

Abstract
Mutations in the gene encoding the transient receptor potential vanilloid member 4 (TRPV4), a Ca2+ permeable nonselective cation channel, cause TRPV4‐related disorders. TRPV4 is widely expressed in the brain; however, the pathogenesis underlying TRPV4‐mediated Ca2+ deregulation in neurodevelopment remains unresolved and an effective therapeutic strategy remains to be established. These issues were addressed by isolating mutant dental pulp stem cells from a tooth donated by a child diagnosed with metatropic dysplasia with neurodevelopmental comorbidities caused by a gain‐of‐function TRPV4 mutation, c.1855C > T (p.L619F). The mutation was repaired using CRISPR/Cas9 to generate corrected isogenic stem cells. These stem cells were differentiated into dopaminergic neurons and the pharmacological effects of folic acid were examined. In mutant neurons, constitutively elevated cytosolic Ca2+ augmented AKT‐mediated α‐synuclein (α‐syn) induction, resulting in mitochondrial Ca2+ accumulation and dysfunction. The TRPV4 antagonist, AKT inhibitor, or α‐syn knockdown, normalizes the mitochondrial Ca2+ levels in mutant neurons, suggesting the importance of mutant TRPV4/Ca2+/AKT‐induced α‐syn in mitochondrial Ca2+ accumulation. Folic acid was effective in normalizing mitochondrial Ca2+ levels via the transcriptional repression of α‐syn and improving mitochondrial reactive oxygen species levels, adenosine triphosphate synthesis, and neurite outgrowth of mutant neurons. This study provides new insights into the neuropathological mechanisms underlying TRPV4‐related disorders and related therapeutic strategies.
Keywords: calcium, dental pulp stem cells, mitochondria, transient receptor potential vanilloid member 4, α‐synuclein
In metatropic dysplasia‐dopaminergic neurons (MD‐DNs) without folic acid (FA) supplementation, TRPV4 mutation (p.L619F) accelerated Ca2+ entry and activated AKT‐mediated SNCA/α‐synuclein (α‐syn) induction, resulting in mitochondrial Ca2+ overload and impaired neurite development. FA attenuated mitochondrial Ca2+ accumulation along with α‐syn downregulation without affecting cytosolic Ca2+ levels and effectively ameliorated mitochondrial function and neurite outgrowth in MD‐DNs. FA also downregulated tumor suppressor p53, which is possibly associated with α‐syn downregulation in FA‐supplemented MD‐DNs.
Abbreviations
- ATP
adenosine triphosphate
- CaM
calcium‐calmodulin
- CGI
CpG island
- DNs
dopaminergic neurons
- DPSCs
dental pulp stem cells
- ER
endoplasmic reticulum
- FA
folic acid
- MAM
mitochondria‐associated membrane
- MD
metatropic dysplasia
- MMP
mitochondrial membrane potential
- MOM
mitochondrial outer membrane
- NURR1
nuclear receptor related 1
- PI3K
phosphatidylinositol‐3 kinase
- PITX3
pituitary homeobox 3
- ROS
reactive oxygen species
- SHEDs
stem cells from human exfoliated deciduous teeth
- siRNA
small interfering RNA
- TH
tyrosine hydroxylase
- TRPV4
transient receptor potential vanilloid member 4
- VDAC1
voltage‐dependent anion channel 1
- α‐syn
alpha‐synuclein
1. INTRODUCTION
Transient receptor potential vanilloid 4 (TRPV4) is a nonselective Ca2+‐permeable cation channel primarily expressed in the plasma membrane 1 , 2 , 3 and its major physiological function is regulating responses to various stimuli, including heat, mechanical force, osmotic pressure, and arachidonic acid metabolites. 4 Cytosolic free Ca2+ that passes through TRPV4 regulates multiple downstream Ca2+‐dependent cellular signaling pathways. 5 TRPV4 broadly expressed in whole tissues is vital for the development and activity of the central nervous system, wherein Ca2+‐dependent events are crucial. 6 Various diseases are caused by the deregulation of TRPV4 function, representative of TRPV4‐related channelopathy. 7 , 8 Functional analyses using agonists and antagonists for wild‐type TRPV4 channels have revealed that excessive Ca2+ entry contributes to pathological conditions via the deregulation of Ca2+‐dependent signaling pathways. 9 Furthermore, functional analysis of disease‐associated TRPV4 mutants has revealed that they promote enhanced Ca2+ entry into cells, regardless of ligand stimulation. 10 , 11 , 12 However, the pathological association between TRPV4 mutants and neuronal development is not fully understood yet mainly because extensively analyzing live patients' brains is limited by ethical and technical issues, and the experimental models carrying disease‐associated TRPV4 mutants are insufficiently available.
Human teeth contain various types of multipotent mesenchymal stem cells. 13 Stem cells from human exfoliated deciduous teeth (SHEDs) and dental pulp stem cells (DPSCs) are derived from deciduous 14 and permanent teeth, 15 respectively, and can exhibit partially different properties. However, they can differentiate into dopaminergic neurons (DNs) in vitro due to their shared neural crest origin. 16 , 17 , 18 , 19 The brain's dopaminergic system modulates broad brain functions, including attention and reward, and dopaminergic dysfunction is associated with many neuropsychiatric disorders. 20 , 21 , 22 , 23 SHEDs and DPSCs from patients diagnosed with neurodevelopmental disorders are promising as disease‐specific cellular models for studying dopaminergic system‐related neuropathology. 24 Previously, DPSCs were isolated from a patient with metatropic dysplasia (MD) and multiple neuropsychiatric symptoms caused by a gain‐of‐function mutation of TRPV4, c.1855C > T (p.L619F). 25 This mutation was corrected using CRISPR/Cas9, and corrected isogenic DPSCs (cMD‐DPSCs) were thus generated. 25 Patient‐derived mutant DPSCs differentiated into DNs (MD‐DNs) without undergoing cell death. 26 However, mutant TRPV4‐mediated constitutive Ca2+ entry damaged mitochondrial function through mitochondrial Ca2+ overload and oxidative stress, leading to impaired neurite development in MD‐DNs compared to that in cMD‐DNs. 26
However, the exact mechanisms involved in mitochondrial Ca2+ overload in MD‐DNs remain unclear. Furthermore, effective treatments for neuropsychiatric symptoms of TRPV4‐related channelopathy have not been well‐established. Pharmacological compounds directly targeting TRPV4 are promising potential therapeutic candidates. 9 Contrastingly, folic acid (FA) is a well‐established and widely recommended natural supplement for pregnant women 27 that prevents birth defects, including neural tube defects. Although the exact mechanisms of its action are not fully understood yet, FA exerts its effects via multiple mechanisms, including epigenetic regulation as a methyl donor in one‐carbon metabolism, mitochondrial activation, and direct reactive oxygen species (ROS) scavenging. 28 , 29 , 30 , 31 In the present study, the neurodevelopmental defects in MD‐DNs were further tested using FA, and the neuropathological association between TRPV4‐mediated Ca2+ deregulation and Ca2+ overload‐induced mitochondrial pathology was examined based on the pharmacological effects of FA.
2. MATERIALS AND METHODS
2.1. DPSC isolation and culture
Experiments using human samples were reviewed and approved by the Kyushu University Institutional Review Board for Human Genome/Gene Research (permission number: 678‐03) and were conducted following the Declaration of Helsinki. Written informed consent was obtained from the patient's guardian.
DPSCs were isolated from a 14‐year‐old boy diagnosed with MD carrying a de novo mutation in TRPV4, c.1855C > T (p.L619F). 25 CRISPR/Cas9 was used to repair the mutation to obtain corrected DPSCs as previously described. 25
DPSC culture, differentiation into DNs, and the analyses were performed based on previously described protocols. 25 , 26 , 30 DPSCs were cultured in Alpha Modification of Eagle's medium (Nacalai Tesque) containing 15% fetal bovine serum (Sigma‐Aldrich), 100 μM L‐ascorbic acid 2‐phosphate (Wako Pure Chemical Industries, Osaka, Japan), 250 μg/mL fungizone (Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (Nacalai Tesque) at 37°C and 5% CO2.
2.2. DPSCs differentiating into DNs
Differentiation of DPSCs to DNs was performed using a two‐step procedure. In total, 1 × 105 DPSCs were seeded onto a six‐well plate and incubated overnight. Subsequently, the cells were cultured in first‐step medium, Dulbecco's modified Eagle medium (Nacalai Tesque) supplemented with 20 ng/mL epidermal growth factor (Peprotech), 20 ng/mL basic fibroblast growth factor (Peprotech), and 1% N2 supplement (Thermo Fisher Scientific) for 2 days. The medium was then replaced with second‐step medium, neurobasal media (Thermo Fisher Scientific) supplemented with 2% B27 supplement (Thermo Fisher Scientific), 1 mM dibutyryladenosine 3,5‐cyclic monophosphate (Nacalai Tesque), 0.5 mM 3‐isobutyl‐1‐methylxanthine (Sigma‐Aldrich), and 200 μM ascorbic acid (Nacalai Tesque). Then, the cells were cultured for 5 days. In addition, 50 μM FA (Wako Pure Chemical Industries), 0.1 μM pan‐AKT inhibitor AKT inhibitor VIII (Cayman, Ann Arbor), or 0.1 μM TRPV4 antagonist GSK2193874 (Cayman) were added from the second‐step.
2.3. RNA interference to knock down α‐synuclein (α‐syn) and p53 expression
The first‐step medium was replaced by the second‐step medium after the first step of DN differentiation, and small interfering RNA (siRNA) transfection was performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific). The siRNA sequences were as follows: α‐syn sense 5′‐GGA GCA GAA AGC AAU CGA UTT‐3′ and antisense 5′‐AUC GAU UGC UUU CUG CUC CTT‐3′; p53 sense 5′‐GAA AUU UGC GUG UGG AGU ATT‐3′ and antisense 5′‐UAC UCC ACA CGC AAA UUU CTT‐3′. The control siRNA was purchased from Sigma‐Aldrich, Burlington, MA, USA, SIC001‐10NMOL.
2.4. Immunocytochemistry and analyses of DN neurite outgrowth and branching
DNs were cultured on cover glasses and fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 10 min. Subsequently, the cells were permeabilized with 0.1% Triton X‐100 for 5 min. The cells were blocked with 2% bovine serum albumin in phosphate‐buffered saline (PBS) for 20 min and incubated with one of the following primary antibodies for 90 min: anti‐Tom20 (#sc‐17764, Santa Cruz Biotechnology), anti‐tyrosine hydroxylase (TH; #66334‐1‐Ig; Proteintech, IL, USA), anti‐α‐syn (#10842‐1‐AP; Proteintech), and anti‐β‐tubulin III antibodies (#T8578; Sigma‐Aldrich). The cells were subsequently incubated with Alexa Fluor‐conjugated secondary antibodies (Thermo Fisher Scientific). After staining with secondary antibodies in the dark for 60 min, the nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI; Dojindo). The cover glass was subsequently mounted on a slide using the ProLong Diamond mounting medium (Thermo Fisher Scientific). Fluorescence images were captured using a Nikon C2 confocal microscope (Nikon).
β‐Tubulin III‐ and DAPI‐stained pictures were acquired to measure the maximum neurite length and the total number of neurite branches in DNs, and 30 cells of each case were analyzed using the neurite outgrowth module of MetaMorph (version 7.8; Molecular Devices).
2.5. Measurement of mitochondrial localization of α‐syn
Tom20‐ and α‐syn‐stained pictures were acquired. Tom20 and α‐syn colocalization was analyzed based on Mander's coefficient using ImageJ software version 1.53 with JACop plugin. In total, 12 pictures from three experiments were analyzed in each case.
2.6. Measurement of mitochondrial membrane potential (MMP)
The cells were incubated with 1 μM JC‐1 (Wako Pure Chemical Industries) for 10 min to measure the MMP. They were then treated with TrypLE Express to detach them from the culture plates, and the JC‐1 red and green signals were measured using a FACSCalibur instrument (BD Biosciences). The medians of the red and green fluorescence signals were measured using Cell Quest version 3.3, and the ratio of red/green fluorescence was calculated.
2.7. Measurement of intracellular adenosine triphosphate (ATP) levels
DNs were harvested in ice‐cold PBS. Subsequently, the adenosine triphosphate (ATP) levels in DNs were measured using the CellTiter‐Glo Luminescent Cell Viability Assay (Promega), according to the manufacturer's instructions. The luminescent signals were detected using a SpectraMax iD3 plate reader (Molecular Devices).
2.8. Measurement of ATP synthase activity
The cells were collected by centrifugation at 800 g for 5 min at 4°C. The cells were subsequently resuspended in PBS and frozen at −20°C. The cells were thawed and the ATP synthase activity was measured using ATP synthase Specific Activity Microplate Assay Kit (Abcam), according to the manufacturer's instructions. The absorbance was measured using a SpectraMax iD3 plate reader (Molecular Devices).
2.9. Measurement of mitochondrial reactive oxygen species (ROS) levels
Mitochondrial ROS levels were determined using flow cytometry as previously described. 30 The cells were incubated with 5 μM of MitoSOX Red (Thermo Fisher Scientific) for 30 min and were subsequently treated with TrypLE Express (Thermo Fisher Scientific) to detach them from the culture plate. The fluorescence signal of 10,000 cells was measured using a FACSCalibur instrument (BD Biosciences). The medians of the fluorescence signals were measured using Cell Quest (version 3.3; BD Biosciences).
2.10. Measurement of cytosolic and mitochondrial calcium levels
DNs were incubated with 0.05% Pluronic F‐127 (Sigma‐Aldrich), 500 μM probenecid (Sigma‐Aldrich), 1 μM Fluo‐4 AM for 1 h (for detection of cytosolic calcium; Thermo Fisher Scientific), or 10 μM Rhod‐2 for 45 min (for detection of mitochondrial calcium; Dojindo) in Hanks' balanced salt solution. Rhod2 detects mitochondrial Ca2+ without damaging mitochondria in this condition. 30 The fluorescent signals were measured using a SpectraMax iD3 plate reader.
2.11. Quantitative reverse transcription polymerase chain reaction (qRT‐PCR)
The total RNA was extracted from cells using an RNAeasy Mini Kit (Qiagen). First‐strand cDNA was synthesized using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). The primer sequences were as follows: SNCA (NM_000345.4): 5′‐CTT GCC TTC AAG CCT TCT GC‐3′ (forward) and 5′‐ACC ACA CTG TCG TCG AAT GG‐3′ (reverse); p53 (NM_000546.6): 5′‐CAC ATG ACG GAG GTT GTG AG‐3′ (forward) and 5′‐ACA CGC AAA TTT CCT TCC AC‐3′ (reverse); RPL13A (NM_001270491.2): 5′‐GCT GTG AAG GCA TCA ACA TTT‐3′ (forward) and 5′‐CAT CCG CTT TTT CTT GTC GTA‐3′ (reverse); and 18S rRNA (NM_001348076.2): 5′‐CGG CTA CCA CAT CCA AGG AA‐3′ (forward) and 5′‐GCT GGA ATT ACC GCG GCT‐3′ (reverse). Quantitative real‐time PCR was performed using the GoTaq qPCR Master Mix (Promega) and analyzed using the QuantStudio 3 Real‐Time PCR System (Thermo Fisher Scientific). The relative expressions of the target genes were analyzed using the comparative threshold cycle method via normalization against the RPL13A levels (for SNCA) or the 18S rRNA levels (for p53).
2.12. Western blotting
The cells were lysed with the sample buffer (62.5 mM Tris–HCl buffer [pH 6.8] containing 2% sodium dodecyl sulfate [SDS], 5% β‐mercaptoethanol, and 10% glycerol) and incubated at 95°C for 5 min. The proteins in the cell lysates were electrophoresed using SDS‐polyacrylamide gel electrophoresis, and immunoblotting was performed using the following antibodies: anti‐α‐syn (#10842‐1‐AP; Proteintech), anti‐AKT (#2920S; Cell Signaling Technology), anti‐Phospho‐AKT Ser473 (#66444‐1‐Ig; Proteintech), anti‐Phospho‐GSK3β Ser9 (#67558‐1‐Ig; Proteintech), anti‐β‐actin (#66009‐1‐Ig; Proteintech), horseradish peroxidase (HRP)‐linked goat polyclonal anti‐mouse IgG (#7076S; Cell Signaling Technology), and HRP‐linked goat polyclonal anti‐rabbit IgG (#7074S; Cell Signaling Technology).
The immunoreactive bands were detected using ECL Prime (Cytiva) and analyzed using LAS‐1000 plus (FujiFilm) and Image Gauge (FujiFilm). α‐Tubulin was considered the internal control. To normalize the protein levels, the chemiluminescent signals of the objective proteins were divided by that of β‐actin.
2.13. Statistical analyses
Statistical analyses were performed using one‐way ANOVA with Prism9 (GraphPad). Values are presented as the mean ± standard error of the mean (SEM) and p < 0.05 indicates statistical significance.
3. RESULTS
3.1. FA improved the outgrowth of neurites, levels of mitochondrial Ca2+, and mitochondrial function of patient‐derived DNs
The effects of FA supplementation on the developmental defects in MD‐DNs were evaluated. FA improved neurite outgrowth and branch formation in MD‐DNs to levels similar to those in untreated cMD‐DNs (Figure 1A,B). The transcription factors NURR1 and PITX3 are essential for DN differentiation, and their mRNA expression and downstream target TH were comparable between the two groups, unaffected by FA (Figure S1A). Furthermore, TRPV4 mutation and FA did not affect transient dopamine secretion in response to potassium chloride (KCl) stimulation (Figure S1B), suggesting that accelerated Ca2+ entry by mutant TRPV4 supported, rather than interfered with, the secretion of KCl‐induced transient dopamine.
FIGURE 1.
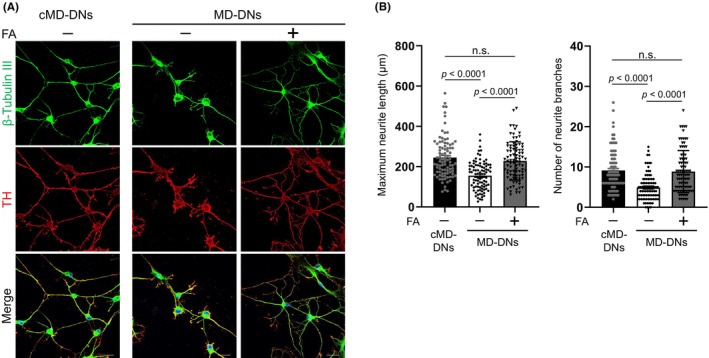
Effect of folic acid (FA) on neurite outgrowth of patient‐derived dopaminergic neurons (MD‐DNs). Dental pulp stem cells were differentiated into DNs in the absence or presence of FA. (A) DNs were visualized by immunofluorescence microscopy using anti‐β‐tubulin III and anti‐TH antibodies. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI). Scale bars = 20 μm. (B) Maximum neurite length and the total number of branches per DN were measured. The mean ± standard error of the mean (SEM) was calculated after analyzing 30 cells from three independent experiments. DNs, dopaminergic neurons; FA, folic acid; MD, metatropic dysplasia; n.s., not significant.
Next, we investigated the effects of FA on regulating Ca2+ entry caused by a gain‐of‐function mutation of TRPV4. In the presence of FA, MD‐DNs maintained high cytosolic Ca2+ levels, but exhibited reduced mitochondrial Ca2+ levels that were comparable to those of untreated cMD‐DNs (Figure 2A). Consistent with the normalized mitochondrial Ca2+ levels, mitochondrial ROS and MMP in FA‐treated MD‐DNs were also reduced and were comparable to those in cMD‐DNs without FA (Figure 2B; Figures S2 and S3). Additionally, the FA exhibited positive effects on mitochondrial ATP synthase activity and ATP levels in MD‐DNs (Figure 2B).
FIGURE 2.
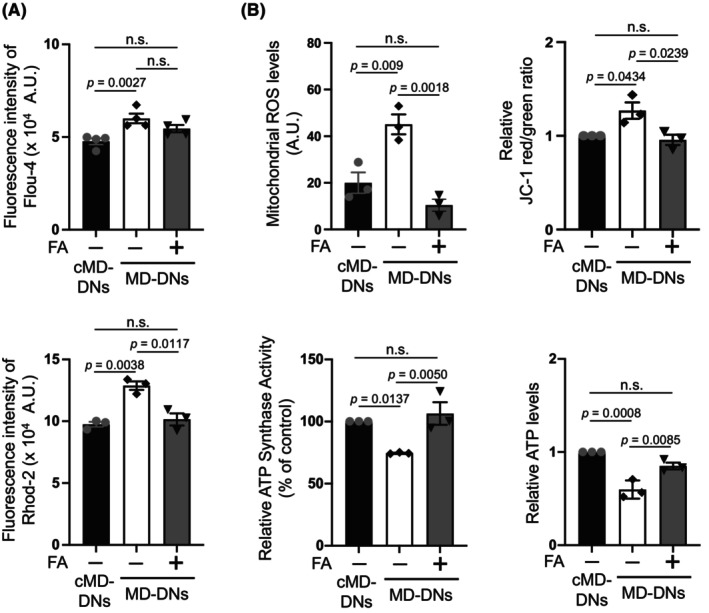
Effects of folic acid (FA) on mitochondrial Ca2+ levels and function in patient‐derived dopaminergic neurons (MD‐DNs). (A) Cytosolic (upper panel) and mitochondrial (lower panel) Ca2+ levels in DNs were measured using Fluo‐4 AM and Rhod2‐AM, respectively. The mean ± standard error of the mean (SEM) was obtained from four (cytosolic Ca2+) or three (mitochondrial Ca2+) independent experiments. (B) Mitochondrial reactive oxygen species (ROS), membrane potential, adenosine triphosphate (ATP) synthase activity, and ATP levels were measured. The mean ± SEM was obtained from three independent experiments. n.s., not significant.
Thus, FA specifically ameliorated the constitutive accumulation of mitochondrial Ca2+ mediated by mutant TRPV4 to improve mitochondrial function and neurite outgrowth in MD‐DNs.
3.2. Mitochondrial Ca2+ accumulation is associated with α‐syn upregulation in patient‐derived DNs
The possible mechanisms underlying the specific reduction in mitochondrial Ca2+ levels by FA in MD‐DNs were elucidated by focusing on α‐syn because it can localize to the mitochondria 32 and may increase Ca2+ permeability of voltage‐dependent anion channel (VDAC) on the mitochondrial outer membrane (MOM). 33 qRT‐PCR and western blotting revealed that the α‐syn transcript and whole protein were increased in MD‐DNs, and FA reduced their levels, similar to those in untreated cMD‐DNs (Figure 3A; Figure S4). Consistent with these alterations, FA reduced the immunofluorescence signal intensity of α‐syn compared to that of mitochondria in MD‐DNs (Figure 3B; Figure S5). Thus, increased mitochondrial α‐syn possibly causes mitochondrial Ca2+ accumulation in MD‐DNs, and FA might potentially target α‐syn to ameliorate mitochondrial Ca2+ accumulation.
FIGURE 3.
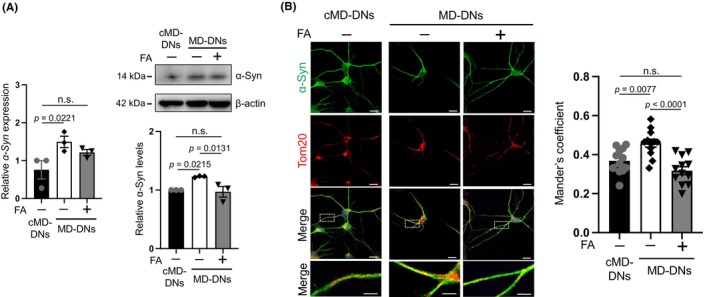
Effects of folic acid (FA) on α‐synuclein (α‐syn) downregulation in patient‐derived dopaminergic neurons (MD‐DNs). Dental pulp stem cells were differentiated into DNs in the absence or presence of FA. (A) α‐syn expression in DNs was measured by quantitative reverse transcription polymerase chain reaction (qRT‐PCR) and western blotting. The mean ± standard error of the mean (SEM) was obtained from three independent experiments. n.s., not significant. (B) Immunofluorescence microscopy was used to visualize DNs using anti‐α‐syn and anti‐Tom20 antibodies, a mitochondrial marker. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI). Scale bars = 20 μm. Boxed regions on the merged images are shown at a greater magnification in the right panels. Scale bars = 5 μm. Tom20 and α‐syn colocalization were analyzed through Mander's coefficient. Fifteen DNs were analyzed in each case. The mean ± SEM was obtained from three independent experiments. In total, 12 pictures from three experiments were analyzed in each case.
siRNA was used to knock down α‐syn to further examine the association between α‐syn and mitochondrial Ca2+ levels in MD‐DNs. siRNA reduced the protein expression levels of α‐syn in MD‐DNs to levels comparable to that in untreated cMD‐DNs (Figure 4A; Figure S6). Under this condition, mitochondrial Ca2+ levels, MMP, and mitochondrial ROS levels in MD‐DNs were reduced and were comparable to those in untreated cMD‐DNs (Figure 4B; Figures S7 and S8).
FIGURE 4.
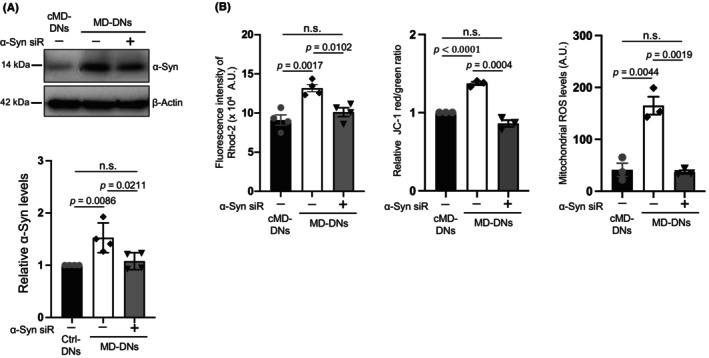
Knockdown effects of α‐synuclein (α‐syn) on mitochondrial Ca2+ levels and function in patient‐derived dopaminergic neurons (MD‐DNs). Dental pulp stem cells were transfected with negative control‐ and α‐syn‐siRNA (α‐syn‐siR) and differentiated into DNs. (A) α‐syn expression in DNs was measured by western blotting. The mean ± standard error of the mean (SEM) was obtained from four independent experiments. (B) Mitochondrial Ca2+, membrane potential, and ROS levels were measured. The mean ± SEM was obtained from four (mitochondrial Ca2+) or three (membrane potential and ROS levels) independent experiments. n.s., not significant.
Thus, mitochondrial Ca2+ accumulation is associated with α‐syn upregulation in MD‐DNs, and FA may regulate mitochondrial Ca2+ levels via α‐syn downregulation.
Mitochondrial Ca2+ is primarily transported from the endoplasmic reticulum (ER) at the proximal contact region between mitochondria and ER, the mitochondria‐associated ER membrane (MAM), via the Ca2+ transport machinery that includes inositol 1,4,5‐trisphosphate receptors (IP3R) on the ER membrane and VDAC on the MOM. 34 , 35 FA effects on ER Ca2+ levels were examined given that the ER Ca2+ levels are closely related to the mitochondrial Ca2+ levels. ER Ca2+ levels were not clearly elevated in the untreated MD‐DNs compared to that in the untreated cMD‐DNs, which were not affected by FA (Figure S9A). Additionally, TRPV4 mutation or FA supplementation did not affect the mitochondria and ER contact region and VDAC1 protein expression (Figures S9B,C and S10). Thus, ER Ca2+ levels were unaffected by TRPV4 mutation and FA might not contribute to regulating ER Ca2+ levels and the MAM containing the Ca2+ transport machinery.
3.3. Upregulation of α‐syn was associated with Ca2+‐dependent AKT activation in patient‐derived DNs
AKT has been previously shown to upregulate α‐syn. 36 The possible mechanisms of increased α‐syn in MD‐DNs were clarified by examining the effects of a pan‐AKT inhibitor, AKT inhibitor VIII, which was added in the second‐step medium of DN induction. The AKT inhibitor (0.1 μM) downregulated α‐syn in MD‐DNs to levels similar to that in untreated cMD‐DNs (Figure 5A). GSK3β phosphorylated at serine 9 residue (as a positive control for AKT activation) was increased in MD‐DNs, which was decreased by the AKT inhibitor (Figure 5A; Figure S11). Considering that GSK3β is a well‐established target of AKT, 37 these data revealed the constitutive activation of AKT in MD‐DNs and the successful effect of the AKT inhibitor. Under these conditions, mitochondrial Ca2+ levels were decreased without affecting cytosolic Ca2+ levels (Figure 5B). To investigate the role of α‐syn and AKT in mitochondrial Ca2+ accumulation in MD‐DNs, we examined α‐syn expression and mitochondrial Ca2+ levels at the early time points after induction of DN differentiation. Two days after the second‐step medium change with the AKT inhibitor of DN induction, α‐syn was upregulated, but mitochondrial Ca2+ levels were not yet elevated in MD‐DNs; therefore, AKT inhibitor was observed to have no effect (Figures S12–S14). These data suggested that AKT was constitutively activated in MD‐DNs, contributing to α‐syn upregulation and mitochondrial Ca2+ accumulation was ongoing during DN differentiation.
FIGURE 5.
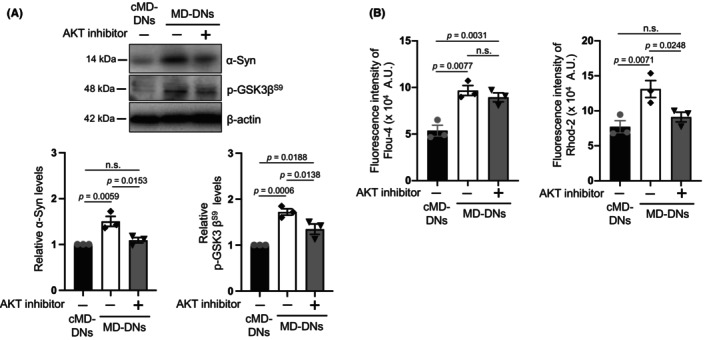
Effects of AKT inhibitor on α‐synuclein (α‐syn) expression and mitochondrial Ca2+ levels in patient‐derived dopaminergic neurons (MD‐DNs). Dental pulp stem cells were differentiated into DNs in the absence or presence of AKT inhibitor VIII. (A) Levels of α‐Syn and phosphorylated GSK3β in DNs were measured using western blotting. The mean ± standard error of the mean (SEM) was obtained from three independent experiments. (B) Cytosolic (left panel) and mitochondrial (right panel) Ca2+ in DNs were measured using Fluo‐4 AM and Rhod2‐AM, respectively. The mean ± SEM was obtained from three independent experiments. n.s., not significant.
The effects of the selective TRPV4 antagonist GSK2193874 38 were examined to clarify the relationship between constitutive AKT activation in MD‐DNs and a gain‐of‐function mutation in TRPV4. GSK2193874 (0.1 μM) suppressed both cytosolic and mitochondrial Ca2+ levels in MD‐DNs to the same extent as those in untreated cMD‐DNs (Figure 6A). As serine 473 of AKT needs to be phosphorylated for its full activation, in these conditions, the protein expression of AKT phosphorylated at serine 473 residue (p‐AKT S473) was examined. 39 MD‐DNs showed increased p‐AKT S473, which was reduced by the TRPV4 antagonist to an extent similar to that in untreated cMD‐DNs (Figure 6B; Figure S15). Expression of α‐syn in MD‐DNs was also reduced by GSK2193874, paralleling the alteration in expression of p‐AKT S473 (Figure 6B; Figure S15). In contrast to the inhibitory effect of GSK2193874 on AKT phosphorylation, FA did not affect the levels of p‐AKT S473 (Figure 6C; Figure S16). Therefore, AKT in MD‐DNs might be constitutively activated by accelerated Ca2+ entry into cells via mutant TRPV4, and Ca2+‐related AKT activation might not be the target of FA.
FIGURE 6.
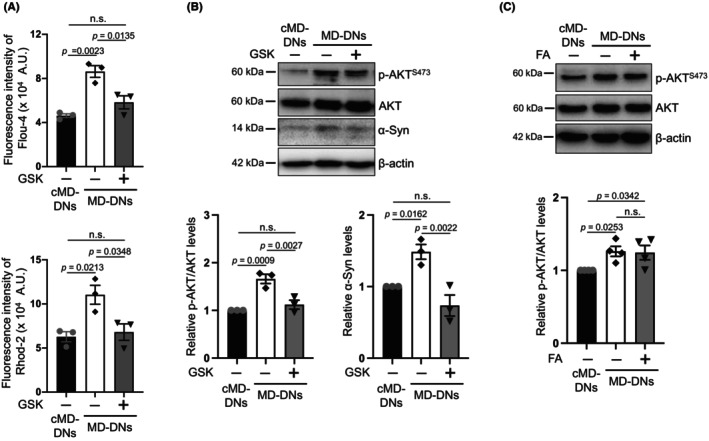
TRPV4 antagonist, but not folic acid (FA), blocked Ca2+ entry and AKT phosphorylation in patient‐derived dopaminergic neurons (MD‐DNs). Dental pulp stem cells were differentiated into DNs in the absence or presence of TRPV4 antagonist GSK2193874 (GSK) or FA. (A) Cytosolic (upper panel) and mitochondrial (bottom panel) Ca2+ levels in DNs were measured using Fluo‐4 AM and Rhod2‐AM, respectively. The mean ± standard error of the mean (SEM) was obtained from three independent experiments. (B) AKT with Ser‐473 phosphorylation (p‐AKT S473) and α‐syn levels of DNs in the absence or presence of GSK were measured using western blotting. The mean ± SEM was obtained from three independent experiments. (C) p‐AKT S473 levels of DNs in the absence or presence of FA were measured using western blotting. The mean ± SEM was obtained from four independent experiments. n.s., not significant.
3.4. FA suppressed p53 expression contributing to α‐syn downregulation in patient‐derived DNs
FA produces S‐adenosylmethionine, the universal methyl donor for methylation reactions, via one‐carbon metabolism and contributes to the epigenetic regulation of gene expression, including CpG methylation. 31 CpG methylation status in the regulatory regions of SNCA is involved in the transcriptional regulation of α‐syn expression. 40 However, no significant difference was detected among the methylation statuses of the CpG island (CGI) in the first intron of SNCA 41 , 42 , 43 for cMD‐DNs, MD‐DNs, and FA‐supplemented MD‐DNs (Figure S17; Table S1).
The tumor suppressor p53 is a transcription factor that binds to the promoter region of SNCA to upregulate its expression. 44 Additionally, p53 expression is suppressed in the kidneys of rats fed an FA‐supplemented diet. 45 In the present study, the p53 expression levels were investigated as a possible mechanism underlying α‐syn downregulation by FA. No difference was observed between the levels of p53 transcripts for cMD‐DNs and MD‐DNs under untreated conditions (Figure 7A). Compared to that in the untreated groups, p53 in MD‐DNs treated with FA was downregulated (Figure 7A). To further investigate the possible role of p53 in α‐syn upregulation in MD‐DNs, p53 was silenced by siRNA (Figure 7B). qRT‐PCR analysis indicated that α‐syn was downregulated in MD‐DNs by p53 silencing (Figure 7B). These data suggested that p53 can potentially be targeted by FA for α‐syn downregulation in MD‐DNs.
FIGURE 7.
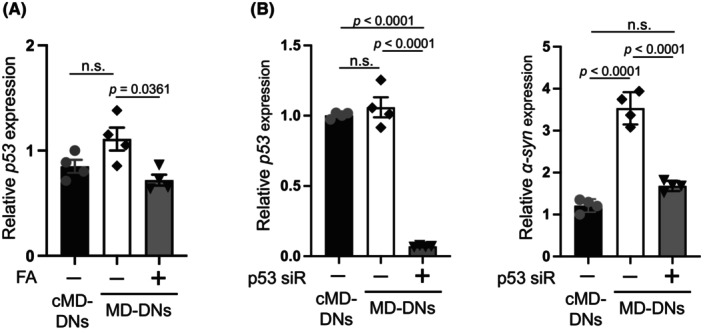
Expression of p53 was suppressed by folic acid (FA), which is related to the expression of α‐syn in patient‐derived dopaminergic neurons (MD‐DNs). (A) Dental pulp stem cells were differentiated into DNs in the absence or presence of FA. p53 expression was measured by quantitative reverse transcription polymerase chain reaction (qRT‐PCR). The mean ± standard error of the mean (SEM) was obtained from four independent experiments. n.s., not significant. (B) Dental pulp stem cells were transfected with negative control‐ and p53‐siRNA (p53‐siR) and differentiated into DNs. p53 and α‐syn expression was measured by qRT‐PCR. The mean ± standard error of the mean (SEM) was obtained from four independent experiments. n.s., not significant.
4. DISCUSSION
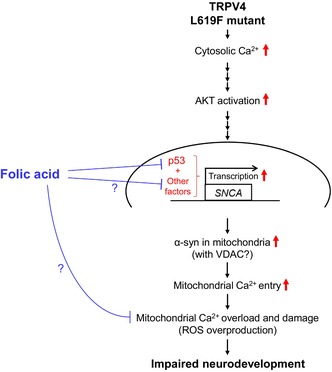
The pathological roles of the mutant TRPV4 in neurodevelopment and effective pharmacological intervention are not fully understood yet. In this study, these issues were addressed using DPSCs with neural plasticity obtained from a patient carrying a gain‐of‐function mutation of TRPV4. Based on the examined pharmacological effects of FA, we further analyzed the pathological relationship between TRPV4‐mediated Ca2+ deregulation and mitochondrial dysfunction in MD‐DNs. In MD‐DNs without FA supplementation, accelerated Ca2+ entry activated AKT‐mediated α‐syn induction, resulting in mitochondrial Ca2+ overload. FA attenuated mitochondrial Ca2+ accumulation along with α‐syn downregulation without affecting cytosolic and ER Ca2+ levels and effectively ameliorated mitochondrial function and neurite outgrowth in MD‐DNs. FA also downregulated p53, which is possibly associated with α‐syn downregulation in FA‐supplemented MD‐DNs. A schematic of the tentative pathway hypothesized by the current study is shown in Figure 8.
FIGURE 8.
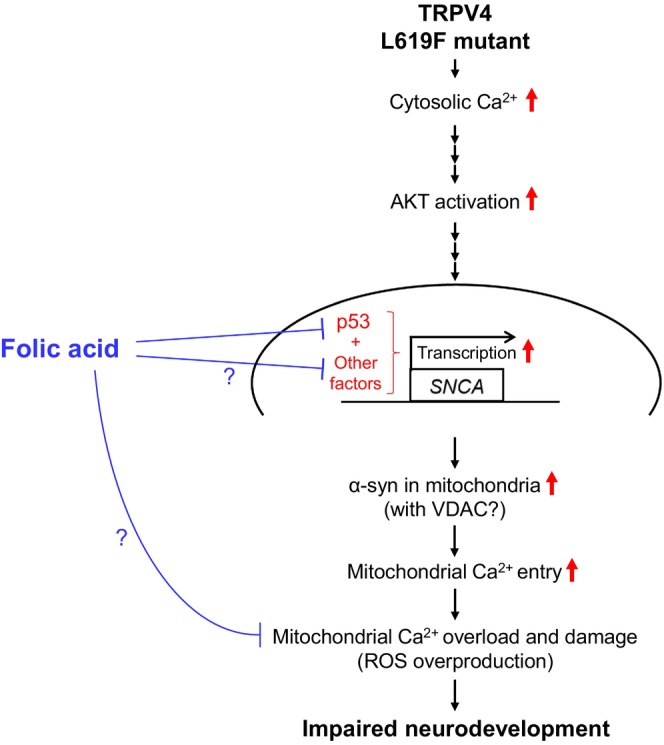
Schematic of the tentative pathway hypothesized in the current study.
TRPV4 participates in the Ca2+‐dependent AKT pathway in the central nervous system, which is either positively 46 , 47 , 48 or negatively 49 , 50 , 51 regulated depending on the cell types and pathological conditions. However, the physiological and pathological roles of the TRPV4‐mediated AKT pathway during neurodevelopment is not fully understood yet. In this study, a constitutive elevation of cytosolic Ca2+ levels was found along with the upregulation of p‐AKT S473 in MD‐DNs compared to that in cMD‐DNs. However, the TRPV4 antagonist GSK2193874 suppressed cytosolic Ca2+ and p‐AKT S473 levels in MD‐DNs. Serine 473 phosphorylation in the hydrophobic C‐terminal regulatory domain following threonine 308 phosphorylation in the catalytic domain is required for complete AKT activation. 52 Our data suggest that the constitutive elevation of cytosolic Ca2+ via the mutant TRPV4 enhanced complete AKT activation. However, the molecular mechanisms underlying Ca2+‐dependent AKT activation in MD‐DNs remain elusive. Phosphatidylinositol‐3 kinase (PI3K)/AKT pathway is the canonical pathway for AKT activation 52 and is activated in nonneuronal cells stimulated with TRPV4 agonist. 53 , 54 , 55 Calcium–calmodulin (Ca2+–CaM) cascade is the noncanonical pathway for Ca2+‐dependent AKT activation. 56 , 57 , 58 In certain squamous cell carcinomas with high TRPV4 expression, calcium/calmodulin‐dependent protein kinase II promotes tumor growth and is involved in Ca2+‐dependent AKT activation. 59 Although further analysis is required for the involvement of these molecules, a gain‐of‐function TRPV4 mutation c.1855C > T (p.L619F) might cause constitutive AKT activation in a Ca2+‐dependent manner during neurodevelopment.
AKT has diverse cellular effects depending on the physiological and pathological conditions in different cell types. One of the downstream targets of AKT is α‐syn. 36 In MD‐DNs, both the mRNA and protein of α‐syn were upregulated, which was reversed by an AKT inhibitor, suggesting that AKT was responsible for inducing α‐syn in MD‐DNs. Moreover, the data using an AKT inhibitor indicated that activated AKT might contribute to the mitochondrial Ca2+ accumulation in MD‐DNs. Mitochondrial Ca2+ accumulation is caused by excessive Ca2+ entry and/or impaired Ca2+ efflux. 60 The primary Ca2+ entry site of mitochondria is VDAC and the mitochondrial calcium uniporter (mtCU) in the outer and inner membranes, respectively. Various other additional molecules are involved in regulating Ca2+ entry at these channels. 60 A part of α‐syn localizes to the mitochondria and regulates mitochondrial functions. 32 Mitochondrial α‐syn contributes to Ca2+ transportation by supporting the ER–mitochondria contact 32 and/or by interacting with VDAC to facilitate Ca2+ entry. 33 The current data revealed that the VDAC1 expression and mitochondria–ER contact area were unaffected by the TRPV4 mutant. However, immunofluorescence analysis suggested an increase in mitochondrial α‐syn in correlation with increased whole α‐syn in MD‐DNs. Additionally, the α‐syn knockdown was effective in reducing mitochondrial Ca2+ levels in MD‐DNs. Thus, constitutive AKT activation might enhance α‐syn induction to promote α‐syn/VDAC1‐mediated Ca2+ entry and participate in mitochondrial Ca2+ overload in MD‐DNs.
Mitochondrial Ca2+ overload damages mitochondrial function via multiple mechanisms, including ROS overproduction, which result in various adverse effects on cellular events. 61 , 62 , 63 This is possibly a mechanism involved in the developmental defects of MD‐DNs. 26 In the current study, FA was tested as a potential therapeutic candidate targeting mitochondrial factors to ameliorate the developmental defects of MD‐DNs. 28 , 29 , 30 , 31 FA normalized mitochondrial Ca2+, ROS levels, and MMPs, and restored ATP levels, improving neurite outgrowth in MD‐DNs. Additionally, FA was effective in reducing mitochondrial α‐syn in correlation with the downregulation of whole α‐syn. However, cytosolic and ER Ca2+ levels as well as VDAC1 expression were unaffected by FA supplementation. Considering the regulatory role of α‐syn in mitochondrial Ca2+ transportation, the pharmacological effects of FA may include ameliorating mitochondrial Ca2+ overload by suppressing Ca2+ entry into mitochondria via α‐syn downregulation in a cytosolic/ER Ca2+‐independent manner.
This study proposed that a potential mechanism underlying α‐syn downregulation by FA might involve the tumor suppressor p53. Multiple candidate factors of epigenetic, cis, and trans elements that regulate α‐syn transcription have been investigated. 64 p53, a trans elements, is a transcriptional regulator that directly binds to the promoter region of SNCA and upregulates its expression. 44 Conversely, FA supplementation suppresses p53 expression through epigenetic modification in the rat kidney in vivo, which is possibly associated with one‐carbon metabolism. 45 In the present study, p53 transcripts were downregulated in FA‐treated MD‐DNs compared to those in untreated MD‐DNs. Furthermore, p53 siRNA potently suppressed p53 transcripts in MD‐DNs compared with those in untreated groups, and was effective in α‐syn downregulation in MD‐DNs. However, α‐syn expression levels of p53 siRNA‐treated MD‐DNs remained comparable to those of untreated cMD‐DNs, suggesting the involvement of p53‐independent pathways in α‐syn suppression in MD‐DNs. The molecular mechanisms underlying FA‐mediated α‐syn downregulation have not been comprehensively elucidated in this study; p53 might be a potential target of FA that may contribute to α‐syn downregulation.
This study has many limitations. First, the signaling pathways involved in Ca2+‐dependent AKT activation and α‐syn induction in MD‐DNs are not fully elucidated. The involvement of PI3K and CaM‐related signaling molecules and genetic/epigenetic factors regulating SNCA require further clarification. Second, the exact mechanisms underlying TRPV4‐mediated mitochondrial Ca2+ overload remain elusive. Considering the multiple pathways downstream of AKT and FA, α‐syn‐independent factors may be involved in mitochondrial Ca2+ accumulation in MD‐DNs. Further experiments, including the overexpression of α‐syn in MD‐DNs in conjunction with either AKT inhibitor or FA treatment, are needed for better comprehension. Additionally, TRPV4 is possibly recruited to the mitochondria to regulate Ca2+ entry. 65 Third, the exact mechanisms underlying the pharmacological effects of FA on mitochondria and neurite development in MD‐DNs are not fully elucidated yet. FA‐induced alterations of the epigenetic modification of SNCA and expression of transcription factors need to be further analyzed. Fourth, the mechanisms underlying the neuropathology of the dopaminergic system in TRPV4‐related MD are not fully understood yet. Analyses of the neural distribution of the dopaminergic system, dopamine levels, and α‐syn expression in the brain are required to elucidate the TRPV4‐related dopaminergic phenotype in patients.
In conclusion, accelerated cytosolic Ca2+ entry because of the gain‐of‐function mutation c.1855C > T (p.L619F) in TRPV4 contributed to AKT/α‐syn‐mediated mitochondrial Ca2+ overload and dysfunction during DN development. FA normalized mitochondrial Ca2+ levels via transcriptional repression of α‐syn without affecting cytosolic/ER Ca2+ levels and AKT activation, improving mitochondrial function and neurite outgrowth in MD‐DNs. Thus, the patient's teeth‐derived stem cells are a promising cellular model for elucidating the mechanisms underlying the neuropathology of TRPV4‐related MD and for testing the pharmacological effects of potential therapeutic agents.
AUTHOR CONTRIBUTIONS
K. Masuda and H. Kato conceived and designed the research; X. Sun, J. Kong, and S. Dong performed the experiments, acquired and analyzed the data; H. Sato, Y. Hirofuji, Y. Ito, and L. Wang were involved in data interpretation and discussion; T.A. Kato, M. Torio, Y. Sakai, and S. Ohga supervised the research; K. Masuda and S. Fukumoto administered the research. All authors were involved in drafting and revising the manuscript.
DISCLOSURES
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
Supporting information
Data S1.
ACKNOWLEDGMENTS
We thank the members of the Department of Pediatric Dentistry and Special Needs Dentistry at Kyushu University Hospital for their valuable suggestions, technical support, and materials. We appreciate the technical assistance that was provided by the Research Support Center at the Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences. This work was supported by the Japan Society for the Promotion of Science [KAKENHI; grant numbers, JP19K10387, JP19K10406, JP21K17163, JP23K09417, and JP23K09439].
Sun X, Kong J, Dong S, et al. TRPV4‐mediated Ca2+ deregulation causes mitochondrial dysfunction via the AKT/α‐synuclein pathway in dopaminergic neurons. FASEB BioAdvances. 2023;5:507‐520. doi: 10.1096/fba.2023-00057
Xiao Sun and Jun Kong have contributed equally to this work and are therefore co‐first authors.
Contributor Information
Satoshi Fukumoto, Email: fukumoto@dent.kyushu-u.ac.jp.
Keiji Masuda, Email: kemasuda@dent.kyushu-u.ac.jp.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Liedtke W, Choe Y, Martí‐Renom MA, et al. Vanilloid receptor‐related osmotically activated channel (VR‐OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695‐702. [DOI] [PubMed] [Google Scholar]
- 3. Wissenbach U, Bödding M, Freichel M, Flockerzi V. Trp12, a novel Trp related protein from kidney. FEBS Lett. 2000;485:127‐134. [DOI] [PubMed] [Google Scholar]
- 4. Garcia‐Elias A, Mrkonjić S, Jung C, Pardo‐Pastor C, Vicente R, Valverde MA. The TRPV4 channel. Handb Exp Pharmacol. 2014;222:293‐319. [DOI] [PubMed] [Google Scholar]
- 5. Voets T, Prenen J, Vriens J, et al. Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem. 2002;277:33704‐33710. [DOI] [PubMed] [Google Scholar]
- 6. Kumar H, Lee SH, Kim KT, Zeng X, Han I. TRPV4: a sensor for homeostasis and pathological events in the CNS. Mol Neurobiol. 2018;55:8695‐8708. [DOI] [PubMed] [Google Scholar]
- 7. Verma P, Kumar A, Goswami C. TRPV4‐mediated channelopathies. Channels. 2010;4:319‐328. [DOI] [PubMed] [Google Scholar]
- 8. Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14:152‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grace MS, Bonvini SJ, Belvisi MG, McIntyre P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol Ther. 2017;177:9‐22. [DOI] [PubMed] [Google Scholar]
- 10. Krakow D, Vriens J, Camacho N, et al. Mutations in the gene encoding the calcium‐permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet. 2009;84:307‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Camacho N, Krakow D, Johnykutty S, et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A. 2010;152a:1169‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PloS One. 2011;6:e19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang GT, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res. 2009;88:792‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miura M, Gronthos S, Zhao M, et al. SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A. 2003;100:5807‐5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci U S A. 2000;97:13625‐13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Sha XJ, Li GH, et al. Comparative characterization of stem cells from human exfoliated deciduous teeth and dental pulp stem cells. Arch Oral Biol. 2012;57:1231‐1240. [DOI] [PubMed] [Google Scholar]
- 17. Kanafi M, Majumdar D, Bhonde R, Gupta P, Datta I. Midbrain cues dictate differentiation of human dental pulp stem cells towards functional dopaminergic neurons. J Cell Physiol. 2014;229:1369‐1377. [DOI] [PubMed] [Google Scholar]
- 18. Fujii H, Matsubara K, Sakai K, et al. Dopaminergic differentiation of stem cells from human deciduous teeth and their therapeutic benefits for parkinsonian rats. Brain Res. 2015;1613:59‐72. [DOI] [PubMed] [Google Scholar]
- 19. Majumdar D, Kanafi M, Bhonde R, Gupta P, Datta I. Differential neuronal plasticity of dental pulp stem cells from exfoliated deciduous and permanent teeth towards dopaminergic neurons. J Cell Physiol. 2016;231:2048‐2063. [DOI] [PubMed] [Google Scholar]
- 20. Klein MO, Battagello DS, Cardoso AR, Hauser DN, Bittencourt JC, Correa RG. Dopamine: functions, signaling, and association with neurological diseases. Cell Mol Neurobiol. 2019;39:31‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang M, Ling KH, Tan JJ, Lu CB. Development and differentiation of midbrain dopaminergic neuron: from bench to bedside. Cells. 2020;9:1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Areal LB, Blakely RD. Neurobehavioral changes arising from early life dopamine signaling perturbations. Neurochem Int. 2020;137:104747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prakash N. Developmental pathways linked to the vulnerability of adult midbrain dopaminergic neurons to neurodegeneration. Front Mol Neurosci. 2022;15:1071731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masuda K, Han X, Kato H, et al. Dental pulp‐derived mesenchymal stem cells for modeling genetic disorders. Int J Mol Sci. 2021;22:2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nonaka K, Han X, Kato H, et al. Novel gain‐of‐function mutation of TRPV4 associated with accelerated chondrogenic differentiation of dental pulp stem cells derived from a patient with metatropic dysplasia. Biochem Biophys Rep. 2019;19:100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun X, Kato H, Sato H, et al. Impaired neurite development and mitochondrial dysfunction associated with calcium accumulation in dopaminergic neurons differentiated from the dental pulp stem cells of a patient with metatropic dysplasia. Biochem Biophys Rep. 2021;26:100968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fekete K, Berti C, Cetin I, Hermoso M, Koletzko BV, Decsi T. Perinatal folate supply: relevance in health outcome parameters. Matern Child Nutr. 2010;6(Suppl 2):23‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joshi R, Adhikari S, Patro BS, Chattopadhyay S, Mukherjee T. Free radical scavenging behavior of folic acid: evidence for possible antioxidant activity. Free Radic Biol Med. 2001;30:1390‐1399. [DOI] [PubMed] [Google Scholar]
- 29. Ormazabal A, Casado M, Molero‐Luis M, et al. Can folic acid have a role in mitochondrial disorders? Drug Discov Today. 2015;20:1349‐1354. [DOI] [PubMed] [Google Scholar]
- 30. Sun X, Dong S, Kato H, et al. Mitochondrial calcium‐triggered oxidative stress and developmental defects in dopaminergic neurons differentiated from deciduous teeth‐derived dental pulp stem cells with MFF insufficiency. Antioxidants. 2022;11:1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bekdash RA. Methyl donors, epigenetic alterations, and brain health: understanding the connection. Int J Mol Sci. 2023;24:2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vicario M, Cieri D, Brini M, Calì T. The close encounter between alpha‐Synuclein and mitochondria. Front Neurosci. 2018;12:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosencrans WM, Aguilella VM, Rostovtseva TK, Bezrukov SM. α‐Synuclein emerges as a potent regulator of VDAC‐facilitated calcium transport. Cell Calcium. 2021;95:102355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3‐sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744‐747. [DOI] [PubMed] [Google Scholar]
- 35. Lee S, Min KT. The Interface between ER and mitochondria: molecular compositions and functions. Mol Cells. 2018;41:1000‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gandelman M, Dansithong W, Kales SC, et al. The AKT modulator A‐443654 reduces α‐synuclein expression and normalizes ER stress and autophagy. J Biol Chem. 2021;297:101191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature. 1995;378:785‐789. [DOI] [PubMed] [Google Scholar]
- 38. Cheung M, Bao W, Behm DJ, et al. Discovery of GSK2193874: an orally active, potent, and selective blocker of transient receptor potential Vanilloid 4. ACS Med Chem Lett. 2017;8:549‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser‐473 kinase as DNA‐dependent protein kinase. J Biol Chem. 2004;279:41189‐41196. [DOI] [PubMed] [Google Scholar]
- 40. Schaffner SL, Kobor MS. DNA methylation as a mediator of genetic and environmental influences on Parkinson's disease susceptibility: impacts of alpha‐Synuclein, physical activity, and pesticide exposure on the epigenome. Front Genet. 2022;13:971298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation regulates alpha‐synuclein expression and is decreased in Parkinson's disease patients' brains. J Neurosci. 2010;30:6355‐6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsumoto L, Takuma H, Tamaoka A, et al. CpG demethylation enhances alpha‐synuclein expression and affects the pathogenesis of Parkinson's disease. PloS One. 2010;5:e15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Wang X, Li R, et al. A DNA methyltransferase inhibitor, 5‐aza‐2′‐deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson's disease‐related genes in dopaminergic neurons. CNS Neurosci Ther. 2013;19:183‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duplan E, Giordano C, Checler F, Alves da Costa C. Direct α‐synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener. 2016;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He X, Xie Z, Dong Q, Li J, Li W, Chen P. Effect of folic acid supplementation on renal phenotype and Epigenotype in early weanling intrauterine growth retarded rats. Kidney Blood Press Res. 2015;40:395‐402. [DOI] [PubMed] [Google Scholar]
- 46. Lu KT, Huang TC, Tsai YH, Yang YL. Transient receptor potential vanilloid type 4 channels mediate Na‐K‐Cl‐co‐transporter‐induced brain edema after traumatic brain injury. J Neurochem. 2017;140:718‐727. [DOI] [PubMed] [Google Scholar]
- 47. Ou‐Yang Q, Li B, Xu M, Liang H. TRPV4 promotes the migration and invasion of glioma cells via AKT/Rac1 signaling. Biochem Biophys Res Commun. 2018;503:876‐881. [DOI] [PubMed] [Google Scholar]
- 48. Huang T, Xu T, Wang Y, et al. Cannabidiol inhibits human glioma by induction of lethal mitophagy through activating TRPV4. Autophagy. 2021;17:3592‐3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jie P, Hong Z, Tian Y, et al. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 2015;6:e1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hong Z, Tian Y, Qi M, et al. Transient receptor potential Vanilloid 4 inhibits γ‐aminobutyric acid‐activated current in hippocampal pyramidal neurons. Front Mol Neurosci. 2016;9:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tian Y, Qi M, Wang Z, et al. Activation of transient receptor potential Vanilloid 4 impairs the dendritic Arborization of newborn neurons in the hippocampal dentate gyrus through the AMPK and Akt signaling pathways. Front Mol Neurosci. 2017;10:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xie R, Xu J, Xiao Y, et al. Calcium promotes human gastric cancer via a novel coupling of calcium‐sensing receptor and TRPV4 channel. Cancer Res. 2017;77:6499‐6512. [DOI] [PubMed] [Google Scholar]
- 54. Nam S, Gupta VK, Lee HP, et al. Cell cycle progression in confining microenvironments is regulated by a growth‐responsive TRPV4‐PI3K/Akt‐p27(Kip1) signaling axis. Sci Adv. 2019;5:eaaw6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meng C, Xia Q, Wu H, et al. Photobiomodulation with 630‐nm LED radiation inhibits the proliferation of human synoviocyte MH7A cells possibly via TRPV4/PI3K/AKT/mTOR signaling pathway. Lasers Med Sci. 2020;35:1927‐1936. [DOI] [PubMed] [Google Scholar]
- 56. Soderling TR. The Ca‐calmodulin‐dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232‐236. [DOI] [PubMed] [Google Scholar]
- 57. Gocher AM, Azabdaftari G, Euscher LM, et al. Akt activation by Ca(2+)/calmodulin‐dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J Biol Chem. 2017;292:14188‐14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tokumitsu H, Sakagami H. Molecular mechanisms underlying Ca(2+)/Calmodulin‐dependent protein kinase kinase signal transduction. Int J Mol Sci. 2022;23:11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujii S, Tajiri Y, Hasegawa K, et al. The TRPV4‐AKT axis promotes oral squamous cell carcinoma cell proliferation via CaMKII activation. Lab Invest. 2020;100:311‐323. [DOI] [PubMed] [Google Scholar]
- 60. Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev. 2022;102:893‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love‐hate triangle. Am J Physiol Cell Physiol. 2004;287:C817‐C833. [DOI] [PubMed] [Google Scholar]
- 62. Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baev AY, Vinokurov AY, Novikova IN, Dremin VV, Potapova EV, Abramov AY. Interaction of mitochondrial calcium and ROS in neurodegeneration. Cells. 2022;11:706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guhathakurta S, Bok E, Evangelista BA, Kim YS. Deregulation of α‐synuclein in Parkinson's disease: insight from epigenetic structure and transcriptional regulation of SNCA. Prog Neurobiol. 2017;154:21‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kanta Acharya T, Kumar A, Kumar Majhi R, et al. TRPV4 acts as a mitochondrial Ca(2+)‐importer and regulates mitochondrial temperature and metabolism. Mitochondrion. 2022;67:38‐58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.