synuclein activity. Nat. Chem. Biol 16, 653–659 (2020). doi: 10.1038/s41589-020-0480-6;pmid: 32152544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seuma M, Faure AJ, Badia M, Lehner B, Bolognesi B, The genetic landscape for amyloid beta fibril nucleation accurately discriminates familial Alzheimer’s disease mutations. eLife 10, e63364 (2021). doi: 10.7554/eLife.63364; pmid: 33522485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.A. 0. Giacomelli et al. , Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet 50, 1381–1387 (2018). doi: 10.1038/s41588-018-0204-y; pmid: 30224644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Starita LM et al. , Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 200, 413–422 (2015). doi: 10.1534/genetics.ll5.175802; pmid: 25823446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jones EM et al. , Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. eLife 9, e54895 (2020). doi: 10.7554/eLife.54895; pmid: 33084570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Amorim CEG et al. , The population genetics of human disease: The case of recessive, lethal mutations. PLOS Genet. 13, el006915 (2017). doi: 10.1371/journal.pgen.l006915; pmid: 28957316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quintáns B, Ordóñez-Ugalde A, Cacheiro P, Carracedo A, Sobrido MJ, Medical genomics: The intricate path from genetic variant identification to clinical interpretation. Appl. Transl. Genomics 3, 60–67 (2014). doi: 10.1016/j.atg.2014.06.001; pmid: 27284505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martin AR et al. , PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet 51, 1560–1565 (2019). doi: 10.1038/S41588-019-0528-2; pmid: 31676867 [DOI] [PubMed] [Google Scholar]
- 109.Thormann A et al. , Flexible and scalable diagnostic filtering of genomic variants using G2P with Ensembl VEP. Nat. Commun 10, 2373 (2019). doi: 10.1038/S41467-019-10016-3;pmid: 31147538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuderna LF et al. , A global catalog of whole-genome diversity from 233 primate species bioRxiv 2023.05.02.538995 [Preprint] (2023); doi: 10.1101/2023.05.02.538995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.C. A. Cassa et al. , Estimating the selective effects of heterozygous protein-truncating variants from human exome data. Nat. Genet 49, 806–810 (2017). doi: 10.1038/ng.3831; pmid: 28369035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tyner C et al. , The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017). pmid: 27899642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kent WJ et al. , The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). doi: 10.1101/gr.229102; pmid: 12045153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH; UniProt Consortium, UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015). doi: 10.1093/bioinformatics/btu739; pmid: 25398609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sali A, Blundell TL, Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815 (1993). doi: 10.1006/jmbi.l993.1626; pmid: 8254673 [DOI] [PubMed] [Google Scholar]
- 116.Pasumarthi RK et al. , TF-Ranking: Scalable TensorFlow Library for Learning-to-Rank. Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2970–2978 (2019). [Google Scholar]
- 117.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH, UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23,1282–1288 (2007). doi: 10.1093/bioinformatics/btm098; pmid: 17379688 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.