

Abstract
Somatic mosaicism is a known cause of neurological disorders, including developmental brain malformations and epilepsy. Brain mosaicism is traditionally attributed to post-zygotic genetic alterations arising in fetal development. Here we describe post-zygotic rescue of meiotic errors as an alternate origin of brain mosaicism in patients with focal epilepsy who have mosaic chromosome 1q copy number gains. Genomic analysis showed evidence of an extra parentally-derived chromosome 1q allele in the resected brain tissue from 5 of 6 patients. This copy number gain is observed only in patient brain tissue, but not in blood or buccal cells, and is strongly enriched in astrocytes. Astrocytes carrying chromosome 1q gains exhibit distinct gene expression signatures and hyaline inclusions, supporting a novel genetic association for astrocytic inclusions in epilepsy. Further, these data demonstrate an alternate mechanism of brain chromosomal mosaicism, with parentally derived copy number gain isolated to brain, reflecting rescue in other tissues during development.
Keywords: Brain mosaicism, neurodevelopment, cerebral cortex, meiotic nondisjunction, single-cell, epilepsy, hyaline astrocytic inclusions
The human body is a genetic mosaic wherein distinct genotypes emerge from various cell lineages, a phenomenon traditionally attributed to post-zygotic errors in mitosis that arise in progenitor cells during embryonic development(1). Brain mosaicism is a known cause of neurological disorders, including developmental brain malformations and epilepsy(2). Notably, mosaic copy number gains of the entire q-arm of chromosome 1 have been identified in brain tissue from pediatric patients with severe neurodevelopmental delay, unilateral polymicrogyria or focal cortical dysplasia, and early-onset focal epilepsy(3–6). Although it has been presumed that these alterations are post-zygotic due to their mosaic allele fraction and absence from blood samples, their origin has not been investigated. We characterized surgically resected brain tissue from six pediatric epilepsy patients and identified somatic mosaic copy number gains of chromosome 1q (Table 1). The six patients were identified in a total cohort of 196 patients with focal epilepsy and brain malformations. All six patients initially presented at young age with infantile spasms and progressed to intractable focal epilepsy requiring surgical treatment. Resected tissue from all six patients showed evidence of cortical malformation and hyaline astrocytic inclusions in the cortex – a relatively rare neuropathological feature previously reported in a subset of epilepsy patients, but for which no genetic etiology has been determined(7, 8).
Table 1.
Patient Summary.
| Patient ID | Sex | Onset Age | Initial Presentation | MRI | Surgery | Engel Class | Neuropathology | Brain regions studied |
|---|---|---|---|---|---|---|---|---|
| 1a | F | 4 months | Focal seizures followed by infantile spasms | Right hemisphere encephalomalacia | Right functional hemispherectomy | IA | FCDIc; Hyaline astrocytic inclusions | Right inferior posterior frontal gyrus and right temporal lobe lateral cortex |
| 2a | F | 6 months | Infantile spasms | Temporal FCD | Right functional hemispherectomy | IA | FCDIa; Hyaline astrocytic inclusions | Right frontal cortex and right superior temporal gyrus |
| 3a | M | 2 months | Infantile spasms | Left frontal PMG; equivocal right frontal PMG | Left functional hemispherotomy | IIIA | FCDIc; Hyaline astrocytic inclusions | Frontoparietal operculum and parahippocampus |
| 4b | M | 2 months | Infantile spasms; intractable focal motor seizures | FCD; Regional areas of PMG | Right hemispherotomy | IA | FCDIc; Hyaline astrocytic inclusions | Right frontal and temporal opercula, amygdala, hippocampus, insula |
| 5b,c | M | 4 months | Infantile spasms followed by onset of focal seizures | Normal | Left temporal lobectomy and parieto-occipital disconnection | IIIA | FCD; Hyaline astrocytic inclusions | Left temporal and parietal lobe |
| 6c | F | 7 months | Infantile spasms followed by focal seizures | FCD | Focal resection (twice) | III | FCD IIa; Hyaline astrocytic inclusions | Left frontal lobe |
Identifying brain mosaicism
We first identified brain-specific mosaicism by performing whole exome sequencing of resected brain and blood samples for each patient (Fig.1a). Copy number analysis revealed a clear, non-integer (i.e., mosaic) gain spanning almost the entire length of the long arm of chromosome 1 (1q21-q44) that was present in brain tissue, but not in blood, from all six patients (Fig.1b). The allele fraction in all patients was estimated at <30%. This mosaic alteration has previously been associated with early-onset epilepsy in the setting of focal brain malformation(3–6, 9, 10). No other potentially causal copy number or sequence variants were observed to explain our patients’ clinical presentation, except for a PTEN somatic variant in Patient 6 that was previously described, but not thought to be necessarily sufficient to explain the patient’s malformation and overall condition(6). To determine more precisely the chromosome 1q copy number, we performed interphase fluorescent in situ hybridization (FISH) on patient brain tissue sections with probes targeting both chromosomes 1q and 1p. Five of six patients had tissue available for FISH. Four of those five patients had evidence of mosaic chromosome 1q tetrasomy, which was further confirmed in two patients via single cell copy number profiling (Fig.1c–d; Extended Data Fig. 1a), comparable to an independent previously reported case(9). One patient (Patient 2) had evidence of mosaic chromosome 1q trisomy.
Fig. 1. Brain mosaic copy number gain of chromosome 1q.
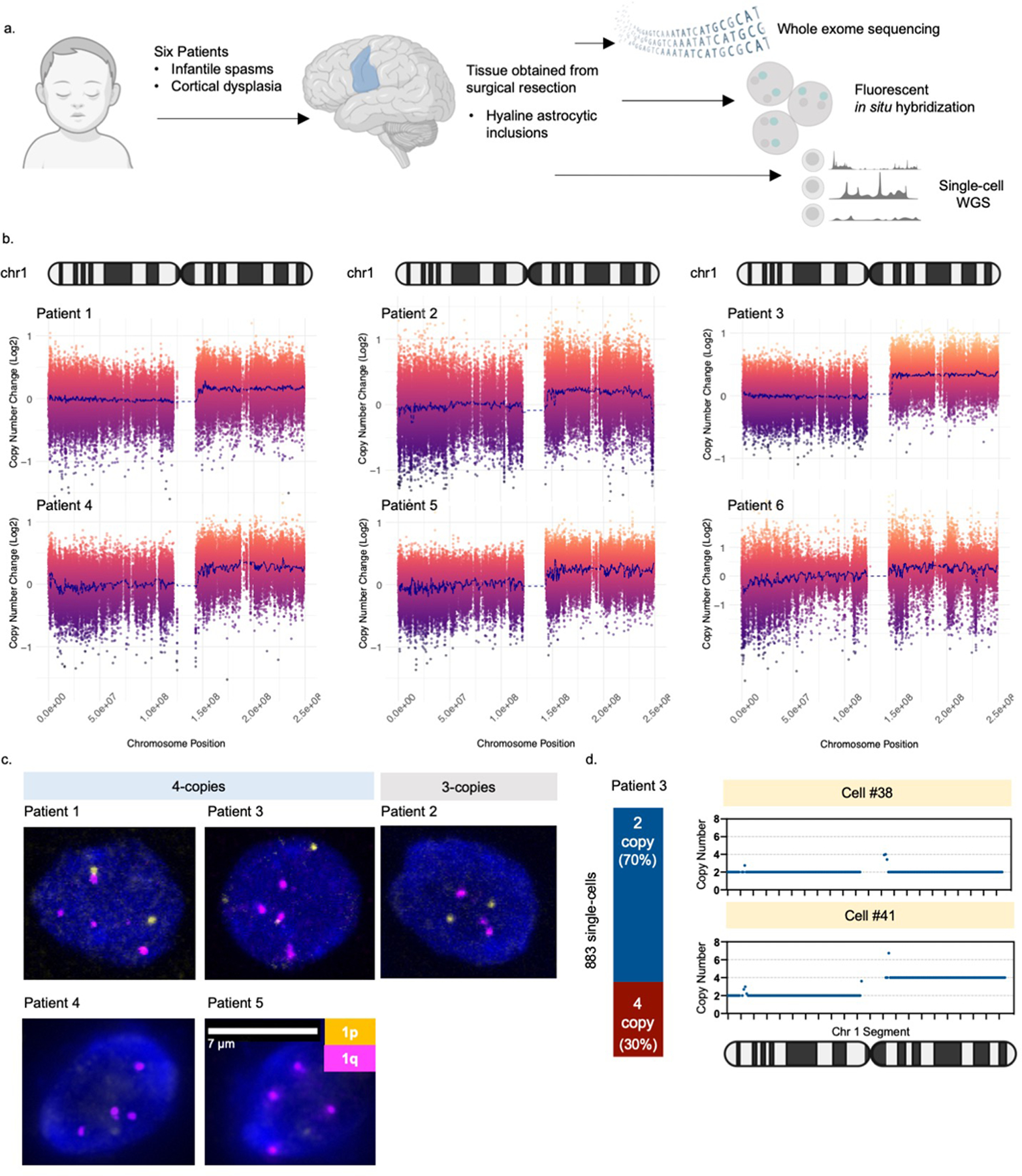
(A) Six patients included in the study met the following criteria: infantile spasms progressing to intractable focal epilepsy, cortical dysplasia, and hyaline astrocytic inclusions. Tissue obtained from surgical resection was evaluated for mosaicism using whole exome sequencing, fluorescent in situ hybridization (FISH), and single-cell whole genome sequencing. (B) Copy number analysis of exome sequencing data comparing brain tissue to a matched blood sample from each patient. Each patient had a non-integer (i.e., mosaic) gain encompassing almost the entire long arm of chromosome 1 (1q21-q44). (C) Representative examples of FISH analysis of patient brain cells showing three or four copies of chromosome 1q present in each cell. Cells with 1q gain were observed on at least 3 slides per patient. (D) Copy number profiling of single-nuclei from Patient 3 brain tissue confirming the presence of chromosome 1q tetrasomy in approximately 30% of nuclei.
To evaluate specificity of the presence of 1q copy number gains, we also evaluated exome sequence data from 342 controls obtained from the North American Brain Expression Consortium (NABEC – dbGAP Study Accession: phs001300.v1.p1), and 15 whole genome-sequenced controls available from the Brain Somatic Mosaicism Network (National Institute of Mental Health Data Archive). None were found to have evidence of a 1q copy number gain, which further supports that the chromosomal anomaly is not common in brain tissue of the general population.
Meiotic origin of mosaicism
Further analysis of exome sequencing data, prior to filtering out high-frequency population variants (see methods), from brain and blood samples revealed that all five patients harboring chromosome 1q tetrasomy had hundreds of brain-restricted single-nucleotide variants on chromosome 1q present at mosaic allele fraction (Fig. 2a, Supplementary Data 1). High-depth targeted sequencing of a subset of these variants (>50,000x sequence depth) revealed no evidence of these variants in patient blood or buccal DNA. However, the variants were present at heterozygous levels (50% allele frequency) in blood or buccal DNA from the corresponding karyotypically-normal mother of all three patients for whom parental samples were available (Fig. 2b; Extended Data Fig. 2a–c, Supplementary Data 2–4). The base change at each variant position matched the maternal allele (Fig. 2c), suggesting that the extra copies of chromosome 1q arose in a maternal gamete during meiosis and were subsequently lost from embryonic cell lineages forming blood and buccal tissue, while being retained in the neural cell lineage. Patient 2—the only patient in whom the 1q gain exists as trisomy—had no evidence of a third haplotype in brain tissue, suggesting the 1q gain arose by a different genetic mechanism, likely as a post-zygotic mitotic error. We then examined exome sequencing data for a cohort of six pediatric patients with central nervous system tumors with chromosome 1q gains under the presumption that chromosomal alterations in tumors originate in mitosis. Consistent with our hypothesis, we observed no evidence of single nucleotide variants (SNVs) representing an additional chromosome 1q allele in any of these tumors (Extended Data Fig. 3a–b).
Fig. 2. Chromosome 1q gain originates in maternal meiosis.
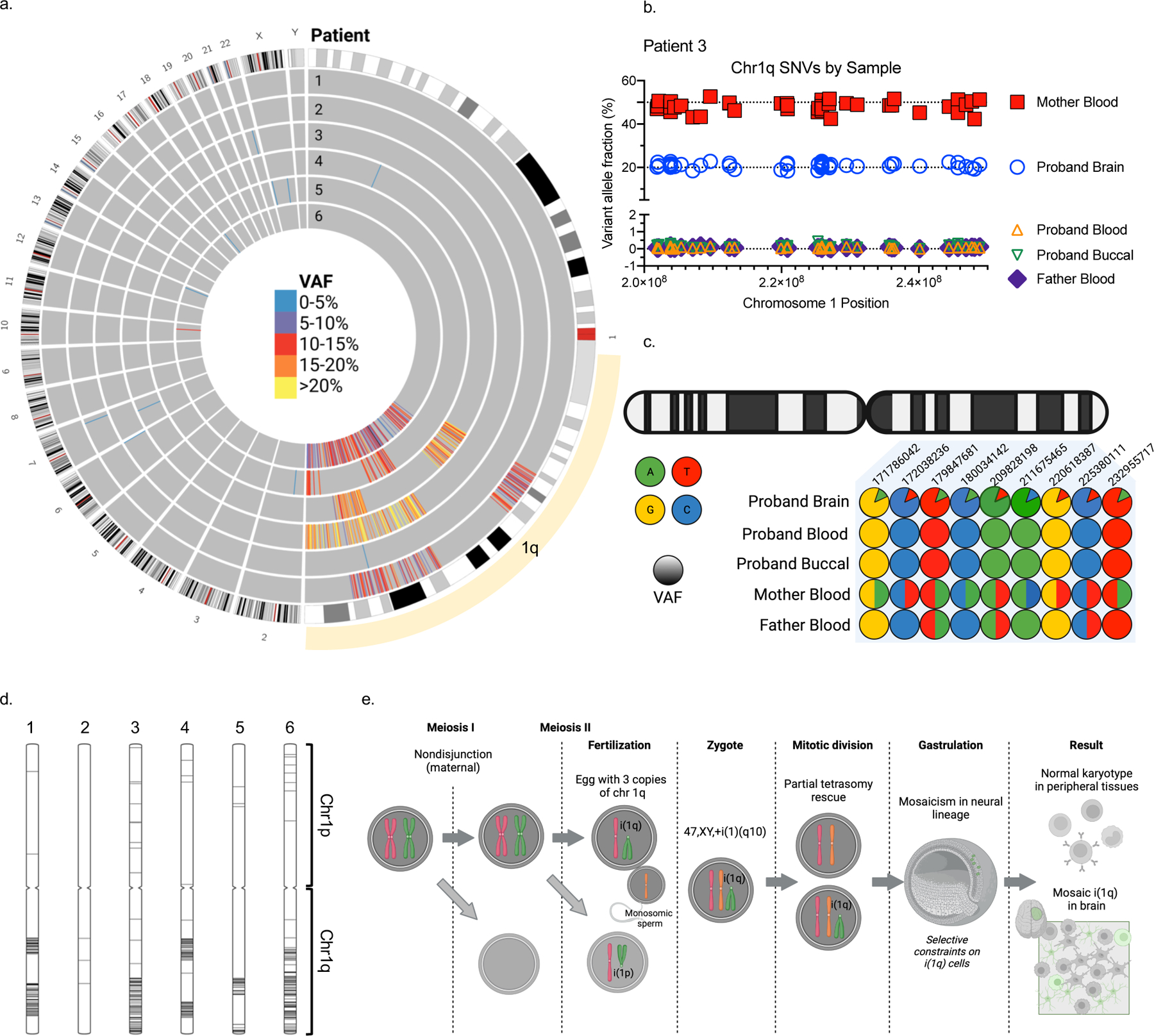
(A) A high number of brain mosaic SNVs were observed on chromosome 1q in five out of six patients. (B) Variant allele fractions for SNVs found on chromosome 1q in Patient 3 brain, blood, buccal, and parental blood samples, demonstrating that these SNVs represent an extra chromosome 1q allele of maternal origin. (C) Illustrative depiction of representative chromosome 1q SNVs from Patient 3 showing a maternal origin of the brain mosaicism. (D) Position of chromosome 1q mosaic SNVs visualized on ideograms for each patient. (E) Hypothetical model for brain mosaicism arising from a maternal meiotic I non-disjunction event with centric mis-division in meiosis II, followed by rescue in non-neural cell lineages.
To resolve the structure of the chromosome 1q gain, which may inform the mechanism by which it occurred in development, we performed optical genome mapping and/or long-read sequencing on brain tissue DNA from three representative patients: Patients 1 and 3 who had evidence of chromosome 1q tetrasomy arising in meiosis, and Patient 2 who had 1q trisomy of possible mitotic origin. The structure of the gain in Patient 2 appeared to be an inverted duplication [der(1)t(1;1)(q21,q44)] as supported by both optical mapping and long-read coverage of the breakpoint (Extended Data Fig. 4a–b). This further supports a post-zygotic mitotic origin of the gain as there is no known mechanism of meiotic rescue for this type of structural variant that leads to mosaicism. On the other hand, the structure of the chromosome 1q gains in Patients 1 and 3 was not resolved by either method. We checked the long-read sequence data from both patients for the presence of the breakpoint observed in Patient 2, but no breakpoint was detected. The absence of a clear breakpoint suggests that the extra 1q copies could exist as an isochromosome, which would not be detected by either approach due to inability to read through the centromere with our current methodologies. No material was available for karyotype analysis.
Next, we examined the position of marker SNVs for the extra chromosome 1q alleles across the length of chromosome 1q. The presence of additional haplotypes near the centromeres would suggest an origin in meiosis I resulting in transmission of a complete maternal homolog, whereas additional haplotypes restricted to the telomeres with evidence of crossover sites would suggest an origin with sister chromatids in meiosis II(11). The positions of marker SNVs in our five patients were mostly distal from the centromere with evidence of crossover (Fig. 2d). These findings suggest a hypothetical model in which an isochromosome could arise from meiotic I nondisjunction followed by centric mis-division during meiosis II, during which one parental chromosome 1 centromere failed to separate properly. The isochromosome could be lost during a subsequent mitotic division in the embryo, via mitotic nondisjunction, tetrasomy rescue, or anaphase lag, thus leading to both disomic and tetrasomic cell lineages(11, 12)(Fig. 2e).
Cell type distribution and function
To assess the distribution of the chromosome 1q gain among cell types in affected brain tissue, we performed genotype analysis from single-nuclei transcriptomes (Extended Data Fig. 5a–b). We profiled approximately 42,000 nuclei from brain tissue of five different patients via single-nuclei RNA-sequencing (Fig.3a–b) and inferred the presence of the chromosome 1q gain by quantifying gene expression based on genomic coordinates, as previously published(13). Chromosome 1 disomic reference nuclei were designated from adjacent unaffected brain tissue when available or from the microglial cell cluster as a comparator from a distinct developmental lineage. Nuclei containing the chromosome 1q gain were identified among multiple cell types, with a strong enrichment in astrocytes in all six patients (Fig. 3c–d, Extended Data Fig. 6a–b). Astrocytes of both genotypes—normal and 1q gain—showed clear expression of canonical astrocytic marker genes (Extended Data Fig. 6c). As our single-nuclei dataset represents a heterogeneous mixture of cell types with unique gene expression profiles, we tested the robustness of our finding by comparing only astrocytes from Patient 3 affected brain tissue to astrocytes from Patient 3 unaffected adjacent brain tissue, which clearly identified the chromosome 1q gain in affected tissue but not unaffected (Extended Data Fig. 6d). To further verify this finding experimentally, we enriched astrocytic nuclei from Patient 3 brain tissue by performing fluorescence-activated nuclei sorting for PAX6 expression, as has been previously done in epileptic brain tissue(14). Then we performed deep targeted sequencing of mosaic variant positions representative of the maternal 1q allele. In agreement with our single-nuclei results, PAX6+ nuclei had an increased representation of the 1q gain compared to either PAX6- nuclei or bulk brain tissue (Extended Data Fig. 6e).
Fig. 3. 1q copy number gain enriched in astrocytes.
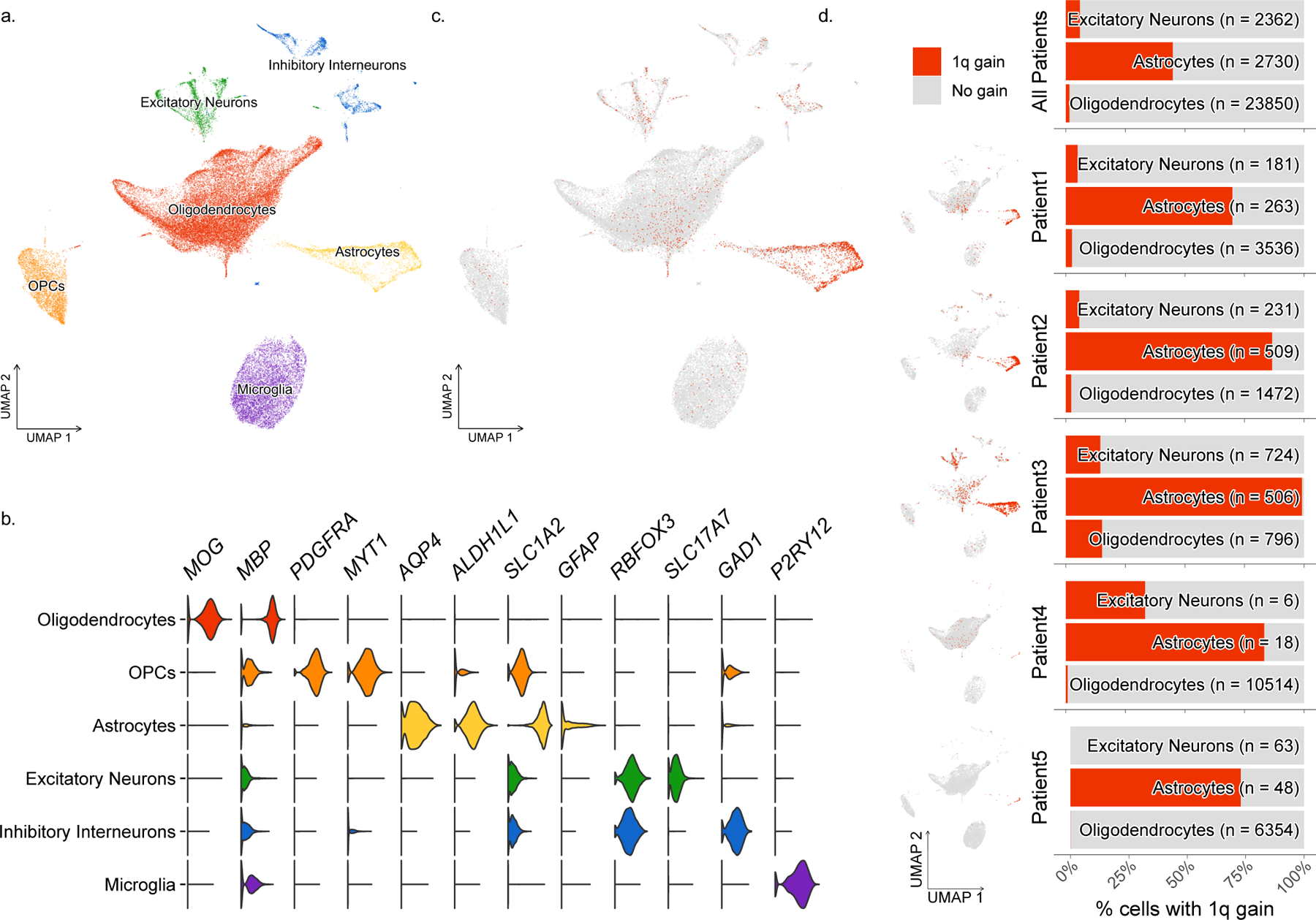
(A) Cell type clusters identified in single-nuclei RNA-sequencing data from 5 patient’s brain samples. (B) Gene expression of representative cell type markers for each cluster. (C) Nuclei expressing the chromosome 1q gain visualized by cluster. (D) Enrichment of chromosome 1q gain in astrocytes in all 5 patients.
To understand the functional importance of mosaic chromosome 1q gain in brain cells, we performed differential gene expression analysis using our single-nuclei RNA-sequencing dataset. We compared nuclei with and without the 1q gain across various cell types, focusing on astrocytes given the unique pathology observed in this patient group (Fig. 4a). Assigning differentially expressed genes into functional gene ontology enrichment annotations revealed categories of biological processes that may be disease-relevant. Astrocytes expressing the 1q gain showed dysregulation of cell-cell adhesion processes and synaptic transmission compared to normal astrocytes (Fig. 4b). Several key cell adhesion molecules were downregulated in astrocytes with the 1q gain (Fig. 4c), similar to other brain malformation conditions(15), whereas genes involved in synaptic transmission, including astrocytic receptors reportedly altered in epilepsy(16), were mostly upregulated (Fig. 4d). All six patients had evidence of hyaline astrocytic inclusions, a relatively rarely reported pathological feature that has not been observed in other genetic subtypes of focal epilepsy. Though the relative proportion of astrocytic inclusions varied among the cases, the cells were similar in appearance with a round to ovoid nucleus with pale or open nuclear chromatin, surrounded by cytoplasmic glassy pink hyaline inclusion(s), and situated in cortical gray matter often within a lacunar-like space (Fig. 4e). Protoplasmic astrocytes are present at synaptic junctions where they regulate structure and function(17). Our results suggest that these unique astrocytes associated with chromosome 1q gains could play a role in seizure propagation given their distinct gene expression signatures.
Fig. 4. 1q copy number gain has cell-type-specific effects.
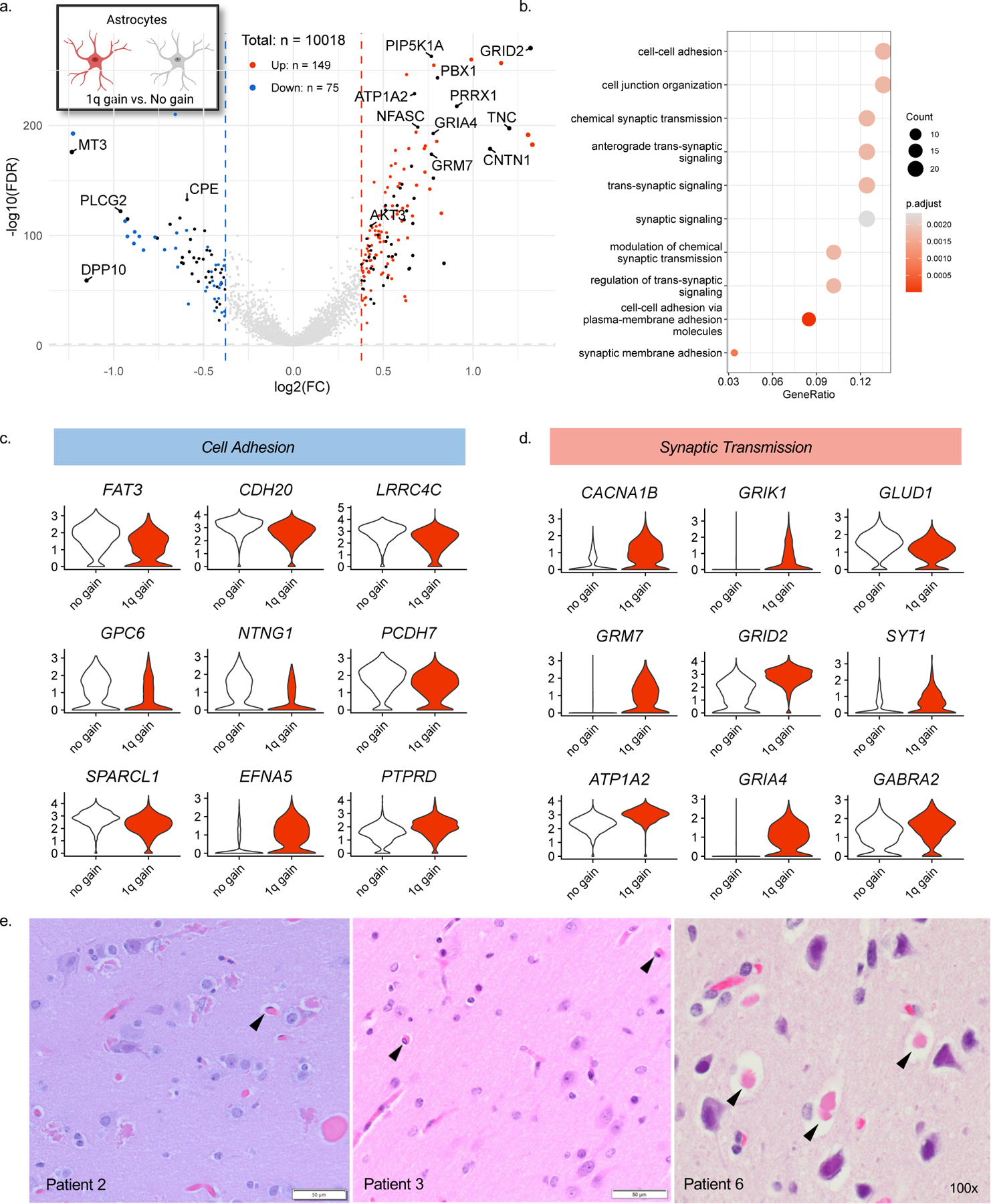
(A) Volcano plot showing differentially expressed genes in astrocytes containing the chromosome 1q gain vs. euploid astrocytes. Black points represent genes located on chromosome 1q. (B) Enriched GO terms for 1q gain astrocytes derived from an over-representation test performed with the enrichGO function of clusterprofiler R package with FDR p-value adjustment. (C) Differentially expressed genes related to cell adhesion and (D) synaptic transmission. (E) Representative H&E stains on FFPE tissues demonstrating hyaline astrocytic inclusions identified in at least two slides per patient upon board-certified neuropathological review.
Given the neuronal abnormalities observed in our patients, we also compared excitatory neurons with and without the 1q gain (Extended Data Fig. 7a) and identified gene ontologies related to neuron projection development and synapse organization (Extended Data Fig. 7b). Differentially expressed genes common to both comparisons (i.e., for astrocytes and excitatory neurons) were predominantly (81.5%) upregulated genes located on chromosome 1q (Extended Data Fig. 7c; Supplementary Data 5). These genes are likely upregulated by virtue of the copy number increase itself.
Discussion
Brain mosaicism is an important explanation for neurodevelopmental disorders and has been typically attributed to post-zygotic genetic variants acquired during development. The exception has been the acquisition of variants in adult life that are thought to contribute to degenerative diseases such as Alzheimer disease(18); this too involves post-zygotic, post-developmental somatic mutation giving rise to mosaic variants. In contrast, our findings point to the presence of a large chromosomal copy number gain that is present only in brain tissue due to post-zygotic loss of maternally derived meiotic errors that remove the copy number gain from other lineages, hence making these copy number gains undetectable in blood (leukocytes) and buccal epithelial cells. Our findings represent a novel mechanism through which brain mosaicism arises in development. In patients with chromosome 1q gains originating in maternal meiosis, the alteration was propagated to the neural lineage while being lost from lineages forming blood and buccal tissues, thus generating brain mosaicism. Partial ‘tetrasomy rescue’ could explain this finding and has been reported to occur for parentally-derived chromosomal abnormalities in previously described case reports(11, 12, 19). Early developmental bottlenecks within the first few cell divisions may provide an opportunity for aneuploidy rescue in the embryo(20). If this rescue occurs for most cell lineages except for the neural lineage, then the alteration could be ultimately restricted to the brain. Chimerism is an unlikely explanation here, as the maternal marker alleles on chromosome 1q were the only alleles that did not identically match the patients’ constitutional DNA.
We attempted to resolve the structure of the chromosome 1q gain by optical genome mapping and long-read sequencing. The patient with mosaic 1q trisomy had a clear inverted duplication of chromosome 1q supported by evidence from both technologies. For the patients with mosaic tetrasomy 1q, the fact that we could not resolve any clear breakpoints leads us to believe the structure may be an isochromosome, in which two q-arms are joined by a centromere. Formation of an isochromosome in development could have occurred at several stages. One possibility, which we favor, is that an isochromosome could form wholly in meiosis by non-disjunction followed by centromeric misdivision, and is subsequently rescued during one of the early mitotic divisions of the embryo (Fig. 2e). As an alternative, the isochromosome could arise in the early cleavage-stage embryo; non-disjunction in meiosis may create a zygote trisomic for chromosome 1 and during the first few cleavages, an isochromosome could form through centromeric misdivision leading to i(1q) and i(1p) lineages (Extended Data Fig. 8a). This model requires non-survival of the i(1p) lineage, as we saw no evidence of i(1p) in our patients. Finally, the isochromosome could arise in the mother during gametogenesis; a maternal mitotic error during oogenesis may have produced an oocyte with i(1q), which was then fertilized and propagated to the embryo (Extended Data Fig. 8b). This model requires a genetically abnormal oocyte to survive through meiosis and fertilization. Of note, there was no history of maternal infertility or recurrent pregnancy losses.
The presence of the chromosome 1q gain in brain tissue, while being absent from blood or buccal cells, underscores the fact that human development represents a complex interplay of developmental bottlenecks and proliferation advantages that are not yet fully understood. In one case report describing a fetus with mosaic chromosome 1q trisomy, the level of mosaicism ranged from 4–93% of cells depending on the tissue studied(21). We cannot rule out the presence of the chromosome 1q gains from tissues that we were unable to study; however, none of our patients showed any non-neurological clinical signs or symptoms. Constitutive 1q trisomy is associated with lethality or very early death, so we are confident that the majority of tissues other than brain do not contain the copy number gain. Moreover, the 1q gain was focally restricted within the brain itself, as in two cases (Patients 1 and 3) there was tissue adjacent to the lesion in which 1q gain was not detected by exome sequencing. In a reported case of fetal teratoma, a maternally-derived isochromosome 1q was observed in the tumor but was absent from skin fibroblasts, demonstrating a precedent for tissue-specific mosaicism arising from a meiotic error(22). Recognizing a meiotic origin for mosaicism has important implications for genetic counseling and recurrence risk. If the chromosome 1q gain is present in additional tissues, then patients could be at increased risk of certain cancers, though it has not been studied in this patient population. Furthermore, women who have a trisomic pregnancy related to increased predisposition to meiotic nondisjunction are at increased risk of recurrence(23).
Enrichment of the chromosome 1q gain in astrocytes suggests that it may confer a proliferative advantage in this cell type. This is supported by the observation that chromosome 1q gain is a common cytogenetic abnormality observed in many brain tumors of glial cell origin, including pediatric high-grade astrocytomas and ependymomas(24). Particularly in ependymomas, which arise from glial stem or progenitor cells, chromosome 1q gain is the most common genomic alteration and often there are no other chromosome imbalances detected - suggesting 1q gain is sufficient for the initiation of tumor formation in this cell type(25–28). Moreover, long-term cultured human neural stem cells are susceptible to recurrent duplications of chromosome 1q, which increase their proliferation rate, but reduce their ability to differentiate into neurons, which may explain the relatively small proportion of neurons compared to astrocytes affected in our cohort(29, 30). A related possibility is that certain cell types, such as neurons, are more susceptible to death as a result of chromosome 1q gain compared to other cell types. Chromosome 1q gain was observed in other neural cell types to a lesser degree, including microglia which arise from a distinct lineage compared to other brain cell types. Depending on when a potential tetrasomy rescue event occurred, the chromosome 1q gain could have persisted into progenitors of multiple lineages, but may have been more compatible with survival in one lineage over the other, giving rise to unequal distributions of affected cells.
Consistent with observed enrichment of the 1q gains in astrocytes, all six patients in this study had evidence of cytoplasmic hyaline astrocytic inclusions, a rarely observed neuropathological feature that has been reported in epilepsy(7, 8, 31). Although we did not perform any co-staining in this study, prior studies demonstrated that the cells with hyaline inclusions express astrocytic markers, including S100 beta, GFAP, and GLT-1(7, 8, 32). In two cases, the hyaline astrocytic inclusions were observed and reported on as a distinctive pathology in resected brain tissue from a cohort of 30 pediatric epilepsy cases without any prior knowledge of genetic findings(7). These two cases were negative for Filamin A staining, in contrast to astrocytic inclusions observed in Aicardi syndrome(32). FLNA was expressed at low levels in our snRNA-seq data, so we could not compare expression levels between astrocytes with or without a 1q gain. These cytoplasmic eosinophilic inclusions in astrocytes have been reported in individuals with epilepsy although this is the first report to our knowledge recognizing an association of this pathology with 1q copy number gains. The association of chromosome 1q gain with hyaline astrocytic inclusions provides new directions for understanding how cell type-specific effects contribute to 1q pathophysiology. Future studies will determine the specificity of the association between the presence of hyaline inclusions and mosaic 1q gain, including assessments of whether 1q copy number gains are present in the brain tissue of individuals with Aicardi syndrome when astrocytic inclusions are detected. Although we did not directly test for the presence of 1q gain in astrocytes with inclusions, according to our single-nuclei data the majority of astrocytes are positive for the gain, despite astrocytic inclusions being quite rare among them. This makes it likely that 1q copy number gains are found in both ‘normal’ astrocytes and the rarer astrocytes with inclusions. If true, this would mean that additional factors contribute to the formation of astrocytic inclusions besides just the genetic alteration. Nevertheless, an observation of astrocytic inclusions upon neuropathology review will suggest a possible mosaic 1q copy number gain. A recent example in which genetics have similarly informed pathology is SLC35A2 mosaicism and its association with MOGHE – a previously unrecognized pathological finding with apparently specific genetic association(33).
Further, a genetic finding of brain mosaic 1q gain may inform the clinical course and prognostics. All six patients in this study had unifying features, including infantile spasms and cortical malformation; however, MRI findings varied among individuals (Extended Data Figure 9). Notably, the outcome also varied, as 3 patients were seizure-free following surgery but 3 continued to have recurrent seizures. Future studies will further specify the clinical features and outcomes associated with chromosome 1q gain.
Altogether, our observations demonstrate a novel mechanism for the origin of brain chromosomal mosaicism and link brain mosaic chromosome 1q gain to a distinct clinical phenotype including early-onset focal epilepsy, cortical malformation, and cytoplasmic hyaline astrocytic inclusions.
Methods:
Patient Samples
Individuals who underwent surgical resection of brain tissue or tumor tissue were enrolled from Nationwide Children’s Hospital (NCH), The University of Texas Health Science Center at Houston (UTH), and Boston Children’s Hospital (BCH). Written informed consent was obtained from parents/legal guardians according to IRB-approved studies at the respective institutions. Initial clinical and genetic characterization for Patients 1–3 was published by Bedrosian et al.(5) and for Patients 4–6 by Lai et al.(6) Formalin-fixed paraffin-embedded brain tissue sections underwent evaluation by a board-certified neuropathologist. Blood, buccal swab, and surgically resected brain or tumor tissue samples were collected as applicable and stored at –80°C prior to use. DNA was extracted from frozen samples using Qiagen AllPrep DNA Kit (NCH) or Qiagen DNeasy Blood & Tissue Kits (UTH and BCH).
Exome sequencing & CNV/variant calling
For Patients 1–3, and 6, DNA isolated from blood and brain tissue was prepared for enhanced exome sequencing using NEBNext Ultra II FS DNA Library Prep Kit (New England Biolabs, Ipswich, MA). IDT xGen Exome Research Panel v2.0 enhanced with the xGenCNV Backbone Panel-Tech Access (Catalog # 10005152, Integrated DNA Technologies, Coralville, IA) was used for target enrichment by hybrid capture. Final libraries were sequenced on an Illumina NovaSeq 6000 to generate paired-end 151-bp reads. For Patients 4–5, DNA samples were prepared for sequencing using Nimblegen SeqCap EZ V3.0 (Catalog # 06465692001, Roche) and were sequenced on Illumina HiSeq2000, HiSeq2500, or NovaSeq 6000 to generate paired-end 151-bp reads. The Churchill pipeline was used for alignment to human reference genome build GRCh38 and secondary analysis(34). Somatic variants were then filtered based on the following characteristics: MuTect2 = PASS, GATK quality score ≥30, depth of sequencing ≥10 reads, alternate allele reads ≥5, VAF ≥2%, and gnomAD WGS and WES population frequency <1%. Variants passing all criteria were then manually reviewed in Integrated Genomics Viewer (IGV) to assess for strand bias, read-end bias, or other indications of sequencing artifacts. VarScan2 and GATK v.4.1.9 were used to assess CNVs and loss of heterozygosity across all chromosomes(35)
Fluorescent in situ hybridization
Dual-color FISH detection of the chromosome 1q gain on 5-µm paraffin-embedded tissue sections (Patients 1–3) was performed using the Vysis IntelliFISH™ 1q21 CKS1B/1p32 CDKN2C Probe Kit (Abbott Laboratories). The following protocol was optimized to allow for reliable FISH analysis on formalin-fixed tissue: The slides were deparaffinized in CitriSolv™, dehydrated in a series of ethanol dilutions, and allowed to airdry before incubation in 0.2N hydrochloric acid at room temperature for 20 minutes. The slides were then rinsed in purified water and 2X saline sodium citrate (SSC) buffer and incubated in the Vysis IntelliFISH™ Pretreatment SSC Solution (Vysis, Cat # 08N85–05) at 80°C for 40 minutes. The resulting tissues were digested in a solution of 75mg Protease IV (Vysis, Cat # 08N85–010) and 50mL Protease Buffer (Vysis, Cat # 31–806014) at 37°C for 25 minutes and dehydrated in 70, 85, and 100% ethanol before airdrying. The hybridization mixture contained 1μL of 1q21 CKS1B SpectrumOrange/1p32 CDKN2C SpectrumGreen probes (Vysis, Cat # 08N78–020), 12μL of Hybridization Buffer (Vysis, Cat # 08N87–001), and 2μL of nuclease-free water. Automated co-denaturation of the slides was performed at 73°C for 5 minutes. The slides were hybridized at 37°C for 3 hours on a RapidFISH™ instrument (Boekel Scientific). After hybridization, the slides were washed and ReadyProbes™ Tissue Autofluorescence Quenching Kit was used according to the manufacturer’s instruction (Invitrogen, Cat # R37630) to reduce unwanted autofluorescence. 10uL of DAPI I counterstain (1000ng/mL) (Vysis, Cat # 06J49–001) was applied to the slides along with coverslips and the slides were stored at −20°C until imaging. FISH was performed on frozen tissue specimens (Patients 4–5) after fixing slides in 4% formaldehyde in PBS at 0°C for 15 minutes immediately after sectioning. Pretreatment for these slides began with a 0.2N hydrochloric acid incubation at room temperature for 20 minutes. Pretreatment remained the same with protease digestion occurring for 18 minutes. The following denaturation and hybridization steps remained the same. After applying coverslips, FISH signals were recorded on a Nikon AX R Confocal equipped with NIS-Elements™ processing software version 5.42.03. The chromosome 1q gain was visually evaluated by counting the number of punctate 1q signals (green) in relation to 1p signals (red) co-localized to well-defined nuclei.
Single-nuclei copy number profiling
Frozen brain tissue was dissociated, and single nuclei were isolated by fluorescence-activated nuclei sorting(36). For Patient 3, approximately 1,000 nuclei were captured using the Chromium Single Cell CNV kit (10x Genomics) and prepared for single nuclei DNA-sequencing according to the manufacturer’s protocol. The final library was sequenced on NovaSeq6000 to achieve a minimum of 750,000 read pairs per cell. Demultiplexing, read alignment, cell calling, and copy number estimation were performed using 10x Genomics cellranger-dna-1.1.0.
Due to discontinuation of the 10x Genomics CNV product, Patient 1 cells were processed by an alternate method. 24 single nuclei were sorted into a plate, and nuclei lysis and DNA amplification were performed using ResolveDNA® whole genome amplification kit (BioSkryb Genomics). To screen the 24 nuclei and identify which had chr1q gain, targeted sequencing of known marker alleles representing the extra 1q material was performed (see methods and Extended Data Fig. 2a). DNA from three nuclei with the chr1q gain and one without were prepared for whole genome sequencing using 500ng input. DNA was fragmented using Covaris® ME220 focused-ultrasonicator with the following settings: target peak (350 base pair); duration (30 seconds); peak power (75 watts); duty factor (20%); and cycles per burst (1000). Sheared DNA was then purified using 0.6X SPRIselect, followed by end-repair, dA-tailing, and adapter ligation using NEBNext Ultra II DNA Library Prep kit reagents (New England Biolabs). Final libraries were pooled and sequenced on NovaSeq 6000 to aim for 10X WGS coverage per sample. The Churchill pipeline was used for alignment to human reference genome build GRCh38 and secondary analysis(34). GATK were used for CNV calling, using an unaffected nucleus (i.e., without the chr1q gain) as a comparator(37). The CNV plot in the unaffected nucleus was generated by comparing to a ‘panel of normals’ comprised of WGS blood samples from 30 healthy donors.
Oncoscan array
DNA samples were prepared for microarray-based whole-genome copy number evaluation using the OncoScan™ CNV Assay kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Cat # 902695). Data were visualized and CNV plots were generated using Chromosome Analysis Suite software (Version 4.3).
High-depth targeted sequencing of 1q variants
Targeted sequencing was performed using a custom hybridization capture panel of 211 probes (IDT xGen, Supplementary Data 6) designed to capture various regions along chromosome 1q. The panel probes were designed to capture 120-bp regions surrounding variants of interest representative of the mosaic, non-constitutional chr1q gain in two of the patients; approximately 100 variants from each patient were targeted. DNA (50ng input) from proband blood, buccal, and brain, maternal and paternal blood, and human reference DNA samples (Coriell Institute) were prepped for sequencing using SRSLY® NanoPlus library kit and ForShear™ enzymatic fragmentation kit following manufacturer’s protocol (Claret Bioscience). Hybridization capture was performed according to the manufacturer (Integrated DNA Technologies), and final libraries were pooled in batches of six and sequenced on Illumina MiniSeq with attempted average coverage of 1,500-fold per variant of interest. In addition to hybridization capture targeted sequencing, amplicon-based sequencing was performed on selected variants to obtain high-depth (>50,000x coverage) (38). Briefly, sequence-specific primers (Supplementary Data 6) were used to amplify 200-bp products containing chr1q mosaic, maternal variants, and the products were prepped for next-generation sequencing using NEBNext Ultra II DNA Library Prep kit reagents (New England Biolabs). Final libraries were pooled and sequenced on the iSeq 100 to aim for >50,000x coverage. The raw, deduplicated BAM files were used with SAMtools mpileup and mpileup2cns VarScan (version 2.4.4) commands to generate read counts for the chr1q variants of interest(35, 39).
Long-read sequencing
HiFi SMRTbell libraries were prepared from 4.0 µg of unsheared gDNA, with a mode size of 14.1 kb, according to the PacBio HiFi Express Template Prep Kit 3.0 protocol (PN 102-390-900 Version 01 [April 2022]). The Sage Science BluePippin system with the 0.75% Agarose Gel Cassette was used to remove molecules below 10 kb from the final SMRTbell library. The resulting size selected SMRTbell library was assessed for DNA concentration by Qubit 1x dsDNA HS and size by TapeStation 4200 Genomic ScreenTape. The required final library metadata were entered into the PacBio SMRT Link Sample Setup (version 11.1.0.156728) to calculate a per sample protocol for complexing using the Sequel II Binding Kit 3.2. The complexed library was sequenced across two 8M SMRT Cells with the Sequel II Sequencing Plate 2.0 chemistry, using adaptive loading with an on-plate loading concentration of 55 pM. Sequence data collection involved a 2-hour pre-extension followed by a 30-hour movie time on the Sequel IIe instrument. Primary analysis included the PacBio HiFi Mapping application within SMRT Analysis v.11.1.0.166339 for alignment to reference build GRCh38, revealing a mean coverage of 16x sequencing depth. HiFi read alignment was visualized in Integrative Genomics Viewer to assess reads for structural variation.
Optical genome mapping
Brain tissue from two patients was sent to Bionano Genomics for optical genome mapping (OGM) to resolve the structure of the chromosome 1q gain. Briefly, high molecular weight genomic DNA was isolated from frozen tissue, prepped for OGM, and loaded onto the Saphyr instrument. Samples were processed and visualized with Solve 3.7/Access 1.7 in reference to the GRCh38 reference genome. Samples were filtered with the manufacturer’s recommended settings and restricted to variants that are absent in the Bionano control structural variant database.
Single-nuclei transcriptomics
Frozen brain tissue was dissociated by mechanical disruption using a dounce homogenizer (36). Nuclei were washed and stained with Hoechst 33342 dye, filtered through a 30 µm mesh filter, and resuspended in PBS for analysis on a BigFoot cell sorter (ThermoFisher). Samples were gated on Hoechst-positive nuclei, debris was excluded using forward and side scatter pulse area parameters (FSC-A and SSC-A), and then aggregates were excluded using pulse width (FSC-W and SSC-W). Purified nuclei were sorted directly into 10x Genomics reaction buffer and processed according to the manufacturer protocol for Chromium Next GEM Single-Cell 3’-Reagent Kit v.3.1. Libraries were sequenced on an Illumina NovaSeq 6000 instrument to generate paired-end sequencing data with a minimum of 50,000 reads per cell. Data pre-processing, including read alignment to the GRCh38 transcriptome, filtering, barcode counting and UMI counting, were performed using 10x Genomics CellRanger v.6.0 software following the default parameters, except for the inclusion of intronic transcripts present in nucleic pre-mRNAs.
Downstream analysis was performed using Seurat v.4 for R(40). Only genes that were expressed in at least three nuclei, as well as cells that expressed at least 200 unique genes and less than 5% mitochondrial transcripts were considered for further analysis. Normalization, variance-stabilization, and integration of feature-barcode matrices were performed using the SCTransform function in Seurat. Dimensionality reduction was performed using principal component analysis (PCA) and the resulting principal components were used to calculated the UMAP coordinates of each cell. The distance matrix was then organized into a K-nearest neighbor graph (KNN) and partitioned into clusters using the original Louvain algorithm. Nuclear doublets were detected via DoubletFinder v2.0 and doublets were removed from the dataset, as well as clusters containing greater than 10% doublets(41). Cell types were annotated based on expression of known markers with reference to the Allen Brain Map Human M1 Cortex Dataset.
Single cells with chr1q gain were identified using inferCNV R package (inferCNV of the Trinity CTAT Project(13) - https://github.com/broadinstitute/inferCNV). For Patients 1 and 3, brain tissues were available wherein the chr1q gain was not identified from exome sequencing copy number variant calling and as such, these tissues were used as a “normal” comparator for the affected (i.e., chr1q-gain) brain tissue when running inferCNV. For patients without adjacent brain tissue lacking the chr1q gain, microglial cells within the single nuclei data set were used as the “normal” comparator cells because the chr1q gain was not identified in Patients 1 or 3 in this specific cell type. The chr1q status for each cell (i.e., gain or no gain) and the corresponding single cell barcodes were obtained from the run.final.infercnv_obj@expr.data by summing expression values from all chr1q genes and identifying the outliers (Q3 + 1.5[interquartile range; IQR]). The resultant chr1q gain status was added to the Seurat object’s metadata slot by joining based on cell barcode.
Differential expression analysis was performed for mosaic vs. wild-type cells using MAST(42). Gene ontology term enrichment was determined for differentially expressed genes with a fold-change magnitude of greater than 1.3 using ClusterProfiler(43).
Fluorescence-activated nuclei sorting
Nuclei were isolated from fresh frozen tissue samples using the Chromium Nuclei Isolation Kit (10X Genomics #1000493). Astrocytic nuclei were enriched using a PAX6 antibody (Novus Biologicals #NBP2–34705APC) at 1:200 followed by incubation for 1 hour at 4°C with agitation. All nuclei were then stained with DAPI (ThermoFisher Scientific #EN62248), and all DAPI+/PAX6+ and DAPI+/PAX6- nuclei were gated and sorted using a Bigfoot Cell Sorter running Sasquatch 1.19.3 software (ThermoFisher Scientific). DNA was extracted from sorted nuclei using QIAamp DNA Micro Kit (Qiagen). The extracted DNA was used for high-depth targeted sequencing (described above) of two chr1q variants representative of the chr1q gain.
Extended Data
Extended Data Fig. 1.
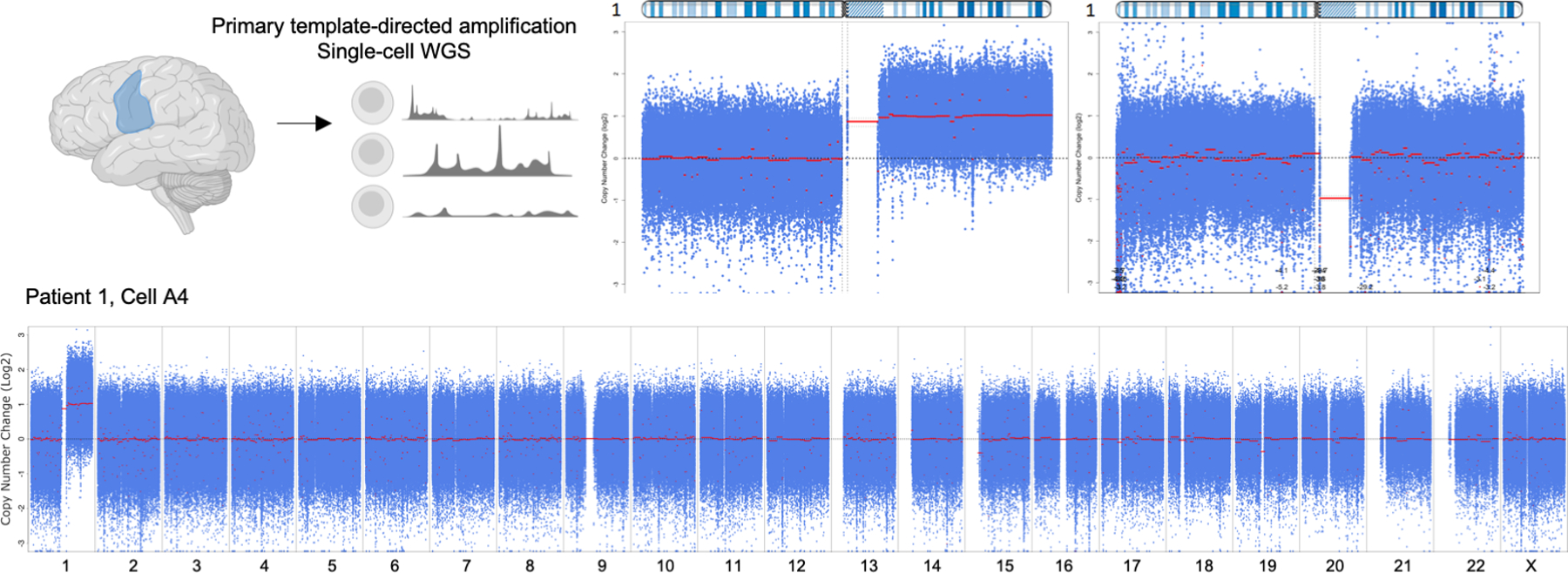
(A) Whole genome sequencing of single-nuclei from Patient 1 brain tissue confirming the presence of chromosome 1q tetrasomy. Chromosome 1 copy number data is shown for representative nuclei with and without 1q tetrasomy (top) and the entire genome is shown (bottom) for the cell with the 1q gain.
Extended Data Fig. 2.
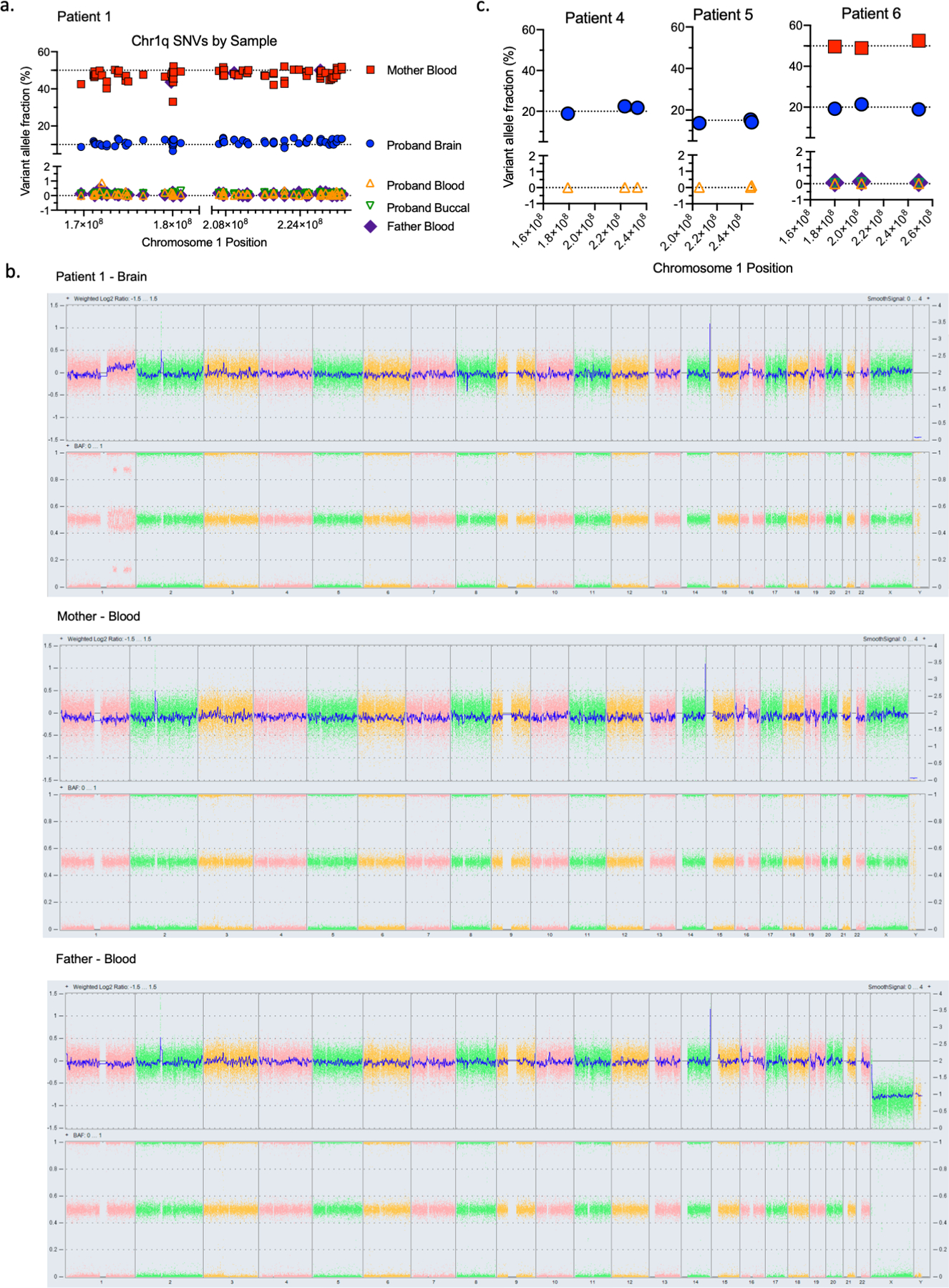
(A) Variant allele fractions for SNVs found on chromosome 1q in Patient 1 brain, blood, buccal, and parental blood samples, demonstrating that these SNVs represent an extra chromosome 1q allele of maternal origin. (B) Oncoscan array results for Patient 1 trio demonstrating a chromosome 1q gain in proband brain tissue but no gain observed in blood samples from either parent, showing the parents are karyotypically normal. (C) Variant allele fractions for a small number of representative chromosome 1q SNVs in available tissues from Patients 4, 5, and 6.
Extended Data Fig. 3.
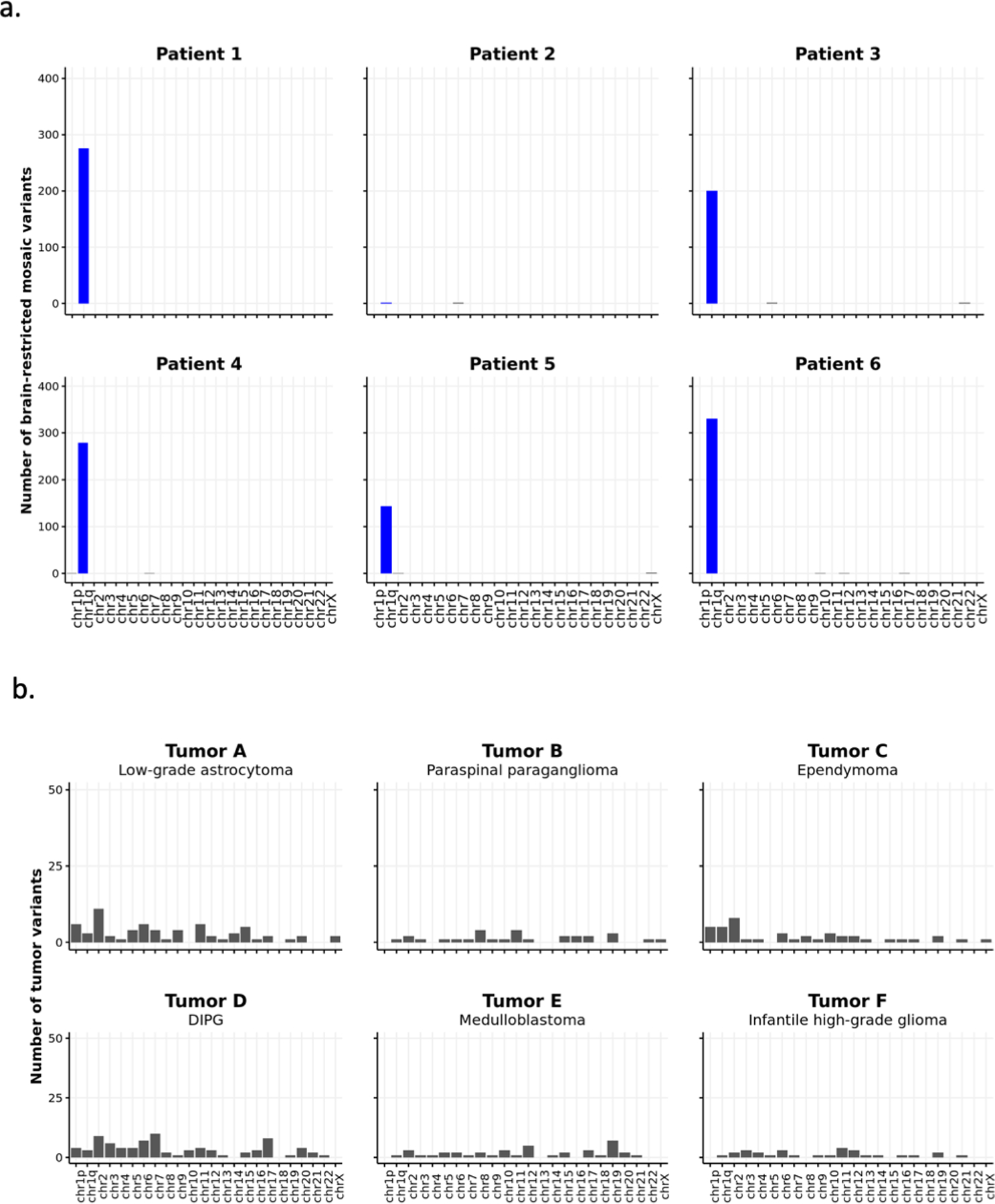
(A) Number of brain-restricted mosaic variants identified from exome sequencing of resected brain tissue versus blood samples from epilepsy Patients 1–6. (B) Somatic variants identified from tumor-normal exome sequencing of various pediatric CNS tumors with chromosome 1q gains. No enrichment of variants on chromosome 1q is observed.
Extended Data Fig. 4.
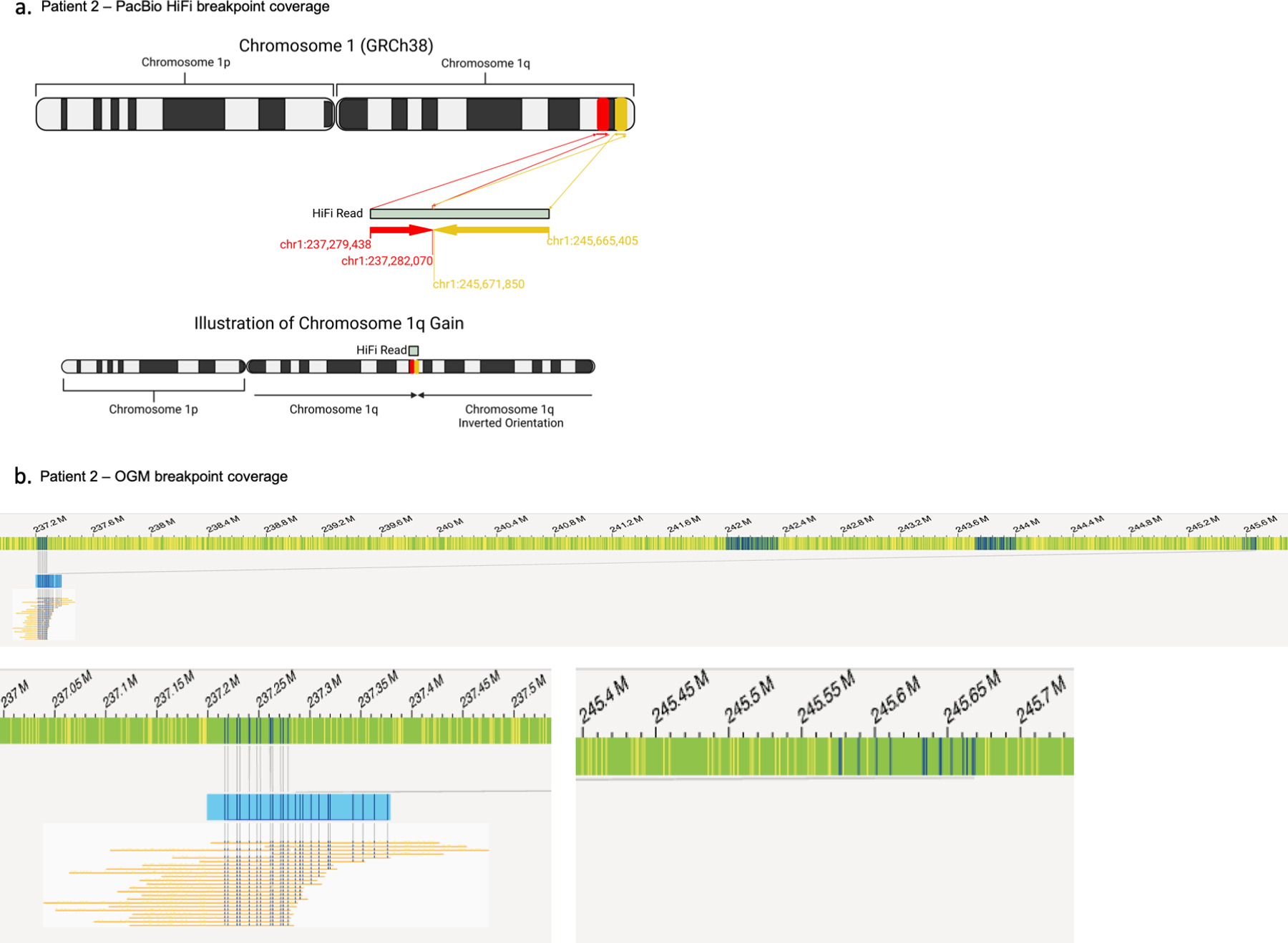
(A) A breakpoint-spanning read from PacBio HiFi long-read sequencing of Patient 2 brain DNA identifies the structure of the gain as an inverted duplication of chromosome 1q. (B) Optical genome mapping coverage of the breakpoint.
Extended Data Fig. 5.
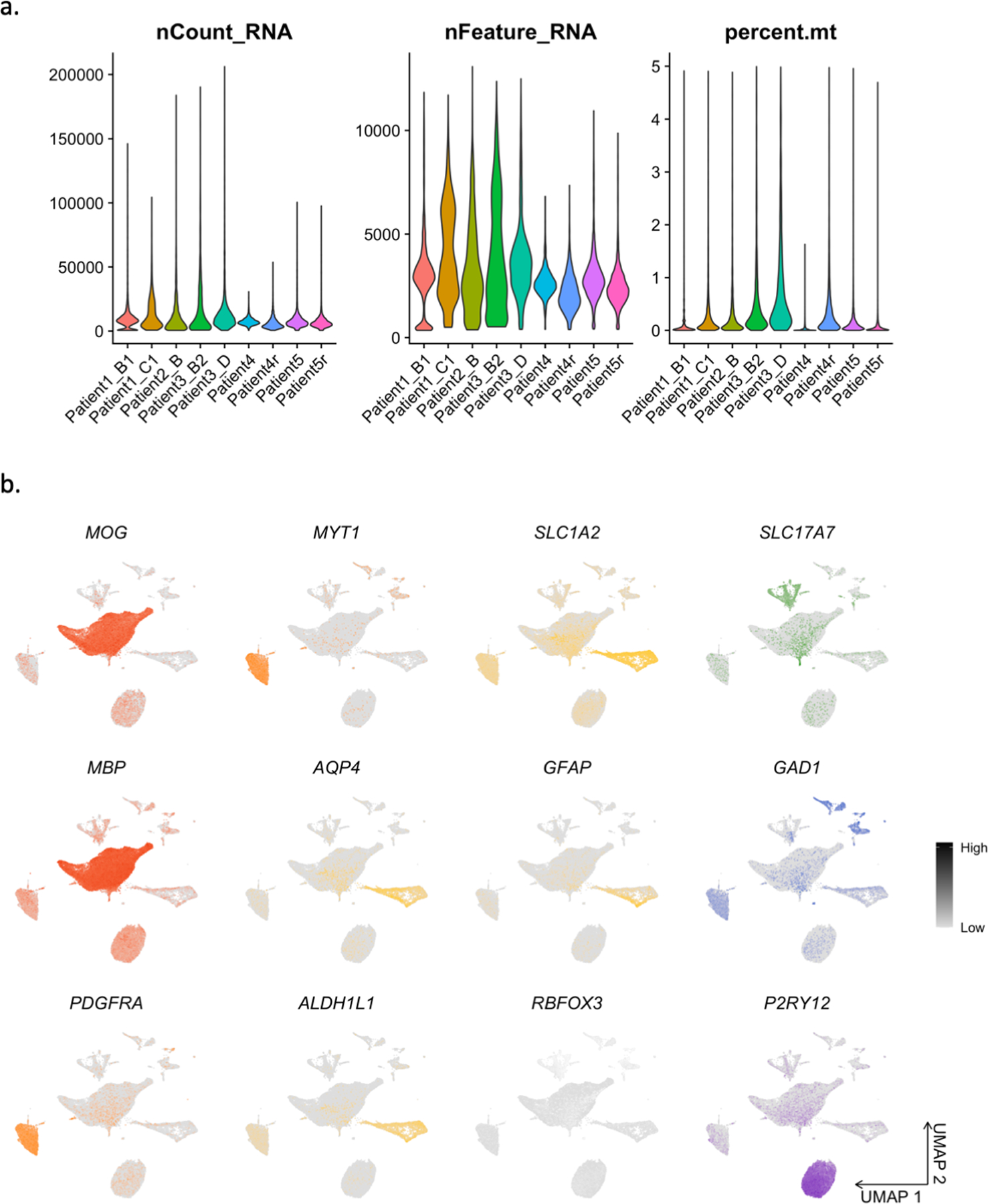
(A) Single-nuclei RNA-seq quality control metrics: number of reads per nucleus, number of genes per nucleus, and percent mitochondrial reads. (B) Feature plots showing expression of cell type markers.
Extended Data Fig. 6.
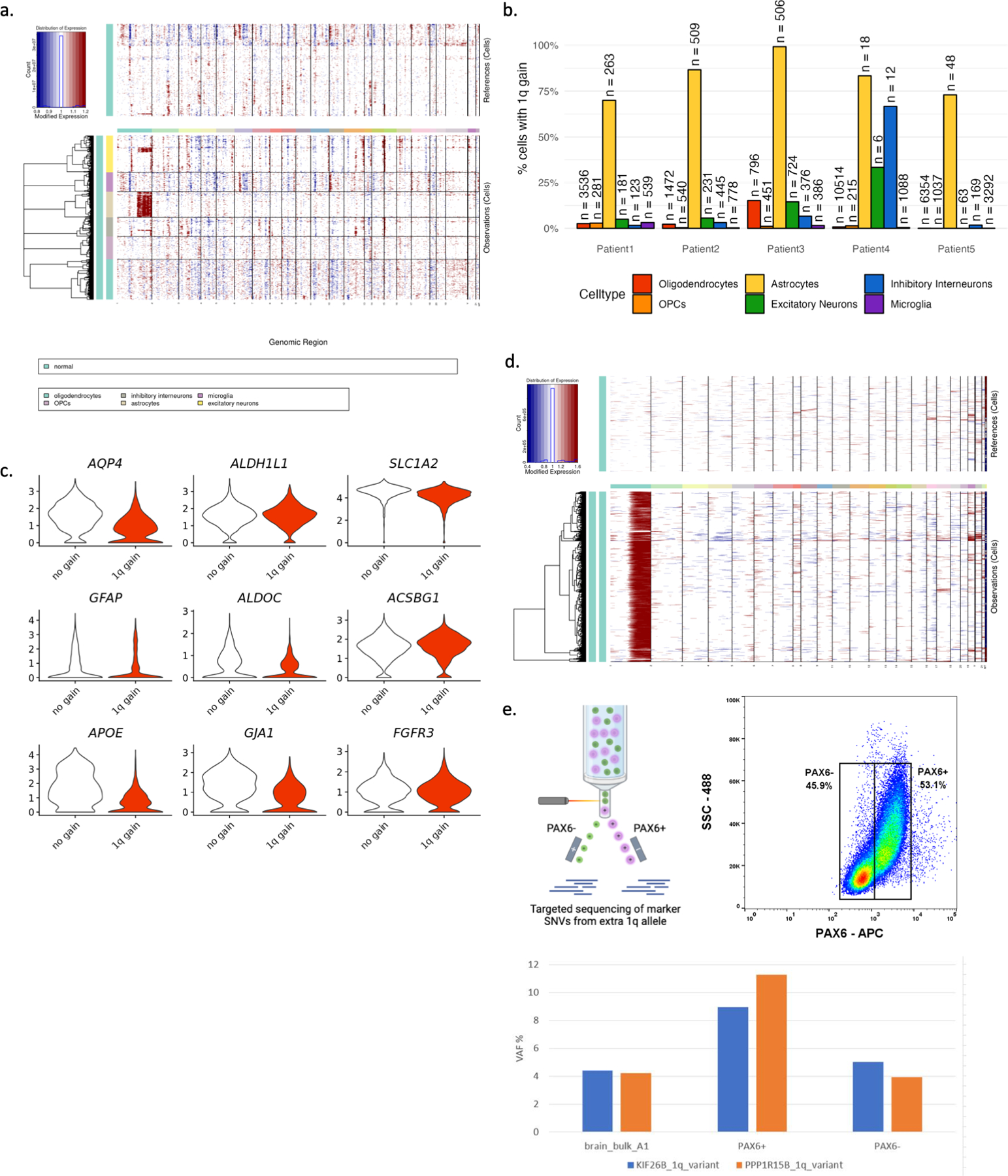
(A) InferCNV output for Patient 3 demonstrating the chromosome 1q gain enriched in astrocytes. (B) Percent of cells with chromosome 1q gain depicted by patient and by cell type. (C) Astrocytes with and without the chromosome 1q gain highly express astrocytic marker genes. (D) InferCNV output comparing Patient 3 astrocytes from affected brain tissue to Patient 3 astrocytes from unaffected adjacent brain tissue. (E) Fluorescence activated nuclei sorting based on PAX6-APC signal to enrich astrocytes followed by targeted sequencing of chromosome 1q marker SNVs in Patient 3. Astrocyte enriched cell population has increased representation of the chromosome 1q gain compared to bulk cells or PAX6-negative cells.
Extended Data Fig. 7.
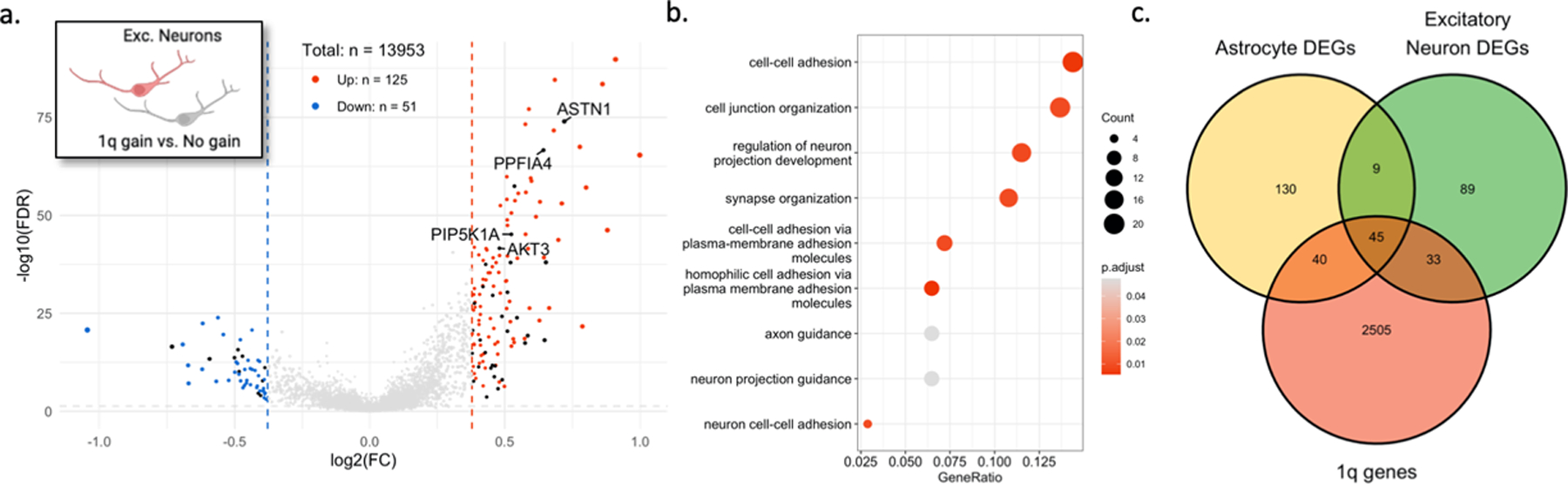
(A) Volcano plot showing differentially expressed genes in excitatory neurons with the chromosome 1q gain vs. those without. Black points represent genes located on chromosome 1q. (B) Enriched GO terms for 1q gain excitatory neurons. (C) Venn diagram showing overlap of differentially expressed genes with genes located on chromosome 1q.
Extended Data Fig. 8.
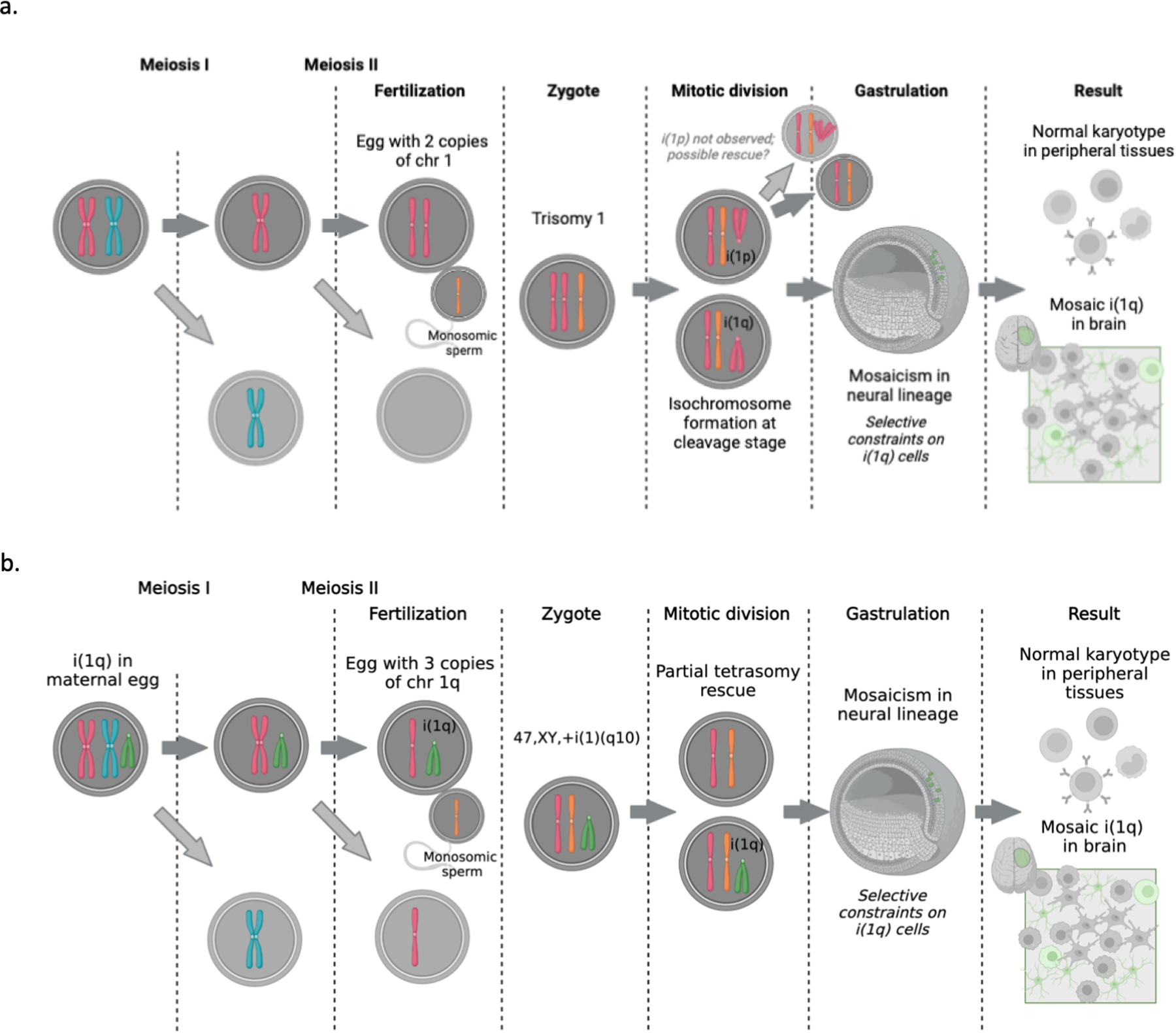
(A) A potential model of mosaic isochromosome 1q formation in the early cleavage-stage embryo. (B) An alternate model of mosaic isochromosome 1q formation originating in an oocyte.
Extended Data Fig. 9.
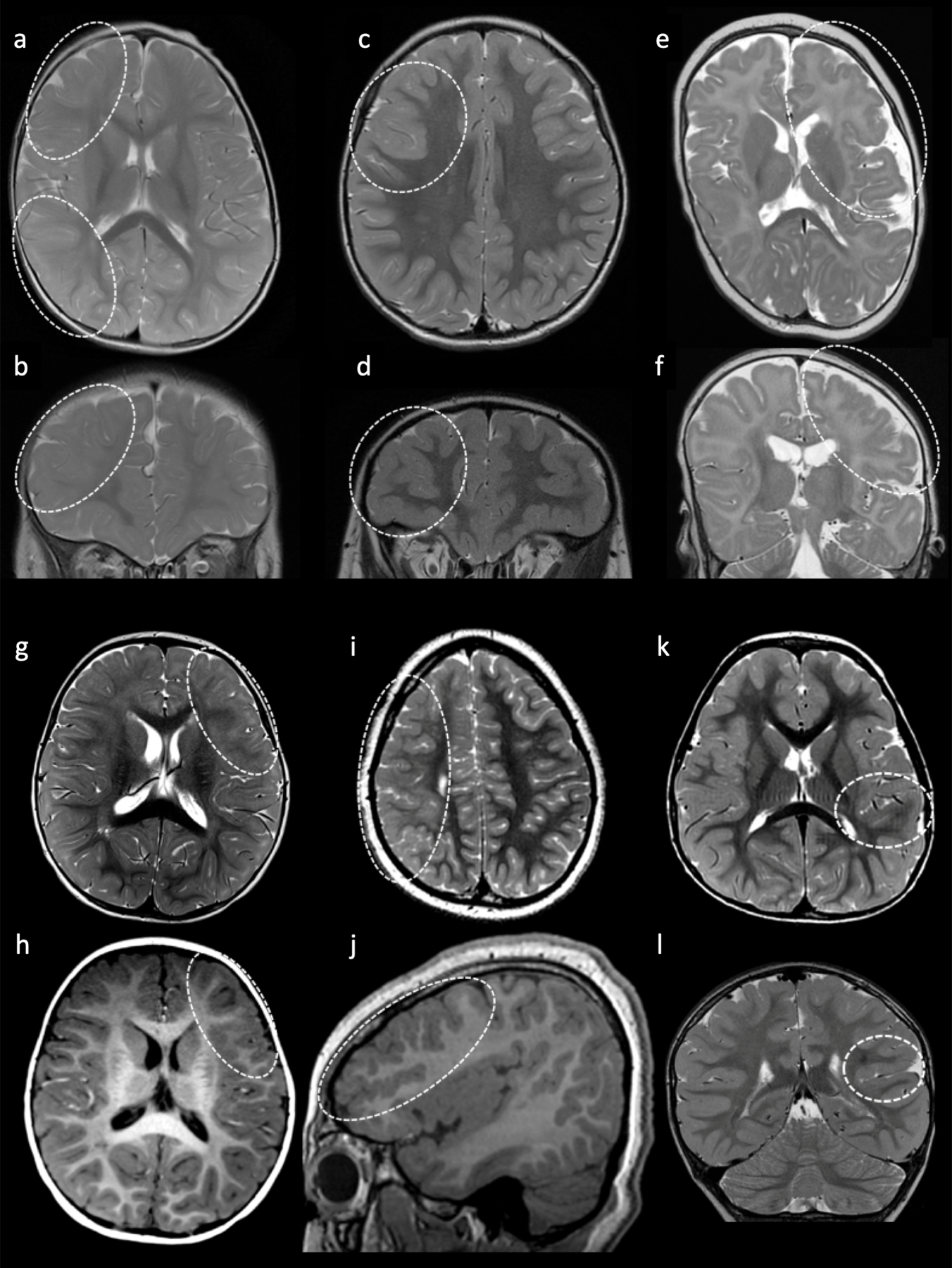
Brain MRIs from subjects with 1q duplications. Axial (A) and coronal (B) T2 images of Patient 1 at 13 months demonstrates hazy T2 prolongation involving the right frontal and parietal lobes (dashed circles). Axial (C) and coronal (D) T2 images of Patient 2 at 5 years of age demonstrate a dysplastic right frontal sulcus (dashed circles). Axial (E) and coronal (F) T2 images of Patient 3 at 9 months of age demonstrate polymicrogyria in the right frontal lobe and insula (dashed circle). Axial T1 (G) and axial T2 (H) images of Patient 6 at 14 months demonstrates hazy T1/T2 prolongation involving the inferior left frontal lobe (dashed circles). Axial T2 (I) and sagittal T1 (J) images of Patient 4 at 9 years of age demonstrate generalized decrease in size of the right hemisphere with a large frontoparietal area of dysplastic cortex (hazy T2 prolongation and increased gyral frequency indicated by dashed circles). Axial T2 (K) and coronal T2 (L) images of Patient 5 at 5 years of age demonstrate subtle T2 prolongation in the left supramarginal and superior temporal gyri (dashed circles).
Supplementary Material
Acknowledgments:
We would like to thank the patients and families for contributing their specimens to advance our understanding of the genetic bases of epilepsy-associated brain malformations. The authors would also like to thank Dr. Richard K. Wilson and the Genomic Services Laboratory and Computational Genomics Group at Nationwide Children’s Hospital. The work was funded by the Nationwide Innovation Fund, L. B. Research and Education Foundation (NS), The Ohio State University Medical Scientist Training Program – T32 Training Grant – T32GM139784 (NS), NIH-NINDS (R01NS094596) to ELH, and NIH-NINDS (R01NS129784) to TAB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Some figures were created in BioRender. Data and/or research tools used in the preparation of this manuscript were obtained from the National Institute of Mental Health (NIMH) Data Archive (NDA). NDA is a collaborative informatics system created by the National Institutes of Health to provide a national resource to support and accelerate research in mental health. Dataset identifier(s): 2962. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or of the Submitters submitting original data to the NDA.
Footnotes
Code Availability:
R scripts to analyze the data and generate the figures are publicly available on Github: https://github.com/bedrosian-lab/1q_mosaicism
Competing Interests Statement: Authors declare that they have no competing interests.
Data Availability:
The single nuclei RNA sequencing data presented in this publication has been deposited in NCBI Gene Expression Omnibus and is accessible through GEO Series accession number GSE221849 (GEO Accession viewer (nih.gov)). Exome sequencing data is available either through dbGAP (phs002128.v1.p1) or BioProject (PRJNA1017875). Capture and targeted sequencing results are available as supplementary data. Exome sequence data from 342 controls was obtained from the North American Brain Expression Consortium (NABEC – dbGAP Study Accession: phs001300.v1.p1), and 15 whole genome-sequenced controls available from the Brain Somatic Mosaicism Network (National Institute of Mental Health Data Archive).
Human reference genome build GRCh38.p12 is available from: Index of /genomes/all/GCF/000/001/405/GCF_000001405.26_GRCh38 (nih.gov)
The Allen Brain Map Human M1 Cortex Dataset is available from: https://portal.brain-map.org/atlases-and-data/rnaseq/human-m1-10x
References:
- 1.Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, Riley-Gillis B, Sestan N, Urban AE, Abyzov A, Vaccarino FM. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 2018;359(6375):550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci 2018;21(11):1504–14. [DOI] [PubMed] [Google Scholar]
- 3.Kobow K, Jabari S, Pieper T, Kudernatsch M, Polster T, Woermann FG, Kalbhenn T, Hamer H, Rossler K, Muhlebner A, Spliet WGM, Feucht M, Hou Y, Stichel D, Korshunov A, Sahm F, Coras R, Blumcke I, von Deimling A. Mosaic trisomy of chromosome 1q in human brain tissue associates with unilateral polymicrogyria, very early-onset focal epilepsy, and severe developmental delay. Acta Neuropathol 2020;140(6):881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012;74(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedrosian TA, Miller KE, Grischow OE, Schieffer KM, LaHaye S, Yoon H, Miller AR, Navarro J, Westfall J, Leraas K, Choi S, Williamson R, Fitch J, Kelly BJ, White P, Lee K, McGrath S, Cottrell CE, Magrini V, Leonard J, et al. Detection of brain somatic variation in epilepsy-associated developmental lesions. Epilepsia 2022;63(8):1981–97. [DOI] [PubMed] [Google Scholar]
- 6.Lai D, Gade M, Yang E, Koh HY, Lu J, Walley NM, Buckley AF, Sands TT, Akman CI, Mikati MA, McKhann GM, Goldman JE, Canoll P, Alexander AL, Park KL, Von Allmen GK, Rodziyevska O, Bhattacharjee MB, Lidov HGW, Vogel H, et al. Somatic variants in diverse genes leads to a spectrum of focal cortical malformations. Brain 2022;145(8):2704–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer GM, Vaziri Fard E, Shah MN, Patel RP, Von Allmen G, Ballester LY, Bhattacharjee MB. Filamin A-negative hyaline astrocytic inclusions in pediatric patients with intractable epilepsy: report of 2 cases. J Neurosurg Pediatr 2020;26(1):38–44. [DOI] [PubMed] [Google Scholar]
- 8.Hedley-Whyte ET, Goldman JE, Nedergaard M, Friedman A, Han X, Schmidt RE, Powers JM. Hyaline protoplasmic astrocytopathy of neocortex. J Neuropathol Exp Neurol 2009;68(2):136–47. [DOI] [PubMed] [Google Scholar]
- 9.Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, Walsh CA. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep 2015;10(4):645. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Rivera JA, Leu C, Macnee M, Khoury J, Hoffmann L, Coras R, Kobow K, Bhattarai N, Perez-Palma E, Hamer H, Brandner S, Rossler K, Bien CG, Kalbhenn T, Pieper T, Hartlieb T, Butler E, Genovese G, Becker K, Altmuller J, et al. The genomic landscape across 474 surgically accessible epileptogenic human brain lesions. Brain 2023;146(4):1342–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 2010;19(7):1263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robberecht C, Voet T, Utine GE, Schinzel A, de Leeuw N, Fryns JP, Vermeesch J. Meiotic errors followed by two parallel postzygotic trisomy rescue events are a frequent cause of constitutional segmental mosaicism. Mol Cytogenet 2012;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pai B, Tome-Garcia J, Cheng WS, Nudelman G, Beaumont KG, Ghatan S, Panov F, Caballero E, Sarpong K, Marcuse L, Yoo J, Jiang Y, Schaefer A, Akbarian S, Sebra R, Pinto D, Zaslavsky E, Tsankova NM. High-resolution transcriptomics informs glial pathology in human temporal lobe epilepsy. Acta Neuropathol Commun 2022;10(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satz JS, Ostendorf AP, Hou S, Turner A, Kusano H, Lee JC, Turk R, Nguyen H, Ross-Barta SE, Westra S, Hoshi T, Moore SA, Campbell KP. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci 2010;30(43):14560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verhoog QP, Holtman L, Aronica E, van Vliet EA. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front Neurol 2020;11:591690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saint-Martin M, Goda Y. Astrocyte-synapse interactions and cell adhesion molecules. FEBS J 2022. [DOI] [PubMed]
- 18.Miller MB, Huang AY, Kim J, Zhou Z, Kirkham SL, Maury EA, Ziegenfuss JS, Reed HC, Neil JE, Rento L, Ryu SC, Ma CC, Luquette LJ, Ames HM, Oakley DH, Frosch MP, Hyman BT, Lodato MA, Lee EA, Walsh CA. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 2022;604(7907):714–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CP, Su YN, Chern SR, Tsai FJ, Wu PC, Chen HE, Chiang SS, Wang W. Mosaic tetrasomy 12p with discrepancy between fetal tissues and extraembryonic tissues: molecular analysis and possible mechanism of formation. Taiwan J Obstet Gynecol 2010;49(2):235–8. [DOI] [PubMed] [Google Scholar]
- 20.Coorens THH, Oliver TRW, Sanghvi R, Sovio U, Cook E, Vento-Tormo R, Haniffa M, Young MD, Rahbari R, Sebire N, Campbell PJ, Charnock-Jones DS, Smith GCS, Behjati S. Inherent mosaicism and extensive mutation of human placentas. Nature 2021;592(7852):80–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eggermann T, Gamerdinger U, Knopfle G, Patzold U, Gembruch U, Hansmann M, Schwanitz G, Hansmann D. Mosaic Trisomy 1q Due to a de novo Translocation in a Foetus with Early Developmental Abnormalities (Karyotype 46,XY, der(14),t(1;14)(p11;p11.2)/46,XY) Delineation of Parent and Cell Stage of Origin. International Journal of Human Genetics 2008;8(4):317–23. [Google Scholar]
- 22.Scheres JM, de Pater JM, Stoutenbeek P, Wijmenga C, Rosenberg C, Pearson PL. Isochromosome 1q as the sole chromosomal abnormality in two fetal teratomas. Possible trisomic or tetrasomic zygote rescue in fetal teratoma with an additional isochromosome 1q. Cancer Genet Cytogenet 1999;115(1):1–10. [DOI] [PubMed] [Google Scholar]
- 23.Warburton D, Dallaire L, Thangavelu M, Ross L, Levin B, Kline J. Trisomy recurrence: a reconsideration based on North American data. Am J Hum Genet 2004;75(3):376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Struski S, Doco-Fenzy M, Cornillet-Lefebvre P. Compilation of published comparative genomic hybridization studies. Cancer Genet Cytogenet 2002;135(1):63–90. [DOI] [PubMed] [Google Scholar]
- 25.Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, Ramaswamy V, Hendrikse LD, Kumar S, Mack SC, Lee JJY, Fong V, Juraschka K, Przelicki D, Michealraj A, Skowron P, Luu B, Suzuki H, Morrissy AS, Cavalli FMG, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019;572(7767):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward S, Harding B, Wilkins P, Harkness W, Hayward R, Darling JL, Thomas DG, Warr T. Gain of 1q and loss of 22 are the most common changes detected by comparative genomic hybridisation in paediatric ependymoma. Genes Chromosomes Cancer 2001;32(1):59–66. [DOI] [PubMed] [Google Scholar]
- 27.Carter M, Nicholson J, Ross F, Crolla J, Allibone R, Balaji V, Perry R, Walker D, Gilbertson R, Ellison DW. Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br J Cancer 2002;86(6):929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Warnatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015;27(5):728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varela C, Denis JA, Polentes J, Feyeux M, Aubert S, Champon B, Pietu G, Peschanski M, Lefort N. Recurrent genomic instability of chromosome 1q in neural derivatives of human embryonic stem cells. J Clin Invest 2012;122(2):569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehrjardi NZ, Molcanyi M, Hatay FF, Timmer M, Shahbazi E, Ackermann JP, Herms S, Heilmann-Heimbach S, Wunderlich TF, Prochnow N, Haghikia A, Lampert A, Hescheler J, Neugebauer EAM, Baharvand H, Saric T. Acquisition of chromosome 1q duplication in parental and genome-edited human-induced pluripotent stem cell-derived neural stem cells results in their higher proliferation rate in vitro and in vivo. Cell Prolif 2020;53(10):e12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minagawa M, Shioda K, Shimizu Y, Isshiki T. Inclusion bodies in cerebral cortical astrocytes: a new change of astrocytes. Acta Neuropathol 1992;84(1):113–6. [DOI] [PubMed] [Google Scholar]
- 32.Van den Veyver IB, Panichkul PP, Antalffy BA, Sun Y, Hunter JV, Armstrong DD. Presence of filamin in the astrocytic inclusions of Aicardi syndrome. Pediatr Neurol 2004;30(1):7–15. [DOI] [PubMed] [Google Scholar]
- 33.Barba C, Blumcke I, Winawer MR, Hartlieb T, Kang HC, Grisotto L, Chipaux M, Bien CG, Hermanovska B, Porter BE, Lidov HGW, Cetica V, Woermann FG, Lopez-Rivera JA, Canoll PD, Mader I, D’Incerti L, Baldassari S, Yang E, Gaballa A, et al. Clinical Features, Neuropathology, and Surgical Outcome in Patients With Refractory Epilepsy and Brain Somatic Variants in the SLC35A2 Gene. Neurology 2023;100(5):e528–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References:
- 34.Kelly BJ, Fitch JR, Hu Y, Corsmeier DJ, Zhong H, Wetzel AN, Nordquist RD, Newsom DL, White P. Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol 2015;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22(3):568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koboldt DC, Miller KE, Miller AR, Bush JM, McGrath S, Leraas K, Crist E, Fair S, Schwind W, Wijeratne S, Fitch J, Leonard J, Shaikhouni A, Hester ME, Magrini V, Ho ML, Pierson CR, Wilson RK, Ostendorf AP, Mardis ER, et al. PTEN somatic mutations contribute to spectrum of cerebral overgrowth. Brain 2021. [DOI] [PMC free article] [PubMed]
- 37.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller KE, Koboldt DC, Schieffer KM, Bedrosian TA, Crist E, Sheline A, Leraas K, Magrini V, Zhong H, Brennan P, Bush J, Fitch J, Bir N, Miller AR, Cottrell CE, Leonard J, Pindrik JA, Rusin JA, Shah SH, White P, et al. Somatic SLC35A2 mosaicism correlates with clinical findings in epilepsy brain tissue. Neurol Genet 2020;6(4):e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184(13):3573–87 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 2019;8(4):329–37 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, Fu X, Liu S, Bo X, Yu G. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2(3):100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single nuclei RNA sequencing data presented in this publication has been deposited in NCBI Gene Expression Omnibus and is accessible through GEO Series accession number GSE221849 (GEO Accession viewer (nih.gov)). Exome sequencing data is available either through dbGAP (phs002128.v1.p1) or BioProject (PRJNA1017875). Capture and targeted sequencing results are available as supplementary data. Exome sequence data from 342 controls was obtained from the North American Brain Expression Consortium (NABEC – dbGAP Study Accession: phs001300.v1.p1), and 15 whole genome-sequenced controls available from the Brain Somatic Mosaicism Network (National Institute of Mental Health Data Archive).
Human reference genome build GRCh38.p12 is available from: Index of /genomes/all/GCF/000/001/405/GCF_000001405.26_GRCh38 (nih.gov)
The Allen Brain Map Human M1 Cortex Dataset is available from: https://portal.brain-map.org/atlases-and-data/rnaseq/human-m1-10x