

Abstract
The stability of the gut microenvironment is inextricably linked to human health, with the onset of many diseases accompanied by dysbiosis of the gut microbiota. It has been reported that there are differences in the microbial community composition between patients and healthy individuals, and many microbes are considered potential biomarkers. Accurately identifying these biomarkers can lead to more precise and reliable clinical decision-making. To improve the accuracy of microbial biomarker identification, this study introduces WSGMB, a computational framework that uses the relative abundance of microbial taxa and health status as inputs. This method has two main contributions: (1) viewing the microbial co-occurrence network as a weighted signed graph and applying graph convolutional neural network techniques for graph classification; (2) designing a new architecture to compute the role transitions of each microbial taxon between health and disease networks, thereby identifying disease-related microbial biomarkers. The weighted signed graph neural network enhances the quality of graph embeddings; quantifying the importance of microbes in different co-occurrence networks better identifies those microbes critical to health. Microbes are ranked according to their importance change scores, and when this score exceeds a set threshold, the microbe is considered a biomarker. This framework’s identification performance is validated by comparing the biomarkers identified by WSGMB with actual microbial biomarkers associated with specific diseases from public literature databases. The study tests the proposed computational framework using actual microbial community data from colorectal cancer and Crohn’s disease samples. It compares it with the most advanced microbial biomarker identification methods. The results show that the WSGMB method outperforms similar approaches in the accuracy of microbial biomarker identification.
Keywords: biomarker, co-occurrence network, gut microbiome, graph representation learning, signed graph
INTRODUCTION
Microbial communities colonize the human body in large numbers, and mounting evidence indicates that these microbiotas play a significant role in human health and disease [1]. The gut harbors the richest microbial populations and significantly influences host physiological health. Extensive research has linked changes in these microbial communities to the onset and progression of numerous diseases, such as autoimmune diseases [2], metabolic anomalies leading to obesity and type 2 diabetes [3] and gastrointestinal cancers like colorectal cancer (CRC) [4]. Therefore, elucidating the associations between microbiomes and diseases can aid in the prevention, diagnosis and prognosis of diseases. Due to the development of high-throughput metagenomic sequencing technologies and the continuous enhancement of microbial databases, researchers can now identify and annotate a greater variety of species in the human gut with increased accuracy. Given the close relationship between the human microbiome and health, microbiome-based predictive analytics aim to use microbial compositions to predict host phenotypes or other clinical outcomes. Microbiome analysis typically employs cost-effective 16S rRNA gene-targeted sequencing methods to obtain reads, which are inputted into bioinformatics pipelines. These pipelines cluster the raw sequencing data to generate operational taxonomic units (OTUs) at specified taxonomic levels. Lastly, these OTUs are classified against publicly available microbial species databases to produce final microbial species composition data tables, where the rows represent samples, and the columns are the microbial abundance matrix at a specific taxonomic level, indicating the quantity of each species in the samples.
Biomarkers are biological indicators that mark changes, or potential changes, in an individual’s biological state, such as genes expressed at different levels and microbes with abundance variations. Accurately identifying biomarkers can increase the precision of clinical diagnoses and prognostic outcomes while enhancing the comprehensive understanding of diseases [5, 6]. Previous studies primarily used correlation analysis methods to identify differentially abundant microbial groups between disease and control groups as biomarkers. However, due to small sample sizes or the complexity of diseases, methods based on abundance difference analysis sometimes yield contradictory results [7]. These discrepancies may arise from limited sample sizes or the multifaceted nature of the disease under investigation.
Machine learning methods, with their discriminatory solid power, have been widely applied in recent years to studies on the correlation between the microbiome and diseases. The abundance of microbial groups is used as feature input into machine learning models to establish mappings with outcome variables. Some methods rank the importance of microbes by measuring the impact of microbial abundance levels on the predictive outcomes or other significance metrics, thereby selecting microbes that have a significant effect on outcome variables to serve as biomarkers for specific diseases [8]. This process is known as ‘feature selection’ in machine learning. Although selecting important microbes as biomarkers based on their impact on outcome variables is viable, this method only focuses on the impact of individual microbial abundance changes on predictive outcomes, overlooking the influence of other microbial groups within the community. Microbes in the human gut interact extensively with each other, not existing as isolated entities but engaging in various interactions such as mutual symbiosis and competition, thus forming multiple gut micro-ecological environments that profoundly affect human health [9]. Current studies have proven that many microbial abundance levels differ between disease and control group samples [10, 11]. However, the lack of understanding of these microbes’ interactions within the community hinders their use as biomarkers for disease diagnosis and prognostic prediction.
Graph-based learning methods have been extensively applied to mine the interactions among different entities. For instance, DeepTraSynergy [12] utilizes various input data, including drug–target interactions, protein–protein interactions and cell–target interactions, to predict the synergistic effects of drug combinations in cancer therapy. TripletMultiDTI [13] introduces a novel multimodal approach to learning and predicting drug–target interactions, facilitating efficient and convenient drug discovery. To better understand the interactions within microbial communities and identify microbial biomarkers closely associated with disease onset, we have constructed microbial co-occurrence networks using rich microbiome data, obtaining further microbial insights through methods that quantify the importance of microbial nodes. In graph theory, this study views such correlation networks as a weighted signed graph, where nodes represent microbial taxa, and edges signify interactions between them. The sign of an edge indicates positive or negative correlations, potentially denoting competition, or mutual symbiosis among microbes. In contrast, the absolute value of the edge’s weight correlates with the interaction strength between microbial taxa. Many studies have also shown that microbial co-occurrence networks improve the identification of disease-related biomarkers [14, 15], providing more reliable guidance for clinical decision-making. The primary steps of WSGMB involve learning the holistic graph representation of disease and control microbial co-occurrence networks and applying it to graph classification tasks. This paper aims to investigate a practical computational framework to identify microbial biomarkers through weighted signed graph classification tasks. To achieve this goal, we generated numerous microbial co-occurrence networks representing diseased and healthy samples using the SparCC (Sparse Correlations for Compositional Data) [16] method. We proposed WSGMB (Weight Signed Graph Convolutional Neural Network for Microbial Biomarker Identification), a novel computational framework for identifying microbial biomarkers based on weighted signed graphs. This method evaluates the importance of bacteria in microbiome co-occurrence networks across different health states by identifying nodes and links that significantly influence the prediction of microbial co-occurrence network categories, thereby characterizing potential microbial biomarkers for specific diseases. We applied WSGMB to real datasets, demonstrating its remarkable ability to identify microbial biomarkers. Our contributions are summarized as follows:
We studied the role and importance of microbes in health and disease co-occurrence networks, measuring the changes in importance, which serves as the basis for biomarker identification. This method helps to filter out the key bacteria that drive the transition from health to disease, providing a reference for clinical diagnosis and prognosis.
We formulated the original problem as a weighted signed graph classification task and proposed a graph neural network-based method, WSGMB, to address it. In WSGMB, a novel convolutional layer was designed for message passing in weighted signed graphs, improving the extraction of structural and weight information from the charts.
WSGMB learns the structural information of microbial co-occurrence networks and the patterns of interactions among microbes, training the graph classification model in an end-to-end manner. By disrupting the connections of specific nodes to others, it computes their impact on the predictive performance of the graph classification model, determining the importance of nodes for health and disease networks.
MATERIALS
Dataset
In this paper, we compiled the necessary dataset for our experiment from the gutMDisorder [17] database, which provides data on the abundance of gut microbiota based on human disease phenotypes and the microbial taxa related to phenotypes. Initially, we downloaded human gut microbiome relative abundance OTUs tables from three CRC studies and two CD studies (see Supplementary Material Table 1) from gutMDisorder, where taxa were aggregated at the genus level, retaining only taxa present in more than 20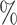 of the samples and with relative abundance greater than 0.0001. Subsequently, we divided the resulting relative abundance OTUs tables into control and case sub-tables. Then, using the SparCC method, we inferred the correlations among microbial taxa by selecting an equal number of samples from both control and disease groups for correlation analysis, thereby generating many microbial co-occurrence networks related to phenotypes. To avoid biases due to data imbalance, we collected an equal number of microbial co-occurrence networks, 500 from both diseased and healthy populations, resulting in control and case networks that serve as input for the weighted signed graph classification algorithm.
of the samples and with relative abundance greater than 0.0001. Subsequently, we divided the resulting relative abundance OTUs tables into control and case sub-tables. Then, using the SparCC method, we inferred the correlations among microbial taxa by selecting an equal number of samples from both control and disease groups for correlation analysis, thereby generating many microbial co-occurrence networks related to phenotypes. To avoid biases due to data imbalance, we collected an equal number of microbial co-occurrence networks, 500 from both diseased and healthy populations, resulting in control and case networks that serve as input for the weighted signed graph classification algorithm.
Problem description
The microbial co-occurrence network is used for analyzing the interactions among microbial communities and can be considered a weighted binary sign network (weighed signed graph) 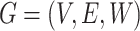 , where
, where 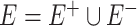 , representing the edges connecting nodes.
, representing the edges connecting nodes. 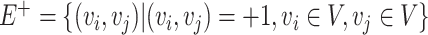 is the set of positive correlation connections among microbial taxa,
is the set of positive correlation connections among microbial taxa, 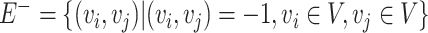 is the set of negative correlation connections, with
is the set of negative correlation connections, with 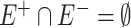 .
. 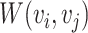 is the weight of the edge, indicating the strength of the correlation between microbial nodes
is the weight of the edge, indicating the strength of the correlation between microbial nodes 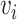 and
and 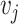 .
.
Although many microbial taxa have been found to be associated with intestinal diseases, due to the complexity of the microbiome, analysis methods based on microbial abundance differences can only identify a few microbial biomarkers. The microbial co-occurrence network provides interaction information between microbes. To understand the differential microbial interactions in healthy and disease networks, we aim to comprehend the interaction patterns of microbes under different health conditions from the weighted signed graph G, identify microbes that undergo significant role changes in the transition from health to disease and propose them as microbial biomarkers for the corresponding conditions.
METHODS
Proposed computational framework
We propose the computational framework WSGMB, which primarily comprises three parts: the construction of microbial co-occurrence networks, weighted signed graph classification and node importance calculation. Firstly, the microbial abundance tables at a specific taxonomic level are divided into OTU subtables corresponding to case and control samples based on phenotypic results, which can also include other variables of interest. For example, we can study microbial interactions at different stages of cancer development. To simplify the model, this study is used solely for binary outcome analysis, that is, healthy and diseased phenotypes. Due to the limitations of sample collection and detection techniques, microbial abundance data may exhibit substantial differences between samples. To eliminate this effect, the abundance of individual microbial taxa is generally divided by the total species abundance obtained from sample sequencing, resulting in a table of species relative abundance for subsequent experimental analysis. Next, the microbial co-occurrence networks inferred by the SparCC method are input into our proposed weighted signed graph classification algorithm WSGCNN (Weight Signed Graph Convolutional Neural Network). This algorithm aims to distinguish healthy and diseased microbial co-occurrence networks by learning the rich interaction information between microbes and the structural information of the co-occurrence network. To measure the impact of nodes on the network, we perturb the network by disrupting the edges connected to specific nodes and calculate the node’s importance to the microbial co-occurrence network. Microbes that exhibit significant differences in role importance between healthy and diseased networks are considered potential biomarkers. The workflow of our proposed method is illustrated in Figure 1.
Figure 1.

Overview of WSGMB, a computational framework based on graph neural networks for the classification of microbial co-occurrence networks and for identifying biomarkers. The input consists of a microbial abundance table divided by phenotype, with the illustration showing data from diseased and healthy samples. An equal number of samples are then extracted from both diseased and healthy groups, and the SparCC is used to infer microbial co-occurrence networks related to the phenotype. These networks serve as training and testing data for the WSGCNN algorithm, which learns the latent graph features associated with the phenotype in the network. Finally, the potential biomarkers are identified by measuring the differences in the importance of nodes across various networks.
Microbial co-occurrence network classification
To classify weighted signed graphs, we propose a new weighted signed undirected graph convolutional neural network model (WSGCNN) that achieves end-to-end learning of node embeddings for weighted signed graphs, ultimately obtaining graph embedding vectors for graph classification, as depicted in Figure 2. In the microbial co-occurrence network, the positivity or negativity and the weights of the edges represent different microbial interaction relationships and semantic information, hence different neighbors of a node should be treated distinctly. We divided the neighbors into two sets based on the sign of the edges, namely, positive neighbors and negative neighbors, and used two aggregators to aggregate neighbor information. Two aggregators were used to assimilate this neighbor information. A learnable parameter is added to the vector obtained for each aggregator, which is then concatenated with the original vector of the node. After processing by a Multi-Layer Perceptron (MLP), a new vector representation of the node is obtained.
Figure 2.

General structure of WSGCNN. The input microbial co-occurrence network first undergoes graph convolution operation through multiple message passing layers, where the nodes aggregate information from different neighbors to update their own features. Then the nodes are sorted using the GSAPool pooling layer, and the whole graph features are obtained after pooling and feature concatenation. Finally, the features are extracted using traditional one-dimensional convolution for learning and predicting network types. The color of the nodes represents the feature information.
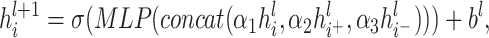 |
(1) |
where 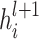 and
and 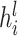 denote the embedding vectors of node
denote the embedding vectors of node 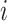 at layers
at layers 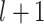 and
and 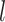 , respectively.
, respectively. 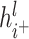 and
and 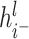 represent the node representations after aggregating positive and negative neighbor information,
represent the node representations after aggregating positive and negative neighbor information, 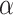 is a learnable parameter and
is a learnable parameter and 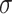 is an activation function. For the aggregation operation of the positive and negative neighbors of a node, we first divide them into two sets based on the sign of the connection, and then use the Softmax method to normalize the weights of the edges:
is an activation function. For the aggregation operation of the positive and negative neighbors of a node, we first divide them into two sets based on the sign of the connection, and then use the Softmax method to normalize the weights of the edges:
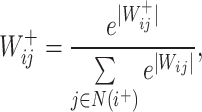 |
(2) |
where 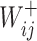 represents the weight of the edges between node
represents the weight of the edges between node 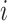 and its positive neighbor node
and its positive neighbor node 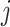 . Similarly, the weight of the edges between node i and its negative neighbors can be determined. The vector of node i after aggregating information from positive neighbors is
. Similarly, the weight of the edges between node i and its negative neighbors can be determined. The vector of node i after aggregating information from positive neighbors is
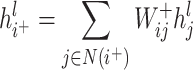 |
(3) |
Given the sparse nature of microbial co-occurrence networks, characterized by a relatively small scale of nodes and edges, previous studies have proven that better graph classification results are obtained using a global pooling architecture [18] when the network is small [19]. Therefore, we chose this graph classification framework to categorize weighted signed graphs. In graph classification, to reduce the number of parameters, enhance learning efficiency and minimize overfitting, it is necessary to perform pooling on the graph neural network. This study adopts GSAPool (Graph Self-Adaptive Pooling) [20], a graph pooling method based on Top-k Selection Pooling. Its advantage lies in considering the topological features and intrinsic features of nodes to rank them comprehensively. Moreover, before discarding the unselected nodes, it aggregates their parts to use all nodes’ feature information fully, generating more representative graph embedding vectors. However, GSAPool is designed for hierarchical pooling architecture [21], as depicted in Figure 3, and we only retained the part that computes the importance scores for node ranking to be used in the global pooling architecture:
Figure 3.
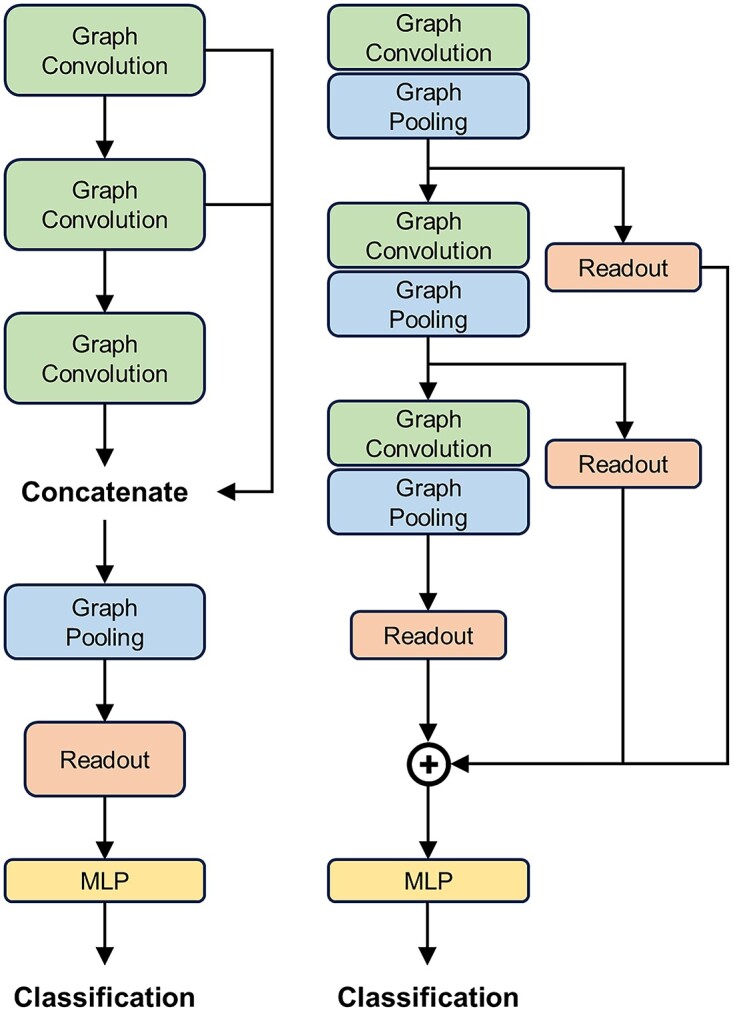
The global pooling architecture (left) and the hierarchical pooling architecture (right).
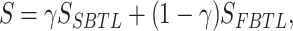 |
(4) |
where the weight 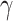 is a hyperparameter that the user can specify;
is a hyperparameter that the user can specify; 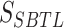 denotes the score of the node’s topological structure feature, while
denotes the score of the node’s topological structure feature, while 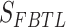 represents the score of the node’s feature. We rank the nodes based on their final scores and select the top k nodes as the pooling result. To measure the importance of each node in the graph, here, k is taken as the total number of nodes, which means we only rank the nodes without conducting a pooling operation.
represents the score of the node’s feature. We rank the nodes based on their final scores and select the top k nodes as the pooling result. To measure the importance of each node in the graph, here, k is taken as the total number of nodes, which means we only rank the nodes without conducting a pooling operation.
To obtain the embedding of the entire graph for the classification of microbial co-occurrence networks, it is necessary to aggregate the features of the nodes in the graph into a fixed-size vector representation, a step known as the graph readout operation in graph classification. Here, we employ a one-dimensional convolutional layer and a max pooling layer to extract the graph feature information, obtaining a fixed-size graph embedding vector. Finally, a fully connected layer and a Softmax layer are used to achieve the predictive results. We use the degree of each node as its initial feature matrix  for two primary reasons: (i) our understanding of microbes is limited, and we cannot obtain information beyond abundance, and (ii) it can test the learning capability of our proposed WSGCNN on the interaction information of microbial community co-occurrence networks. The selection of hyperparameters used in the study can be found in the supplementary materials. Furthermore, we used the Python-based DGL (Deep Graph Library) [22] to implement the WSGCNN algorithm for weighted signed graph classification.
for two primary reasons: (i) our understanding of microbes is limited, and we cannot obtain information beyond abundance, and (ii) it can test the learning capability of our proposed WSGCNN on the interaction information of microbial community co-occurrence networks. The selection of hyperparameters used in the study can be found in the supplementary materials. Furthermore, we used the Python-based DGL (Deep Graph Library) [22] to implement the WSGCNN algorithm for weighted signed graph classification.
Measuring microbial status from network classification
The selection of hyperparameters used in the study can be found in the supplementary materials. Clinical studies may be more concerned with identifying critical microbes and their interactions that drive the shift from a healthy to a diseased state in patients [23]. Using these microbes as biomarkers for diseases can facilitate clinical interventions and treatments. To discern the differences in microbial taxa between case networks and control networks and to describe the significance of microbes during the onset of specific diseases, we employed an intuitive method of ‘kicking out’ nodes from the graph and observed its impact on the predictive probability of the classification task for the co-occurrence network. Specifically, after completing the model prediction task of graph classification, we removed the edges associated with specific nodes from the graph. These modified graphs, now lacking specific node information, were reintroduced into the model for prediction. Then, we calculated the rate of change in prediction accuracy of the new graph relative to the original graph as an importance score for the nodes:
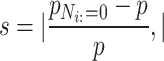 |
(5) |
where 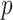 denotes the predictive probability of the original graph classification and
denotes the predictive probability of the original graph classification and 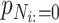 represents the new predictive probability of graph classification obtained after removing the edges connected to node
represents the new predictive probability of graph classification obtained after removing the edges connected to node 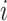 .
.
To measure the differences in the status of various microbes within healthy and diseased networks, we calculated the importance scores for each node in the two types of co-occurrence networks, then subtracted one score from the other and took the absolute value to obtain the ‘difference score’ for each node:
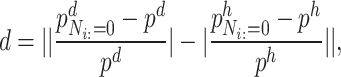 |
(6) |
where 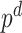 and
and 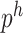 , respectively, denote the classification probabilities for the disease and health networks. By applying the above process to every node in the graph, nodes with higher scores indicate a greater likelihood of being the key bacteria driving the transition to a healthy state.
, respectively, denote the classification probabilities for the disease and health networks. By applying the above process to every node in the graph, nodes with higher scores indicate a greater likelihood of being the key bacteria driving the transition to a healthy state.
Statistical analysis
All statistical analyses were conducted in R version 4.3.1 and visualizations were generated using the ‘ggplot2’ package. P-values less than 0.05 were considered to indicate statistical significance. For each microbiome study, microbial taxa were aggregated at the genus level, retaining only those taxa with a relative abundance greater than 0.0001 and present in at least 20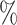 of the samples. The co-occurrence network of microbes was constructed using SparCC. The absolute values of the correlation coefficients of all nodes were calculated, and after subtracting the autocorrelation, the mean was computed. This average was used as a threshold for determining correlations between nodes. The topological coefficients of the network were calculated using the ‘igraph’ R package. For the nodes within the co-occurrence network, we calculated the mean and interquartile range (IQR) and displayed these using box plots.
of the samples. The co-occurrence network of microbes was constructed using SparCC. The absolute values of the correlation coefficients of all nodes were calculated, and after subtracting the autocorrelation, the mean was computed. This average was used as a threshold for determining correlations between nodes. The topological coefficients of the network were calculated using the ‘igraph’ R package. For the nodes within the co-occurrence network, we calculated the mean and interquartile range (IQR) and displayed these using box plots.
EXPERIMENTS
Microbial co-occurrence network threshold selection
The selection of the threshold affects the topological structure of the microbial network. When the threshold is fixed at 0.2, there is a tendency for the topological parameters of the three studies to decrease with the increase in the number of samples in the study queue (Figure 4A). When we fix the number of samples in the study queue, the network topological parameters show the same trend of decrease with the increasing threshold, indicating that choosing a fixed threshold may not correctly reflect the interaction relationships of the microbiome co-occurrence network (Figure 4B). When the selected threshold is high, only a tiny portion of nodes and edges are retained in the network, and most nodes will be discarded, which will significantly affect our fair analysis of all nodes.
Figure 4.

A The average degree distribution of microbial networks constructed based on three CRC studies with different sample sizes. B The edges distribution of microbial networks constructed based on three CRC studies at different thresholds.
To dynamically adjust the threshold to mitigate the impact of changes in sample quantity on the microbial network structure, we use the average of the absolute values of other correlation coefficients in the correlation network, excluding autocorrelation, as the threshold. The advantage of this approach is that even if the number of samples in the research queue changes, the microbial network can still maintain similar topological parameters.
Graph classification model selection
The structure of graphs is distinct from that of images and typically unsuited for deeper network architectures, as they can lead to excessive smoothing of node features. Conversely, shallow networks may need to learn accurate node features [24]. Thus, selecting an appropriate network architecture is critical for graph classification tasks. Given that microbial co-occurrence networks have fewer nodes, we manually choose the number of weighted signed graph convolutional layers that are most suitable for the classification model. Specifically, we begin by exploring with a single graph convolutional layer, then progressively increasing the number of layers until the model’s classification performance no longer improves.
We compared our proposed Weighted Signed Graph Convolutional Neural Network (WSGCNN) with several baseline methods to examine the efficacy of our approach. Since we describe microbial co-occurrence network classification as a weighted signed graph classification task, we selected a representative signed graph embedding model, SNEA [25]. Another method we consider is the classic graph classification approach DGCNN [26], for which we adopted the GraphSAGE [27] convolutional layers.
To investigate the importance of the individual components of WSGCNN, we designed the following variants:
WSGCNN-Weight: Only considers the weights of connections between nodes, disregarding the sign of the edges.
WSGCNN-Sign: Only considers the sign of the edges between nodes, treating the weight of every edge in the graph as equal to 1.
Using CRC1 research as an example, we tested the five methods mentioned above (test results for CRC2 and CRC3 can be found in Supplementary Material Tables 2 and 3). Each method was run 10 times with 100 epochs each, using different random data splits (80-20 training-validation) for model training. We calculated several evaluation metrics: the area under the ROC curve (AUC), Specificity (SP), Precision (PR) and Recall (RE) for binary classification of graphs.
The results in Table 1 indicate that our proposed WSGCNN surpasses the baseline across all metrics, achieving a prediction accuracy of 100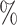 , demonstrating its powerful capability in classifying microbial co-occurrence networks. The DGCNN method, utilizing GraphSAGE as the message-passing layer, can learn the topological features of microbial networks and has also achieved a high prediction accuracy. In contrast, the predictive capability of the attention-based signed graph embedding method SNEA ranks last among the five methods, which may be due to the inapplicability of the balance theory used by the SNEA method to microbial co-occurrence networks. Comparisons between the WSGCNN’s two variants, WSGCNN-Weight and WSGCNN-Sign, and the WSGCNN itself reveal that the type of interactions between microbes can provide additional biological information, enabling the model to achieve higher classification accuracy. As supplementary information, the strength of microbial interactions is highly subtle, yet it still manages to enhance the predictive performance of the model.
, demonstrating its powerful capability in classifying microbial co-occurrence networks. The DGCNN method, utilizing GraphSAGE as the message-passing layer, can learn the topological features of microbial networks and has also achieved a high prediction accuracy. In contrast, the predictive capability of the attention-based signed graph embedding method SNEA ranks last among the five methods, which may be due to the inapplicability of the balance theory used by the SNEA method to microbial co-occurrence networks. Comparisons between the WSGCNN’s two variants, WSGCNN-Weight and WSGCNN-Sign, and the WSGCNN itself reveal that the type of interactions between microbes can provide additional biological information, enabling the model to achieve higher classification accuracy. As supplementary information, the strength of microbial interactions is highly subtle, yet it still manages to enhance the predictive performance of the model.
Table 1.
The performances of different graph classification methods
| Message passing layers | DGCNN | SNEA | WSGCNN-Weight | WSGCNN-Sign | WSGCNN | |
|---|---|---|---|---|---|---|
| AUC | 1 | 0.9465 | 0.8115 | 0.9705 | 0.9930 | 0.9875 |
| 2 | 0.9910 | 0.9325 | 0.9775 | 0.9930 | 1.0000 | |
| 3 | 0.9755 | 0.9420 | 0.9845 | 0.9940 | 0.9955 | |
| SP | 1 | 0.9650 | 0.8120 | 0.9750 | 0.9910 | 0.9880 |
| 2 | 0.9880 | 0.9330 | 0.9750 | 0.9950 | 1.0000 | |
| 3 | 0.9800 | 0.9520 | 0.9800 | 0.9970 | 0.9990 | |
| PR | 1 | 0.9659 | 0.8160 | 0.9752 | 0.9912 | 0.9881 |
| 2 | 0.9882 | 0.9359 | 0.9753 | 0.9950 | 1.0000 | |
| 3 | 0.9800 | 0.9514 | 0.9805 | 0.9970 | 0.9990 | |
| RE | 1 | 0.9640 | 0.8110 | 0.9660 | 0.9950 | 0.9870 |
| 2 | 0.9940 | 0.9320 | 0.9800 | 0.9910 | 1.0000 | |
| 3 | 0.9710 | 0.9320 | 0.9890 | 0.9910 | 0.9920 |
Note: Bold indicates the best prediction results.
Identification of biomarkers in CRC and CD networks
To identify bacteria associated with diseases, we collected OTUs tables of microbial relative abundances from three CRC studies and two Crohn’s disease (CD) studies. We generated 500 case networks and 500 control networks for each survey. We then retrieved 66 CRC-related and 61 CD-related bacteria from the gutMDisorder database. We found, through comparison, that the degree of marked bacteria was almost higher than that of unmarked bacteria in both CRC disease and control networks (Figure 5A), and the same result was observed in CD (Supplementary Figure 3A). This suggests that marked bacteria play an important role in microbial co-occurrence networks. Therefore, assessing the status changes of bacteria in different networks using the WSGMB method to identify disease-related bacteria could be an effective strategy.
Figure 5.

A Average degree of nodes in 500 co-occurrence networks for cases and controls from three CRC studies. Orange represents microbial markers in the gutMDisorder database, and blue represents other bacteria. B Prediction power of three methods in three CRC studies. The red AUC value shows the best prediction of each study across three methods.
We evaluated the accuracy of the WSGMB method using 66 known CRC-related bacteria and 61 known CD-related bacteria and selected two other methods, CACONET [28] and NetMoss [29], for comparison. CACONET is also a graph neural network-based method for assessing node importance, which posits that marked bacteria are only important in case networks. The NetMoss method identifies biological markers associated with various diseases by evaluating the role changes of bacteria in microbial case and control network modules. However, this method is mainly designed for integrated microbial networks; for ease of comparison, we applied it only to microbial networks of individual studies.
The microbial biomarkers of the three CRC and CD studies were identified using the three methods, respectively. The WSGMB method performed the best among the three, achieving an Area Under the Curve (AUC) of over 70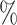 in each CRC study (Figure 5B) and an AUC of over 65
in each CRC study (Figure 5B) and an AUC of over 65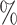 in the two CD studies (Supplementary Figure 3B). We observed that WSGMB predicts better when the network topological characteristics of marked and unmarked bacteria are significantly different. By focusing solely on significant microbes within case networks, the CACONET method may overlook the changes in bacteria between healthy and case networks, leading to poor predictive performance. The NetMoss method, when applied to individual CRC and CD microbial networks, failed to identify the most marked bacteria, and did not demonstrate the excellent predictive performance seen in integrated networks. The WSGMB method is more sensitive to recognizing status changes of bacteria across different networks due to its ability to utilize the interaction information between microbes, thus identifying more marked bacteria. Notably, some bacterial genera such as Acinetobacter, Lachnospira and Phascolarctobacterium, which have been proven to be closely related to human health and disease (Supplementary Figure 2), had relatively high differential scores. Previous research has shown that Lachnospira’s fermentation of pectin to produce short-chain fatty acids [30] plays a key role in preventing CRC [31], which may explain their high differential scores.
in the two CD studies (Supplementary Figure 3B). We observed that WSGMB predicts better when the network topological characteristics of marked and unmarked bacteria are significantly different. By focusing solely on significant microbes within case networks, the CACONET method may overlook the changes in bacteria between healthy and case networks, leading to poor predictive performance. The NetMoss method, when applied to individual CRC and CD microbial networks, failed to identify the most marked bacteria, and did not demonstrate the excellent predictive performance seen in integrated networks. The WSGMB method is more sensitive to recognizing status changes of bacteria across different networks due to its ability to utilize the interaction information between microbes, thus identifying more marked bacteria. Notably, some bacterial genera such as Acinetobacter, Lachnospira and Phascolarctobacterium, which have been proven to be closely related to human health and disease (Supplementary Figure 2), had relatively high differential scores. Previous research has shown that Lachnospira’s fermentation of pectin to produce short-chain fatty acids [30] plays a key role in preventing CRC [31], which may explain their high differential scores.
DISCUSSION
Although several machine learning methods have been developed for screening potential microbial biomarkers, they rely solely on the abundance characteristics of bacteria. The difficulty in sample collection and the complexity of processing mean that most studies analyze very few samples, often far fewer than the number of microbial features, which significantly limits the capabilities of machine learning. Many microbes inhabit the human gut, forming a complex community structure, with species often engaged in mutualistic symbiosis or competition, creating tightly connected co-occurrence networks. Therefore, focusing only on the abundance of specific microbes does not provide a complete view of the community, nor does it comprehensively assess the roles these microbes play within the health and disease network. Unlike methods that link the abundance of individual microbial groups with phenotypic outcomes, our proposed framework WSGMB classifies co-occurrence networks constructed for microbial populations, revealing differences in the status of specific bacteria across various networks by disrupting microbial interactions.
Another relevant work using graph neural networks to identify essential microbes is CACONET. In comparison with CACONET, several points need to be clarified. First, CACONET employs traditional graph classification methods without considering the signs and weights of the network edges; our computational framework has designed WSGCNN for weighted signed graphs to fully utilize the information of connection signs and weights, with innovative pooling methods yielding superior classification predictions. Secondly, in constructing co-occurrence networks, CACONET chose a uniform correlation coefficient cut-off threshold, which may not retain true microbial interactions as microbial interactions tend to weaken with an increase in the sample size of study cohorts. Our approach averages the correlation coefficients, retaining edges stronger than the average connection strength, thus eliminating network structure changes due to varying sample sizes. Lastly, our microbial biomarker identification strategy assesses the significance of microbes within both healthy and disease networks, reflecting the status changes of specific microbes in the community from health to disease; CACONET, on the other hand, only focuses on the importance of microbes to disease networks, overlooking their impact on healthy networks.
Our proposed Weighted Signed Graph Convolutional Neural Network (WSGCNN) outperforms the DGCNN method used in CACONET on real datasets, demonstrating superior capability in classifying co-occurrence networks. This also indirectly indicates the method’s ability to extract unique graph features from weighted signed graphs. To differentiate the contribution of microbes in predicting network categories, we disrupted the interactions of the same bacteria in both healthy and disease networks. We calculated the change in prediction accuracy for both networks, suggesting that focusing on the importance of bacteria in the network may be a reasonable strategy to distinguish disease-related bacteria from others.
CRC is a highly prevalent gastrointestinal disease, often accompanied by dysbiosis of intestinal microbial populations [32]. The gut microbes linked to this cancer are considerably affected by environmental determinants such as dietary habits [33] and lifestyle, resulting in pronounced variances across different regions and ethnic groups [34].This variation poses challenges for early clinical screening of CRC biomarkers based on microbial abundance data and often leads to contradictory results among different microbiome studies. We tested our method using data from three CRC microbiome studies. We found that the WSGMB method could identify more biomarkers than previous methods, demonstrating the advantages of a network-based approach in identifying disease microbial biomarkers. The results also suggest that interactions among microbes may be a unique feature of co-occurrence networks, and disrupting these interactions could be a new avenue for clinical treatment. In the field of graph machine learning, it is usually necessary to assign original features to the nodes in the graph before performing the message-passing step in the graph neural network model training. However, the current understanding of microbes is unclear, preventing the accurate representation of individual microbial features. We used the degree of each node as its original feature, which may not fully represent the feature information of the node, thus limiting the predictive capability of the graph classification model. In the future, as research advances, establishing a feature database for microbes will undoubtedly enhance the ability of graph neural networks to discriminate microbial co-occurrence networks, thereby increasing the accuracy of microbial biomarker identification.
LIMITATIONS
This study has three crucial potential limitations. First, a fully accurate categorization of the microbiome co-occurrence networks has not yet been achieved. This is because we only used one topological feature—the degree of the nodes—when designing the initial characteristics of the microbial nodes without providing additional unique information inherent to the microbes themselves. Using only one topological feature to develop significantly limits the learning capabilities of the weighted signed graph neural networks regarding the features of the microbiome co-occurrence networks. It may affect the assessment of the importance of microbial nodes. Secondly, the number of samples used in this study is limited. Due to the complex process of collecting and analyzing gut microbiota, most microbiome studies struggle to cover many samples. The limited number of samples may result in some microbial interactions not being fully reflected and hence overlooked. Finally, because only the impact of single microbes on the co-occurrence network was considered, the magnitude of the designed node difference scores in this study is relatively small, which may affect the determination of important microbes.
In our future work, we plan to establish a database with a larger number of samples to enhance the outcomes of our research and to fully exploit the characteristic information of the microbes themselves to improve the predictive performance of the weighted signed graph neural networks. Moreover, future research will explore the effects of various microbial interactions on human health to enable more precise identification of microbial biomarkers.
CONCLUSION
We propose a computational framework that employs a novel weighted signed graph neural network to identify microbial biomarkers for intestinal diseases through a microbiome co-occurrence network constructed from gut microbiome data. By leveraging the correlations among microbes, we created disease and control microbiome co-occurrence networks based on health status. We designed a message-passing layer for this weighted signed graph to extract potential microbial interaction information within the co-occurrence network, achieving precise classification of the microbiome co-occurrence network. Subsequently, by disrupting the connections of specific microbial nodes in the co-occurrence network and calculating their impact on the classification accuracy of the two types of co-occurrence networks, we determined the importance differences of nodes across various networks, thereby identifying whether microbial nodes serve as biomarkers. Encouraging results were obtained in studies of CRC and CD, with a significant improvement in the identification accuracy of microbial biomarkers compared with competitive methods.
From our analysis, we can infer that the proposed weighted signed graph neural network and computational framework capture the interaction information among microbes effectively. Quantifying the changes in microbial node importance characterizes the differences between microbes in disease and control samples, and these methods contribute to the increased accuracy of microbial biomarker identification. In the future, we hope to apply the WSGMB to the identification tasks of microbial biomarkers for multiple phenotypes rather than being limited to the current binary phenotypes.
The WSGMB method significantly enhances the capability to identify network-based microbial biomarkers. With the development of more accurate correlation inference methods and interpretable graph neural networks, this computational framework will aid in our understanding of the link between human microbiome interactions and health. By considering microbial interactions on top of individual microbial abundance studies, we can discover more overlooked details, which may play a crucial role in disease clinical screening and treatment plan design.
Key Points
We examine microbial roles within health-disease co-occurrence networks for biomarker identification. This strategy aids in pinpointing pivotal bacteria that catalyze the transition from a healthy to a diseased state, offering valuable insights for clinical diagnosis and prognosis.
Recasting the primary problem as a weight-signed graph classification task and unveiling WSGMB, a graph neural network-based solution. Within WSGMB, a novel convolutional layer facilitates efficient message passing for weighted signed graphs, optimizing the extraction of both structural and weight attributes from such graphs.
Ensuring WSGMB’s capability to train graph classification models end-to-end, leveraging microbial co-occurrence network structural information and microbial interaction patterns. The framework also quantifies node significance in health and disease networks by altering specific node connections and calculating their impact on the predictive performance of the graph classification model.
Supplementary Material
ACKNOWLEDGMENTS
We thank the anonymous reviewers for their valuable suggestions.
Author Biographies
Shuheng Pan is a master’s student at Tsinghua University. His research interests include deep learning, graph neural networks and bioinformatics.
Xinyi Jiang is a master’s student at Tsinghua University. Her research interests include gut microbiome, biomaterials and tissue engineering.
Kai Zhang is a professor in the institute of data and information at Tsinghua Shenzhen International Graduate School. He works on artificial intelligence and big data methods for microbiomics and pharmacokinetics.
Contributor Information
Shuheng Pan, Institute of Data and Information, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518005, China.
Xinyi Jiang, Institute of Data and Information, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518005, China.
Kai Zhang, Institute of Data and Information, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518005, China.
DATA AVAILABILITY
The datasets were derived from the following sources in the public domain, the CRC OTUs tables and related microbes from http://bio-annotation.cn/gutMDisorder/browse.dhtml. The implementation of WSGMB and the preprocessed data is available at https://github.com/panshuheng/WSGMB/tree/master.
AUTHOR CONTRIBUTIONS STATEMENT
K.Z. conceived the project, S.P. conducted the experiments, S.P. and X.J. analyzed the results and wrote the manuscript.
References
- 1. Aggarwal N, Kitano S, Ying GR, et al. Microbiome and human health: current understanding, engineering, and enabling technologies. Chem Rev 2022;123(1):31–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaheen WA, Quraishi MN, Iqbal TH. Gut microbiome and autoimmune disorders. Clin Exp Immunol 2022;209(2):161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartstra AV, Bouter KEC, Bäckhed F, Nieuwdorp M. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care 2015;38(1):159–65. [DOI] [PubMed] [Google Scholar]
- 4. Thomas RM. Role of bacteria in the development of colorectal cancer. Clin Colon Rectal Surg 2023;36(02):105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shahanavaj K, Gil-Bazo I, Castiglia M, et al. Cancer and the microbiome: potential applications as new tumor biomarker. Expert Rev Anticancer Ther 2015;15(3):317–30. [DOI] [PubMed] [Google Scholar]
- 6. Li L, Kang Y. The gut microbiome and autoimmune hepatitis: implications for early diagnostic biomarkers and novel therapies. Mol Nutr Food Res 2023;2300043. [DOI] [PubMed] [Google Scholar]
- 7. Hawinkel S, Mattiello F, Bijnens L, Thas O. A broken promise: microbiome differential abundance methods do not control the false discovery rate. Brief Bioinform 2019;20(1):210–21. [DOI] [PubMed] [Google Scholar]
- 8. Cui C, Song Y, Mao D, et al. Predicting the postmortem interval based on gravesoil microbiome data and a random forest model. Microorganisms 2022;11(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Praagh JB, Havenga K. What is the microbiome? A description of a social network. Clin Colon Rectal Surg 2023;36(02):091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peterson SN, Snesrud E, Liu J, et al. The dental plaque microbiome in health and disease. PloS One 2013;8(3):e58487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhuang H, Cheng L, Wang Y, et al. Dysbiosis of the gut microbiome in lung cancer. Front Cell Infect Microbiol 2019;9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rafiei F, Zeraati H, Abbasi K, et al. Deeptrasynergy: drug combinations using multimodal deep learning with transformers. Bioinformatics 2023;39(8):btad438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dehghan A, Razzaghi P, Abbasi K, Gharaghani S. Tripletmultidti: multimodal representation learning in drug-target interaction prediction with triplet loss function. Expert Syst Appl 2023;120754. [Google Scholar]
- 14. Naqvi A, Rangwala H, Keshavarzian A, Gillevet P. Network-based modeling of the human gut microbiome. Chem Biodivers 2010;7(5):1040–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aogáin MM, Narayana JK, Tiew PY, et al. Integrative microbiomics in bronchiectasis exacerbations. Nat Med 2021;27(4):688–99. [DOI] [PubMed] [Google Scholar]
- 16. Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol 2012;8(9):e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qi C, Cai Y, Qian K, et al. Gutmdisorder v2. 0: a comprehensive database for dysbiosis of gut microbiota in phenotypes and interventions. Nucleic Acids Res 2023;51(D1):D717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilmer J, Schoenholz SS, Riley PF, et al. Neural message passing for quantum chemistry. In: International conference on machine learning. PMLR, 2017, 1263–72. [Google Scholar]
- 19. Lee J, Lee I, Kang J. Self-attention graph pooling. In: International conference on machine learning. PMLR, 2019, 3734–43. [Google Scholar]
- 20. Zhang L., Wang X., Li H., Zhu G., Shen P., Li P., Xiaoyuan L., Shah Syed Afaq Ali, and Bennamoun M.. Structure-feature based graph self-adaptive pooling. In Proceedings of The Web Conference 2020, pages 3098–104, 2020.
- 21. Cangea C, Veličković P, Jovanović N, et al. Towards sparse hierarchical graph classifiers. arXiv preprint arXiv:181101287 2018. [Google Scholar]
- 22. Wang M, Da Zheng ZY, Gan Q, et al. Deep graph library: a graph-centric, highly-performant package for graph neural networks. arXiv preprint arXiv:190901315 2019. [Google Scholar]
- 23. Kneis B, Wirtz S, Weber K, et al. Colon cancer microbiome landscaping: differences in right-and left-sided colon cancer and a tumor microbiome-ileal microbiome association. Int J Mol Sci 2023;24(4):3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Liu J, Li Y. Automatic search of architecture and hyperparameters of graph convolutional networks for node classification. Appl Intell 2023;53(9):11104–19. [Google Scholar]
- 25. Huang J., Shen H., Liang H., and Cheng X.. Signed graph attention networks. In Artificial Neural Networks and Machine Learning–ICANN 2019: Workshop and Special Sessions: 28th International Conference on Artificial Neural Networks, Munich, Germany, September 17–19, 2019, Proceedings 28, pages 566–77. Springer, 2019. [Google Scholar]
- 26. Zhang M., Cui Z., Neumann M., and Chen Y.. An end-to-end deep learning architecture for graph classification. In Proceedings of the AAAI conference on artificial intelligence, 32, 2018. [Google Scholar]
- 27. Hamilton W, Ying Z, Leskovec J. Inductive representation learning on large graphs. Advances in neural information processing systems 2017;30. [Google Scholar]
- 28. Yuanwei X, Nash K, Acharjee A, Gkoutos GV. Caconet: a novel classification framework for microbial correlation networks. Bioinformatics 2022;38(6):1639–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao L, Zhang F, Zhao F. Large-scale microbiome data integration enables robust biomarker identification. Nat Comput Sci 2022;2(5):307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu Z, Lei ZY, Zeng WZ, Yang JF. Effect of tea polysaccharides on faecal microbiota and their short-chain fatty acid metabolic products. Int Food Res J 2023;30(1). [Google Scholar]
- 31. Hinnebusch BF, Meng S, Wu JT, et al. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr 2002;132(5):1012–7. [DOI] [PubMed] [Google Scholar]
- 32. Yang J, Wei H, Zhou Y, et al. High-fat diet promotes colorectal tumorigenesis through modulating gut microbiota and metabolites. Gastroenterology 2022;162(1):135–49. [DOI] [PubMed] [Google Scholar]
- 33. Veettil SK, Wong TY, Loo YS, et al. Role of diet in colorectal cancer incidence: umbrella review of meta-analyses of prospective observational studies. JAMA Netw Open 2021;4(2):e2037341–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cai J-A, Zhang Y-Z, En-Da Y, et al. Gut microbiota enterotypes mediate the effects of dietary patterns on colorectal neoplasm risk in a chinese population. Nutrients 2023;15(13):2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets were derived from the following sources in the public domain, the CRC OTUs tables and related microbes from http://bio-annotation.cn/gutMDisorder/browse.dhtml. The implementation of WSGMB and the preprocessed data is available at https://github.com/panshuheng/WSGMB/tree/master.