

Abstract
Drugs acting at the opioid receptor family are clinically used to treat chronic and acute pain, though they represent the second line of treatment behind GABA analogs, antidepressants and SSRI’s. Within the opioid family mu and kappa opioid receptor are commonly targeted. However, activation of the mu opioid receptor has side effects of constipation, tolerance, dependence, euphoria, and respiratory depression; activation of the kappa opioid receptor leads to dysphoria and sedation. The side effects of mu opioid receptor activation have led to mu receptor drugs being widely abused with great overdose risk. For these reasons, newer safer opioid analgesics are in high demand. For many years a focus within the opioid field was finding drugs that activated the G protein pathway at mu opioid receptor, without activating the β-arrestin pathway, known as biased agonism. Recent advances have shown that this may not be the way forward to develop safer analgesics at mu opioid receptor, though there is still some promise at the kappa opioid receptor. Here we discuss recent novel approaches to develop safer opioid drugs including efficacy vs bias and fine-tuning receptor activation by targeting sub-pockets in the orthosteric site, we explore recent works on the structural basis of bias, and we put forward the suggestion that Gα subtype selectivity may be an exciting new area of interest.
Keywords: Opioid, Mu, Kappa, Gα-subtype bias, Selectivity, Efficacy
1. Introduction
Chronic pain affects millions of people, in fact, the CDC estimates that in 2021 20.9% of adults in the United States (U.S.) experienced chronic pain [1]. The primary medications to combat this are agonists acting at the mu opioid receptor (MOR), however, this comes with its own issues as, whilst MOR activation gives potent analgesia, side effects include tolerance, dependence, constipation, euphoria and respiratory depression [2–4]. The combination of these side effects gives MOR agonists a high abuse liability and risk of overdose, which has directly contributed to the opioid epidemic in the U.S. at present [5].
On a molecular level, activation of opioid receptors triggers conformational rearrangements that lead to interactions with G protein heterotrimers and subsequent activation and dissociation of the G protein subunits. Following G protein activation, the receptor is phosphorylated and β-arrestins are recruited, leading to receptor internalization [6].
Work in the early 2000′s where β-arrestin was reported to mediate the unwanted side effects of MOR activation [7–9] shifted the opioid field towards the investigation of agonists that activate MOR through the G protein pathway without activating β-arrestin, known as bias or functional selectivity. After two decades of research, there are many contradictory findings on putative G protein biased MOR agonists and in fact, the “β-arrestin mediates negative side effects” paradigm has been refuted in several studies [10–12]. Now, the field is shifting once again, with new promising works focusing on bias at the kappa opioid receptor (KOR), low efficacy at MOR leading to a wider therapeutic window [13], [14], engagement of the sodium binding site using bitopic ligands and bias between Gα subtypes. Here, we discuss new developments in the opioid field with a focus on efficacy and bias at MOR and KOR.
2. Is bias or intrinsic efficacy more important for determining the safety profile of opioid agonists?
In this review we will refer to residues using Ballesteros-Weinstein numbering of residues [15], shown in superscript numbers; Ballesteros-Weinstein numbering allows for comparison of residues between transmembrane (TM) domains. The first number refers to the TM, the second number is how close the residue is to the most conserved residue in the TM, which is given the number 50.
2.1. Comparing bias and intrinsic efficacy
A series of studies conducted in the early 2000′s showed that, in mice lacking β-arrestin2, morphine induced lower levels of constipation and respiratory depression whilst retaining analgesic effects [7–9]. These studies lead to the hypothesis that β-arrestin mediated the “bad” side effects of MOR drugs whilst the G protein pathway mediated the “good” effects. Since then, the opioid field has been dominated by the search for MOR G protein biased agonists on the hypothesis that preferential activation of the G protein pathway at MOR would lead to safer analgesics. However, in more recent years this hypothesis has been challenged by multiple independent studies, which found that in the absence of β-arrestin2 the negative side effects of MOR agonists remain[10–12]. In light of this, the field has shifted to other avenues to determine safter MOR analgesics, one such idea is that lower efficacy drugs represent a safer route to MOR mediated analgesia.
Efficacy is the ability of an agonist to activate a receptor and produce a response, after binding [16]. The idea that low efficacy can determine a safer side effect profile at MOR has been studied using agonists with a range of intrinsic efficacy [13]. In vitro signalling was explored to determine possible bias then analgesia and respiratory depression was determined in vivo. Decreasing efficacy has been shown to correlate with an increasing therapeutic window, indicating that MOR agonists with lower efficacy will have less overdose liability whilst maintaining analgesic effects [13]. This study suggested that bias was not in fact related to therapeutic window, however, it should be noted that the calculations behind this study have been challenged, with the suggestion that a degree of G protein bias is required for an improved side effect profile [17].
Mixed MOR/KOR partial agonists MP1207 and MP1208 display decreased efficacy for G protein activation and minimal β-arrestin recruitment; in vivo, these molecules lead to analgesia with less respiratory depression and condition placed preference/aversion (CPP/CPA) relative to full efficacy ligands [18]. This gives further evidence that low efficacy is key for a safer side effect profile of opioids, however,t must be noted that polypharmacology and not bias alone may play a role here.
At the other end of the efficacy scale, a MOR superagonist, isotonitazene, has also been investigated recently and may provide clarity on the action of super potent, highly efficacious MOR agonists [19]. Isotonitazene, whilst not biased, does show extreme potency and efficacy for MOR G protein and β-arrestin2 pathways, relative to DAMGO. In a whole animal, isotonitazene induced greater respiratory depression with a longer duration of action, compared to DAMGO and fentanyl. This supports the idea that higher efficacy agonists have a narrower therapeutic window.
2.2. New low efficacy agonists and putative G protein biased agonists
An analog of mitragynine [20], derivative SC13, has been shown to have 60% efficacy in a Gαi1 MOR BRET assay, relative to DAMGO, and 10% efficacy in an unamplified Nb33 assay measuring activate state human and mouse MOR, with minimal β-arrestin1/2 efficacy [21]. In mice, SC13 displayed dose dependent antinociception, with similar potency to morphine; at ED80 doses SC13 displayed no CPP/CPA, at ED80 doses SC13 displayed no inhibition of gastrointestinal passage and at 15-fold higher than ED50 doses SC13 displayed no respiratory depression or hyperlocomotion. An analog of 7-hydroxymitragynine (7-OH) mitragynine (a mitragynine derivative), 11-F-OH, was tested in a cAMP and Nb33 BRET assay [22], 11-F-OH was found to have < 10% efficacy for Nb33 recruitment relative to DAMGO and ~20% efficacy for mouse MOR inhibition of cAMP accumulation. In mice, 11-F-OH displayed analgesia with similar potency as 7-OH and morphine, but with dramatically lower efficacy (~40%). These data on SC13 and 11-F-OH point to a narrow window whereby MOR efficacy around 10–20% can lead to morphine-like analgesia without unwanted side effects, but a decrease below 10% efficacy leads to a greatly attenuated analgesic effect.
The study of MOR G protein biased agonists is perhaps made more complicated by the lack of clarity on putative G protein biased agonists; well-known agonists PZM21, TRV130 and SR-17018 all display very little β-arrestin2 recruitment, but their level of G protein efficacy seems to change between different studies which makes it unclear if these agonists are indeed G protein biased or if it is purely their low efficacy leading to decreased β-arrestin recruitment [13], [23–27]. The range of efficacy values for these drugs is likely impacted by the assay used, the level of amplification and the receptor reserve[28]. In a recent study of TRV130, receptor reserve was depleted with pre-incubation with irreversible antagonist β-funaltrexamine, and TRV130 was revealed to be a partial agonist for cAMP accumulation [29]. PZM21 and TRV130 have been shown to display analgesia in vivo with lower levels of respiratory depression relative to morphine and fentanyl [23,26], however in another study, PZM21 was suggested to depress respiration to the same level as morphine [25], PZM21 has also been suggested to have a safer effect profile on account of low intrinsic efficacy [13]. TRV130, also referred to as oliceradine, has been approved by the food and drug administration (FDA) for intravenous (IV) injection in pain management [30], however, the most recent data from phase III trials does show low incidence of respiratory depression in postsurgical pain patients [31].
Recently, PZM21 has been further investigated using cryo-EM structural analysis whereby the structure of PZM21 bound to mouse MOR (mMOR) in complex with the Gαi1 heterotrimer was solved [32] (PZM21-MOR-Gi-scFv complex; PDB: 7SBF). It was shown that the thiophenylalkyl moiety strongly interacts with the lipophilic vestibules (formed by TM2, TM3 and extracellular loop (ECL) 1) with residues VAL1433.28, ILE1443.29, TRP133ECL1 and ASN1272.63 (Fig. 1a). PZM21 was originally thought to be a key new drug candidate, however, alongside the previously discussed issues, its occupancy in CNS is reported to be moderate when systematically administrated in mice [26] and moreover, this analog is not free from causing tolerance, withdrawal symptoms [33] and abuse liability [34]. Nonetheless, the cryo-EM structure of PZM21 has been used to design novel analogs based on initial molecular modelling. Among the newly proposed compounds, FH210, with a suitable bioisosteric replacement of the thiophenylalkyl moiety to naphthyl substituted acryl amide (Fig. 1c), improves lipophilicity (FH210: logP 4.5 and PZMZ21: clogP 3.2). The corresponding cryo-EM of FH210 (FH210-MOR-Gi-scFv complex; PDB:7SCG) retains the conserved ionic interaction (salt bridge) between the ammonium group and ASP1473.32 and a conventional hydrogen bond within carboxamide NH and TYR3267.43, which is also displayed by PZM21. The most significant observation was the proximity of the phenol hydroxy group to HIS2976.52 as previously detected in MOR bound structures of PZM21 [32], DAMGO [35], BU72 [36] and β-FNA [37]. Apart from the fundamental interactions, the cryo-EM structure of FH210 highlights an extra 31 Å2 contact area within the lipophilic vestibule incorporating TM2, TM3, ECL1 and ECL2 (hydrophobic interaction with naphthyl substituent). This allows additional interactions with ASP216ECL2, CYS217ECL2, TRP133ECL1 and ASN1272.63 (Fig. 1b). Structural analyses with these templates provide innovative information for understanding the binding mechanism of low efficacy, low β-arrestin recruiting agonists and thus open opportunity to design novel safer analgesics.
Fig. 1.
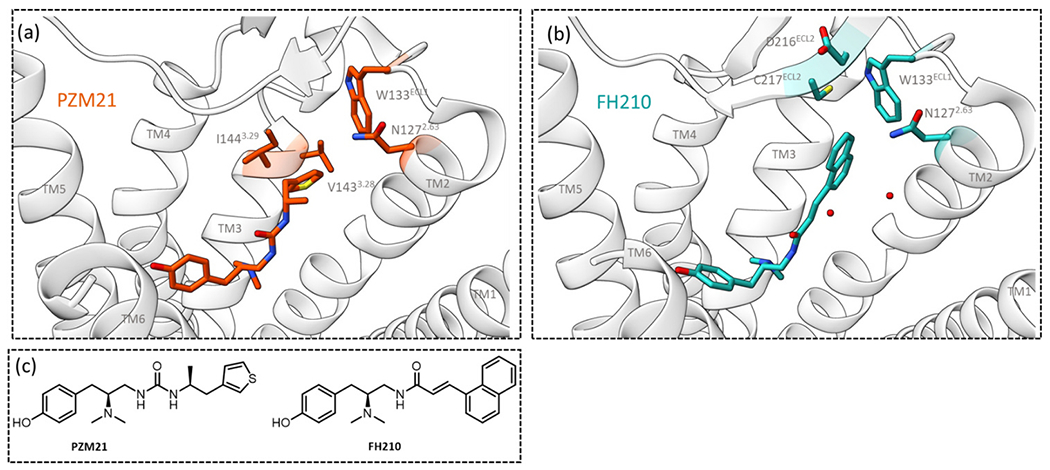
Images of cryo-EM structures of MOR bound to (a) PZM21 and (b) analogue FH210. Key residues interacting with each ligand are highlighted and labelled. TMs have been labelled, TM7 is not shown, two water molecules are included in the FH210 structure. Images were made in ChimeraX from the cryo-EM structures obtained from the protein databank for PZM21 (PDB: 7SBF) and FH210 (PDB: 7SGC). (c) Chemical structures of PZM21 and its modified analog with a naphthyl substituted acryl amide, FH210.
2.3. Is G protein bias at KOR still an important area to explore?
The concept of G protein bias at MOR leading to decreased side effects has been refuted, as negative effects remain in vivo even after the knockout of β-arrestin2 [10–12,38,39]. However, G protein bias and decreased β-arrestin signalling may still remain a viable avenue at KOR. Activation of KOR by full agonist U50,488 has been shown to activate p38 mitogen activated protein kinase (MAPK) via a GPCR kinase (GRK) 3 and β-arrestin dependent pathway [40], subsequently, this KOR activated p38 MAPK pathway has been shown to mediate KOR CPA [41,42]. Further to this, antipruritic effects brought about by KOR agonists have been suggested to act without involvement of the β-arrestin pathway [43], in fact, the G protein biased drug nalfurafine is used clinically in the treatment of itch [44], and shows a promising separation of CPA from KOR therapeutic effects, in animals. Multiple G protein biased KOR drugs have been characterised and investigated in vivo to explore the role of G proteins vs β-arrestins. G protein biased drug RB-64 displays analgesia without sedation or anhedonia-like effects, however the drug still induced CPA, further to this, full agonists U69,593 and salvinorin A both induced CPA in wild type and β-arrestin2 KO mice in this work. 6′-GNTI has been shown to induce potent analgesia in a rat model of thermal allodynia [45] and is a partial agonist at KOR GαoB with an absence of β-arrestin2 recruitment [46], 6′-GNTI shows no CPA in animals. Triazole 1.1 is another KOR G protein biased agonist, which, in mice, induced antinociception and antipruritic effects, without affecting locomotor activity and in rats gave analgesia without dysphoria [47]. HS666 is a KOR G protein biased agonist that gives analgesia without altering locomotor activity or producing CPA [48]. Therefore, G protein bias at KOR may still yield analgesia without negative side effects, but this area needs to be further explored, there are several agonists that can be used as tools to investigate this.
2.4. Bias and Fentanyls
Fentanyl (Fig. 2a) is a potent synthetic MOR agonist, generally used as an adjunct to anesthesiology or in patches and lozenges for breakthrough pain [49–51]. Whilst fentanyl is widely used in a clinical setting, it is also a drug of abuse due to its classic MOR side effects. Fentanyl-related overdose deaths have been increasing sharply in recent years, in the U.S. there were over 70,000 fentanyl-related overdose deaths in 2021 alone [5].
Fig. 2.
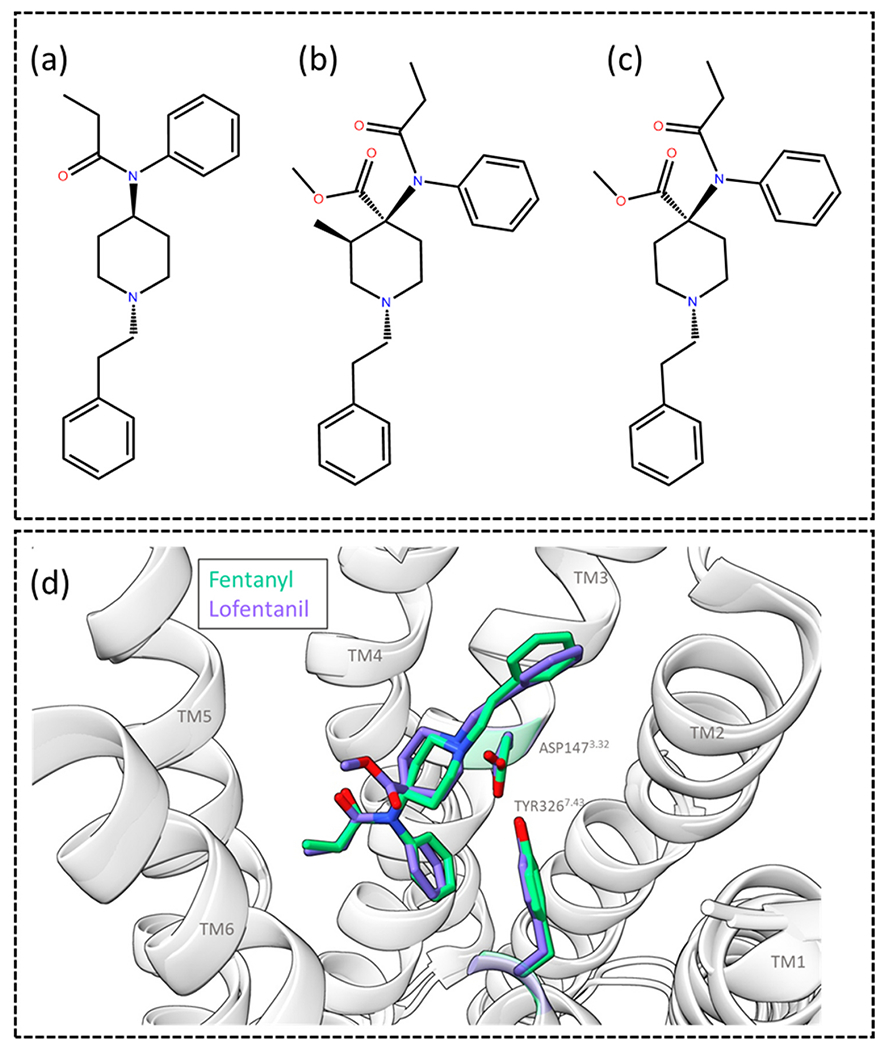
Molecular structures of (a) fentanyl, (b) carfentanil and (c) lofentanil. (d) Image of the superimposed MOR bound fentanyl and lofentanil cryo-EM structures, displaying how the two analogues overlap in the binding site. ASP1473.32 and TYR3267.43 are shown and labelled, the TMs are labelled, the top of TM7 is removed for clarity. Images were made in ChimeraX from the cryo-EM structures obtained from the protein databank for MOR bound to fentanyl (PDB: 8EF5) and lofentanil (PDB: 7T2H).
Due to the risk posed by fentanyl, understanding its pharmacology has become an important area of investigation [52]. Fentanyl is 50–100 times more potent than morphine in vivo [53] and is selective for MOR [54]. A structural basis for fentanyl’s increased potency over morphine has been explored; in a comparison of the cryo-EM structures of fentanyl and morphine bound to human MOR (hMOR), it was observed that fentanyl forms π-π stacking with residues TRP2956.48 and TYR3287.43 and forms additional interactions in a TM2/3 subpocket [27]. In fact, mutation of residues within this subpocket had a greater effect on fentanyl’s potency, compared to morphine [27]. π-π interaction is a strong noncovalent interaction between the π bonds of aromatic rings [55]. This type of van der Waals force usually occur between two or more aromatic rings that are parallel to each other. In proteins amino acids such as phenylalanine, tryptophan and tyrosine can form π- π interactions with aromatic attachment in the ligand [56,57]. These interactions are very imperative for protein structure and protein–ligand binding. The distance between two interacting centroids is 3.5 Å – 4.5 Å [57].
In 2017 fentanyl was first suggested to display bias for β-arrestin2, and this was given as a reason for its increased risk of respiratory depression [58]. However, since then multiple studies have been conducted exploring the potential β-arrestin2 bias of fentanyl, with no general consensus, in fact fentanyl has been shown to have either no bias or even G protein bias in multiple independent studies [13], [21], [59–62]. This wide range of fentanyl pharmacology given by different studies is perhaps due to the stage of the signalling cascade that the signal is read at, degree of amplification, receptor reserve and the method of bias calculation [16], [28], [52]. Fentanyl signalling at a range of Gα subtypes has been studied, fentanyl displays a potency range from 0.9 nM to 19 nM in the order Gαz > Gαi2> GαoA ≈ GαoB > Gαi1 > Gαi3; fentanyl recruited β-arrestin1/2 at a level comparable to DAMGO [24].
Fentanyl has many structural analogs, which each have unique pharmacology and varying degrees of potency and efficacy. One fentanyl analog is particularly interesting due to its extreme potency; carfentanil (Fig. 2b) has been described as a weapon of mass destruction and has been implicated in multiple overdose deaths in the last 10 years [63], [64]. Carfentanil has recently been shown to have potent β-arrestin2 bias and high efficacy for the β-arrestin2 pathway, relative to DAMGO [61]. Carfentanil’s β-arrestin2 bias was shown to lead to potent cell surface receptor loss and rapid desensitization, the in vivo effects of this bias remain to be explored.
Lofentanil (Fig. 2c) is another highly potent fentanyl analogue, lofentanil activates both the G protein and β-arrestin pathways and, in fact, displays bias for β-arrestin1/2 relative to DAMGO and in comparison to Gαi1, Gαi2, Gαi3, GαoA and GαoB [65]. A cryo-EM structure of lofentanil in mMOR has been solved and can be compared to fentanyl; lofentanil forms a salt bridge with ASP1473.32, as well as further interactions with MET1513.36, TRP2936.48, ILE2966.51, ILE3227.39 and TRP3267.43 [65], fentanyl and carfentanil have also been shown to form similar interactions [27], [61]. Lofentanil and fentanyl both bind with their phenethyl moieties in a subpocket made up of TM2/3, and display π-π stacking with TYR3267.43 (Fig. 2d), in the cryo-EM structures residue GLN1242.60 forms an interaction with TYR3267.43. In molecular dynamics (MD) simulations of the lofentanil cryo-EM structure, residue TYR1282.64 also binds and stabilized GLN1242.60, allowing it to interact with TYR3267.43 [65].
2.5. Bias displayed by endogenous opioid peptides
Understanding bias and receptor selectivity on an endogenous level is important as it gives more information on how these phenomena exist naturally. Endogenous peptides such as endomorphin-1, leu-enkephalin, β-endorphin and dynorphin 1–17 display a range of efficacy and potency for Gα subtypes and β-arrestins at MOR (Fig. 3a-d). Dynorphin 1–17 displays low potency or efficacy for β-arrestin1/2 whilst retaining efficacy and potency at a range of Gα subtypes (Fig. 3d) [24].
Fig. 3.
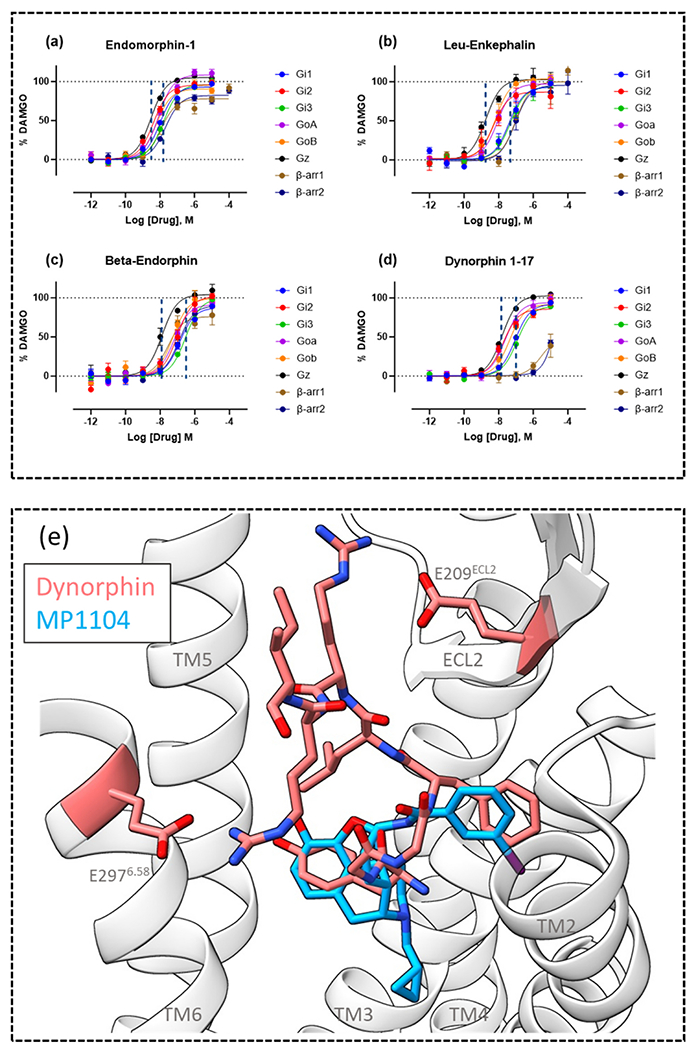
Concentration-response curves of endogenous peptides (a) endomorphin-1, (b) leu-enkephalin, (c) beta-endorphin and (d) dynorphin 1–17. Concentration-response curves were generated from MOR TRUPATH BRET and a MOR β-arrestin1/2 recruitment BRET assay. (e) Image of the superimposed poses of the cryo-EM structures of KOR bound to endogenous peptide dynorphin and small molecule MP1104, ribbons from the dynorphin bound KOR structure are shown, with residues E209ECL2 and E2976.58 highlighted and labelled. TMs are labelled, TM7 has been removed for clarity. Images were made in ChimeraX from the cryo-EM structures obtained from the protein databank for dynorphin (PDB: 8F7W) and MP1104 (PDB: 6B73).
Material from: Faouzi et al. [24].
Bias for different pathways has been determined for several endogenous peptides at opioid receptors; endomorphin-2 has been shown to display β-arrestin2 bias over G protein at MOR [66], this endogenous peptide behaves similarly to β-arrestin2 biased synthetic agonist carfentanil [61] but signals with lower efficacy. Peptide α-neoendorphin displays bias for adenylyl cyclase inhibition over β-arrestin 2 recruitment [67], whilst endomorphin-1 is biased for β-arrestin2 vs β-arrestin1. Bias of endogenous peptides has also been shown as differential based on the opioid receptor subtype, the peptide BAM 18 is unbiased at KOR and DOR but displays β-arrestin2 bias at MOR whilst met-enkephalin RF displays G protein bias at MOR, β-arrestin2 bias at DOR and no bias at KOR [68]. Given that these are all naturally occurring peptides it gives evidence that different degrees of pathway bias at different opioid receptors is an important mechanism for modulating opioid receptor response and helps to explain the presence of many endogenous opioid peptides.
2.6. How do endogenous peptides display opioid subtype selectivity?
In addition to displaying varying degrees of pathway bias, endogenous peptides also display selectivity for opioid receptor subtypes; understanding this phenomenon can help aid the design of future selective opioid ligands. Cryo-EM structures of the opioid receptor family bound by endogenous opioid peptides have been solved [69]. In a comparison of the structure of KOR bound to endogenous peptide dynorphin and the structure of KOR bound to small molecule agonist MP1104 (Fig. 3e), which displays pan opioid activity [18], [70], there were key interactions between dynorphin and KOR that do not occur between MP1104 and KOR. Dynorphin forms salt bridges with residues GLU209ECL2 and GLU2976.58 and mutation of either of these residues leads to a decreased ability of dynorphin to activate KOR. Moreover, when KOR, MOR and DOR were compared, differences in the extracellular regions and at the top of TM6/7 were highlighted as potential selectivity filters between the receptor subtypes [69]. In particular, K3056.58 in MOR was indicated as incompatible with dynorphin binding and mutation to GLU (the residue in position 6.58 in KOR) greatly increased the ability of dynorphin to activate MOR. The reverse of this was also explored, with TYR3137.36 (a TRP in MOR) indicated as creating a steric clash with endomorphin and deltorphin, preventing their binding to KOR. Conversely, there is a conserved hydrophobic region in the orthosteric binding site of MOR, DOR and KOR which allows for activation of all receptor subtypes by the YGGF opioid motif.
3. Structural mechanisms of bias
3.1. Is TM6/7 the key to determining bias?
Bias at opioid receptors has been largely explored in the context of searching for novel G protein biased agonists at MOR or KOR, however, the underlying mechanism of bias is not fully understood. Many studies have been conducted to investigate how bias is mediated on a molecular and structural level.
Multiple studies using MDs have suggested that MOR exists in a different state when bound to biased or unbiased agonists, it has been proposed that the receptor exists in four distinct states during activation and that morphine and TRV130 (putatively G protein biased, though contested) stabilize distinct conformational states [71]. In another study where MDs were generated for agonists that either fully recruit β-arrestin2 or show minimal recruitment, distinct receptor conformations were again observed; agonists that recruit β-arrestin2 displayed inward movements at the top of TM6, outward movements at the top of TM7 and no movement of helix 8, whilst agonists that displayed minimal β-arrestin2 recruitment displayed outward movements at the top of TM6, inward movements at the top of TM7 and rearrangements at helix 8 [72]. Additional MD work indicated that receptor conformation rearrangements were different between morphine and PZM21 [73], which display different levels of β-arrestin recruitment, though both display less than standard agonist DAMGO [24]. Similarly, in another MD study, agonist morphine was compared with TRV130 to find that allosteric coupling between the orthosteric binding site and the intracellular side of the receptor was distinct between the two ligands [74].
One mechanism that has been proposed by multiple groups and shown in multiple class A GPCRs is the differential interaction with TM6 and TM7 by biased and unbiased drugs. In 5-HT2b and β2 adrenoceptor an inward shift in TM7 with minimal TM6 movement has been tied to β-arrestin2 bias [75], [76]. In a study of low efficacy agonist mitragynine pseudoindoxyl (MP) and β-arrestin biased lofentanil, cryo-EM structures of the two ligands were solved and probed to understand lofentanil’s β-arrestin bias [65]. MDs revealed two alternative conformations of TM7; when lofentanil was bound to MOR, TM7 displayed a rotation and inward movement, this was not seen for MP, which recruits minimal β-arrestin [77], these two TM7 (TYR3267.43) conformations were determined to be a canonical and alternative receptor state (Fig. 4). The alternative TM7 conformation was found to be controlled by the formation of a hydrogen bond between GLN1242.60 and TYR3267.43 [65]. Mutation of GLN1242.60A reduces β-arrestin recruitment for both DAMGO and lofentanil, diminishes 100-fold potency for DAMGO, and decreases Gαi1 response more than half for MP.
Fig. 4.
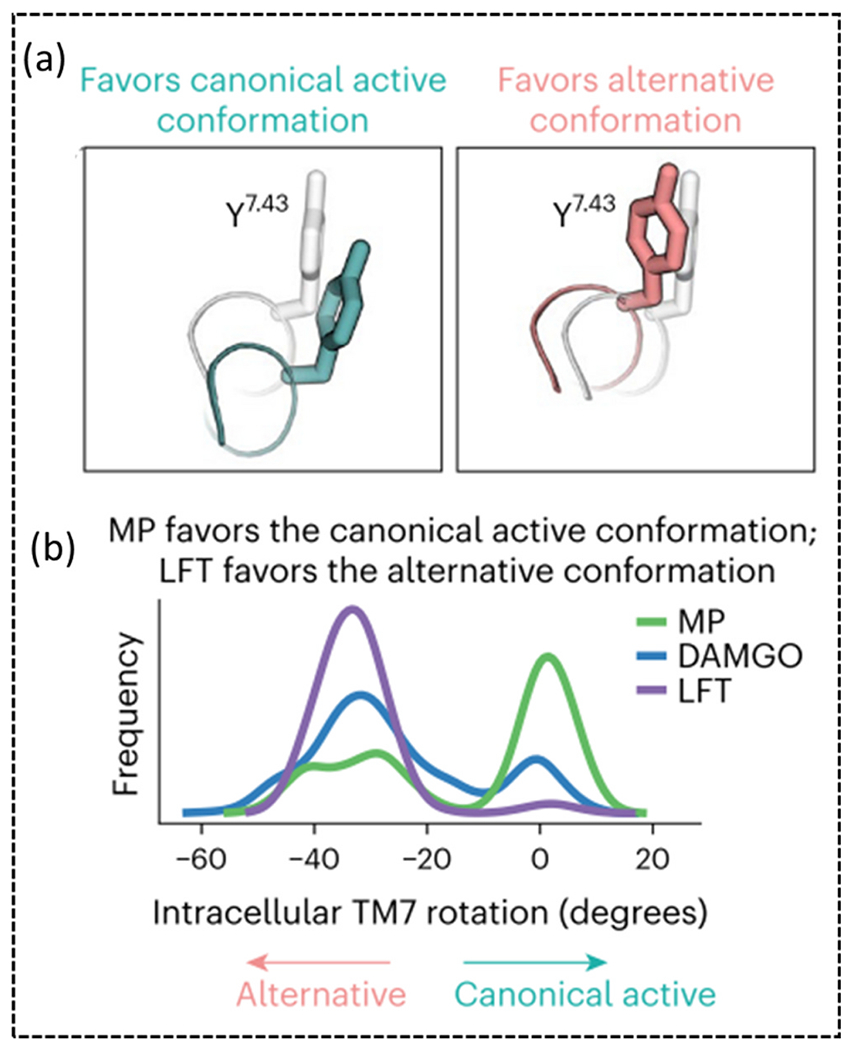
(a) Image of TYR3267.43 in the canonical and alternative conformation. (b) Plot showing the frequency of the intracellular TM7 rotation in MD simulations with MP, DAMGO and lofentanil (LFT).
Material from Qu et al. [65].
Within TM7, the conformation of the residue in position 7.43, which in many class A GPCRs is a tyrosine, has been tied to β-arrestin2 bias in multiple studies [78–80]. Mutation of TYR3267.43 to PHE in MOR has different effects on different agonists; DAMGO and met-enkephalin display a decrease in potency and efficacy for both G protein activation and β-arrestin2 recruitment, whilst endomorphin-1 and endomorphin-2 display increased potency and efficacy for β-arrestin2 recruitment [81]. In the previously mentioned lofentanil MP study, a TYR3267.43PHE mutation reduced the maximal β-arrestin2 recruitment by 94% and 57% for DAMGO and lofentanil, respectively, whilst Gαi1 activation was decreased by 68%, 36% and 13% for MP, DAMGO and lofentanil respectively. The β-arrestin effect is more detectable for DAMGO and MP, where the mutation (TYR3267.43PHE) disrupts a direct H-bond interaction by losing the phenolic OH between ASP1473.32, TYR3267.43 and GLN1242.60. Whereas, for lofentanil the π-π interaction is less harmed, and it is reflected in the corresponding signaling [65]. In KOR, mutation of TYR3207.43 to LEU leads to a decrease in potency of the novel ligand MP1104 for cAMP and a decrease in potency and efficacy for β-arrestin2 recruitment [82]. Together there is compelling evidence that interaction with TYR7.43 has a key involvement in determining β-arrestin signalling.
Other TM7 residues have also been implicated in bias, between receptors MOR and KOR the residue in position 7.35 is different; 7.35 is a TRP and TYR, in MOR and KOR, respectively. A ligand named 3′-iodobenzoylnaltrexamide (IBNtxA [83]) is balanced at KOR but displays G protein bias at MOR; when TYR3127.35 is mutated to TRP in KOR, making it MOR-like, IBNtxA displays G protein bias. This study suggests that the residue in position 7.35 may be involved in the level of β-arrestin2 signalling in MOR and KOR [82].
Single particle Cryo-EM has been used to generate structures of hMOR bound to DAMGO, fentanyl, morphine, SR17018, PZM21 and TRV130 [27]. Comparison of these structures revealed that SR17018 and PZM21, which both display minimal β-arrestin2 recruitment, form no contacts with TM7 and unstable contacts with TM6; TRV130, which retains a small amount of β-arrestin2 recruitment, displays hydrophobic interactions with TM6/7 which are far weaker than those observed for fentanyl. Mutation of residues within TM6/7 resulted in minimal changes in G protein activation, and large decreases in β-arrestin recruitment, further supporting the hypothesis that TM6/7 interactions are required for β-arrestin recruitment [27].
There is mounting evidence that interactions with TM7 and the conformation of TM7 is a key mechanism for determining β-arrestin bias, and to some extent G protein bias, when TM7 is not engaged.
In a study of the mechanism of bias at KOR, a crystal structure of KOR in complex with G protein biased agonist nalfurafine, bound by nb39, was solved. In MDs, GLN1152.60 formed favorable interactions with nalfurafine, and clashes with WMS-X600 (a β-arrestin biased agonist), in vitro mutation of GLN1152.60ASN reduced the potency of nalfurafine but increased the potency of WMS-X600 [84]. Nalfurafine destabilised the bond between LYS227539and GLU2976.58, mutation of LYS2275.39 to ALA increased G protein coupling and reduced β-arrestin recruitment for nalfurafine, suggesting that this bridge is important for β-arrestin recruitment. Further to this, the position of residue TRP2876.48 was shown to be distinct for β-arrestin-biased agonist WMS-X600. Mutation of TRP2876.48 to ALA had a greater effect on the potency of β-arrestin2 recruitment for WMS-X600 compared to U50,488, the same effect was observed for nalfurafine, though this residue has been tied to general receptor efficacy in the past in MOR and in other class A GPCRS [35], [85–89].
4. Targeting receptor sub-pockets to modulate efficacy and bias
4.1. Modulating receptor efficacy and bias by targeting distinct orthosteric sub-pockets
In order to design opioid drugs that may give low efficacy which, as discussed above, could lead to favorable side effect profiles, it is important to understand the mechanisms by which agonists modulate efficacy and bias.
As mentioned in the previous section, a recent study was conducted to compare super potent highly efficacious fentanyl analogue lofentanil (Fig. 5a) and MP, which is a minor metabolite of kratom derived indole alkaloid mitragynine (Fig. 5a). MP has low efficacy for G protein activation, minimal β-arrestin signalling (Fig. 5b) and a subsequently improved pharmacological profile [20], [65], [90]. MP is a key agonist to study as it is reported to exhibit slower antinociceptive tolerance than that of morphine and has reduced respiratory depression at equianalgesic doses when compared with morphine [20], [77]. Lofentanil has been shown to have higher efficacy at all Gα subtypes and β-arrestins compared to MP. In this 2022 study, two cryo-EM structures of lofentanil (Fig. 5c) and MP (Fig. 5d), bound to MOR in complex with a Gαi1 heterotrimer, were reported and compared to a previously published DAMGO MOR structure [35]. Both structurally distinguishable MOR ligands (MP and lofentanil) suitably fit in an identical orthosteric pocket formed by TM2/3/5/6/7. They also preserved the conventional salt bridge interaction between receptor ASP1473.32 and protonatable amine (NH+). Similar to the phenol portion of DAMGO [35], MP and lofentanil share a common binding pocket (central pocket, cp) majorly formed by the TM5-TM6 region; the anilide moiety from lofentanil and β-methoxyacrylate tail from MP are oriented in this common cp (Fig. 5e, f). Lofentanil and MP stabilize the cp by creating hydrophobic interactions with MET1513.36, TRP2936.48, ILE2966.51, ILE3227.39 and TYR3267.43 through anilide and β-methoxyacrylate moieties respectively. However, there were also dissimilar structural arrangements between the ligands, the elongated 1-phenethyl portion of lofentanil orients itself in a hydrophobic subpocket (sp1) formed by TM1/2 and ECL1/2, whilst the low efficacy ligand, MP extends its 9-methoxyaromatic ring of the indole moiety towards an extracellular outlet involving TM1, TM2 and TM7 and thus creates a unique subpocket 2 (sp2) (Fig. 5e, f). The occupation of this sp2 pocket by MP and lack of occupation of the sp1 pocket is believed to be the rationale for MP’s low intrinsic efficacy at G-protein and β-arrestin signaling pathways compared to lofentanil and DAMGO [65]. These findings highlight that MOR ligands that signal with large differences in efficacy may be interacting with distinct orthosteric sub-pockets.
Fig. 5.
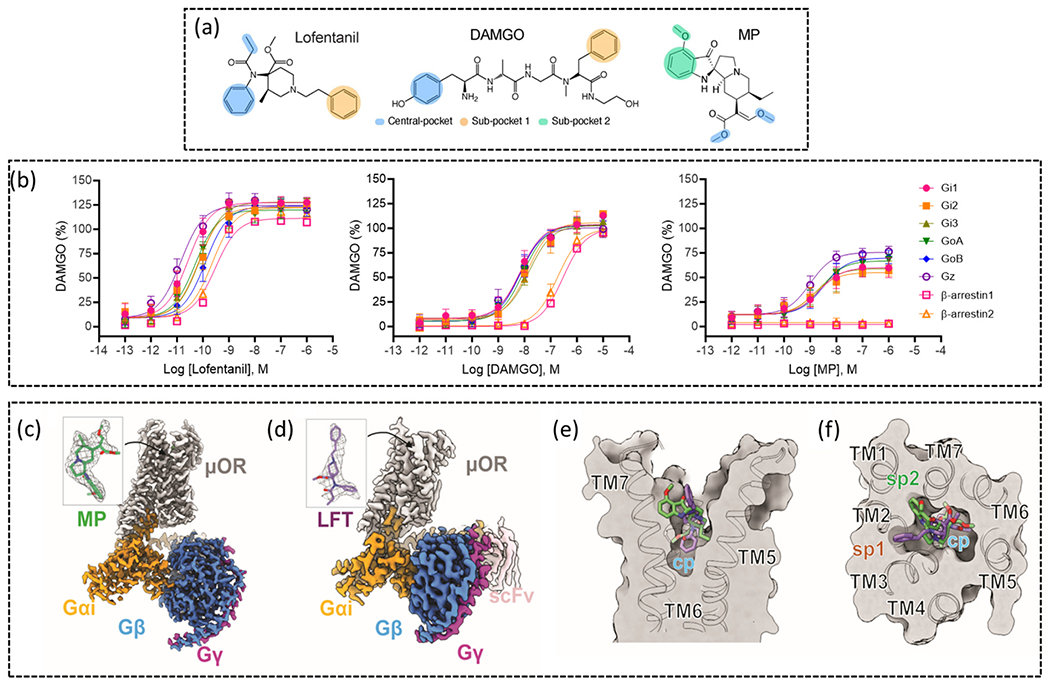
(a) Molecular structures of lofentanil, DAMGO and MP, moieties are colour coded to indicate central and sub-pocket binding. (b) Concentration-response curves of MOR activation by lofentanil, DAMGO and MP, data was generated using TRUPATH BRET and a β-arrestin1/2 BRET assay. (c) Cryo-EM map of MOR bound to MP and in complex with Gαi1βγ (PDB: 7T2G). (d) Cryo-EM map of MOR bound to lofentanil (LFT) in complex with Gαi1βγ stabilized by ScFv16 (PDB: 7T2H). (e) Image showing the side-view of the receptor with MP and LFT occupying the central pocket. (f) Image from the top-view of the receptor showing the overlay of MP and LFT and the occupancy of the two sub-pockets.
Material from Qu et al. [65].
Interaction between ligands and distinct pockets in the orthosteric site leading to differential signalling has also been investigated using mixed MOR/KOR agonists. A novel structure-based hypothesis in functional selectivity was established for putative KOR and MOR biased ligands in the morphinan template. The reported active state crystal structure of KOR with the ligand MP1104 [82] (PDB: 6B73) offers opportunity to better understand the structural differences behind distinguishable pharmacology in biased and unbiased ligands [70], [82]. MP1104 behaves as a full agonist at both hMOR and KOR and robustly engages β-arrestin2 for both (Fig. 6a). Modelling of ligand receptor interaction indicates that the 6β-amidophenyl part orients itself towards the TM2/3 region (Fig. 6e), which is possibly a major switch towards β-arrestin2 recruitment [89]. Next, an analog of MP1104, MP1202 was synthesized; whereby the 7,8 double bind in ring C of the morphinan template was reduced. [18] (Fig. 6d). Unfortunately, MP1202 was incapable of distinguishing G protein recruitment over β-arrestin2 at KOR (Fig. 6b). Through crystallography open database and quantum mechanics energy calculations, it was confirmed that the unsaturated ring of MP1104 is preferred to be in a boat conformation in its co-crystal structure at KOR. Yet more, the docking study in the crystal structures with MP1202 indicates similar orientation, in this way, MP1202 contacts the TM2-TM3 region at KOR. Docking analysis of 6′-GNTI, which is partial agonist in KOR for G protein activation and acts as a potent anticonvulsant and antiseizure candidate [46], [91–93], indicated its engagement at the acidic TM5-ECL2 region via a guanidino group. This TM5-ECL2 region behaves as a distinct sub-pocket at the orthosteric binding site for controlling opioid receptor functional selectivity. Considering the presence of acidic residues ASP2235.35 and GLU209ECL2 in TM5-ECL2 region, the next chemical modification was performed by introducing a polar and/or charged moiety at the 6β-amidophenyl arm (Fig. 6d). Modifications of compound MP1202 to introduce more polar residues such as methylamino and methyl guanidino substitutions generated compounds MP1207 and MP1208 which have partial agonism for KOR alongside greatly reduced β-arrestin recruitment (Fig. 6c). MP1207/08 show effective analgesia with annulated CPP/CPA as well as reduced respiratory depression [18]. Supportively, an MD study of the crystal structure of MP1104 also indicated formation of a stable salt bridge interaction between amino and guanidine moieties with negatively charged ASP2235.35 and GLU209ECL2 in the TM5-ECL2 region, which is achieved through the favored chair conformation (Fig. 6 f, g). Overall, this structure-based drug design points out fundamental involvement of two important sub-pockets in the orthosteric binding site. One of them is the TM2/3 subpocket (hydrophobic vestibule) which can lead to either β-arrestin2 bias or unbiased signaling depending upon receptor variety. The other polar TM5-ECL2 region at the entrance of the orthosteric site is responsible for making ligand G-protein bias by controlling annulated β-arrestin2 recruitment. A similar pattern was seen at MOR as well (not discussed because of lack of space). Overall, these findings provide an inherent mechanistic vista for understanding binding implication in balanced/biased agonism with improved pharmacology.
Fig. 6.
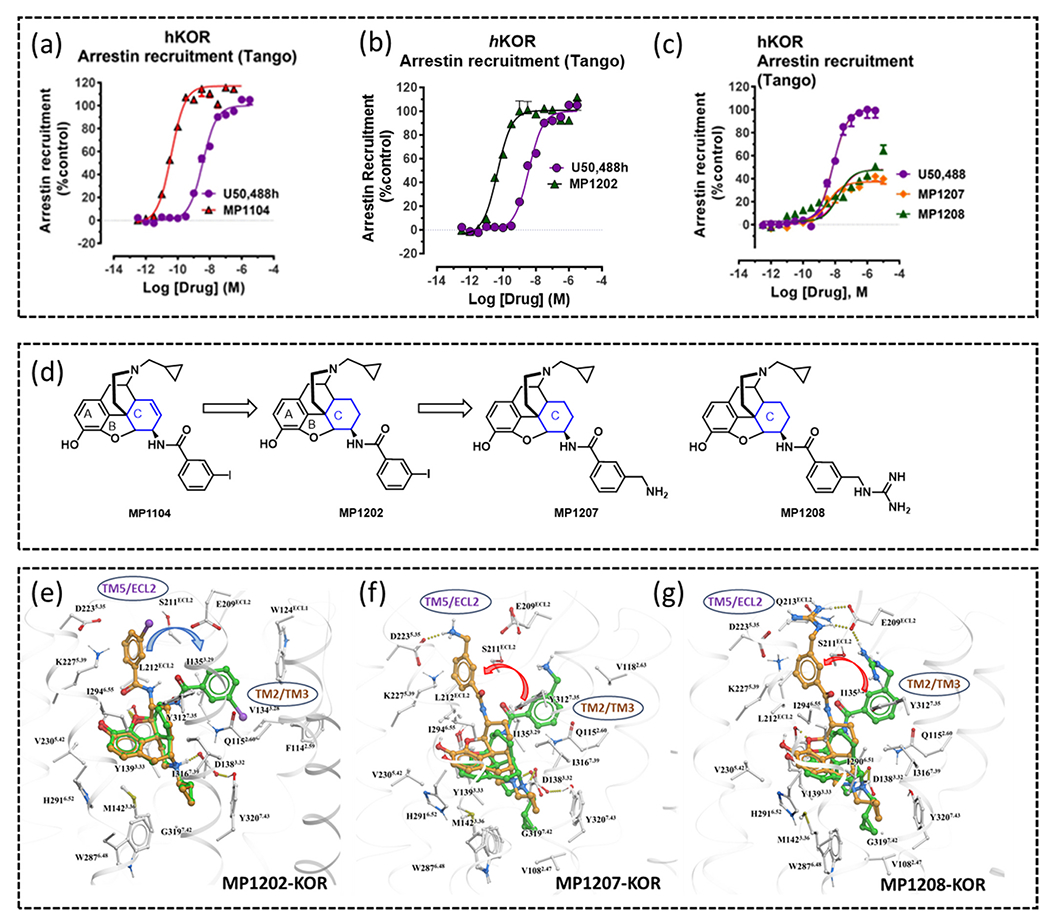
β-arrestin recruitment of (a) MP1104, (b) MP1202 and (c) MP1207/MP1208 measured using a TANGO assay and using human KOR, highlights the decreased β-arrestin recruitment of MP1207/MP1208. (d) Chemical modifications on MP1104 for development of biased analogues. (e) MP1202 ring C favors the boat conformation (green stick) at KOR and resides in a region between TM2-TM3, (f) MP1207 and (g) MP1208 ring C favors a chair conformation (brown stick) at KOR and resides in theTM5-ECL2 region.
Material from Uprety et al. [18].
4.2. Engaging the sodium binding site with bitopic ligands
The role of sodium in allosteric modulation was first documented in 1973 [94]. Binding of a sodium ion (Na+) in class A GPCRs has significant impact for enhancing antagonist binding and reducing agonist binding affinity. Thus, targeting this pocket with suitable ligands attracts the attention for developing low efficacy agonists. The agonist and antagonist distinguishing effect is very much sodium dependent. Lithium for example does not drive the receptor into its inactive form, other mono and divalent ions fail to separate their corresponding opioid activation [95]. During the last decade, progress of several high-resolution crystal structures of different GPCRs and their complexes have enabled the identification and probing of the mechanism of the ion binding site. These include the crystal structures of adenosine A2A receptor (PDB: 4EIY [96], 3EML [97]), β1 adrenoceptor (PDB: 4BVN [98], 4AMJ [99], 2Y02 [100], 3ZPR [101], 5A8E [102]), β2 adrenoceptor (PDB: 2RHJ [103]), protease activated PAR1 (PDB: 3VW7 [104]), delta opioid receptor (PDB: 4N6H [105], 4RWD [106]), and MOR (PDB: 4DKL[37]). These structures disclose a significant role of the binding of a sodium ion in controlling receptor functional selectivity and receptor conformational changes. Interestingly, only the sodium site is found to be conserved in all class A GPCRs and plays a crucial role in ligand binding affinity [107], [108]. A key finding is the presence of conserved aspartic acid residue, ASP2.50 in this water rich allosteric sodium binding pocket. Most interestingly, the mutation of ASP2.50 into either alanine or asparagine eliminates the influential effect of sodium on agonist and antagonist binding. Further studies reveal that this allosteric pocket has significant role in controlling agonist binding affinity, [108–110], mediation of receptor conformation changes [96], and regulation of bias GPCR signaling (towards either G protein or β-arrestin) [105], [111].
A novel approach to modulating receptor activation is the development of bitopic ligands engaging the sodium site in MOR. These bitopic ligands were computationally designed by replacing the phenyl ring of fentanyl with flexible alkyl chain (amide N of fentanyl and carboxy of ASP1142.50 distance is 13 Å) (Fig. 7a). The hypothesis was to occupy the orthosteric site (by the fentanyl scaffold) and to stretch out to the allosteric pocket (through a flexible alkyl linker) and interact with highly conserved acidic residue ASP2.50 through a positively charged guanidino group or amino group [24] (Fig. 7b). Structural activity relationship (SAR) analysis of a small library of compounds in this series suggested the length of the linker as well as appropriate war head (guanidino or amino) was key to engaging the Na+ binding site. The amino warhead containing bitopics were less active compared to the guanidino group-based warhead (Fig. 8a). The greater basicity of the guanidino group compared to the amino group and greater potential to form H-bonding with other polar residues and waters in this pocket was believed to be the reasons for the greater binding affinity as well as potency of the guanidino bitopics. The length of the linker was critical in determining the signaling properties of bitopics. While the C3 alkyl chain was too short, the C11 linker was too long to engage the ASP2.50 residue. Cryo-EM of the C5 and C6 linker-based analogs named C5 guano (Fig. 7c) and C6 guano (Fig. 7f) shed light into the effects of linker length on efficacy and Gα-subtype selectivity. C5 guano was found to have higher efficacy at all Gα-subtypes and no selectivity across subtypes (Fig. 8c). C6 guano on the other hand showed lower efficacy at all subtypes and, unlike established MOR controls, showed lower Gz efficacy (Fig. 8c). Both ligands showed lower efficacy at both β-arrestin subtypes compared to the parent template, fentanyl as well as DAMGO (Fig. 8c). Cryo-EM structures revealed that C5 guano was unable to form a strong salt bridge interaction with ASP2.50, being 4 Å away (Fig. 7d, e), whilst C6 guano formed a strong salt bridge being, 3 Å away from ASP2.50 (Fig. 7g, h). This is suggestive that occupying the Na+site controls G-protein efficacy and Gα-subtype selectivity.
Fig. 7.
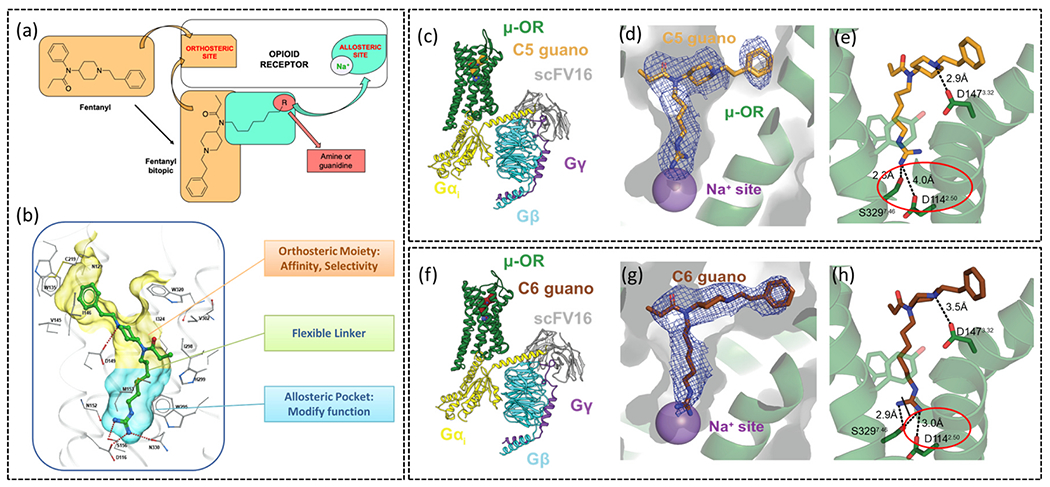
(a) Diagram detailing the design of bitopic ligands on the fentanyl backbone. (b) Diagram of the bitopic ligand occupying the orthosteric and allosteric pockets in MOR. (c) Cryo-EM structure of MOR bound to bitopic ligand C5 guano, in complex with Gαi1βγ stabilised by ScFv16 (PDB: 7U2L) (d) C5 guano reaches the sodium binding site and (e) interacts with residues in the allosteric sodium binding site. (f) Cryo-EM structure of C6 guano bound to MOR, in complex with Gαi1βγ stabilised by ScFv16 (PDB: 7U2K). (g) C6 guano is able to reach the sodium allosteric site and (h) is close to residues in this site.
Material from: Faouzi et al. [24].
Fig. 8.
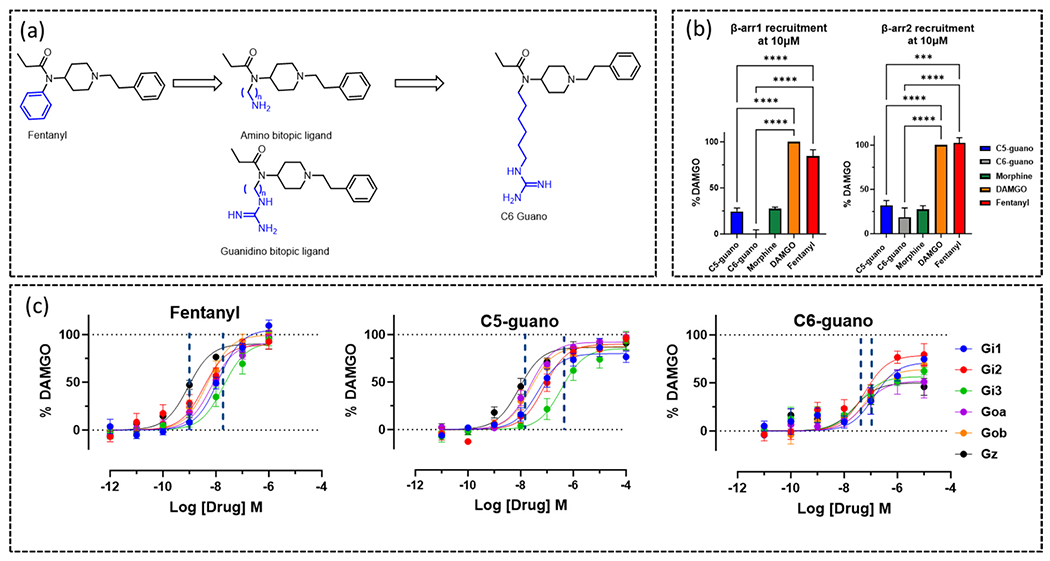
(a) Chemical modification on fentanyl for development of lead bitopic ligands. (b) β-arrestin1 and β-arrestin2 recruitment to MOR measured for 10 μM agonist, using a BRET assay. (c) TRUPATH BRET concentration-response curves of G protein activation at MOR for fentanyl, C5 guano and C6 guano.
Material from: Faouzi et al. [24].
C6 guano displayed high selectivity for specific receptor binding (against DOR, KOR, alpha1A and alpha2A) and for functional assays (against DOR, KOR). C6 guano showed analgesia against multiple pain models, for example neuropathic pain, chronic injury and inflammatory pain. C6 guano has no respiratory depression, unlike morphine, and shows no aversion effects. C6 guano results in no hyperlocomotion when exposed by 300 nmol dose, whereas morphine does initiate from 100 nmol. As we will discuss in the following section, this novel bitopic ligand was able to distinguish not only G protein over β-arrestin1/2, but also displayed a degree of efficacy-based differences when screened at the full range of Gα subtypes.
5. G protein Gα subtype selectivity
5.1. Evidence for Gα subtypes modulating different effects in vivo
An interesting new hypothesis in the opioid field, and indeed at multiple other class A GPCRs, is the idea that selectivity for different Gα subtypes may lead to different effects in vivo and in the case of MOR and KOR could be a new avenue for the development of safer analgesics. MOR, in addition to Gαi1 and Gαi2 (most drugs are screened at these two subtypes) can also couple to Gαi3, GαoS, GαoB and Gαz KOR can couple to these isoforms as well as Gαgustducin. The predominant isoforms in brain are Gαo and Gαz [112–114]. There is some evidence that different Gα subtypes may mediate different effects in vivo; when Gα subtypes were decreased individually by the introduction of antisense oligonucleotides in mice, the analgesic effects of endomorphin-1 and endomorphin-2were most affected by a reduction in Gαi1 and Gαi3, whilst decrease of Gαi2 affected the analgesia induced by DAMGO, morphine and endomorphin-1 [115], [116]. In the same study, Gαz knockout affected analgesia from all agonists studied whilst GαoA/B affected none. Further studies have also shown that different Gα subtypes are important in the supraspinal analgesia brought about by several opioid agonists [117], it has also been suggested that spinal and supraspinal analgesia given by the same ligand may be mediated by different Gα subtypes [118]. Subtype Gαz has been studied by multiple groups; deletion of Gαz in mice has been shown to lead to a reduction in morphine analgesia [119], as well as a development of a greater degree of tolerance compared to morphine with a faster onset of action and decreased lethality [120], [121]. GαoA/B has also been found to be important in analgesia, as a reduction in mice led to decreased supraspinal antinociception in response to morphine, methadone and nalbuphine [122]. These studies focused on analgesia and tolerance, it would be interesting to explore how individual Gα subtypes may play a role in other MOR side effects such as constipation, euphoria and perhaps most importantly, respiratory depression. There is also very little in vivo data on the role of Gα subtypes at KOR as many of these focus on MOR agonists. In addition to work on opioids, Gα subtype selectivity at the adenosine A1 receptor has been explored, the ligand BnOCPA was determined to have high selectivity for GαoB and subsequent analgesia without harmful side effects such as bradycardia and cardiorespiratory depression [123].
5.2. Agonists that activate opioid receptors with Gα subtype selectivity
The study of activation of different Gα subtypes has been made possible by the TRUPATH BRET platform [124] which is now widely used. The ability to test this range has highlighted that many previously characterized drugs do display different efficacy and potency for the range of Gα subtypes. Lower efficacy agonists in particular seem to give a wider range of potency and efficacy for different Gα subtypes [24], [65], [125], presumably due to differences in receptor reserve and efficacy being more evident with partial agonists, particularly when overexpression systems are used.
Novel agonist and mitragynine metabolite 7-OH [90], [126] has been shown to signal at MOR through Gα subtypes with a varying degree of efficacy. 7-OH showed highest efficacy for signaling through Gαz and GαoA with very little efficacy for activation of Gαi1 and Gαi2. When tested in vivo, 7-OH displayed respiratory depression similar to morphine, hyperlocomotion and CPP [24]. The novel bitopic ligand C6 guano (discussed more extensively above) was shown at MOR to display higher efficacy for Gαi1 and Gαi2, in vivo this compound gave antinociception without hyperlocomotion, respiratory depression or CPP/CPA [24].
At KOR, agonists U50,488, nalfurafine, salvinorin A and WMS-X600 all display highest potency at Gαz [84], [125]. Pentazocine has been shown to be a partial agonist at Gαi1 and a full agonist through Gαz, similarly, MOR antagonist naltrexone exhibits very low partial agonism at Gαi1 at KOR but is able to achieve 60% of U50,488 efficacy when signalling through Gαz [125].
5.3. How does one receptor activate Gα subtypes differentially?
The question arises, how is this differential potency and efficacy achieved by agonists signalling through different Gα subtypes at the same receptor? The structural mechanism for this has been explored using cryo-EM structures of KOR in complex with Gαi1, GαoA, Gαz and Gαgustducin, bound by two different ligands, which display similar efficacy but varying potency for the Gα subtypes [127] (Fig. 9a–d). The interaction site between KOR and G proteins is made up of the intracellular side of KOR and the αN and α5 helices of the Gα subunit. GαoA interacted with KOR primarily through the α5 helix whilst Gαi1, GαoA, and Gαgustducin interacted via both α5 and αN, and different positioning of the αN helix was observed (Fig. 9e, f). Residue ARG1563.50 was highlighted as a key residue for the interaction of KOR and G protein, this residue appeared to be most important for the interaction with Gαgustducin. Another residue that is involved in the interaction between KOR and Gα is ASN3368.49 (a helix 8 KOR residue that interacts with α5), which, when mutated to alanine leads to a small rightward shift in potency for Gαi1, GαoA, and Gαz alongside an abolishment in the coupling of Gαgustducin.
Fig. 9.
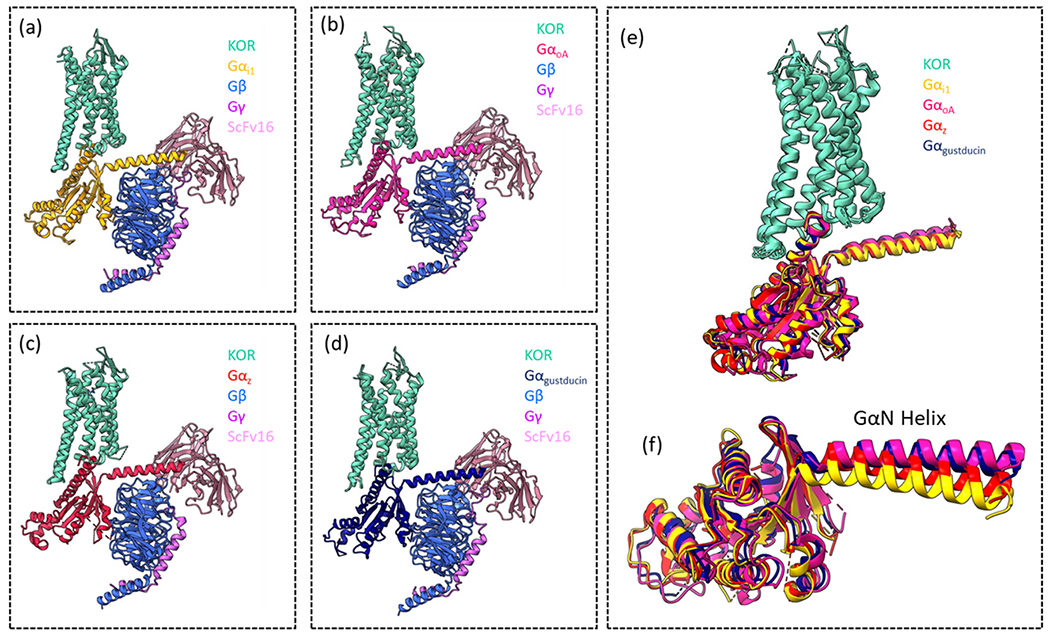
Images showing the cryo-EM structures of KOR in complex with (a) Gαi1βγ (PDB: 8DzP), (b) αoAβγ PDB: 8DZQ), (c) αzβγ (PDB: 8DZQ) and (d) Gαgustducinβγ (PDB: 8DZR). (e) Image of all four structures superimposed, showing only the receptor and Gα subunits, colour-coded. (f) All four Gα subunits superimposed, highlighting the different positioning of the GαN helix.
These structures and observations were first published in Han et al. [127].
5.4. Calculating bias factors from previously published TRUPATH BRET data
For this review, we used previously published concentration-response curves for a range of agonists, where TRUPATH BRET was used to investigate Gα subtypes at MOR, and we calculated bias factors. Data was first re-fit to the Black-Leff operational model [128] to generate log(τ/KA), also known as the transduction ratio. Bias factors were then calculated using DAMGO as a standard agonist (as it activates Gα subtypes through MOR with similar potency), this generated Δlog(τ/KA), all other Gα subtypes were then compared to the activation of MOR through Gαi1 to generate ΔΔlog(τ/KA). This method used for calculating bias is previously published [18], [61], [129], [130].
Interestingly, of the range of agonists we tested, all appeared to have some degree of bias for one or more of the Gα subtypes compared to Gαi1 and relative to DAMGO. Morphine, a prototypical morphinan agonist showed bias for Gα subtypes GαoA and Gαz (Fig. 10a). Carfentanil, which was previously shown to be biased for β-arrestin2 [61], also displayed bias for GαoA relative to DAMGO and compared to Gαi1 (Fig. 10b). We investigated two endogenous peptides, leu-enkephalin and beta-endorphin, leu-enkephalin displayed bias for GαoB, GαoA and Gαz (Fig. 10c) whilst beta-endorphin displayed bias for Gz (Fig. 10d). We also carried out these calculations for two low efficacy agonists, PZM21 and TRV130, which are reputed to display G protein bias over β-arrestin (though this finding has been contested). PZM21 appears to have bias for Gαi2, GαoB and Gαz over Gαil (Fig. 10e) whilst TRV130 has bias for Gαi2 and, of all the agonists studied here, displayed the largest bias for GαoB, GαoA and Gαz (Fig. 10f). This was of particular interest as these putative G protein biased agonists have been tested thoroughly against Gαi subtypes, with far less exploration of Gαo and Gαz effects. These findings highlight the ability of different, structurally distinct agonists, with differing efficacies, to display bias between Gα subtypes (compared to Gαil and relative to DAMGO). The paradigm of G protein vs β-arrestin is far more complex than initially thought, and these calculations highlight that the full array of Gα subtypes should be considered when a new agonist is introduced.
Fig. 10.
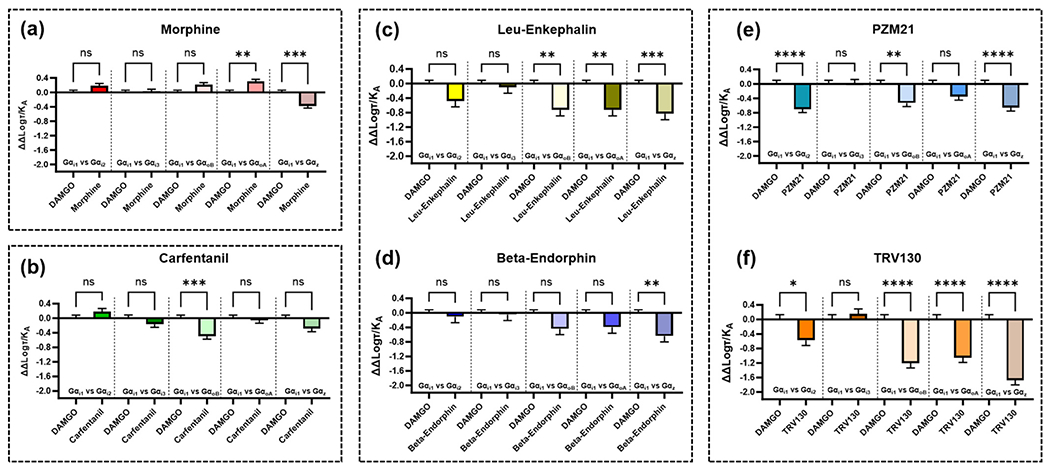
Graphs showing the ΔΔlog(τ/KA) values as calculated by fitting concentration-response curves obtained from Faouzi et al. [24] to the operational model and comparing first to DAMGO and then between Gαil and the other Gα isoforms measured. Bias calculations were carried out for (a) morphine, (b) carfentanil, (c) leu-enkephalin, (d) beta-endorphin, (e) PZM21 and (f) TRV130. Error bars show the pooled SEM for the ΔΔlog(τ/KA) values. Statistical significance was determined using a one-way ANOVA with šídák post-test to compare between each pair as indicated by pairwise comparisons, (ns = no significance, * p < 0.05; ** p < 0.006; *** p < 0.0002; **** p < 0.0001.
6. Future directions and conclusions
Here we have discussed new advances in the field of opioid pharmacology, with a focus on bias and efficacy.
There is growing evidence that G protein bias at MOR may not be as therapeutically revolutionary as first thought [10–12], and instead low efficacy for both the G protein and β-arrestin pathway may be required for safer analgesia when activating MOR [13]. Indeed, many low efficacy MOR agonists have been shown to have favorable effect profiles in vivo [18], [21], [24], [65]. Novel approaches to achieve MOR partial agonism have also been investigated, including engagement of the sodium binding site using bitopic ligands, by which addition of a guanidine linker to potent, widely abused, fentanyl turns it into a partial agonist that displays no respiratory depression or CPP [24]. However, there is evidence that this window for efficacy that provides analgesia without negative side effects may be narrow and efficacy measurements for partial agonists must therefore be thoroughly tested with multiple assays [21], [22]. KOR also presents a possible avenue for designing new safer analgesics, there is still evidence at KOR that bias for the G protein pathway over β-arrestin may lead to analgesia with reduced side effects [40–42], [84].
Perhaps the most interesting new area to be explored recently is Gα subtype specificity, both MOR and KOR activation can lead to signalling through multiple Gα subtypes, at MOR there is evidence that different side effects could be mediated by different Gα subtypes [115–122], though this is not fully elucidated and warrants further investigation. In this review we investigated existing data detailing activation of MOR through the Gα subtype range and we calculated bias between Gα subtypes. Our calculations indicate that several MOR agonists may be displaying previously unknown Gα subtype bias, which brings a new level of complexity to the G protein vs β-arrestin paradigm, and we theorize that, selectivity between individual Gα subtypes may be a new area worth exploring. The evidence that putative G protein biased agonists TRV130 and PZM21 may be signalling with very different potency at different Gα subtypes perhaps adds some clarity on why different groups often find very varied signalling patterns and bias from these agonists, especially when some groups have used assays where specific Gα subtypes (usually Gαi1 or Gαi2) are measured, whilst other groups use downstream assays such as inhibition of cAMP, where the endogenously expressed Gα subtypes are all included.
It is interesting to note that pain is a complex condition; pain can occur acutely, or chronically (lasting more than 3 months) and pain can originate from different sources, for instance neuropathic pain which originates from neural tissue, cancer pain or post-surgical pain [131]. Opioids are often used in many pain situations such as fentanyls used in the clinic for short-acting relief of acute pain [50], or the prescription of opioids for chronic pain, which is considered to be the route of the opioid epidemic in the U.S. Side effects of respiratory depression and euphoria occur rapidly, whilst tolerance and dependence can develop over time [132], and there is evidence that these effects are not all mediated by the same pathway [10–12]. It is therefore important to use these novel approaches to determine new opioid analgesics to decrease the side effect profile in both the context of acute and chronic pain. It is also possible that there may not be a “one fit for all” analgesic that is efficacious in the treatment of all types of pain, it is key to test analgesics in multiple models of acute nociception and chronic pain and there is also merit in developing analgesics for very specific pain contexts. Modulating opioid receptor activation through different Gα subtypes may be a viable method for the generation of analgesics for specific pain contexts as there is evidence that different Gα subtypes are key in the analgesia brought about by existing opioid analgesics [115], [117], the question now is, are these Gα subtypes differentially involved in specific side effects of opioids or even types of pain?
Given the range of novel approaches discussed in this review, it does beg the question, which approach is best? Whilst the approaches do range, from G protein bias at KOR, bitopic ligands engaging the sodium allosteric site, partial agonists, Gα subtype specificity and targeting distinct receptor sub-pockets, there does appear to be a theme of low efficacy. Evidence points to an efficacy window whereby receptor activation of either MOR or KOR, can lead to analgesia with decreased side effects relative to agonists with higher efficacy [13], [18], [21], [24]. However, given the broadness of pain, as discussed above, it is possibly better to have multiple different approaches to this problem, and it may be the case that one approach could be highly efficacious for one pain context and less effective in another. The field is moving away from β-arrestin vs. G protein bias and towards a wide range of novel methods for safer opioid analgesics.
Acknowledgements
SM is supported by NIH grants RO1DA057790RO1 and startup funds from the Department of Anesthesiology, Washington University School of Medicine.
Footnotes
CRediT authorship contribution statement
Nokomis Ramos-Gonzalez: Writing – original draft, Writing – review & editing, Visualization, Formal analysis. Barnali Paul: Writing – original draft, Visualization. Susruta Majumdar: Conceptualization, Writing – review & editing, Visualization, Supervision and Funding.
Declaration of Competing Interest
S.M. is a co-founder of Sparian biosciences. S.M. has patents on mitragynine and related molecules.
References
- [1].Rikard SM, Strahan AE, Schmit KM, Guy GP Jr, Chronic pain among adults — United States, 2019–2021, MMWR Morb. Mortal. Wkly. Rep vol. 72 (2023) 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pasternak G, Childers S, Pan Y, Emerging insights into Mu opioid pharmacology, Handb. Exp. Pharm vol. 258 (2020) 89–125. [DOI] [PubMed] [Google Scholar]
- [3].Pasternak G, Pan Y, Mu opioids and their receptors: evolution of a concept, Pharm. Rev vol. 65 (2013) 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Varga BR, Streicher JM, Majumdar S, Strategies towards safer opioid analgesics-a review of old and upcoming targets, Br. J. Pharm vol. 180 (7) (2023) 975–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].CDC, National Center for Health Statistics. U.S. overdose deaths in 2021 increased half as much as in 2020 – but are still up 15%. [Google Scholar]
- [6].Al-Hasani R, Bruchas MR, Molecular mechanisms of opioid receptor-dependent signaling and behaviour, Anesthesiology vol. 115 (6) (2011) 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Raehal K, Walker J, Bohn L, Morphine side effects in beta-arrestin 2 knockout mice, J. Pharm. Exp. Ther vol. 314 (2005) 1195–1201. [DOI] [PubMed] [Google Scholar]
- [8].Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG, Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence, Nature vol. 408 (2000) 720–723. [DOI] [PubMed] [Google Scholar]
- [9].Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT, Enhanced morphine analgesia in mice lacking beta-arrestin 2, Science vol. 286 (1999) 2495–2498. [DOI] [PubMed] [Google Scholar]
- [10].He L, Gooding S, Lewis E, Felth L, Gaur A, Whisder J, Pharmacological and genetic manipulations at the μ-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice, Neuropsychopharmacology vol. 46 (2021) 2241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kliewer A, et al. , Phosphorylation-deficient G-protein-biased μ opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects, Nat. Comm vol. 10 (367) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kliewer A, et al. , Morphine-induced respiratory depression is independent of β-arrestin 2 signalling, Br. J. Pharmacol vol. 177 (2020) 2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gillis A, et al. , Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists, Sci. Signal vol. 13 (2020). [DOI] [PubMed] [Google Scholar]
- [14].Paul B, Sribhashyam S, Majumdar S, Opioid signaling and design of analgesics, Prog. Mol. Biol. Transl. Sci vol. 195 (2023) 153–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ballesteros JA, Weinstein H, Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors, Methods Neurosci vol. 25 (1995) 366–428. [Google Scholar]
- [16].Kelly E, Efficacy and ligand bias at the μ-opioid receptor, Br. J. Pharmacol vol. 169 (2013) 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stahl E, Bohn L, Low intrinsic efficacy alone cannot explain the improved side effect profiles of new opioid agonists, Biochemistry vol. 61 (2022) 1923–1935, 10.1126/scisignal.aaz3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Uprety R, et al. , Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site, Elife vol. 10 (2021) 1–58, 10.7554/eLife.56519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Malcolm NJ, et al. , Mu-opioid receptor selective superagonists produce prolonged respiratory depression, iScience vol. 26 (7) (2023), 107121, 10.1016/j.isci.2023.107121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chakraborty S, Majumdar S, Natural products for the treatment of pain: chemistry and pharmacology of Salvinorin A, mitragynine, and collybolide, Biochemistry vol. 60 (18) (2021) 1381–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chakraborty S, et al. , A novel mitragynine analog with low-efficacy mu opioid receptor agonism displays antinociception with attenuated adverse effects, J. Med Chem vol. 64 (18) (2021) 13873–13892, 10.1021/acs.jmedchem1cOl273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bhowmik S, et al. , Site selective C–H functionalization of Mitragyna alkaloids reveals a molecular switch for tuning opioid receptor signaling efficacy, Nat. Commun vol. 12 (1) (2021), 10.1038/s41467-021-23736-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].DeWire SM, et al. , A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine, J. Pharm. Exp. Ther vol. 344 (3) (2013) 708–717. [DOI] [PubMed] [Google Scholar]
- [24].Faouzi A, et al. , Structure-based design of bitopic ligands for the μ-opioid receptor, Nature vol. 613 (7945) (2022) 767–774, 10.1038/s41586-022-05588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hill R, et al. , The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception, Br. J. Pharmacol vol. 175 (2018) 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Manglik A, et al. , Structure-based discovery of opioid analgesics with reduced side effects, Nature vol. 537 (2016) 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhuang Y, et al. , Molecular recognition of morphine and fentanyl by the human μ-opioid receptor, Cell vol. 185 (23) (2022) 4361–4375.e19, 10.1016/j.cell.2022.09.041. [DOI] [PubMed] [Google Scholar]
- [28].Kelly E, Conibear A, Henderson G, Biased agonism: lessons from studies of opioid receptor agonists, Annu. Rev. Pharmacol. Toxicol vol. 63 (2023) 491–515, 10.1146/annurev-pharmtox-052120. [DOI] [PubMed] [Google Scholar]
- [29].Singleton S, Baptista-Hon D, Edelsten E, McCaughey K, Camplisson E, Hales T, TRV130 partial agonism and capacity to induce anti-nociceptive tolerance revealed through reducing available μ-opioid receptor number, Br. J. Pharm vol. 178 (2021) 1855–1868. [DOI] [PubMed] [Google Scholar]
- [30].Viscusi ER, A critical review of oliceridine injection as an IV opioid analgesic for the management of severe acute pain, Expert Rev. Neuro Ther vol. 22 (6) (2022) 419–426. [DOI] [PubMed] [Google Scholar]
- [31].Brzezinski M, et al. , Low Incidence of opioid-induced respiratory depression observed with oliceridine regardless of age or body mass index: exploratory analysis from a phase 3 open-label trial in postsurgical pain, Pain. Ther vol. 10 (2021) 457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang H, et al. , Structure-based evolution of G protein-biased μ-opioid receptor agonists, Angew. Chem. - Int. Ed vol. 61 (26) (2022), 10.1002/anie.202200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kudla L, et al. , Functional characterization of a novel opioid, PZM21, and its effects on the behavioural responses to morphine, Br. J. Pharm vol. 176 (23) (2019) 4434–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ding H, et al. , Antinociceptive, reinforcing, and pruritic effects of the G-protein signalling-biased Mu opioid receptor agonist PZM21 in non-human primates, Br. J. Anaesth vol. 125 (4) (2020) 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Koehl A, et al. , Structure of the μ-opioid receptor-Gi protein complex, Nature vol. 558 (7711) (2018) 547–552, 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Huang W, et al. , Structural insights into μ-opioid receptor activation, Nature vol. 7565 (2015) 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Manglik A, et al. , Crystal structure of the μ-opioid receptor bound to a morphinan antagonist, Nature vol. 485 (7398) (2012) 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kelly E, Conibear A, Henderson G, Biased agonism: lessons from studies of opioid receptor agonists, Annu Rev. Pharm. Toxicol vol. 63 (2023) 491–515. [DOI] [PubMed] [Google Scholar]
- [39].Faouzi A, Varga BR, Majumdar S, Biased opioid ligands, Molecules vol. 25 (18) (2020), 10.3390/molecules25184257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bruchas MR, Macey TA, Lowe JD, Chavkin C, Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes, J. Biol. Chem vol. 281 (26) (2006) 18081–18089, 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bruchas MR, et al. , Stress-induced p38 mitogen-activated protein kinase activation mediates κ-opioid-dependent dysphoria, J. Neurosci vol. 27 (43) (2007) 11614–11623, 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Land BB, et al. , Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking, PNAS vol. 106 (45) (2009) 19168–19173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morgenweck J, Frankowski KJ, Prisinzano TE, Aubé J, Bohn LM, Investigation of the role of βarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus, Neuropharmacology vol. 99 (2015) 600–609, 10.1016/j.neuropharm.2015.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Inui S, Nalfurafine hydrochloride to treat pruritus: a review, Clin. Cosmet. Invest. Dermatol vol. 11 (8) (2015) 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Berg KA, et al. , Allosteric interactions between delta- and kappa-opioid receptors in peripheral sensory neurons, Mol. Pharmacol vol. 81 (2012) 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rives ML, Rossillo M, Liu-Chen LY, Javitch JA, 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment, J. Biol. Chem vol. 287 (32) (2012) 27050–27054, 10.1074/jbcCl12.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brust TF, et al. , Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria, Sci. Signal vol. 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Spetea M, et al. , Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice, Br. J. Pharm vol. 174 (15) (2017) 2444–2456, 10.llll/bph.13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Burns SM, Cunningham CW, Mercer SL, DARK classics in chemical neuroscience: Fentanyl, ACS Chem. Neurosci vol. 9 (2018) 2428–2437. [DOI] [PubMed] [Google Scholar]
- [50].Peng PWH, Sandler AN, A review of the use of fentanyl analgesia in the management of acute pain in adults, Anesthesiology vol. 90 (1999) 579–599. [DOI] [PubMed] [Google Scholar]
- [51].Stanley TH, The fentanyl story, J. Pain vol. 15 (12) (2014) 1215–1226. [DOI] [PubMed] [Google Scholar]
- [52].Kelly E, Sutcliffe K, Cavallo D, Ramos-Gonzalez N, Alhosan N, Henderson G, The anomalous pharmacology of fentanyl, Br. J. Pharmacol (2023). [DOI] [PubMed] [Google Scholar]
- [53].Hill R, Santhakumar R, Dewey W, Kelly E, Henderson G, Fentanyl depression of respiration: comparison with heroin and morphine, Br. J. Pharm vol. 177 (2020) 254–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Comer SD, Cahill CM, Fentanyl: receptor pharmacology, abuse potential, and implications for treatment, Neurosci. Biobehav Rev vol. 106 (2019) 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Johnson DW, Hof F, Aromatic interactions: frontiers in knowledge and application. Monographs in Supramolecular Chemistry, The Royal Society of Chemistry, Cambridge, 2017. [Google Scholar]
- [56].McGaughey GB, Gagné M, Rappé AK, π-Stacking interactions, J. Biol. Chem vol. 273 (25) (1998) 15458–15463. [DOI] [PubMed] [Google Scholar]
- [57].Martinez CR, Iverson BL, Rethinking the term “Pi-stacking”, Chem. Sci vol. 3 (7) (2012) 2191. [Google Scholar]
- [58].Schmid CL, Kennedy NM, Ross NC, Cameron MD, Bannister TD, Bohn LM, Bias factor and therapeutic window correlate to predict safer opioid analgesics, Cell vol. 171 (2017) 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Burgueño J, et al. , A complementary scale of biased agonism for agonists with differing maximal responses, Sci. Rep vol. 7 (2017) 153–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crowley RS, Riley AP, Alder AF, Anderson RJ III, Luo D, et al. , Synthetic studies of neoclerodane diterpenes from salvia divinorum: design, synthesis, and evaluation of 322 analogues with improved potency and g-protein activation bias at the μ-opioid receptor, ACS Chem. Neurosci vol. 11 (2020) 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ramos-Gonzalez N, et al. , Carfentanil is a β-arrestin-biased agonist at the μ opioid receptor, Br. J. Pharm (2023), 10.llll/bph.16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rivero G, et al. , Endomorphin-2: a biased agonist at the μ-opioid receptor, Mol. Pharm vol. 82 (2) (2012) 178–188, 10.1124/mol.112.078659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].George AV, Lu JJ, Pisano MV, Metz J, Erickson TB, Carfentanil—an ultra potent opioid, Am. J. Emerg. Med vol. 28 (4) (2010) 530–532. [DOI] [PubMed] [Google Scholar]
- [64].Zawilska JB, Kuczyńska K, Kosmal W, Markiewicz K, Adamowicz P, Carfentanil – from an animal anesthetic to a deadly illicit drug, Forensic Sci. Int vol. 320 (2021). [DOI] [PubMed] [Google Scholar]
- [65].Qu Q, et al. , Insights into distinct signaling profiles of the μOR activated by diverse agonists, Nat. Chem. Biol (2022), 10.1038/s41589-022-01208-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rivero G, et al. , Endomorphin-2: a biased agonist at the μ-opioid receptor, Mol. Pharm vol. 82 (2) (2012) 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Thompson GL, Lane JR, Coudrat T, Sexton PM, Christopoulos A, Canals M, Biased agonism of endogenous opioid peptides at the μ-opioid receptor, Mol. Pharm vol. 88 (2) (2015) 335–346, 10.1124/mol.115.098848. [DOI] [PubMed] [Google Scholar]
- [68].Gomes I, et al. , Biased signaling by endogenous opioid peptides, PNAS vol. 117 (21) (2020) 11820–11828, 10.1073/pnas.2000712117/-/DCSupplemental. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang Y, et al. , Structures of the entire human opioid receptor family, Cell vol. 186 (2) (2023) 413–427.e17, 10.1016/j.cell.2022.12.026. [DOI] [PubMed] [Google Scholar]
- [70].Váradi A, et al. , Synthesis and characterization of a dual kappa-delta opioid receptor agonist analgesic blocking cocaine reward behavior, ACS Chem. Neurosci vol. 6 (11) (2015) 1813–1824, 10.1021/acschemneur-o.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kapoor A, Martinez-Rosell G, Provasi D, De Fabritiis G, Filizola M, Dynamic and kinetic elements of μ-opioid receptor functional selectivity, Sci. Rep vol. 7 (1) (2017), 10.1038/s41598-017-11483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cong X, et al. , Molecular insights into the biased signaling mechanism of the μ-opioid receptor, Mol. Cell vol. 81 (20) (2021) 4165–4175.e6, 10.1016/j.molcel.2021.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Liao S, Tan K, Floyd C, Bong D, Pino MJ, Wu C, Probing biased activation of mu-opioid receptor by the biased agonist PZM21 using all atom molecular dynamics simulation, Life Sci. vol. 269 (. 2021), 10.1016/j.lfs.2021.119026. [DOI] [PubMed] [Google Scholar]
- [74].Schneider S, Provasi D, Filizola M, How oliceridine (TRV-130) binds and stabilizes a μ-opioid receptor conformational state that selectively triggers G protein signaling pathways, Biochemistry vol. 55 (46) (2016) 6456–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, et al. , Structural determinants of 5-HT2B receptor activation and biased agonism, Nat. Struct. Mol. Biol vol. 25 (9) (2018) 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K Biased signaling pathways in 㯂-adrenergic receptor characterized by 19F NMR, Science (1979) vol. 335 (6072) (2012) 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Váradi A, et al. , Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit μ-Arrestin-2, J. Med. Chem vol. 59 (18) (2016) 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Suomivuori C, et al. , Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor, Science vol. 367 (6480) (2020) 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wingler LM, et al. , Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR, Science vol. 367 (6480) (2020) 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Shao Z, et al. , Identification and mechanism of G protein-biased ligands for chemokine receptor CCR1, Nat. Chem. Biol vol. 18 (2022) 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hothersall JD, et al. , Residues W320 and Y328 within the binding site of the μ-opioid receptor influence opiate ligand bias, Neuropharmacology vol. 118 (2017) 46–58, 10.1016/j.neuropharm.2017.03.007. [DOI] [PubMed] [Google Scholar]
- [82].Che T, et al. , Structure of the nanobody-stabilized active state of the kappa opioid receptor, Cell vol. 172 (1–2) (2018) 55–67.e15, 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Grinnell S, et al. , Synthesis and characterization of azido aryl analogs of IBNtxA for radio-photoaffinity labeling opioid receptors in cell lines and in mouse brain, Cell Mol. Neurobiol vol. 41 (5) (2021) 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].El Daibani A, et al. , Molecular mechanism of biased signaling at the kappa opioid receptor, Nat. Commun vol. 14 (1) (2023), 1338, 10.1038/s41467-023-37041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wess J, Nanavati S, Vogel Z, Maggio R, Functional role of proline and tryptophan residues highly conserved among G protein-coupled receptors studied by mutational analysis of the m3 muscarinic receptor, EMBO J. vol. 12 (1) (1993) 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].McAllister S, et al. , An aromatic microdomain at the cannabinoid CB(1) receptor constitutes an agonist/inverse agonist binding region, J. Med. Chem vol. 46 (24) (2003) 5139–5152. [DOI] [PubMed] [Google Scholar]
- [87].Singh R, Hurst D, Barnett-Norris J, Lynch D, Reggio P, Guarnieri F, Activation of the cannabinoid CB1 receptor may involve a W6 48/F3 36 rotamer toggle switch. J. Pept. Res vol. 60 (6) (2002) 357–370. [DOI] [PubMed] [Google Scholar]
- [88].Holst B, et al. , A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors, J. Biol. Chem vol. 285 (6) (2010) 3973–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sutcliffe K, Henderson G, Kelly E, Sessions R, Drug binding poses relate structure with efficacy in the μ opioid receptor, J. Mol. Biol vol. 429 (12) (2017) 1840–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chakraborty S, et al. , Oxidative metabolism as a modulator of kratom’s biological actions, J. Med. Chem vol. 64 (22) (2021) 16553–16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ho JH, Stahl EL, Schmid CL, Scarry SM, Aubé J, Bohn LM, G protein signaling–biased agonism at the κ-opioid receptor is maintained in striatal neurons, Sci. Signal vol. 11 (542) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].White KL, et al. , The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo, J. Pharmacol. Exp. Ther vol. 352 (1) (. 2015) 98–109, 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zangrandi L, Burtscher J, MacKay JP, Colmers WF, Schwarzer C, The G-protein biased partial κ opioid receptor agonist 6′-GNTI blocks hippocampal paroxysmal discharges without inducing aversion, Br. J. Pharm vol. 173 (11) (2016) 1756–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pert CB, Pasternak G, Snyder SH, Opiate agonists and antagonists discriminated by receptor binding in brain, Science vol. 182 (4119) (1973) 1359–1361. [DOI] [PubMed] [Google Scholar]
- [95].Pert CB, Snyder SH, Opiate receptor binding of agonists and antagonists affected differentially by sodium, Mol. Pharm vol. 10 (6) (1974) 868–879. [Google Scholar]
- [96].Liu W, et al. , Structural basis for allosteric regulation of GPCRs by sodium ions, Science (1979) vol. 337 (6091) (2012) 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Jaakola VP, et al. , The 2.6 angstrom crystal structure of a human A 2A adenosine receptor bound to an antagonist, Science vol. 322 (5905) (2008) 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Miller-Gallacher JL, et al. , The 2.1 Å resolution structure of cyanopindolol-bound B1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor, PLoS ONE vol. 9 (3) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Warne T, Edwards PC, Leslie AGW, Tate CG, Crystal structures of a stabilized B1-adrenoceptor bound to the biased agonists bucindolol and carvedilol, Structure vol. 20 (5) (2012) 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Warne T, et al. , The structural basis for agonist and partial agonist action on a β1 adrenergic receptor, Nature vol. 469 (7329) (2011) 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Christopher JA, et al. , Biophysical fragment screening of the β1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design, J. Med. Chem vol. 56 (9) (2013) 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sato T, et al. , Pharmacological analysis and structure determination of 7-methylcyanopindolol-bound β1-adrenergic receptor, Mol. Pharm vol. 88 (6) (2015) 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Raymond A, et al. , Combined protein construct and synthetic gene engineering for heterologous protein expression and crystallization using gene composer, BMC Biotechnol. vol. 9 (1) (2009), 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zhang C, et al. , High-resolution crystal structure of human protease-activated receptor 1, Nature vol. 492 (7429) (2012) 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Fenalti G, et al. , Molecular control of δ-opioid receptor signalling, Nature vol. 506 (7487) (2014) 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Fenalti G, et al. , Structural basis for bifunctional peptide recognition at human δ-opioid receptor, Nat. Struct. Mol. Biol vol. 22 (3) (2015) 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC, Allosteric sodium in class a GPCR signaling, Trends Biochem. Sci vol. 39 (5) (2014) 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zarzycka B, Zaidi SA, Roth BL, Katritch V, Harnessing ion-binding sites for GPCR pharmacology, Pharm. Rev vol. 71 (4) (2019) 571–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hu X, Wang Y, Hunkele A, Provasi D, Pasternak GW, Filizola M, Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis, PLoS Comput. Biol vol. 15 (1) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M, Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions, Biochemistry vol. 53 (31) (2014) 5140–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Selvam B, Shamsi Z, Shukla D, Universality of the sodium ion binding mechanism in class A G-protein-coupled receptors, Angew. Chem. Int. Ed vol. 57 (12) (2018) 3048–3053. [DOI] [PubMed] [Google Scholar]
- [112].Hinton D, Blanks J, Fong H, Casey P, Hildebrandt E, Simons M, Novel Localization of a G protein, gaz in neurons of brain and retina, J. Neurosci vol. 70 (8) (1990) 2763–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Fong H, Yoshimoto K, Eversole-Cire P, Simon M, Identification of a GTP-binding protein alpha subunit that lacks an apparent ADP-ribosylation site for pertussis toxin, Proc. Natl. Acad. Sci. USA vol. 85 (9) (1988) 3066–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Asano T, Semba R, Kamiya N, Ogasawara N, Kato K, Go, a GTP-binding protein: immunochemical and immunohistochemical localization in the rat, J. Neurochem vol. 50 (1988) 1164–1169. [DOI] [PubMed] [Google Scholar]
- [115].Sanchez-Blazquez PS, et al. , Endomorphin-1 and Endomorphin-2 show differences in their activation of opioid receptor-regulated G proteins in supraspinal antinociception in mice 1, J. Pharm. Exp. Ther vol. 291 (1) (1999) 12–18. [PubMed] [Google Scholar]
- [116].Raffa R, Martinez R, Connelly C, G-protein antisense oligodeoxyribonudeotides and mu-opioid supraspinal antinociception, Eur. J. Pharm vol. 258 (1994) 5–7. [DOI] [PubMed] [Google Scholar]
- [117].Sénchez-Blézquez P, Gó Mez-Serranillos P, Garzó J, Agonists determine the pattern of G-protein activation in-opioid receptor-mediated supraspinal analgesia, Brain Res. Bull vol. 54 (2) (2001) 229–235. [DOI] [PubMed] [Google Scholar]
- [118].Standifer KM, Rossi GC, Pasternak GW, Differential blockade of opioid analgesia by antisense oligodeoxynucleotides directed against various G protein a subunits, Mol. Pharm vol. 50 (1996) 293–298. [PubMed] [Google Scholar]
- [119].Yang J, et al. , Loss of signaling through the G protein, G z, results in abnormal platelet activation and altered responses to psychoactive drugs, PNAS vol. 97 (18) (2000) 9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hendry IA, et al. , Hypertolerance to morphine in G-deficient mice, Brain Res. vol. 870 (2000) 10–19. [DOI] [PubMed] [Google Scholar]
- [121].Leek KJ, et al. , Deletion of guanine nucleotide binding protein αz subunit in mice induces a gene dose dependent tolerance to morphine, Neuropharmacology vol. 46 (6) (2004) 836–846, 10.1016/j.neuropharm.2003.ll.024. [DOI] [PubMed] [Google Scholar]
- [122].Lamberts JT, Jutkiewicz EM, Mortensen RM, Traynor JR, Mu-opioid receptor coupling to gα o plays an important role in opioid antinociception, Neuropsychopharmacology vol. 36 (10) (2011) 2041–2053, 10.1038/npp.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wall MJ, et al. , Selective activation of Gαob by an adenosine A1 receptor agonist elicits analgesia without cardiorespiratory depression, Nat. Commun vol. 13 (1) (2022), 10.1038/s41467-022-31652-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Olsen RHJ, DiBerto JF, English JG, et al. , TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome, Nat. Chem. Biol vol. 16 (2020) 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Barnett ME, Knapp BI, Bidlack JM, Unique pharmacological properties of the kappa opioid receptor signaling through Gaz as shown with bioluminescence resonance energy transfer, Mol. Pharm vol. 98 (4) (2020) 462–474, 10.1124/mol.120.119404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kruegel A, et al. , 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects, ACS Cent. Sci vol. 5 (6) (2019) 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Han J, et al. , Ligand and G-protein selectivity in the κ-opioid receptor, Nature (2023), 10.1038/s41586-023-06030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Black JW, Leff P, Operational models of pharmacological agonism. Proc. R. Soc. Lond. Ser. B: Biol. Sci vol. 220 (1219) (1983) 141–162. [DOI] [PubMed] [Google Scholar]
- [129].Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S, A simple method for quantifying functional selectivity and agonist bias, ACS Chem. Neurosci vol. 3 (3) (2012) 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kolb P, et al. , Community guidelines for GPCR ligand bias: IUPHAR review 32, Br. J. Pharm vol. 179 (14) (2022) 3651–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bonezzi C, Fornasari D, Cricelli C, Magni A, Ventriglia G, Not all pain is created equal: basic definitions and diagnostic work-up, Paint. Ther vol. 9 (2020) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Benyamin R, et al. , Opioid complications and side effects, Pain. Physician (2008) 105–120. [PubMed] [Google Scholar]