

Summary
The traditional transition metal catalyzed neutral C(sp)-C(sp2) cross-coupling reaction between a nucleophile and an electrophile is a key technique for the formation of carbon-carbon bonds. Herein, we present a general gold-catalyzed oxidative Sonogashira cross-coupling of arylboronic acids and terminal arylalkynes at room temperature with excellent functional-group tolerance and good chemoselectivity. Moreover, our mechanistic studies suggested a third pathway involving a base-assisted transmetalation between the gold(I) catalyst and aryl boronic acid might predominate in our reaction conditions, rather than the previously assumed oxidation of the gold(I) complex or deprotonation of alkynes.
Subject areas: Chemistry, Chemical reaction, Catalysis
Graphical abstract

Highlights
-
•
Gold-catalyzed oxidative Sonogashira cross-coupling
-
•
Room temperature, excellent functional-group tolerance, and good chemoselectivity
-
•
Base-assisted transmetalation between the gold(I) catalyst and aryl boronic acid
Chemistry; Chemical reaction; Catalysis
Introduction
Transition metal catalyzed cross-coupling reactions of organic electrophiles and organometallic nucleophiles have been proven to be particularly versatile,1,2,3 which is demonstrated in several name reactions.4,5,6,7,8,9,10,11 Biaryl alkynes are a crucial class of building blocks in organic synthesis and a common structural component of pharmaceuticals and organic materials. The Sonogashira reaction between hetero(aryl) halides and terminal alkynes remains one of the most popular cross-coupling reactions for the synthesis of biaryl alkynes (Figure 1A).12,13,14,15,16 However, the high catalytic reactivity of palladium (or nickel) and copper catalysts restrains the tolerance of halogen and organosilyl functional groups in these biaryl alkyne products, and makes it very susceptible to homocoupling upon exposure to air. Aside from the traditional neutral C(sp)-C(sp2) cross-coupling reactions, the oxidative cross-coupling of two nucleophiles represents a valuable alternative but a more challenging reaction manifold.17,18
Figure 1.

Design a new cross-coupling strategy for biaryl alkynes
Over the past two decades, homogeneous gold catalysis has been extensively used to efficiently and selectively activate carbon-carbon π-bonds, in particular acetylenes.19,20,21,22,23 Although the higher standard reduction potential of AuIII/AuI than PdII/Pd0 (1.41 and 0.99, respectively), indicates that the oxidative addition of AuI may be thermodynamically more difficult, the transfer of acetylenes in gold redox catalysis has raised significant interest in recent years.24,25,26,27,28 Instead of π-activation and functionalization of the triple bond of alkynes, these gold-catalyzed alkynylation reactions provide new modes of activation for acetylenes and hold the promise of unique reactivities and selectivities that are different from those of other transition metals (e.g., Pd, Ni, Cu).29,30,31,32,33 The gold-catalyzed oxidative coupling has been widely used to extend the scope of the construction of C-C bonds between coupling partners that are not easily accessed using alternative catalysts (Figure 1B).24,25,26,27,28 The insights into elementary conversions at gold and the mechanistic details of these transformations have attracted tremendous research interest.34,35,36,37 Relevant researchers have developed two common reaction modes for gold-catalyzed oxidative cross-coupling. One starts with the deprotonation of a terminal alkyne or nucleophilic addition to form a functionalized gold(I) species (Figure 1B, path a).30,31,38,39,40 The other is the oxidation of gold(I) into gold(III) with external oxidants as the initial step, which has been demonstrated in many reports (Figure 1B, path b).41,42,43,44,45,46,47 The third reaction mode involves a transmetalation between the gold(I) catalyst and organometallic reagents, which appears in a stoichiometric manner, however, it has been less investigated in a catalytic manner in gold chemistry (Figure 1B, path c).48,49,50,51
In 2019, we reported an Au(I)- and Ir(III)-catalyzed alkynylative cyclization of o-alkylnylphenols with iodoalkynes, and a key benzofuranyl gold(I) complex was formed under base conditions and proposed to promote the oxidative addition to iodoalkynes via a photosensitized energy transfer process.52 An early example of gold-catalyzed oxidative Sonogashira cross-coupling reaction of terminal alkynes with arylboronic acids was reported by Zhang, the O/N functionalized terminal alkyl alkynes and silver catalyst were required in this reaction, and moderate yields of aryl alkynes were obtained.42 They proposed that the active cationic gold(I) species were oxidized by Selectfluor to give a gold(III) species, which was based on previous literature, rather than any mechanistical experiments. Due to the continued interest in gold catalysis and alkyne chemistry,53,54,55,56 herein, we present a gold-catalyzed oxidative Sonogashira cross-coupling of arylboronic acids with terminal arylalkynes at room temperature (Figure 1C). This gold-catalyzed oxidative C(sp)-C(sp2) cross-coupling can synthesize biaryl alkyne with excellent functional-group tolerance for downstream diversification, good chemoselectivity, easily available reagents, and very mild conditions, which provides an alternative pathway to traditional neutral C(sp)-C(sp2) cross-coupling reactions between a nucleophile and an electrophile. Moreover, our mechanistic studies suggest that a third pathway involving a base-assisted transmetalation between the gold(I) catalyst and aryl boronic acid as the initial step takes precedence over previously proposed deprotonation of the terminal alkyne or oxidation of the gold(I) complex. The reactive arylgold(I) species, which was generated from the base-assisted transmetalation, has shown potential utilization, such as in photochemistry. Our work will contribute to the generation of new catalytic transformations through gold catalysis and have new applications in organic synthesis, chemical biology, and materials science.
Results and discussion
We initially selected the readily available 4-fluorophenylacetylene (1a) and phenylboronic acid (2a) for cross-coupling under the catalysis of Ph3PAuCl (Table 1). After substantial optimization of the reaction conditions, it was found that the combination of Ph3PAuCl (10 mol %), Selectfluor (2 equiv.), K2CO3 (2 equiv.), and 0.5 mL of MeOH at room temperature for overnight (18 h) gave the best result since an 87% yield of the desired cross-coupling product 3 was obtained (entry 1). Delightedly, reducing the amount of gold to 5 mol % and MeOH to 0.15 mL could also achieve 86% conversion (entry 2). And it showed that the coupling reaction occurs faster in MeOH than in MeCN (entry 3). Other bases, such as CsF or Cs2CO3, were tested, but the yield was not as high as K2CO3 (entries 4 and 5). When the reaction system was exposed to air, the yield just slightly decreased (entry 6). It was worth noting that the biaryl alkyne 3 could be obtained as 66% when reducing the amount of phenylboronic acid (2a) to 1 equiv (entry 7). Finally, control experiments indicated that the reaction cannot work in the absence of K2CO3, a gold catalyst, or an oxidant (entries 8–10).
Table 1.
Variation of conditions
| Entry | Variation of Conditionsa | Yield of 3 |
|---|---|---|
| 1 | As above | 87% |
| 2 | 5 mol % Ph3PAuCl, 0.15 mL MeOH | 86% |
| 3 | MeCN as solvent | 32% |
| 4 | CsF as base | 43% |
| 5 | Cs2CO3 as base | 63% |
| 6 | In air | 81% |
| 7 | 1 eq. PhB(OH)2 | 66% |
| 8 | No K2CO3 | <5% |
| 9 | No Ph3PAuCl | ND |
| 10 | No Selectfluor | ND |

Reaction conditions: 1a (0.2 mmol), 2a (2 eq.), Ph3PAuCl (10 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.5 mL), 18 h, room temperature, isolated yields, n.d. = not detected.
This novel gold-catalyzed oxidative Sonogashira reaction provides a complementary method for obtaining biaryl alkynes in good to excellent yields at room temperature (Figures 2 and 3). Firstly, the scope of terminal arylalkynes was studied in reaction with phenylboronic acid 2a under optimal reaction conditions (Table 1, entry 1). Terminal arylalkynes bearing halogen groups in the para position, such as F, Cl, Br, and I, delivered the desired products 3–6 with excellent yields. The compatibility of halogen substituents provides a promising opportunity for downstream diversification of the coupling reactions and functional-group installation. Other electron-withdrawing groups, like – CF3 and – CN at the para position, furnished products 7 and 8 in almost stoichiometric yields (94% and 99%). The presence of electron-donating groups, such as methyl, ethyl, and isopropyl groups at the para position, could react with phenylboronic acid 2a to obtain the products in high yields (9–11). The arylalkyne bearing a tert-butyl group at the para-position gave the corresponding product 12 in a lower yield (60%). The exciting result was that trimethylsilane substituted arylalkyne could also be coupled with phenylboronic acid 2a to obtain a high yield (13, 93%), which is difficult for traditional-metal cross-coupling, as the reserved trimethylsilane in the product was a valuable group for further elaboration. Other substitution patterns, like meta or ortho, of the aromatic alkynes also afforded the correspondingly desired biaryl alkynes 14–17 in very good yields. Besides terminal aryl alkynes, alkynes bearing phenoxyl, pyridyl, cyclohexenyl, and cyclohexyl groups (18–21) could also be tolerated with this gold-catalyzed oxidative Sonogashira reaction, albeit in significantly lower yields (20–32%).
Figure 2.

Variation of the terminal alkynes
aGeneral substrate scope with respect to terminal alkynes and boronic acids. Reaction conditions: terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (10 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.5 mL), 18 h, isolated yields; bReaction conditions: terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (5 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.15 mL), 18 h, isolated yields.
Figure 3.

Variation of the boronic acids
aGeneral substrate scope with respect to terminal alkynes and boronic acids. Reaction conditions: terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (10 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.5 mL), 18 h, isolated yields; bReaction conditions: terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (5 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.15 mL), 18 h, isolated yields.
Next, this protocol allows efficient alkynylation with a broad range of boronic acids, which are commercially accessible and widely used reagents in Suzuki-Miyaura coupling (Figure 3). The reaction of para methyl, F, Cl, Br, and I substituted phenylboronic acids with p-trifluoromethyl phenylacetylene could give 58–82% yields (22–26). Arylboronic acids bearing halogen groups, such as F or Br in the meta position (27 and 29), or F both in the para and ortho positions (28), afforded corresponding biaryl alkynes in good to excellent yields. The electron-donating substituents, like the isopropyl group in the para position (30) or two methyl groups in the meta positions (31), on the arylboronic acid, also reacted very smoothly. An excellent yield was obtained again when the trimethylsilane substituted arylboronic acid was subjected to this oxidative Sonogashira reaction (32). A promising feature of this catalytic coupling is that the useful halogen and TMS substituents are compatible with the same products (35–38), which is clearly indicative of a flexible gold-catalyzed alkynylation coupling. Changing the CF3 group of phenylacetylene to the F group, arylboronic acids bearing different substituents at the para- or ortho-position provided the corresponding products in good to excellent yield (39–41). And it was found that this cross-coupling is not suitable for boronic acids bearing pyridinyl units or potassium organotrifluoroborates (42–43).
Synthetic utilities
To feature the synthetic robustness of gold-catalyzed oxidative cross-coupling, the reaction conditions were first used in the gram-scale synthesis of the biaryl alkyne 3 in a high yield of 79% (Figure 4A). The tolerated substituted group provides possibilities for the diversification of further cross-coupling products. As illustrated, the use of two different gold catalysts in relay allows for easy access to SCF3 substituted biaryl alkyne 44 (Figure 4B),57 and the downstream Suzuki58 and Sonogashira coupling59 cascade of biaryl alkyne can readily be achieved at the halogen substituents (Figures 4C and 4D). The biaryl alkyne product is a valuable scaffold for further elaboration. For instance, the alkyne moiety can be hydrogenated by formic acid under palladium (0) catalysis to Z-alkene 47 or E-isomer 48 (Figures 4E and 4F).60 Compared with traditional transition metals, gold is generally considered to be green and biocompatible, which could be applied in food additives and pharmaceuticals. Retinoids, which contain the biaryl alkyne motif, are a common active ingredient in many biological processes and are used in medicine.61 This gold-catalyzed flexible cross-coupling protocol was utilized for the synthesis of this valuable retinoid analogue 51 in 67% yield (Figure 4G).
Figure 4.

Synthetic utilities
(A) Terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (10 mol %), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.5 mL), 18 h; (B) AgSCF3 (1.05 eq.), (MeDalphos)AuCl (5 mol %), AgSbF6 (0.2 eq.), DCE (1 mL), 70°C, 1 h; (C) B2pin2 (2 eq.), Pd(PPh3)4 (3 mol %), K3PO4 (2 eq.), MeCN (0.1 M), blue LEDs; (D) TMS acetylene (1.2 eq.), Pd(PPh3)2Cl2 (4 mol %), CuI (2 mol %), Et3N (0.25 M), 80°C; (E) HCO2H (2 eq.), Pd(dba)3 (1 mol %), dppb (4 mol %), dioxane (0.2 mL), 80°C; (F) HCO2H (25% aq., 3 eq.), Pd(dba)3 (1 mol %), dppb (2 mol %), dioxane (0.4 mL), 80°C; (G) standard conditions: terminal alkyne (0.2 mmol), boronic acid (2 eq.), Ph3PAuCl (10 mol%), Selectfluor (2 eq.), K2CO3 (2 eq.), MeOH (0.5 mL), 18 h, room temperature, isolated yields.
Mechanistic studies
Having established that the gold-catalyzed oxidative cross-coupling is facile, we undertook investigations into the mechanism, and three reaction pathways were envisaged (Figure 5). Previous literature offered two possible pathways for this gold-catalyzed alkynylation reaction aided by an external oxidant: formation of a gold(I)-acetylide complex Au-I via deprotonation of the terminal alkyne (Figure 5, path a),38,39,40 or oxidation of gold(I) into gold(III) B with Selectfluor (Figure 5, path b).41,42,43,44,45,46,47 In this work, we postulated a third pathway in which a base-assisted transmetalation between the gold catalyst and arylboronic acid (Figure 5, path c) might predominate in this gold-catalyzed oxidative C(sp)-C(sp2) cross-coupling.
Figure 5.

Three possible reaction pathways
During our ongoing mechanistic studies on reactions, stoichiometric experiments with putative reaction intermediates provide several mechanistic clues (Figure 6). According to the three proposed mechanistic pathways of Figure 5, gold(I) acetylide Au-I of the path a and arylgold(I) Au-II of the path c were first prepared in almost quantitative yield under our reaction conditions, as shown in Figure 6A. Another important point is that there will be no reaction in the absence of a base. Subjecting these two gold intermediates to react with another reaction patterner delivered cross-coupling product 39 and side product biarene 2bb, which indicated that both these two gold intermediates (Au-I and Au-II) are likely involved in the reaction mechanism (Figure 5).
Figure 6.

Stoichiometric and NMR monitoring experiments
(A) Synthesis of Au-I and Au-II.
(B) Coupling experiments with Au-I and Au-II.
(C) Formation of dimer.
(D) Competition experiment.
(E) The reaction catalyzed by gold(III) complex.
(F) NMR monitoring experiment.
Then several control experiments were designed to understand the reaction selectivity of the gold catalyst with terminal alkyne and boronic acid. When the individual terminal alkyne 1a or boronic acid 2b was added to the standard conditions but for a shorter time (0.5 h), indeed, the biarene side product 2bb was isolated in a higher yield than that of the diyne side product 1aa (47% vs. 10%, Figure 6C). An intermolecular competition experiment of the gold catalyst with terminal alkyne 1a and boronic acid 2b further confirmed the above result (Figure 6D), in which the formation of arylgold(I) (Au-II) was significantly higher than that of gold(I) acetylide (Au-I) (88% vs. 7% for 2 h), indicating that the gold catalyst Ph3PAuCl might prefer reacting with boronic acid 2b (Figure 5, path c) than terminal alkyne 1a (Figure 5, path a).
Gold has a high standard reduction potential, and the elementary step called oxidative addition bridges the critical oxidative-reductive cycle. If using the gold(III) catalyst Ph3PAuCl3 instead of the gold catalyst Ph3PAuCl and the oxidant Selectfluor, 13% of the desired cross-coupling product would be obtained as expected (Figure 6E).
As three reaction pathways are proposed in the beginning, the NMR monitoring experiment of the gold catalyst with boronic acid and Selectfluor provides a “discrimination effect” to understand the rest of the reaction mechanism. According to the short-time 31P-NMR monitoring spectrum, boronic acid was identified as the only species reacting with the gold catalyst (Figure 6F) and full conversion from Ph3PAuCl to the arylgold(I) complex (Au-II) will take less than 10 min (Figure 6F, spectra c). Thus, reaction path c might predominate over paths a and b in our reaction conditions (Figure 5).
Based on previous literature48,49,50,51 and mechanism investigations, a plausible mechanism involving base-assisted transmetalation is outlined in Figure 7 for this gold-catalyzed oxidative Sonogashira cross-coupling. In line with previous mechanistic studies of the Au(I)/B transmetalation process, by using of inorganic base and alcohol, the in situ formation of an L-Au-OR complex explains the new C(sp2)−Au bond of Au-II. Followed by the oxidation of Selectfluor to form an arylgold(III) intermediate Int-2, deprotonation of the terminal alkyne to Int-3, and reductive elimination leads to the final cross-coupling product.
Figure 7.

Proposed reaction mechanism
Other working systems
The protocol of base-assisted transmetalation of the arylgold(I) complex is not restricted to boronic acid. Other organometallic reagents, such as organosilane, organostannane, organozinc-based nucleophiles, and Grignard reagent, could undergo oxidative cross-coupling, although they showed lower reactivity (Figure 8A, entries 2–5), as homocoupling products predominated in most of these cases. And it was found that the boronic acid pinacol ester compound also afforded a good yield (entry 6), revealing that organoboron compounds may favor cross-coupling to a greater extent.
Figure 8.

Other applications with the arylgold(I) complex
Photochemical gold catalysis has attracted enormous attention in the past decades. It was found that a direct oxidative addition of haloalkynes to the key arylgold(I) complex was established under blue LEDs, without photocatalyst or heating conditions (Figure 8B).52,62 This photosensitized oxidative addition process revealed the potential utilization of the arylgold(I) complex in photochemistry.
Conclusion
In summary, this work describes a facile gold-catalyzed oxidative Sonogashira cross-coupling reaction at room temperature that was triggered by a base-assisted transmetalation step. Compared with traditional transition-metal catalyzed neutral cross-coupling reactions, this gold-catalyzed oxidative cross-coupling provides an alternative reaction manifold to synthesize biaryl alkyne with excellent functional-group tolerance, good chemoselectivity, and readily available reagents. This kind of biaryl alkyne is easily functionalized and can construct useful bioactive molecular complexes, which is reluctant for traditional transition-metal catalyzed coupling reactions. The insight into elementary conversions at gold and the mechanistic details of these transformations is important but challenging work. Previously, deprotonation of alkynes or oxidation of the gold(I) catalyst was often regarded as two common reaction modes for gold-catalyzed oxidative cross-coupling, our mechanistic studies suggested a third pathway involving a base-assisted transmetalation between the gold(I) catalyst and aryl boronic acid might predominate in our reaction conditions. Our work would facilitate the development of gold redox catalytic transformations and promote more unexpected discoveries in the future.
Limitations of the study
This study is limited to arylboronic acids, and further research is needed on suitable reaction systems for alkyl boric acid and borate substrates.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| phenylboronic acid | Energy Chemical | CAS: 98-80-6 |
| 4-fluorophenylacetylene | Meryer | CAS: 766-98-3 |
| Ph3PAuCl | Macklin | CAS: 14243-64-2 |
| Selectfluor | Meryer | CAS: 140681-55-6 |
| K2CO3 | Meryer | CAS: 584-08-7 |
| MeOH | Energy Chemical | CAS: 67-56-1 |
| MeCN | Energy Chemical | CAS: 75-05-8 |
| CsF | Energy Chemical | CAS: 13400-13-0 |
| Cs2CO3 | Energy Chemical | CAS: 534-17-8 |
| AgSCF3 | Energy Chemical | CAS: 811-68-7 |
| AgSbF6 | Energy Chemical | CAS: 26042-64-8 |
| DCE | Energy Chemical | CAS: 107-06-2 |
| K3PO4 | Energy Chemical | CAS: 7778-53-2 |
| MeCN | Energy Chemical | CAS: 75-05-8 |
| Pd(PPh3)4 | Energy Chemical | CAS: 14221-01-3 |
| Pd(PPh3)2Cl2 | Energy Chemical | CAS: 13965-03-2 |
| CuI | Energy Chemical | CAS: 7681-65-4 |
| Et3N | Energy Chemical | CAS: 121-44-8 |
| Pd(dba)3 | Energy Chemical | CAS: 52409-22-0 |
| CDCl3 | Energy Chemical | CAS:865-49-6 |
| Other | ||
| Silica gel (200-300 mesh) | Taiyuan Kangda | |
| thin layer chromatography using TLC silica gel plates | Taiyuan Kangda | |
| AVIII 600 MHz | Bruker | https://bruker.com |
| HRMS | AGILENT | https://www.agilent.com.cn/ |
| Software and algorithms | ||
| ChemDraw Ultra 16.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Zhonghua Xia (zhonghua.xia@bit.edu.cn).
Materials availability
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.
Data and code availability
-
•
This paper does not report original code.
-
•
Data and code generated in gold-catalyzed oxidative Sonogashira coupling reaction are available within the article and the supplemental experimental procedures or from the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.
Experimental model and subject details
This study did not use experimental models typical in life sciences.
Method details
General information
All reactions involving air sensitive reagents or intermediates were carried out in pre-heated glassware under an argon atmosphere using glovebox. All solvents and commercial reagents were used as purchased from Aladdin, Meryer, Energy Chemical or Macklin. Acetonitrile and methanol were purified by distillation over calcium hydride under dry Argon atmosphere.
Chromatographic purifications of products were accomplished using force-flow chromatography (FC) on Davisil (LC60A) SI 60 Å (40 - 63 μm) silica gel. Thin layer chromatography (TLC) was performed on Adsorbent silica gel F254 precoated plates (0.25 mm). Organic solutions were concentrated under reduced pressure using a rotary evaporator (35°C, <50 torr).
All the 1H, 13C, 19F and 31P NMR spectra were recorded on Bruker Avance 600 spectrometers. All NMR spectra were recorded in CDCl3 at room temperature. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker 600 AVANCE spectrometer (600 MHz). Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker 600 AVANCE spectrometer (151 MHz). Fluorine nuclear magnetic resonance (19F NMR) spectra were recorded on a Bruker 600 AVANCE spectrometer (565 MHz). Phosphorus nuclear magnetic resonance (31P NMR) spectra were recorded on a Bruker 600 AVANCE spectrometer (243 MHz). Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3 = 7.26 ppm). NMR data are represented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in Hertz (Hz), integration. 13C NMR spectra are internally referenced to CDCl3 (77.16 ppm). ESI+-spectra were measured on AGILENT Q-TOF 6520 liquid chromatography-mass spectrometry. EI spectra were obtained using an Thermo Fisher Scientific Exactive GC mass spectrometer.
General procedure (GP1)

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: alkyne 1 (0.2 mmol), boronic acid 2 (2 equiv), Ph3PAuCl (10 mol%, 9.9 mg or 5 mol%, 4.9 mg), Selectfluor (2 equiv, 141.7 mg), K2CO3 (2 equiv, 55.3 mg) and methanol (0.15 or 0.5 mL). The vial was capped with a Teflon septum cap and sealed with electrical tape. The reaction vial was removed from the glove box, set to stir (500 rpm) at room temperature for 18 hours. After the reaction was completed, the reaction was extraction with ethyl acetate and evaporated the organic solvent to obtain the crude product. The crude product was purified by column chromatography to afford the desired product.
Limitation and unsuccessful examples
More examples were tested, and, it showed that this gold-catalyzed oxidative Sonogashira reactions has some limitations.

Gram scale experiments

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (1 equiv, 0.8408 g, 7 mmol), phenylboronic acid 2a (2 equiv, 1.7070 g, 14 mmol), PPh3AuCl (10 mol%, 0.3463 g, 0.7 mmol), Selectfluor (2 equiv, 4.9589 g, 14 mmol), K2CO3 (2 equiv, 1.9349 g, 14 mmol) and methanol (17.5 mL). The vial was capped with a Teflon septum cap and sealed with electrical tape. The reaction vial was removed from the glove box, set to stir (500 rpm) at room temperature for 18 hours. After the reaction was completed, the reaction was extraction with ethyl acetate and evaporated the organic solvent to obtain the crude product. The crude product was purified by column chromatography to afford the desired product 3 (79%, 1.084 g).
Synthesis of (4-(Phenylethynyl)phenyl)(trifluoromethyl)sulfane (44)

An 8-mL reaction vial was loaded with MeDalPhosAuCl (6.5 mg, 5 mol%), AgSCF3 (44.1 mg, 1.05 equiv) and 1 mL DCE. 6 (60.8 mg, 0.2 mmol) was added and the resulting mixture was stirred at room temperature for 1 min. AgSbF6 (13.7 mg, 0.2 equiv) was then added and the reaction was stirred at room temperature for 1 min before being heated to 70°C. Upon completion, the reaction mixture was diluted with 2 mL DCM and then filtered, eluted with mixed solvent (hexane/EtOAc). The filtrate was concentrated and purified with flash chromatography using n-hexane to give the desired product 44 (82%, 45.6 mg).
Synthesis of 4,4,5,5-Tetramethyl-2-(4-(phenylethynyl)phenyl)-1,3,2-dioxaborolane (45)

An 8-mL reaction vial was loaded with 5 (51.2 mg, 0.2 mmol, 1.0 equiv), Pd(PPh3)4 (6.9 mg, 3 mol %), bis(pinacolato)diboron (101.6 mg, 2.0 equiv), K3PO4 (84.9 mg, 2.0 equiv) and dry MeCN (2.0 mL). Then, the reaction mixture was then irradiated in reactor with cooling device using LED (two Kessil A360W/X blue LEDs lights, λmax = 460 nm) for 8 hours. After full conversion, the reaction mixture was transferred into a flask and concentrated in vacuum. Purification of the crude product was achieved by flash column chromatography using petroleum n-hexane/ethyl acetate as eluents on silica gel.
Synthesis of Trimethyl((4-(phenylethynyl)phenyl)ethynyl)silane (46)

A reaction vial was charged with Pd(PPh3)2Cl2 (5.6 mg, 4 mol %), CuI (0.8 mg, 2 mol %), and 5 (51.2 mg, 0.2 mmol) in triethylamine (0.8 mL); then, ethynyltrimethylsilane (23.6 mg, 1.2 mmol) were added. The reaction mixture was stirred at 80°C for 24 h. After the starting material was consumed, the mixture was quenched with saturated NH4Cl solution and extracted with ethyl acetate. The combined organic extracts were washed with water and brine, and dried over anhydrous Na2SO4 and concentrated in vacuo. Then the reaction mixture was filtered through a pad of silica gel and washed with diethyl ether. The solvent was evaporated under the reduced pressure to afford 46 (93%, 50.9 mg) as product.
Synthesis of (Z)-1-chloro-4-styrylbenzene (47)

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. The glass vial was charged with Pd2(dba)3 (1.8 mg, 1 mol%), dppb (3.4 mg, 4 mol%) and 4 (42.4 mg, 0.2 mmol). 0.2 mL of dioxane was subsequently injected. After stirring the mixture at room temperature for 15 min, 16 μL of HCO2H was injected and the reaction was heated to 80°C for 15 h. After removal of the solvent under vacuo, the residues were passed through a short silica chromatography (n-hexane as eluent) to afford 47 (86%, 36.8 mg).
Synthesis of (E)-1-chloro-4-styrylbenzene (48)

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. The glass vial was charged with Pd2(dba)3 (1.8 mg, 1 mol%), dppb (1.7 mg, 2 mol%) and 4 (42.4 mg, 0.2 mmol). 0.4 mL of dioxane was subsequently injected. After stirring the mixture at room temperature for 15 min, 48 μL of aqueous HCO2H (25%) was added. The reaction was heated to 80°C for 2 h, and another 48 μL of aqueous HCO2H (25%) was added. The reaction was heated at 80°C for another 8 h. After removal of the solvent under vacuo, the residues were passed through a short silica chromatography (n-hexane as eluent) to afford 48 (88%, 37.6 mg).
Synthesis of Methyl 4-((5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)ethynyl)benzoate (51)

PdCl2 (35.5 mg,10 mol%), Cu(OAc)2 (39.9 mg, 10 mol%), 6-bromo-1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene (0.5344 g, 2 mmol) and PPh3 (0.2623 g, 0.5 equiv) were placed in a 100 mL round-bottom flask under N2. Dry, degassed triethylamine (50 mL) and TMSA (0.2353 g, 1.2 equiv) was added via syringe. After 18 h at 70°C, triethylamine was evaporated and obtain the product 49 (92%, 0.5229 g) as a viscous, pale yellow oil.
To a solution of 49 (0.2842 g, 1 mmol) in methanol (3 ml), K2CO3 (12.2 mg, 9 mol%) was added. After stirring the solution for 4 h, the filtrate was concentrated and further extracted with anhydrous ether and sodium bicarbonate solution. Then, dried over anhydrous Na2SO4 and purified with flash chromatography using n-hexane to give the desired product 50 (94%, 0.2000 g).
Following general procedure GP1 with 50 (42.4 mg, 0.2 mmol) and 4-(methoxycarbonyl)phenylboronic acid 2n (72.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (n-hexane/ ethyl acetate, 30:1) to afford 51 as yellow solid (46.4 mg, 67%).
Mechanistic studies and stoichiometric experiments
Synthesis of Au-I and Au-II

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (12.1 mg, 0.1 mmol) or 4-tolylboronic acid 2b (13.6 mg, 0.1 mmol), PPh3AuCl (49.5 mg, 1 equiv), K2CO3 (27.6 mg, 2 equiv) and MeOH (0.5 mL). The mixture was stirred at room temperature for 18 h. After the reaction, the solvent is removed by rotary evaporation, and the pure product Au-I (96%, 55.5 mg) and Au-II (97%, 53.4 mg) is obtained by recrystallization with n-hexane.
Coupling experiments with Au-I and Au-II

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: Au-I (57.8 mg, 0.1 mmol) and 4-tolylboronic acid 2b (13.6 mg, 0.1 mmol) or Au-II (55.1 mg, 0.1 mmol) and 4-fluorophenylacetylene 1a (12.1 mg, 0.1 mmol), Selectfluor (70.8 mg, 2 equiv), K2CO3 (27.6 mg, 2 equiv) and MeOH (0.5 mL). The mixture was stirred at room temperature for 18 h. After completed, the crude product is purified with flash chromatography using n-hexane to give the coupling product.
Formation of dimer

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (12.1 mg, 0.1 mmol) or 4-tolylboronic acid 2b (13.6 mg, 0.1 mmol), PPh3AuCl (5.0 mg, 10 mol%), Selectfluor (70.8 mg, 2 equiv.), K2CO3 (27.6 mg, 2 equiv.) and MeOH (0.5 mL). The mixture was stirred at room temperature for 0.5 h and 2 h. After quenched, the crude product is purified with flash chromatography using n-hexane as eluent to give the homo-coupling product.
Competition experiment

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (12.1 mg, 0.1 mmol), 4-tolylboronic acid 2b (13.6 mg, 0.1 mmol), PPh3AuCl (49.5 mg, 1 equiv), Selectfluor (70.8 mg, 2 equiv), K2CO3 (27.6 mg, 2 equiv) and MeOH (0.5 mL). Add 1,3,5-trimethoxybenzene (7.2 mg, 0.4 equiv.) as internal standard. At 5, 30, 60 and 120 minutes, a small amount of reaction solution was taken out by syringe and do 1H-NMR analysis. The yield was determined by 1H-NMR spectra.
Reaction Catalyzed by Gold(III) Complex

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (12.1 mg, 0.1 mmol), 4-tolylboronic acid 2b (27.2 mg, 2 equiv), PPh3AuCl3 (56.4 mg, 1 equiv), K2CO3 (55.3 mg, 2 equiv) and MeOH (0.5 mL). The mixture was stirred at room temperature for 18 h. After completed, the crude product is purified with flash chromatography using n-hexane to give the coupling products.

To a solution of PPh3AuCl (100 mg, 0.2mmol) in 2 mL of CH2Cl2 was added a solution of PhICl2 (58 mg, 1.0 equiv) in 2 mL of CH2Cl2. After stirring for 20 h at room temperature in the dark, recrystallization in n-hexane to get Ph3PAuCl3 as a yellow powder. 1H NMR (600 MHz, CDCl3) δ 7.76 - 7.66 (m, 9H), 7.57 (m, 6H). 31P NMR (243 MHz, CDCl3) δ 43.6.
Other working systems
With other organometallic nucleophiles

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: 4-fluorophenylacetylene 1a (24.0 mg, 0.2 mmol), organometallic nucleophiles (2 equiv), PPh3AuCl (9.9 mg, 10 mol%), Selectfluor (141.7 mg, 2 equiv), K2CO3 (55.3 mg, 2 equiv) and methanol (0.5 mL). The vial was capped with a Teflon septum cap and sealed with electrical tape. The reaction vial was removed from the glove box, set to stir (500 rpm) at room temperature for 18 hours. After the reaction was completed, the reaction was extraction with ethyl acetate and evaporated the organic solvent to obtain the crude product. The crude product was purified by column chromatography to afford the coupling product.
Application in Photochemistry

A glass vial equipped with a PTFE-coated stir bar was brought into an Ar-filled glove box. To the reaction vial the following were added successively: Au-II (550.1 mg, 0.1 mmol), 1-fluoro-4-(iodoethynyl)benzene or 1-(bromoethynyl)-4-fluorobenzene (1 equiv), methanol (2 mL). The reaction vial was removed from the glove box, and the vial was capped with a Teflon septum cap. Then, the reaction mixture was irradiated with LED (two Kessil A360W/X blue LEDs lights, λmax = 460 nm) with cooling fan to keep the reaction at room temperature for 18 hours. After the reaction was completed, the reaction was extraction with ethyl acetate and evaporated the organic solvent to obtain the crude product. The crude product was purified by column chromatography to afford the coupling products.
Spectroscopic data
1,4-Bis(4-fluorophenyl)buta-1,3-diyne (1aa)

1H NMR (600 MHz, CDCl3) δ 7.53 - 7.50 (m, 4H), 7.06 - 7.01 (m, 4H).
4,4'-Dimethyl-1,1'-biphenyl (2bb)

1H NMR (600 MHz, CDCl3) δ 7.50 (d, J = 8.1 Hz, 4H), 7.25 (d, J = 7.9 Hz, 4H), 2.41 (s, 6H).
1-Fluoro-4-(phenylethynyl)benzene (3)

Following general procedure GP1 with 4-fluorophenylacetylene 1a (24.1 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 3 as white solid (34.1 mg, 87%).
1H NMR (600 MHz, CDCl3) δ 7.57 - 7.47 (m, 4H), 7.36 - 7.34 (m, 3H), 7.09 - 7.01 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 162.6 (d, JC-F = 249.2 Hz), 133.6 (d, JC-F = 7.6 Hz), 131.7, 128.6, 128.5, 123.2, 119.5 (q, JC-F = 3.1 Hz), 115.8 (d, JC-F = 22.7 Hz), 89.2, 88.4. 19F NMR (565 MHz, CDCl3) δ -111.0.
1-Chloro-4-(phenylethynyl)benzene (4)

Following general procedure GP1 with 4-chlorophenylacetylene 1b (27.3 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 4 as white solid (42.3 mg, 95%).
1H NMR (600 MHz, CDCl3) δ 7.57 - 7.55 (m, 2H), 7.49 - 7.47 (m, 2H), 7.39 - 7.33 (m, 5H). 13C NMR (151 MHz, CDCl3) δ 134.4, 132.9, 131.7, 128.8, 128.6, 128.5, 123.1, 121.9, 90.5, 88.4.
1-Bromo-4-(phenylethynyl)benzene (5)

Following general procedure GP1 with 4-bromophenylacetylene 1c (36.2 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 5 as white solid (40.9 mg, 80%).
1H NMR (600 MHz, CDCl3) δ 7.56 - 7.51 (m, 2H), 7.49 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 7.38 - 7.31 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 133.2, 131.8, 131.7, 128.6, 128.5, 123.1, 122.6, 122.4, 90.7, 88.5.
1-Iodo-4-(phenylethynyl)benzene (6)

Following general procedure GP1 with 1-ethynyl-4-iodobenzene 1d (45.6 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 6 as white solid (57.2 mg, 94%).
1H NMR (600 MHz, CDCl3) δ 7.69 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 3.6 Hz, 2H), 7.35 (d, J = 3.5 Hz, 3H), 7.26 - 7.24 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 137.7, 133.2, 131.8, 128.7, 128.5, 123.1, 123.0, 94.2, 91.0, 88.6.
1-(Phenylethynyl)-4-(trifluoromethyl)benzene (7)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 7 as white solid (49.2 mg, 94%).
1H NMR (600 MHz, CDCl3) δ 7.65 - 7.60 (m, 4H), 7.59 - 7.53 (m, 2H), 7.41 - 7.35 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 132.0, 131.9, 130.1 (q, JC-F = 33.2 Hz), 129.0, 128.6, 127.3, 125.5 (q, JC-F = 4.6 Hz), 124.1 (q, JC-F = 273.3 Hz), 122.7, 91.9, 88.1. 19F NMR (565 MHz, CDCl3) δ -62.8.
4-(Phenylethynyl)benzonitrile (8)

Following general procedure GP1 with 4-cyanophenylacetylene 1f (25.4 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 8 as white solid (40.2 mg, 99%).
1H NMR (600 MHz, CDCl3) δ 7.61 - 7.58 (m, 4H), 7.55 - 7.54 (m, 2H), 7.39 - 7.38 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 132.1, 132.0, 131.8, 129.2, 128.5, 128.2, 122.3, 118.5, 1115, 93.8, 87.8.
1-Methyl-4-(phenylethynyl)benzene (9)

Following general procedure GP1 with 4-ethynyltoluene 1g (23.2 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 9 as white solid (31.1 mg, 81%).
1H NMR (600 MHz, CDCl3) δ 7.54 - 7.52 (m, 2H), 7.43 (d, J = 8.1 Hz, 2H), 7.37 - 7.30 (m, 3H), 7.16 (d, J = 7.9 Hz, 2H), 2.37 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 138.5, 131.7, 131.6, 129.2, 128.5, 128.2, 123.6, 120.3, 89.7, 88.9, 21.7.
1-Mthyl-4-(phenylethynyl)benzene (10)

Following general procedure GP1 with 4-ethylphenylacetylene 1h (26.1 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 10 as white solid (37.9 mg, 92%).
1H NMR (600 MHz, CDCl3) δ 7.56 - 7.53 (m, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.38 - 7.32 (m, 3H), 7.20 (d, J = 8.0 Hz, 2H), 2.68 (q, J = 7.6 Hz, 2H), 1.27 (t, J = 7.6 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 144.8, 131.8, 131.7, 128.4, 128.2, 128.1, 123.6, 120.6, 89.7, 88.9, 28.98, 15.5.
1-Isopropyl-4-(phenylethynyl)benzene (11)

Following general procedure GP1 with 4-isopropylphenylacetylene 1i (28.8 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 11 as white solid (44.1 mg, 89%).
1H NMR (600 MHz, CDCl3) δ 7.58 - 7.54 (m, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.39 - 7.33 (m, 3H), 7.24 (d, J = 8.0 Hz, 2H), 2.97 - 2.92 (m, 1H), 1.29 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 149.4, 131.8, 131.7, 128.4, 128.2, 126.6, 123.7, 120.7, 89.7, 88.8, 34.2, 23.9.
1-(Tert-butyl)-4-(phenylethynyl)benzene (12)

Following general procedure GP1 with 4-(tert-butyl)phenylacetylene 1j (31.6 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 12 as white solid (28.1 mg, 60%).
1H NMR (600 MHz, CDCl3) δ 7.55 - 7.52 (m, 2H), 7.48 (d, J = 8.3 Hz, 2H), 7.39 - 7.32 (m, 5H), 1.34 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 151.7, 131.7, 131.5, 128.4, 128.2, 125.5, 123.7, 120.4, 89.7, 88.9, 34.9, 31.3.
Trimethyl(4-(phenylethynyl)phenyl)silane (13)

Following general procedure GP1 with (4-ethynylphenyl)trimethylsilane 1k (34.9 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 13 as white solid (46.5 mg, 93%).
1H NMR (600 MHz, CDCl3) δ 7.56 - 7.54 (m, 2H), 7.51 (s, 4H), 7.37 - 7.33 (m, 3H), 0.29 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 141.2, 133.4, 131.8, 130.8, 128.5, 128.4, 123.7, 123.5, 89.9, 89.7, -1.1.
1-Fluoro-2-(phenylethynyl)benzene (14)

Following general procedure GP1 with 2-fluorophenylacetylene 1l (24.0 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 14 as white solid (33.3 mg, 85%).
1H NMR (600 MHz, CDCl3) δ 7.60 - 7.56 (m, 2H), 7.55 - 7.52 (m, 1H), 7.39 - 7.36 (m, 3H), 7.35 - 7.30 (m, 1H), 7.16 - 7.09 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 162.8 (d, JC-F = 252.2 Hz), 133.6, 131.8, 130.1 (d, JC-F = 7.6 Hz) 128.7, 128.5, 119.5 (d, JC-F = 4.6 Hz), 123.0, 115.7 (d, JC-F = 21.2 Hz), 112.1 (d, JC-F = 16.7 Hz), 94.5, 82.8. 19F NMR (565 MHz, CDCl3) δ -109.8.
1-Chloro-2-(phenylethynyl)benzene (15)

Following general procedure GP1 with 2-chlorophenylacetylene 1m (27.3 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 15 as white solid (28.8 mg, 68%).
1H NMR (600 MHz, CDCl3) δ 7.62 - 7.55 (m, 3H), 7.45 - 7.42 (m, 1H), 7.38 - 7.36 (m, 3H), 7.27 - 7.23 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 136.1, 133.4, 131.9, 129.5, 129.4, 128.8, 128.5, 126.6, 123.4, 123.1, 94.7, 86.3.
1-Bromo-2-(phenylethynyl)benzene (16)

Following general procedure GP1 with 2-bromophenylacetylene 1n (36.2 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 16 as white solid (34.3 mg, 67%).
1H NMR (600 MHz, CDCl3) δ 7.66 - 7.56 (m, 4H), 7.41 - 7.36 (m, 3H), 7.33 - 7.28 (m, 1H), 7.20 - 7.17 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 133.3, 132.6, 131.8, 129.5, 128.8, 128.5, 127.1, 125.8, 125.5, 123.0, 94.1, 88.2.
1-Methyl-3-(phenylethynyl)benzene (17)

Following general procedure GP1 with 3-ethynyltoluene 1n (23.2 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 17 as white solid (33.1 mg, 86%).
1H NMR (600 MHz, CDCl3) δ 7.59 - 7.56 (m, 2H), 7.43 - 7.34 (m, 5H), 7.28 (t, J = 7.6 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 2.39 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 138.1, 132.3, 131.7, 129.3, 128.8, 128.5, 128.4, 128.3, 123.5, 123.2, 89.7, 89.2, 21.4.
(3-Phenoxyprop-1-yn-1-yl)benzene (18)

Following general procedure GP1 with phenyl propargyl ether 1p (26.4 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 18 as white solid (13.3 mg, 32%).
1H NMR (600 MHz, CDCl3) δ 7.46 - 7.42 (m, 2H), 7.34 - 7.28 (m, 5H), 7.04 (d, J = 8.6 Hz, 2H), 7.00 (t, J = 7.3 Hz, 1H), 4.92 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 158.0, 132.0, 129.6, 128.8, 128.4, 122.5, 121.6, 115.2, 87.3, 84.1, 56.8.
4-(Phenylethynyl)pyridine (19)

Following general procedure GP1 with 3-ethynylpyridine 1q (20.6 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (n-hexane/ ethyl acetate, 5:1) to afford 19 as white solid (7.5 mg, 21%).
1H NMR (600 MHz, CDCl3) δ 8.62 - 8.60 (m, 2H), 7.58 - 7.54 (m, 2H), 7.42 - 7.38 (m, 5H). 13C NMR (151 MHz, CDCl3) δ 149.5, 132.1, 129.5, 128.7, 125.8, 122.2, 100.1, 94.6, 86.7.
(Cyclohex-1-en-1-ylethynyl)benzene (20)

Following general procedure GP1 with (1-cyclohexenyl)acetylene 1r (21.2 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 20 as white solid (10.6 mg, 29%).
1H NMR (600 MHz, CDCl3) δ 7.45 - 7.40 (m, 2H), 7.29 - 7.26 (m, 3H), 6.22 - 6.19 (m, 1H), 2.25 - 2.21 (m, 2H), 2.16 - 2.13 (m, 2H), 1.71 - 1.66 (m, 2H), 1.64 - 1.60 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 135.3, 131.2, 128.4, 127.9, 123.9, 120.9, 91.4, 86.9, 29.4, 25.9, 22.5, 21.7.
(Cyclohexylethynyl)benzene (21)

Following general procedure GP1 with cyclohexane 1s (21.6 mg, 0.2 mmol) and phenylboronic acid 2a (48.8 mg, 0.4 mmol). The crude product was purified by flash column chromatography (n-hexane/ ethyl acetate, 5:1) to afford 21 as white solid (7.4 mg, 20%).
1H NMR (600 MHz, CDCl3) δ 7.42 - 7.37 (m, 2H), 7.29 - 7.24 (m, 3H), 2.60 - 2.56 (m, 1H), 1.92 - 1.85 (m, 2H), 1.79 - 1.73 (m, 2H), 1.57 - 1.51 (m, 3H), 1.37 - 1.33 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 131.7, 128.3, 127.5, 124.3, 94.6, 80.7, 32.9, 29.8, 26.1, 25.1.
1-Methyl-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (22)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-tolylboronic acid 2b (54.4 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 22 as white solid (42.7 mg, 82%).
1H NMR (600 MHz, CDCl3) δ 7.61 (q, J = 8.5 Hz, 4H), 7.46 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 7.9 Hz, 2H), 2.39 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 139.2, 131.9, 131.8, 129.8 (q, JC-F = 33.2 Hz), 129.4, 127.5, 125.4 (q, JC-F = 3.1 Hz), 124.1 (q, JC-F = 273.3 Hz), 119.6, 92.2, 87.6, 21.7. 19F NMR (565 MHz, CDCl3) δ -62.7.
1-Fluoro-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (23)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-fluorobenzeneboronic acid 2c (56.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 23 as white solid (30.6 mg, 58%).
1H NMR (600 MHz, CDCl3) δ 7.62 (s, 4H), 7.56 - 7.52 (m, 2H), 7.10 - 7.04 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 163.0 (d, JC-F = 250.7 Hz), 133.8 (d, JC-F = 9.1 Hz), 131.89, 130.1 (q, JC-F = 33.2 Hz), 127.1, 125.5(q, JC-F = 4.6 Hz), 124.1 (q, JC-F = 273.3 Hz), 118.8 (d, JC-F = 4.6 Hz), 116.0 (d, JC-F = 22.7 Hz), 90.8, 87.8. 19F NMR (565 MHz, CDCl3) δ -62.9, -112.6.
1-Chloro-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (24)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-chlorophenylboronic acid 2d (62.5 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 24 as white solid (37.8 mg, 71%).
1H NMR (600 MHz, CDCl3) δ 7.62 (s, 4H), 7.48 (d, J = 8.6 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 135.1, 133.1, 132.0, 130.3 (q, JC-F = 33.2 Hz), 129.0, 126.9, 125.5 (q, JC-F = 3.1 Hz), 124.1 (q, JC-F = 273.3 Hz), 121.2, 90.7, 89.0. 19F NMR (565 MHz, CDCl3) δ -62.8.
1-Bromo-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (25)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-bromophenylboronic acid 2e (80.3 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 25 as white solid (43.4 mg, 67%).
1H NMR (600 MHz, CDCl3) δ 7.62 (s, 4H), 7.51 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 133.3, 132.0, 131.9, 130.3 (q, JC-F = 33.2 Hz), 126.9, 125.5 (q, JC-F = 3.1 Hz), 124.1 (q, JC-F = 273.3 Hz), 123.3, 121.7, 90.8, 89.2. 19F NMR (565 MHz, CDCl3) δ -62.8.
1-Iodo-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (26)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-iodophenylboronic acid 2f (99.1 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 26 as white solid (45.4 mg, 61%).
1H NMR (600 MHz, CDCl3) δ 7.72 (d, J = 8.4 Hz, 2H), 7.61 (s, 4H), 7.26 (d, J = 8.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 137.8, 133.3, 132.0, 130.32 (q, JC-F = 33.2 Hz), 126.90, 125.5 (q, JC-F = 3.1 Hz), 124.1 (q, JC-F = 273.3 Hz), 122.21, 95.02, 90.90, 89.44. 19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C15H8F3I [M]+: 371.9623, found 371.9618.
1-Fluoro-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene (27)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 3-fluorobenzeneboronic acid 2g (56.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 27 as white solid (49.1 mg, 93%).
1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 2.6 Hz, 4H), 7.37 - 7.32 (m, 2H), 7.26 - 7.23 (m, 1H), 7.09 - 7.06 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 162.6(d, JC-F = 247.6 Hz), 132.1, 130.4 (q, JC-F = 31.7 Hz), 130.2 (d, JC-F = 9.1 Hz), 127.78 (d, JC-F = 3.1 Hz), 126.77, 125.5(q, JC-F = 4.6 Hz), 124.6(d, JC-F = 10.6 Hz), 124.1 (q, JC-F = 273.3 Hz), 118.7 (d, JC-F = 24.2 Hz), 116.4 (d, JC-F = 21.2 Hz), 90.5, 88.9. 19F NMR (565 MHz, CDCl3) δ -62.85, -112.6. HRMS (EI) calc. for C15H8F4 [M]+: 264.0562, found 264.0553.
2,4-Difluoro-1-((4-(trifluoromethyl)phenyl)ethynyl)benzene (28)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 2,4-difluorophenylboronic acid 2h (63.2 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 28 as white solid (37.8 mg, 67%).
1H NMR (600 MHz, CDCl3) δ 7.63 (q, J = 8.4 Hz, 4H), 7.54 - 7.49 (m, 1H), 6.94 - 6.86 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 163.3 (dd, JC-F = 265.8 Hz), 163.2 (dd, JC-F = 265.8 Hz), 134.5 (dd, JC-F = 3.1 Hz), 132.96, 132.01, 130.5 (q, JC-F = 33.3 Hz), 126.69, 125.4(q, JC-F = 4.6 Hz), 124.1 (q, JC-F = 273.3 Hz), 111.9 (dd, JC-F = 3.1 Hz), 107.8 (dd, JC-F = 3.1 Hz), 104.6 (t, JC-F = 25.7 Hz), 92.77, 84.15.19F NMR (565 MHz, CDCl3) δ -62.9, -105.0, -106.0. HRMS (EI) calc. for C15H7F5 [M]+: 282.0468, found 282.0465.
1-Bromo-3-methyl-5-((4-(trifluoromethyl)phenyl)ethynyl)benzene (29)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 3-bromo-5-methylphenylboronic acid 2i (85.9 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 29 as white solid (63.5 mg, 94%).
1H NMR (600 MHz, CDCl3) δ 7.61 (s, 4H), 7.51 (s, 1H), 7.34 (s, 1H), 7.29 (s, 1H), 2.35 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 140.3, 132.9, 132.1, 131.8, 131.7, 131.2, 130.4 (q, JC-F = 33.3 Hz), 126.9, 125.4 (q, JC-F = 4.6 Hz), 124.9, 123.3 (q, JC-F = 321.6 Hz), 90.5, 88.9, 21.2.19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C16H10BrF3 [M]+: 337.9918, found 337.9914.
1-Isopropyl-4-((4-(trifluoromethyl)phenyl)ethynyl)benzene (30)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-isopropylbenzeneboronic acid 2j (65.6 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 30 as white solid (50.1 mg, 87%).
1H NMR (600 MHz, CDCl3) δ 7.64 - 7.58 (m, 4H), 7.48 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 2.95 - 2.91 (m, 1H), 1.26 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 150.1, 132.0, 131.9, 129.8 (q, JC-F = 31.7 Hz), 127.6, 126.8, 125.4(q, JC-F = 4.6 Hz), 124.2 (q, JC-F = 271.8 Hz), 120.0, 92.2, 87.5, 34.3, 24.0. 19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C18H15F3 [M]+: 288.1126, found 288.1121.
1,3-Dimethyl-5-((4-(trifluoromethyl)phenyl)ethynyl)benzene (31)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 3,5-dimethylphenylboronic acid 2k (60.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 31 as white solid (33.4 mg, 61%).
1H NMR (600 MHz, CDCl3) δ 7.63 - 7.63 (m, 4H), 7.19 (s, 2H), 7.01 (s, 1H), 2.33 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 138.2, 131.9, 130.9, 129.8 (q, JC-F = 33.3 Hz), 129.6, 127.5, 125.4 (q, JC-F = 4.6 Hz), 124.2 (q, JC-F = 271.8 Hz), 122.3, 92.3, 87.5, 21.3. 19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C17H13F3 [M]+: 274.0969, found 274.0966.
Trimethyl(4-((4-(trifluoromethyl)phenyl)ethynyl)phenyl)silane (32)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-(trimethylsilyl)phenylboronic acid 2l (77.6 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 32 as white solid (61.1 mg, 96%).
1H NMR (600 MHz, CDCl3) δ 7.62 (q, J = 8.4 Hz, 4H), 7.52 (s, 4H), 0.29 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 142.1, 133.5, 132.0, 131.0, 130.1 (q, JC-F = 33.2 Hz), 127.4, 125.4 (q, JC-F = 4.6 Hz), 124.1 (q, JC-F = 271.8 Hz), 123.0, 92.1, 88.5, -1.1. 19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C18H17F3Si [M]+: 318.1052, found 318.1046.
4-((4-(Trifluoromethyl)phenyl)ethynyl)-1,1'-biphenyl (33)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-biphenylboronic acid 2m (79.2 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 33 as white solid (51.5 mg, 80%).
1H NMR (600 MHz, CDCl3) δ 7.68 - 7.59 (m, 10H), 7.47 (t, J = 7.5 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 141.8, 140.4, 132.4, 132.0, 130.3 (q, JC-F = 33.2 Hz), 130.0, 128.0, 127.3, 127.2, 125.5 (q, JC-F = 4.6 Hz), 124.1 (q, JC-F = 271.8 Hz), 121.6, 91.9, 88.8. 19F NMR (565 MHz, CDCl3) δ -62.8. HRMS (EI) calc. for C21H13F3 [M]+: 322.0969, found 322.0963.
Methyl 4-((4-(trifluoromethyl)phenyl)ethynyl)benzoate (34)

Following general procedure GP1 with 4-(trifluoromethyl)phenylacetylene 1e (34.1 mg, 0.2 mmol) and 4-(methoxycarbonyl)phenylboronic acid 2n (72.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (n-hexane/ ethyl acetate, 5:1) to afford 34 as white solid (27.4 mg, 45%).
1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 2H), 7.66 - 7.59 (m, 6H), 3.94 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 166.6, 132.1, 131.9, 130.8 (q, JC-F = 33.2 Hz), 130.2, 129.8, 129.0, 127.33, 125.5 (q, JC-F = 4.6 Hz), 124.2 (q, JC-F = 271.8 Hz), 91.0, 90.9, 52.5. 19F NMR (565 MHz, CDCl3) δ -62.9. HRMS (EI) calc. for C17H11F3O2 [M]+: 304.0711, found 304.0707.
(4-((4-Chlorophenyl)ethynyl)phenyl)trimethylsilane (35)

Following general procedure GP1 with (4-ethynylphenyl)trimethylsilane 1k (34.9 mg, 0.2 mmol) and 4-chlorophenylboronic acid 2d (62.5 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 35 as white solid (42.1 mg, 70%).
1H NMR (600 MHz, CDCl3) δ 7.51 - 7.48 (m, 4H), 7.47 - 7.45 (m, 2H), 7.34 - 7.31 (m, 2H), 0.28 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 141.6, 134.4, 133.4, 133.0, 130.8, 128.9, 123.3, 122.0, 90.7, 88.8, -1.1. HRMS (EI) calc. for C17H17ClSi [M]+: 284.0788, found 284.0785.
(4-((4-Fluorophenyl)ethynyl)phenyl)trimethylsilane (36)

Following general procedure GP1 with (4-ethynylphenyl)trimethylsilane 1k (34.9 mg, 0.2 mmol) and 4-fluorobenzeneboronic acid 2c (56.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 36 as white solid (27.9 mg, 52%).
1H NMR (600 MHz, CDCl3) δ 7.53 - 7.49 (m, 6H), 7.05 (t, J = 8.7 Hz, 2H), 0.28 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 162.6 (d, JC-F = 249.2 Hz), 141.3, 133.6 (d, JC-F = 7.6 Hz), 133.4, 131.6, 130.8, 118.6 (d, JC-F = 3.1 Hz), 115.7 (d, JC-F = 21.2 Hz), 89.4, 88.8, -1.1. 19F NMR (565 MHz, CDCl3) δ -111.0.
(4-((3-Bromo-5-methylphenyl)ethynyl)phenyl)trimethylsilane (37)

Following general procedure GP1 with (4-ethynylphenyl)trimethylsilane 1k (34.9 mg, 0.2 mmol) and 3-bromo-5-methylphenylboronic acid 2i (85.9 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 37 as white solid (38.3 mg, 56%).
1H NMR (600 MHz, CDCl3) δ 7.52 - 7.47 (m, 5H), 7.29 (d, J = 10.8 Hz, 2H), 2.33 (s, 3H), 0.28 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 141.6, 140.1, 133.4, 132.3, 131.5, 131.1, 130.8, 125.1, 123.2, 122.1, 90.6, 88.5, 21.1, -1.1. HRMS (EI) calc. for C18H19BrSi [M]+: 342.0439, found 342.0436.
(4-((3-Fluorophenyl)ethynyl)phenyl)trimethylsilane (38)

Following general procedure GP1 with (4-ethynylphenyl)trimethylsilane 1k (34.9 mg, 0.2 mmol) and 3-fluorobenzeneboronic acid 2g (56.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 38 as white solid (27.3 mg, 51%).
1H NMR (600 MHz, CDCl3) δ 7.51 (s, 4H), 7.34 - 7.29 (m, 2H), 7.24 - 7.21 (m, 1H), 7.07 - 7.00 (m, 1H), 0.28 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 162.5 (d, JC-F = 246.2 Hz), 141.7, 133.4, 130.9, 130.1 (d, JC-F = 7.6 Hz), 127.6 (d, JC-F = 3.1 Hz), 125.3 (d, JC-F = 10.6 Hz), 123.2, 118.5 (d, JC-F = 24.2 Hz), 115.7 (d, JC-F = 21.2 Hz), 90.6, 88.6, -1.1. 19F NMR (565 MHz, CDCl3) δ -113.1. HRMS (EI) calc. for C17H17FSi [M]+: 268.1084, found 268.1081.
1-Fluoro-4-(p-tolylethynyl)benzene (39)

Following general procedure GP1 with 4-fluorophenylacetylene 1a (24.1 mg, 0.2 mmol) and 4-tolylboronic acid 2b (54.4 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 39 as white solid (41.2 mg, 98%).
1H NMR (600 MHz, CDCl3) δ 7.52 - 7.47 (m, 2H), 7.41 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 7.9 Hz, 2H), 7.04 (t, J = 8.7 Hz, 2H), 2.37 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 162.5 (d, JC-F = 246.2 Hz), 138.6, 133.5 (d, JC-F = 9.1 Hz), 131.6, 129.3, 120.1, 119.7 (d, JC-F = 3.1 Hz), 115.7 (d, JC-F = 22.7 Hz), 89.4, 87.8, 21.7. 19F NMR (565 MHz, CDCl3) δ -113.3.
1-(Tert-butyl)-4-((4-fluorophenyl)ethynyl)benzene (40)
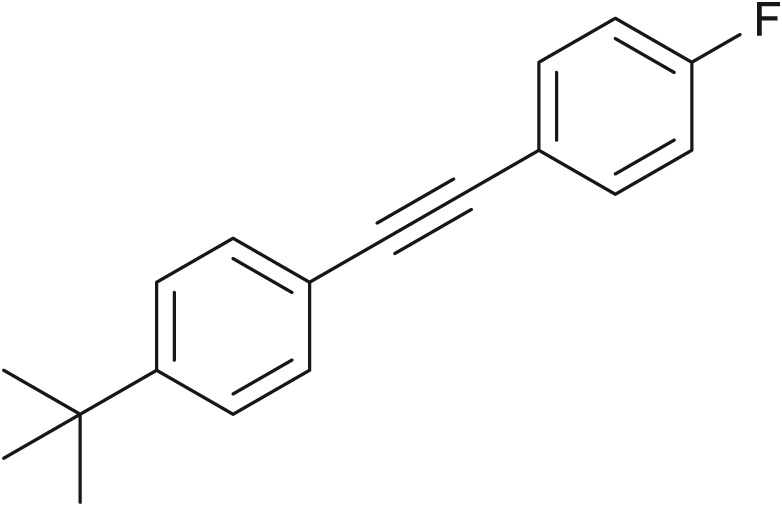
Following general procedure GP1 with 4-fluorophenylacetylene 1a (24.1 mg, 0.2 mmol) and 4-tert-butylphenylboronic acid 2o (71.2 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 40 as white solid (46.4 mg, 92%).
1H NMR (600 MHz, CDCl3) δ 7.53 - 7.45 (m, 4H), 7.38 (d, J = 8.4 Hz, 2H), 7.04 (t, J = 8.7 Hz, 2H), 1.34 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 162.5 (d, JC-F = 246.2 Hz), 151.8, 133.6 (d, JC-F = 7.6 Hz), 131.4, 125.5, 120.2, 119.8 (d, JC-F = 3.1 Hz), 115.7 (d, JC-F = 21.2 Hz), 89.3, 88.8, 34.9, 31.3. 19F NMR (565 MHz, CDCl3) δ -113.0.
1-Fluoro-2-((4-fluorophenyl)ethynyl)benzene (41)

Following general procedure GP1 with 4-fluorophenylacetylene 1a (24.1 mg, 0.2 mmol) and 2-fluorophenylboronic acid 2p (56.0 mg, 0.4 mmol). The crude product was purified by flash column chromatography (100% n-hexane) to afford 41 as white solid (31.7 mg, 74%).
1H NMR (600 MHz, CDCl3) δ 7.59 - 7.48 (m, 3H), 7.32 (q, J = 6.9 Hz, 1H), 7.17 - 7.03 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 162.8 (d, JC-F = 250.7 Hz), 162.7 (d, JC-F = 252.2 Hz), 133.8 (d, JC-F = 7.6 Hz), 133.5 (d, JC-F = 9.1 Hz), 130.2 (d, JC-F = 7.6 Hz), 124.1 (d, JC-F = 4.6 Hz), 119.2 (d, JC-F = 3.1 Hz), 115.8 (d, JC-F = 22.7 Hz), 115.7 (d, JC-F = 22.7 Hz), 111.9 (d, JC-F = 16.7 Hz), 93.4 (d, JC-F = 4.6 Hz), 82.5. 19F NMR (565 MHz, CDCl3) δ -109.9, 110.4.
(4-(Phenylethynyl)phenyl)(trifluoromethyl)sulfane (44)

1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 8.1 Hz, 2H), 7.57 - 7.53 (m, 4H), 7.41 - 7.34 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 136.2, 132.6, 131.9, 130.0 (q, JC-F = 309.1 Hz) 129.0, 128.6, 126.4, 124.3, 122.8, 92.9, 88.2. 19F NMR (565 MHz, CDCl3) δ -42.5. HRMS (EI) calc. for C15H9F3S [M]+: 278.0377, found 278.0370.
4,4,5,5-Tetramethyl-2-(4-(phenylethynyl)phenyl)-1,3,2-dioxaborolane (45)

1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 8.1 Hz, 2H), 7.56 - 7.52 (m, 4H), 7.37 - 7.32 (m, 3H), 1.36 (s, 12H). 13C NMR (151 MHz, CDCl3) δ 134.7, 131.8, 130.9, 128.5, 126.1, 123.3, 100.1, 90.8, 89.7, 84.1, 25.0. HRMS (EI) calc. for C20H21BO2 [M]+: 304.1635, found 304.1632.
Trimethyl((4-(phenylethynyl)phenyl)ethynyl)silane (46)

1H NMR (600 MHz, CDCl3) δ 7.55 - 7.52 (m, 2H), 7.49 - 7.43 (m, 4H), 7.36 - 7.34 (m, 3H), 0.27 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 132.0, 131.8, 131.5, 128.6, 128.5, 123.5, 123.1, 123.0, 104.8, 96.4, 91.4, 89.2, 0.1.
(Z)-1-Chloro-4-styrylbenzene (47)

1H NMR (600 MHz, CDCl3) δ 7.26 - 7.15 (m, 9H), 6.63 (d, J = 12.2 Hz, 1H), 6.54 (d, J = 12.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 137.0, 135.8, 132.9, 131.1, 130.4, 129.1, 129.9, 128.5, 128.4, 127.5.
(E)-1-Chloro-4-styrylbenzene (48)

1H NMR (600 MHz, CDCl3) δ 7.51 (d, J = 7.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.39 - 7.26 (m, 5H), 7.11 - 7.03 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 137.1, 136.0, 133.3, 129.5, 129.0, 128.9, 128.0, 127.8, 127.5, 126.7.
Trimethyl((5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)ethynyl)silane (49)

1H NMR (600 MHz, CDCl3) δ 7.41 (s, 1H), 7.22 (s, 2H), 1.67 (s, 4H), 1.27 (s, 6H), 1.26 (s, 6H), 0.25 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 145.9, 145.0, 130.4, 129.2, 126.6, 120.2, 106.0, 92.8, 35.1, 34.4, 31.9 31.8, 22.8, 14.3, 0.24.
6-Ethynyl-1,1,4,4-tetramethyl-1,2,3,4-tetrahydronaphthalene (50)

1H NMR (600 MHz, CDCl3) δ 7.45 (s, 1H), 7.26 (s, 2H), 3.01 (s, 1H), 1.68 (s, 4H), 1.28 (s, 6H), 1.27 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 146.2, 145.2, 130.7, 129.3, 126.8, 119.2, 84.4, 76.1, 35.1, 35.0, 34.5, 34.3, 31.9, 31.8.
Methyl 4-((5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)ethynyl)-benzoate (51)

1H NMR (600 MHz, CDCl3) δ 8.01 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.49 (s, 1H), 7.30 (s, 2H), 3.93 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 166.8, 146.3, 145.3, 131.6, 130.2, 129.6, 129.3, 128.9, 128.5, 126.9, 119.7, 93.2, 87.8, 52.4, 35.1, 35.0, 34.6, 34.4, 31.9, 31.8.

Au-I: 1H NMR (600 MHz, CDCl3) δ 7.59 - 7.42 (m, 17H), 6.94 (t, J = 8.7 Hz, 2H). 19F NMR (565 MHz, CDCl3) δ -113.4. 31P NMR (243 MHz, CDCl3) δ 42.3.

Au- II:1H NMR (600 MHz, CDCl3) δ 7.61 (m, 6H), 7.48 (m, 11H), 7.13 (d, J = 7.2 Hz, 2H), 2.32 (s, 3H). 31P NMR (243 MHz, CDCl3) δ 43.7.

1H NMR (600 MHz, CDCl3) δ 7.76 - 7.66 (m, 9H), 7.57 (m, 6H). 31P NMR (243 MHz, CDCl3) δ 43.6.
Quantification and statistical analysis
This study did not use statistical analyses typical in life sciences.
Acknowledgments
We thank Prof. Qiang Zhu (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences) and Prof. Honggen Wang (Sun Yat-sen University) for helpful discussions. Financial support from the National Natural Science Foundation of China (22201019 to Z.X.) and the Beijing Institute of Technology Research Fund Program for Young Scholars (grant number: 2020CX04260) is gratefully acknowledged.
Author contributions
L.Z. performed synthetic experiments and mechanistic studies. W.Z., D.L., and R.Y. commented on the final data of the manuscript and contributed to the analysis and interpretation of the data. Z.X. conceived the idea, designed the research, and prepared the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108531.
Supplemental information
References
- 1.Johansson Seechurn C.C.C., Kitching M.O., Colacot T.J., Snieckus V. Palladium-Catalyzed Cross-Coupling: A Historical Contextual Perspective to the 2010 Nobel Prize. Angew. Chem. Int. Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 2.Hazari N., Melvin P.R., Beromi M.M. Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem. 2017;1 doi: 10.1038/s41570-017-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu C., Liu J., Li M.-B., Bäckvall J.E. Palladium-catalyzed oxidative dehydrogenative carbonylation reactions using carbon monoxide and mechanistic overviews. Chem. Soc. Rev. 2020;49:341–353. doi: 10.1039/c9cs00397e. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki A. Cross-coupling reactions of organoboranes: an easy way to construct C-C bonds (Nobel Lecture) Angew. Chem. Int. Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- 5.Negishi E., Zeng X., Tan Z., Qian M., Hu Q., Huang Z. Wiley-VCH; 2004. Palladium or Nickel-Catalyzed Cross-Coupling with Organometals Containing Zinc, Aluminum, and Zirconium: The Negishi Coupling; pp. 815–889. [Google Scholar]
- 6.Devendar P., Qu R.-Y., Kang W.-M., He B., Yang G.-F. Palladium-Catalyzed Cross-Coupling Reactions: A Powerful Tool for the Synthesis of Agrochemicals. J. Agric. Food Chem. 2018;66:8914–8934. doi: 10.1021/acs.jafc.8b03792. [DOI] [PubMed] [Google Scholar]
- 7.Nakao Y., Hiyama T. Silicon-based cross-coupling reaction: an environmentally benign version. Chem. Soc. Rev. 2011;40:4893–4901. doi: 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]
- 8.Denmark S.E., Regens C.S. Palladium-catalyzed cross-coupling reactions of organosilanols and their salts: practical alternatives to boron- and tin-based methods. Acc. Chem. Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamao K. Discovery of the cross-coupling reaction between grignard reagents and C(sp2) halides catalyzed by nickel–phosphine complexes. J. Organomet. Chem. 2002;653:23–26. [Google Scholar]
- 10.Tarahomi M., Alinezhad H., Maleki B. Immobilizing Pd nanoparticles on the ternary hybrid system of graphene oxide, Fe3O4 nanoparticles, and PAMAM dendrimer as an efficient support for catalyzing sonogashira coupling reaction. Appl. Organomet. Chem. 2019;33 [Google Scholar]
- 11.Maleki B., Nejat R., Vahdani Z. Three-dimensional graphene-magnetic Organometallic nanohybrid as High-Performance Visible Light Photocatalyst for the C-C Coupling. Polycycl. Aromat. Comp. 2022;42:3638–3650. [Google Scholar]
- 12.Negishi E.i., Anastasia L. Palladium-catalyzed alkynylation. Chem. Rev. 2003;103:1979–2017. doi: 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]
- 13.Doucet H., Hierso J.-C. Palladium-based catalytic systems for the synthesis of conjugated enynes by Sonogashira reactions and related alkyny-lations. Angew. Chem. Int. Ed. 2007;46:834–871. doi: 10.1002/anie.200602761. [DOI] [PubMed] [Google Scholar]
- 14.Chinchilla R., Nájera C. Chemicals from alkynes with palladium catalysts. Chem. Rev. 2014;114:1783–1826. doi: 10.1021/cr400133p. [DOI] [PubMed] [Google Scholar]
- 15.Wang D., Gao S. Sonogashira coupling in natural product synthesis. Org. Chem. Front. 2014;1:556–566. [Google Scholar]
- 16.Ayogu J.I., Onoabedje E.A. Recent advances in transition metal-catalysed cross-coupling of (hetero)aryl halides and analogues under ligand-free conditions. Catal. Sci. Technol. 2019;9:5233–5255. [Google Scholar]
- 17.Liu C., Zhang H., Shi W., Lei A. Bond formations between two nucleophiles: transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]
- 18.Shi W., Liu C., Lei A. Transition-metal catalyzed oxidative cross-coupling reactions to form C-C bonds involving organometallic reagents as nucleophiles. Chem. Soc. Rev. 2011;40:2761–2776. doi: 10.1039/c0cs00125b. [DOI] [PubMed] [Google Scholar]
- 19.Hashmi A.S.K. Gold-catalyzed organic reactions. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]
- 20.Dorel R., Echavarren A.M. Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 2015;115:9028–9072. doi: 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campeau D., León Rayo D.F., Mansour A., Muratov K., Gagosz F. Gold-catalyzed reactions of specially activated alkynes, allenes, and alkenes. Chem. Rev. 2021;121:8756–8867. doi: 10.1021/acs.chemrev.0c00788. [DOI] [PubMed] [Google Scholar]
- 22.Bandini M. Gold-catalyzed decorations of arenes and heteroarenes with C-C multiple bonds. Chem. Soc. Rev. 2011;40:1358–1367. doi: 10.1039/c0cs00041h. [DOI] [PubMed] [Google Scholar]
- 23.Gorin D.J., Toste F.D. Relativistic effects in homogeneous gold catalysis. Nature. 2007;446:395–403. doi: 10.1038/nature05592. [DOI] [PubMed] [Google Scholar]
- 24.Brand J.P., Li Y., Waser J. Gold-catalyzed alkynylation: acetylene-transfer instead of functionalization. Isr. J. Chem. 2013;53:901–910. [Google Scholar]
- 25.Hopkinson M.N., Gee A.D., Gouverneur V. Au(I)/Au(III) catalysis: an alternative approach for C-C oxidative coupling. Chem. Eur J. 2011;17:8248–8262. doi: 10.1002/chem.201100736. [DOI] [PubMed] [Google Scholar]
- 26.Garcia P., Malacria M., Aubert C., Gandon V., Fensterbank L. Gold-catalyzed cross-couplings: new opportunities for C-C bond formation. ChemCatChem. 2010;2:493–497. [Google Scholar]
- 27.Nijamudheen A., Datta A. Gold-catalyzed cross-coupling reactions: an overview of design strategies, mechanistic studies, and applications. Chem. Eur J. 2020;26:1442–1487. doi: 10.1002/chem.201903377. [DOI] [PubMed] [Google Scholar]
- 28.Bhoyare V.W., Tathe A.G., Das A., Chintawar C.C., Patil N.T. The Interplay of carbophilic activation and Au(I)/Au(III) catalysis: an emerging technique for 1,2-difunctionalization of C-C multiple bonds. Chem. Soc. Rev. 2021;50:10422–10450. doi: 10.1039/d0cs00700e. [DOI] [PubMed] [Google Scholar]
- 29.Brand J.P., Charpentier J., Waser J. Direct alkynylation of Indole and pyrrole heterocycles. Angew. Chem. Int. Ed. 2009;48:9346–9349. doi: 10.1002/anie.200905419. [DOI] [PubMed] [Google Scholar]
- 30.de Haro T., Nevado C. Gold-catalyzed ethynylation of arenes. J. Am. Chem. Soc. 2010;132:1512–1513. doi: 10.1021/ja909726h. [DOI] [PubMed] [Google Scholar]
- 31.Hofer M., de Haro T., Gómez-Bengoa E., Genoux A., Nevado C. Oxidant speciation and anionic ligand effects in the gold-catalyzed oxide tive coupling of arenes and alkynes. Chem. Sci. 2019;10:8411–8420. doi: 10.1039/c9sc02372k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tlahuext-Aca A., Hopkinson M.N., Sahoo B., Glorius F. Dual gold/photoredox-catalyzed C(sp)-H arylation of terminal alkynes with diazonium salts. Chem. Sci. 2016;7:89–93. doi: 10.1039/c5sc02583d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S., Rojas-Martin J., Toste F.D. Visible light-mediated gold-catalysed carbon(sp2)-carbon(sp) cross-coupling. Chem. Sci. 2016;7:85–88. doi: 10.1039/c5sc03025k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang B., Hu M., Toste F.D. Homogeneous gold redox chemistry: organometallics, catalysis, and beyond. Trends Chem. 2020;2:707–720. doi: 10.1016/j.trechm.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harper M.J., Arthur C.J., Crosby J., Emmett E.J., Falconer R.L., Fensham-Smith A.J., Gates P.J., Leman T., McGrady J.E., Bower J.F., Russell C.A. Oxidative addition, transmetalation, and reductive elimination at a 2,2'-bipyridyl-ligated gold center. J. Am. Chem. Soc. 2018;140:4440–4445. doi: 10.1021/jacs.8b01411. [DOI] [PubMed] [Google Scholar]
- 36.Cadge J.A., Sparkes H.A., Bower J.F., Russell C.A. Oxidative addition of alkenyl and alkynyl iodides to a AuI Complex. Angew. Chem. Int. Ed. 2020;59:6617–6621. doi: 10.1002/anie.202000473. [DOI] [PubMed] [Google Scholar]
- 37.Hopkinson M.N., Ross J.E., Giuffredi G.T., Gee A.D., Gouverneur V. Gold-catalyzed cascade cyclization-oxidative alkynylation of allenoates. Org. Lett. 2010;12:4904–4907. doi: 10.1021/ol102061k. [DOI] [PubMed] [Google Scholar]
- 38.Leyva-Pérez A., Doménech-Carbó A., Corma A. Unique distal size selectivity with a digold catalyst during alkyne homocoupling. Nat. Commun. 2015;6:6703. doi: 10.1038/ncomms7703. [DOI] [PubMed] [Google Scholar]
- 39.Peng H., Xi Y., Ronaghi N., Dong B., Akhmedov N.G., Shi X. Gold-catalyzed oxidative cross-coupling of terminal alkynes: selective synthesis of unsymmetrical 1,3-diynes. J. Am. Chem. Soc. 2014;136:13174–13177. doi: 10.1021/ja5078365. [DOI] [PubMed] [Google Scholar]
- 40.Zhang G., Peng Y., Cui L., Zhang L. Gold-catalyzed homogeneous oxidative cross-coupling reactions. Angew. Chem. Int. Ed. 2009;48:3112–3115. doi: 10.1002/anie.200900585. [DOI] [PubMed] [Google Scholar]
- 41.Wang W., Jasinski J., Hammond G.B., Xu B. Fluorine-enabled cationic gold catalysis: functionalized hydration of alkynes. Angew. Chem. Int. Ed. 2010;49:7247–7252. doi: 10.1002/anie.201003593. [DOI] [PubMed] [Google Scholar]
- 42.Qian D., Zhang J. Au(I)/Au(III)-catalyzed Sonogashira-type reactions of functionalized terminal alkynes with arylboronic acids under mild conditions. Beilstein J. Org. Chem. 2011;7:808–812. doi: 10.3762/bjoc.7.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brenzovich W.E., Jr., Benitez D., Lackner A.D., Shunatona H.P., Tkatchouk E., Goddard W.A., III., Toste F.D. Gold-catalyzed Intramolecular aminoarylation of alkenes: C-C bond formation through bimolecular reductive elimination. Angew. Chem. Int. Ed. 2010;49:5519–5522. doi: 10.1002/anie.201002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tkatchouk E., Mankad N.P., Benitez D., Goddard W.A., III, Toste F.D. Two metals are better than one in the gold catalyzed oxidative heteroarylation of alkenes. J. Am. Chem. Soc. 2011;133:14293–14300. doi: 10.1021/ja2012627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ball L.T., Lloyd-Jones G.C., Russell C.A. Gold-catalyzed direct arylation. Science. 2012;337:1644–1648. doi: 10.1126/science.1225709. [DOI] [PubMed] [Google Scholar]
- 46.Ball L.T., Lloyd-Jones G.C., Russell C.A. Gold-catalyzed oxidative coupling of arylsilanes and arenes: origin of selectivity and improved precatalyst. J. Am. Chem. Soc. 2014;136:254–264. doi: 10.1021/ja408712e. [DOI] [PubMed] [Google Scholar]
- 47.Liu K., Li N., Ning Y., Zhu C., Xie J. Gold-catalyzed oxidative biaryl cross-coupling of organometallics. Chem. 2019;5:2718–2730. [Google Scholar]
- 48.Partyka D.V., Zeller M., Hunter A.D., Gray T.G. Relativistic functional groups: aryl carbon-gold bond formation by selective transmeta-lation of boronic acids. Angew. Chem. Int. Ed. 2006;45:8188–8191. doi: 10.1002/anie.200603350. [DOI] [PubMed] [Google Scholar]
- 49.Partyka D.V., Zeller M., Hunter A.D., Gray T.G. Arylgold(I) complexes from base-assisted transmetalation: structures, nmr properties, and density-functional theory calculations. Inorg. Chem. 2012;51:8394–8401. doi: 10.1021/ic3009464. [DOI] [PubMed] [Google Scholar]
- 50.Hashmi A.S.K., Ramamurthi T.D., Rominger F. Synthesis, structure and reactivity of organogold compounds of relevance to homogeneous gold catalysis. J. Organomet. Chem. 2009;694:592–597. [Google Scholar]
- 51.Dupuy S., Crawford L.E., Bühl M., Slawin A.M.Z., Nolan S.P. The role of metal hydroxide complexes in late transition metal-mediated transmetalation reaction: the case of gold. Adv. Synth. Catal. 2012;354:2380–2386. [Google Scholar]
- 52.Xia Z., Corcé V., Zhao F., Przybylski C., Espagne A., Jullien L., Le Saux T., Gimbert Y., Dossmann H., Mouriès-Mansuy V., et al. Photosensitized oxidative addition to gold(I) enables alkynylative cyclization of o-alkylnylphenols with iodoalkynes. Nat. Chem. 2019;11:797–805. doi: 10.1038/s41557-019-0295-9. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L., Wei C., Wu J., Liu D., Yao Y., Chen Z., Liu J., Yao C.-J., Li D., Yang R., Xia Z. Photoinduced inverse sonogashira coupling reaction. Chem. Sci. 2022;13:7475–7481. doi: 10.1039/d2sc01933g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei C., Wu J., Zhang L., Xia Z. Gold(I)-catalyzed selective hydroarylation of indoles with haloalkynes. Org. Lett. 2022;24:4689–4693. doi: 10.1021/acs.orglett.2c01921. [DOI] [PubMed] [Google Scholar]
- 55.Wu J., Wei C., Zhao F., Du W., Geng Z., Xia Z. Gold(I)-catalyzed tandem cyclization/hydroarylation of o-alkynylphenols with haloalkynes. J. Org. Chem. 2022;87:14374–14383. doi: 10.1021/acs.joc.2c01804. [DOI] [PubMed] [Google Scholar]
- 56.Du W., Yang R., Wu J., Xia Z. Flexible synthesis of benzofuranones from ortho-alkynyl phenols or benzofurans. Eur. J. Org. Chem. 2023;26 [Google Scholar]
- 57.Mudshinge S.R., Yang Y., Xu B., Hammond G.B., Lu Z. Gold (I/III)-catalyzed Trifluoromethylthiolation and Trifluoromethylselenolation of organohalides. Angew. Chem. Int. Ed. 2022;61 doi: 10.1002/anie.202115687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao J.-H., Zhou Z.-Z., Zhang Y., Su X., Chen X.-M., Liang Y.-M. Visible-light-mediated borylation of aryl and alkyl halides with a palladium complex. Org. Biomol. Chem. 2020;18:4390–4394. doi: 10.1039/d0ob00028k. [DOI] [PubMed] [Google Scholar]
- 59.Shang Q., Tang H., Liu Y., Yin M., Su L., Xie S., Liu L., Yang W., Chen Y., Dong J., et al. Cu(I) catalysis for selective condensation/bicycloaromatization of two different arylalkynes: direct and general construction of functionalized C-N axial biaryl Compounds. Chem. Sci. 2022;13:263–273. doi: 10.1039/d1sc03865f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen R., Chen T., Zhao Y., Qiu R., Zhou Y., Yin S., Wang X., Goto M., Han L.-B. Facile regio- and stereoselective hydrometalation of alkynes with a combination of carboxylic acids and group 10 transition metal complexes: selective hydrogenation of alkynes with formic acid. J. Am. Chem. Soc. 2011;133:17037–17044. doi: 10.1021/ja2069246. [DOI] [PubMed] [Google Scholar]
- 61.Christie V.B., Barnard J.H., Batsanov A.S., Bridgens C.E., Cartmell E.B., Collings J.C., Maltman D.J., Redfern C.P.F., Marder T.B., Przyborski S., Whiting A. Synthesis and evaluation of synthetic retinoid derivatives as inducers of stem cell differentiation. Org. Biomol. Chem. 2008;6:3497–3507. doi: 10.1039/b808574a. [DOI] [PubMed] [Google Scholar]
- 62.Yang Y., Schießl J., Zallouz S., Göker V., Gross J., Rudolph M., Rominger F., Hashmi A.S.K. Gold-catalyzed C(sp2)-C(sp) coupling by alkynylation through oxidative addition of bromoal-kynes. Chem. Eur J. 2019;25:9624–9628. doi: 10.1002/chem.201902213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper does not report original code.
-
•
Data and code generated in gold-catalyzed oxidative Sonogashira coupling reaction are available within the article and the supplemental experimental procedures or from the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.