

Abstract
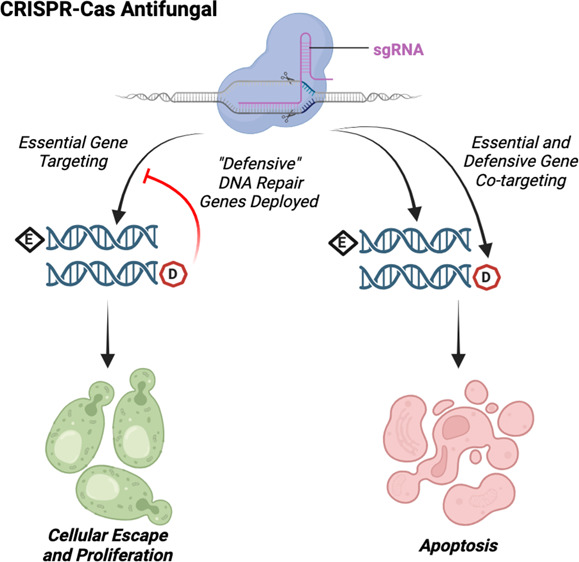
The emergence of virulent, resistant, and rapidly evolving fungal pathogens poses a significant threat to public health, agriculture, and the environment. Targeting cellular processes with standard small-molecule intervention may be effective but requires long development times and is prone to antibiotic resistance. To overcome the current limitations of antibiotic development and treatment, this study harnesses CRISPR-Cas systems as antifungals by capitalizing on their adaptability, specificity, and efficiency in target design. The conventional design of CRISPR-Cas antimicrobials, based on induction of DNA double-strand breaks (DSBs), is potentially less effective in fungi due to robust eukaryotic DNA repair machinery. Here, we report a novel design principle to formulate more effective CRISPR-Cas antifungals by cotargeting essential genes with DNA repair defensive genes that remove the fungi’s ability to repair the DSB sites of essential genes. By evaluating this design on the model fungus Saccharomyces cerevisiae, we demonstrated that essential and defensive gene cotargeting is more effective than either essential or defensive gene targeting alone. The top-performing CRISPR-Cas antifungals performed as effectively as the antibiotic Geneticin. A gene cotargeting interaction analysis revealed that cotargeting essential genes with RAD52 involved in homologous recombination (HR) was the most synergistic combination. Fast growth kinetics of S. cerevisiae induced resistance to CRISPR-Cas antifungals, where genetic mutations mostly occurred in defensive genes and guide RNA sequences.
Keywords: CRISPR, Cas enzymes, Saccharomyces cerevisiae, CRISPR-Cas antifungal, antibiotic resistance, essential genes, defensive genes, essential and defensive gene cotargeting, homologous recombination (HR), nonhomologous end joining (NHEJ)
Fungi represent a particularly resistant class of pathogens that are the source of dysbiosis in a variety of societally relevant hosts and environments.1,2 In humans, fungal pathogens are responsible for over one billion infections and 1.5 million deaths each year with species of Candida, Aspergillus, and Cryptococcus accounting for 90% of fungal-related deaths.3 The prevalence of fungal infections is surging due to an increase in immunocompromised individuals as well as an increase in geographic range and dispersal due to climate change.4 Along with an increased at-risk population, the threat of fungal pathogens is amplified by the rapid emergence of antifungal resistance. Overuse of current antifungals in both medical and agricultural settings has escalated the emergence of antifungal-resistant species.5 Azole resistance is widespread in Candida, Aspergillus, and Cryptococcus species, while echinocandin and polyene resistance is less frequent.5,6 Most notably, Candida auris, a fungal pathogen first identified in Japan in 2009 that since has spread globally with cases rising in parallel with the COVID-19 pandemic, is often multidrug resistant with 90% of isolates being fluconazole resistant and 30% of isolates being resistant to amphotericin B in the United States.7 The increasing threat of antifungal-resistant species and the lack of new antifungal drugs warrant investigation into novel antifungal strategies.
CRISPR-Cas (clustered regularly interspaced short palindromic repeats (CRISPR)-associated) gene editing, a powerful method for knocking out key genes in the growth regulation cycle, has recently shown promise for antimicrobial treatment in bacterial systems.8 Adaptability, specificity, and efficiency in designing guide RNAs (gRNAs) to enable precise gene inactivation make CRISPR-Cas systems ideal for neutralizing antibiotic-resistant and rapidly evolving pathogens. In principle, such CRISPR-Cas systems can also be employed as antifungals. However, the conventional CRISPR-based antimicrobial design that mostly relies on the high lethality of Cas-induced DNA double-strand-breaks (DSBs) faces an additional barrier to neutralizing fungal pathogens because robust eukaryotic DNA repair mechanisms can easily counteract DSB activity, rendering Cas enzymes only mildly toxic, if at all.9 Even in prokaryotes lacking robust DNA repair mechanisms, continuous repair of CRISPR-Cas induced DSBs by homologous recombination (HR) can lead to cell survival.10 Due to this limitation, a new strategy of cotargeting essential genes along with DNA repair defensive genes and ensuring their complete or near-complete knockout is vital to achieve efficacious CRISPR-Cas antifungals but is currently unexplored. Inhibiting HR via the phage Mu-Gam protein in Escherichia coli has been shown to restore CRISPR-Cas killing activity of weak gRNAs and enhance the killing activity of stronger gRNAs, supporting the strategy of cotargeting essential and DNA repair genes for improved CRISPR-Cas antimicrobials.10 Since the cataloged diversity of preferred repair mechanisms shows a dependence on growth stages, systematic characterization of essential and DNA repair gene interactions is critical for the effective design of CRISPR-Cas antifungals but is challenging due to a large combinatorial search of optimal cotargets, especially when facing nonmodel fungal pathogens. Therefore, the use of a model organism to understand the significance of this relationship is essential for formulating effective CRISPR-Cas antifungals.
The yeast Saccharomyces cerevisiae is a widely studied model organism for understanding fungal genetics and biology.11 Due to its highly curated genome, the essentiality of its genes is well documented and much is known of its robust DNA repair pathways and growth cycles, which exhibit many similarities to the clinically relevant pathogenic yeasts of the Candida genus.12 Even though S. cerevisiae is generally considered to be safe, some strains are known to be opportunistic human pathogens that can be deadly to immunocompromised individuals.13 Thus, the established genetics and clinical relevance make S. cerevisiae an ideal model for studying the effectiveness of essential and defensive gene cotargeting as a CRISPR-Cas antifungal strategy.
In this study, we investigated promising CRISPR-Cas systems for use as antifungals by screening multiple target sites across essential genes that are important for cell survival in S. cerevisiae. We demonstrated the enhanced overall lethality of CRISPR-Cas antifungals by cotargeting essential genes with DNA repair defensive genes that removed the ability of the organism to effectively repair the DSB sites. Through modulations that affect cell growth kinetics such as solid versus liquid growth media and cell inoculation, we identified that control of the abundant expression of CRISPR-Cas systems is critical to enhance potency of CRISPR-Cas antifungals while minimizing antifungal resistance. Overall, this study presents a novel design principle for formulating effective CRISPR-Cas antifungals and provides mechanistic insights into the potential rise of antifungal resistance.
Results and Discussion
Cotargeting Essential and Defensive Gene Machinery Is More Effective for CRISPR-Cas Antifungal Formulation
The robust DNA repair mechanisms of eukaryotes present significant hindrance to the development of CRISPR-Cas antifungals (Figure 1A). We hypothesized that cotargeting both essential and defensive gene machinery increases the potency of CRISPR-Cas antifungals (Figure 1B). To test this hypothesis, we investigated the capability of multiplexing gRNAs targeting both essential and defensive gene machinery in S. cerevisiae. We began by choosing a set of 3 DNA repair defensive genes, RAD51, RAD52, and LIF1, that are key elements to either HR or nonhomologous end joining (NHEJ) pathways and a set of 23 essential genes involving replication, transcription, translation, and metabolism (Table S1). We then constructed 55 strains with duet plasmid systems carrying gRNAs targeting 55 essential–defensive (E–D) gene combinations on one plasmid and Cas9 on a second plasmid (Tables S2, S3). While 69 total combinations were possible, only 55 were successfully constructed, and we chose to proceed with just these 55 strains in the interest of rapid screening for top performing candidates. As a control, we also constructed 29 strains carrying gRNAs targeting 23 essential (E), 3 defensive (D), and 3 defensive–defensive (D–D) genes. CRISPR-Cas antifungals in these strains were programmed for activation by galactose induction of Cas9 expression. We next characterized the potency of the CRISPR-Cas antifungals by both a serial dilution growth assay on solid media and in liquid culture where cells would not grow in the presence of galactose if the CRISPR-Cas antifungal treatment was effective.
Figure 1.

Design of CRISPR-Cas antifungals. (A, B) CRISPR-Cas antifungal formulation (A) with essential gene targeting alone and (B) with essential and defensive gene cotargeting. Targeting a single essential gene locus alone is not effective to eliminate cell survival due to active DNA repair machinery. Cotargeting parts of the DNA repair response reduces the cell’s capability to repair legions at essential and defensive sites, increasing lethality via essential gene knockout and DSB-induced apoptosis. (C, D) Heat maps show the effectiveness of (C) single essential or defensive gene targeting and (D) essential and defensive gene cotargeting. Each heat map represents serial dilution (1×, 10×, 100×, and 500×) of cells that were treated with different CRISPR-Cas antifungals, spotted on galactose plates (Cas9 “on”, test) and glucose plates (Cas9 “off”, control), and incubated for 48 h at 28 °C. Noninduced strains grown on glucose plates are indicated in red, while induced Cas9 strains grown on galactose pads are indicated in purple. The top 17 strains chosen for further characterization are marked with a star. Larger colony size is represented by darker coloration. Note that panels B and D show a representative list of characterized strains; the complete list can be found in Figure S1. (E) Gene cotargeting interaction analysis.
In the case of gRNAs targeting single essential or defensive gene loci, we found most strains still exhibited a viable phenotype (Figures 1C and S1) likely because the robust DNA repair machinery of S. cerevisiae repaired any breaks at these loci, resulting in only mild toxicity. In contrast, cotargeting of essential and defensive genes proved more effective at inhibiting growth (Figures 1D and S1). As a control, we also found that dual or multiplex targeting essential genes further increased toxicity but fell short of the efficacy found by cotargeting essential and defensive gene machinery simultaneously, which leaves the cell unable to reliably repair a break at the essential gene sites (Figure S1). We identified 17 strains (Figures 1D and 2), including Vip47 (harboring LIF1, DNA19), Vip48 (LIF1, ACC2), Vip52 (LIF1, RPL10E), Vip54 (LIF1, RPL37A), Vip55 (LIF1, RPS13B), Vip56 (RAD51, DNA43), Vip57 (RAD51, ISP45), Vip59 (RAD51, RPL7), Vip60 (RAD51, RPS21), Vip69 (RAD52, RPL17), Vip73 (RAD52, RPL7), Vip74 (LIF1, RPL1), Vip76 (RAD52, LIG1), Vip81 (RAD51, RPS4), Vip90 (RAD52, RPS4), Vip93 (LIF1, RSP42), and Vip94 (RAD52, RPO22), that exhibited more effective potency of CRISPR-Cas antifungals using the serial dilution growth assay on solid media. Overall, cotargeting both essential and defensive genes is a promising design principle for formulating the most effective CRISPR-Cas antifungals.
Figure 2.

Potency of CRISPR-Cas antifungals and potential rise of antifungal resistance. (A) Slower cell growth on solid media did not induce antifungal resistance. All selected strains carrying the top performing CRISPR-Cas antifungals exhibited loss of growth on galactose (Cas9 “on”) plate. (B) Faster cell growth on liquid media escaped CRISPR-Cas antifungal treatment. Three strains (Vip54, Vip57, and Vip59) exhibited a normal growth phenotype as compared to the control strain (Vip46*); nine strains (Vip47, Vip48, Vip55, Vip56, Vip69, Vip73, Vip74, Vip81, and Vip90) showed a growth lag but started to enter exponential growth after 48-h outgrowth; and the remaining five strains (Vip52, Vip60, Vip76, Vip93, and Vip94) exhibited growth arrest. (C) Comparison of potency between the top CRISPR-Cas antifungals and antibiotic Geneticin (G418). The percentage of viable cells for each strain is presented above each bar. Cell growth on glucose served as a negative control with inactive CRISPR-Cas systems, while cell characterization without a carbon source served as a positive control. In panels B and C, each value is a mean ± 1 standard deviation (n ≥ 2).
To determine which defensive gene is more effective for cotargeting with an essential gene or vice versa, we formulated a gene cotargeting interaction model (eq 3) that utilizes the serial dilution growth data on the solid media for S. cerevisiae under different CRISPR-Cas antifungal treatment. The model calculates an interaction score (−1 ≤ IED ≤ 1) for a defensive and essential gene cotarget. A positive interaction score indicates a synergistic interaction between the cotargets leading to a more lethal phenotype than expected, while a negative score implies an antagonistic interaction. The analysis revealed that strains cotargeting RAD52 with an essential gene had the most synergistic interactions with their essential gene targets (Figure 1E). RAD52 plays a key role in both RAD51-dependent and RAD51-independent HR pathways making RAD52 null mutants particularly susceptible to DSBs.14 Although both are important in HR, cotargeting RAD51 with an essential gene was likely not as synergistic as RAD52 since RAD51-independent HR mechanisms such as single-strand annealing (SSA) or break-induced replication (BIR) could still be utilized by the cells.14 Cotargeting the NHEJ gene LIF1 with an essential gene was also not as synergistic as RAD52 presumably since HR is the predominant DSB repair mechanism in S. cerevisiae.15
Fast Cell Growth Kinetics Induced CRISPR-Cas Antifungal Resistance
We next asked whether cell growth kinetics affected the potency of the CRISPR-Cas antifungals. We hypothesized that the fast-growing cells could escape the treatment, propagate, and eventually dominate the entire culture. To test the hypothesis, we characterized growth of the 17 promising strains carrying the CRISPR-Cas antifungals on solid and liquid media, where cells grow faster in liquid media than in solid media. Vip46 cotargeting defensive genes RAD51 and RAD52 were included as a control due to their lack of toxicity on solid media (Figure 2A). Consistent with the initial screening, none of the strains grew on solid media when the CRISPR-Cas antifungals were activated (Figure 2A). However, only 5 out of the 17 strains, including Vip52, Vip60, Vip76, Vip93, and Vip94, exhibited flat growth curves on liquid media (Figure 2B), suggesting a greater efficacy of CRISPR-Cas growth inhibition. The remaining strains were escapees from the CRISPR-Cas antifungal treatment due to faster growth on liquid than solid media. Using one of the top-performing strains, Vip60 (RAD51, RPS21), in a case study, we found that placement of the induced strain in noninducing media (glucose as carbon source) after 48 h was able to recover growth (Figure S2). In addition, CRISPR-Cas antifungal in Vip60 was active and potent for at least two rounds of culture transfer in liquid media.
To evaluate the potency of CRISPR-Cas antifungals in detail, we compared the cell viability of five high-performing strains under four different growth conditions including no treatment (normal growth on glucose, negative control), carbon starvation (positive control), Geneticin treatment (G418 antibiotic, test case), and CRISPR-Cas antifungals (test case; Figure 2C). Under the normal growth condition on glucose without any treatment, all strains grew quickly but exited exponential growth phase after 48 h with a high count of viable cells but a low percentage of cell viability (19–27%). When subjected to carbon starvation, all strains experienced growth inhibition with a high percentage of cell viability (74–98%) after 48 h, implying that the cells were mostly dormant. In the presence of antibiotic treatment, cells exhibited not only growth inhibition but also a low percentage of cell viability (10–29%). As compared to the carbon starvation scenario, all strains under CRISPR-Cas antifungal treatment showed more severe growth inhibition with a lower percentage of cell viability (25–52%). CRISPR-Cas antifungals in Vip52, Vip60, and Vip76 strains exhibited high potency like G418 antibiotic treatment with low cell count and low percentage of cell viability, while others mostly showed dormant phenotypes (low cell count and high percentage of cell viability) like under carbon starvation. In our design, Vip52 cotargets the defensive gene LIF1 (ligase interacting factor mediating NHEJ in DNA double-strand break repair) and the essential gene RPL10E (ribosomal protein of 60S unit), while Vip60 cotargets the defensive gene RAD51 (radiation sensitive protein mediating HR in DNA double-strand break) and the essential gene RPS21 (ribosomal protein of 40S unit), and Vip76 cotargets the defensive gene RAD52 (radiation sensitive protein mediating HR in DNA double-strand break) and the essential gene LIG1 (tRNA ligase). Even though Vip52, Vip60, and Vip76 are designed to target different gene sets, their potency as antifungals signifies the importance of the essential and defensive gene cotargeting strategy.
Taken altogether, CRISPR-Cas systems can be potentially used as novel antifungals by cotargeting defensive and essential genes. Identifying optimal combinations of the essential and defensive gene targets that outpace the fast cell growth kinetics and cause cell death rather than cellular dormancy will be critical in formulating effective CRISPR-Cas antifungals.
High Cell Density Caused a More Pronounced Community Escape of CRISPR-Cas Antifungal Treatment
To further understand the robustness of growth suppression in the top-performing strains, we investigated the effect of high cell density inoculation on the potency of CRISPR-Cas antifungals. We differentially seeded cultures with either a low inoculation (0.05 OD, 5.5 × 105 cells/mL, as conducted in previous experiments) or a high inoculation cell density (0.2 OD, 2.5 × 106 cells/mL). Without carbon starvation, the increase in cell inoculation resulted in a pronounced increase in growth rates with a shorter lag phase in all media types (Figures 3A–F). While the escape phenomenon (or CRISPR-Cas antifungal resistance) dominated in high cell density inoculation, it was more significant for the antibiotic treatment than for the CRISPR-Cas antifungal treatment. Consistent with growth characterization in solid versus liquid media, the CRISPR-Cas antifungal activities of Vip52, Vip60, and Vip76 also exhibited the best performance with high cell inoculation scenario among the characterized strains (Figure 3G). The resistance to G418 in high cell inoculation, but not in low cell inoculation, was likely because the greater number of cells produced a greater effective concentration of aminoglycoside 3′-phosphotransferases and hence provided the innate resistance via degradation of the antibiotic.1 This broad escape phenomenon induced by a higher initial cell concentration, and therefore higher loading of enzymes responsible for counteracting antimicrobial activity, suggests that ongoing kinetics of the culture is also a major driving factor of the escape from CRISPR-Cas antifungal treatment, in contrast to an initial inhibition/inefficiency of the machinery that eventually allows for escaped cells to proliferate. This escape phenomenon was more pronounced when CRISPR-Cas antifungals were less effective as seen in the nontop-performing strains (Figure S3). Overall, faster growth kinetics in high cell inoculation reduced the potency of CRISPR-Cas antifungals and caused pronounced community escape, as observed in solid versus liquid media. The differential inoculation results underscore the similarity in escape profiles between traditional small-molecule antibiotics and CRISPR-Cas antifungals, requiring a deeper investigation into the roles of cellular repair machinery, stress response mechanisms, and effective CRISPR-Cas systems (Cas enzymes and gRNAs) to this phenotype.
Figure 3.

Higher cell inoculation is more prone to CRISPR-Cas antifungal resistance. (A–F) Effect of cell inoculation concentrations on the potency of CRISPR-Cas antifungals. Five strains (Vip52, Vip60, Vip76, Vip93, and Vip94) harboring the most effective antifungal designs and one control strain (Vip46*) were investigated with an initial OD of either 0.05 or 0.2. Higher concentration of cells at the outset of the experiment overwhelmed the population by decreasing lag time, thereby increasing both antibiotic and CRISPR-Cas antifungal resistance. (G) Viable cell count at 36-h outgrowth confirmed the increase in viable cells in the high inoculation scenario. In panels A–G, each value is a mean ±1 standard deviation (n ≥ 2).
Genotypes of Escape Population Contained Small Mutation Frequency in Targeted Genes and CRISPR-Cas Sequences
To better understand how cells escaped the CRISPR-Cas antifungal treatment, we performed both amplicon and plasmid sequencing for representative strains harboring single- and co-targeting CRISPR-Cas antifungals using a combination of Illumina (MiSeq) and Oxford Nanopore sequencing (Figure 4A). Our results showed that escape populations were primarily composed of cells that have avoided mutation to the targeted genes (Figure 4B,C) as well as any mutations on the plasmid sequence harboring CRISPR-Cas9 sequences (Figure 4D,E). Illumina sequencing of approximately 1 kb PCR-amplified regions around the Cas9 target site on the genome revealed mutation frequencies of less than 9% for all essential genes except for a small deletion (2 bp) in the ACC2 gene in the Vip48 strain (Figures 4B and S4). Mutations in defensive genes occurred at a higher frequency than essential gene mutations but were still relatively rare with no mutations detected in many of the cotargeting sequenced strains (Figure 4B). Mutations in both essential and defensive genes were dominated by large deletions (>20 bp) destroying the target sites and hence the coding sequences of the target genes, implying gene knockouts (Figures 4C and S4). However, as these mutated sequences represent a small minority of the total population, this suggests that the dominance of HR over NHEJ repairs prevents significant edits even at nonessential gene loci. The rapid division of cells with shortened lag phases provides a considerable number of templates for HR machinery to overcome the CRISPR-Cas rate of double-strand breaks, as observed for cultures growing in solid versus liquid media (Figure 2) and high cell inoculation (Figure 3). In the cotargeted strains with higher mutation frequencies in defensive genes, the mutations observed may not have conferred a complete defensive gene knockout, leaving the cells with sufficient DNA repair machinery to escape the CRISPR-Cas antifungal treatment.
Figure 4.

Identification of mutations from representative single- and co-targeted strains that escape from the CRISPR-Cas antifungal treatment. (A) Experimental workflow for sequencing. (B) Genetic mutation frequency of the targeted chromosomal genes. Different colored stacked bars represent different mutations presented as mutation ID numbers in Figures 4C and S4. Reads for defensive genes of Vip55 and Vip56 failed QC. (C) Mutated sequences for the representative strains Vip10, Vip43, and Vip60. PAM sequences are highlighted in blue. Single targeted strains show low levels of mutation with the exception of the defensive genes, in particular RAD51. As seen in the sequence alignments provided for Vip10, Vip43, and Vip60, mutants are dominated by large (>20 bp) deletions, although mutation frequency is low and heterogeneous. Mutated sequences for other characterized strains were presented in Figure S4. (D) Locations of mutations in the gRNA cassettes on extracted gRNA plasmids. (E) Variants by type in gRNA cassettes.
Oxford nanopore resequencing of the Cas9-bearing plasmids extracted from the 17 high-performing strains (average sequencing depth of 20,000×) revealed no detectable rearrangements on the plasmid and no nucleotide variants over a 1% threshold, indicating that, unlike in bacterial pathogens, Cas9 mutation is not the dominant pathway to escape. Illumina amplicon sequencing of the guide cassette from gRNA plasmids revealed high frequencies of mutation at this site of the plasmids (Figure 4D,E). Mutations occurred at various sites along the gRNA operon, including the promoter (7%), scaffold (17%), and target sequence (71%) (Figure 4D). Single nucleotide variants (SNVs) were the most common (60%), but the remaining 40% of mutations were made up of indels and other multinucleotide variants (MNVs), which led to greater loss-of-function (Figure 4E). This sequencing result, consistent with previous studies in other species,16 indicates that the primary route of CRISPR-Cas antimicrobial evasion is the mutation of the gRNA operon on the delivered plasmid. This study shows that Saccharomyces and eukaryotes also more broadly exhibit increased resistance over bacteria due to the robust and redundant DNA repair mechanisms.
Taken together, the competition among kinetics of cell growth, DNA repair, and CRISPR-Cas activity is a key factor in controlling antifungal effectiveness. Increasing the efficiency of gene knockouts through the selection of superior Cas proteins and gRNAs in conjunction with more effective ways to reduce the DNA repair pathway expression will be critical to deploy CRISPR-Cas systems as an antifungal therapy.
Conclusions
The development of novel antifungals is a critical area of concern for public health, agriculture, and the environment. This study presented the design principles and identified key factors and areas of improvement for an effective CRISPR-Cas antifungal formulation. Importantly, the robust DNA repair mechanisms encountered in eukaryotic pathogens must be addressed by means of direct targeting or utilization as a “Trojan Horse” to counteract their role in directing cells to a persister phenotype. This study underscores the importance of such a cotargeting approach of essential and defensive genes to increase CRISPR-Cas antifungal activity. In addition, informed selection of these defensive genes is needed based on the pathogen in question, as certain pathways are favored in different species. Future work should focus on evaluating the temporal dimension to determine means of preventing escaped persister cells from re-colonizing a host. Furthermore, the delivery of CRISPR-Cas systems to fungal pathogens in vivo must be addressed for these systems to function as a viable antifungal strategy.
Materials and Methods
Strains and Plasmids
A complete list of plasmids, strains, and primers used in this study is presented in Tables S2, S3, and S4, respectively. The plasmid vectors p415-GalL-Cas9-CYC1t (Addgene plasmid # 43804) and p426-SNR52p-gRNA.CAN1.Y-SUP4t (Addgene plasmid # 43803) were a gift from George Church and were used as the backbone plasmids for all studies.17 All gRNAs were designed using CASPER.18 The p426 derived vectors containing the gRNA sequences were built via Phusion PCR in a 40 μL reaction using the p426 plasmid as a template, gRNA Gibson primers with homology for the template and the inset, and a generic primer with homology for the plasmid (AT_gRNA_BB_F or AT_gRNA_BB_R). Inserts were constructed using the same template and complementary primers to the backbone (i.e., one unique to a gRNA and one generic). Linker sequences were used to construct an insert with one gRNA sequence followed by the SUP4t terminator, a short spacing sequence, and then another SNR52 promoter with the second gRNA sequence at the end. These inserts were inserted into the p426 backbone using the same Gibson assembly protocol described.19
Transformations were performed with Top10 E. coli using 5 μL of a reacted Gibson Assembly. Upon addition, cells were left on ice for 3 h followed by a 45 s heat shock at 42 °C. After heat shock, cells were inoculated in 1 mL of lysogeny broth (LB) and shaken for 1 h at 37 °C. Cultures were spun down and concentrated for plating on 100 mg/L Amp plates, where they grew overnight at 37 °C. Transformants were confirmed by colony PCR, and plasmids were confirmed by Sanger sequencing following plasmid extraction. Approximately 1 μg of each confirmed plasmid with the Cas9 carrying plasmid was transformed into S. cerevisiae BY4741 using electroporation (2.1 kV, 200 Ω, 25 μF).
Media and Culturing Conditions
The parent strain used in this study was S. cerevisiae BY4741. Strains carrying the Cas9 and gRNA plasmids were cultured in selective media with the appropriate double auxotrophy (SC-Ura–Leu–), the desired carbon source (20 g/L glucose or 20 g/L galactose for induction of Cas9). For agar plate serial dilution assay, strains were cultured in SC-Ura–Leu– plus glucose media overnight at 28 °C until the exponential phase was reached. They were then washed once with PBS, diluted 10×, 100×, and 500× in PBS, 1 μL of sample spotted on the agar plate, and incubated at 28 °C for 48–72 h. For growth assays in broth, culture preparation followed the same incubation followed by PBS wash. Cultures were then diluted with culture media (SC-Ura–Leu– plus carbon source) to the appropriate starting OD (0.05 or 0.2). Strains were cultured in a Duetz plate with a working volume of 500 μL in a maxQ 6000 shaker (Thermo Fisher) set at 28 °C and 400 rpm. OD measurements were taken with a BioTek plate reader.
Gene Cotargeting Interaction Analysis
After 48 h, all dilutions of spotted strains on the solid media were scored on a scale of 0–4 based on spot size with 4 being the largest and 0 being no growth (Figure S1). Assuming the gene cotargeting interaction is additive, the expected interaction score (IEXP) is then quantified according to the following equation:
| 1 |
where i is the dilution index, N is the number of dilutions, SE,i (0 ≤ SE,i ≤ 4) is the score of the single essential gene targeting for dilution i, and SD,i (0 ≤ SD,i ≤ 4) is the score of the single defensive gene targeting for dilution i. The expected score IEXP (0 ≤ SEXP ≤ 4) is then normalized with respect to N and further divided by 2. The observed interaction score (IOBS) for the essential and defensive gene cotargeting is then quantified according to the following equation:
| 2 |
where i is the dilution index, N is the number of dilutions, and SED,i is the score of the essential and defensive gene cotargeting for dilution i. The gene cotargeting interaction score, IED (−1 ≤ IED ≤ 1), is then calculated as follows:
| 3 |
The calculated score is normalized with respect to the maximum spot score. A positive score indicates synergy between the essential and defensive gene cotargets as they were more lethal than expected, whereas a negative score indicates antagonism as the cotargets were less lethal than expected.
Flow Cytometry
To perform viability assessment with flow cytometry, samples were taken and diluted to within a range of OD 0.1–1.0 to ensure accurate counting by the flow cytometer. Samples were then further diluted 20× to a total volume of 200 μL by the Guava ViaCount staining solution (EMD Millipore #4000–0041), mixed by pipetting, and allowed to sit in the dark for 5 min. Samples were taken in a 96-well round-bottom plate and placed in a Guava EasyCyte 6HT flow cytometer. Gating voltages were calibrated on a 1:1 mixture of an exponential phase BY4741 culture and a 20 s microwaved sample of the same strain.
Sequencing to Identify Mutated Sequences
A deep-hybrid sequencing using Illumina MiSeq and Oxford Nanopore was performed to examine both the target chromosomal loci and the plasmids carrying CRISPR-Cas from escaped strains after the CRISPR-Cas antifungal treatment. Briefly, plasmids and genomic DNA from selected strains were extracted 48 h after galactose induction. For examining the target chromosomal loci, about 1 kb around each target locus was PCR amplified from 100 ng of extracted genomic DNA with Phusion Hot Start II DNA Polymerase (Thermo Fisher) using the locus-specific primers found in Table S4. Amplicons were purified using the Omega Biotek E.Z.N.A Cycle Pure Kit (SKU: D6492–01) and pooled in equimolar ratios. For investigating gRNA regions from extracted plasmids, the promoter plus guide RNA region was PCR amplified using primers AT-gRNA-swap_F and AT-gRNA-swap_R, purified, and pooled in equimolar ratios.
For Illumina sequencing of the target chromosomal loci and plasmid gRNA regions, amplicons were further prepared as genomes using the Nextera XT library preparation kit and evaluated on a bioanalyzer for quality control. Pools were then all combined and diluted to 4 nM. Final products were diluted to a final loading concentration of 4 pM, pooled with 20% 10 pM PhiX, and loaded on a Version 3 flow cell reading 275 bp, paired-end, on the Illumina MiSeq at the University of Tennessee Genomics Core.
For Oxford Nanopore sequencing of target chromosomal loci and the extracted Cas9 and gRNA plasmids, amplicons and plasmids were prepared using the LSK109 ligation sequencing kit with the native barcoding kit (EXP-NBD104) and sequenced on a MinION R9.4.1 flow cell with an average sequencing depth of 20,000×. Sequencing data were imported and analyzed in the Qiagen CLC Genomics Workbench 20.0.4. Indels and rearrangements in target loci were analyzed by using the InDels and Structural Variants tool. Variants in the Cas9 plasmids and gRNA regions were analyzed by using the Low Frequency Variant Detection tool with a minimum frequency threshold of 1%.
Acknowledgments
The research was financially supported by the DARPA YFA award and Director Fellowship (D17AP00023). The authors would like to thank Dr. Renee Wegrzyn and the DARPA team for useful discussion during the course of the project.
Data Availability Statement
All data of this study are included in the article and SI Appendix including Figures S1–S4 and Tables S1–S4.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00342.
Screening potency of CRISPR-Cas antifungals; effect of growth adaptation in liquid cultures on potency of CRISPR-Cas antifungal (Vip60); effect of cell inoculation density on escapee proliferation in non-top performing Vip strains; sequence alignments from targeted amplicon sequencing of representative single- and co-targeted strains; gene annotation; list of plasmids; list of strains; list of primers (PDF)
Author Contributions
CTT conceived the research study. CTT and BJM designed the research. BJM, XZ, and JCC performed research; BJM, CC, and CTT analyzed data; and BJM, CC, and CTT wrote the paper.
The views, opinions, and/or findings contained in this article are those of the authors and should not be interpreted as representing the official views or policies, either expressed or implied, of the funding agency.
The authors declare no competing financial interest.
Supplementary Material
References
- Scorzoni L.; de Paula e Silva A. C. A.; Marcos C. M.; Assato P. A.; de Melo W. C. M. A.; de Oliveira H. C.; Costa-Orlandi C. B.; Mendes-Giannini M. J. S.; Fusco-Almeida A. M. Antifungal Therapy: New Advances in the Understanding and Treatment of Mycosis. Front Microbiol 2017, 8, 36. 10.3389/fmicb.2017.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas V. K.; Bushkin G. G.; Bernstein D. A.; Getz M. A.; Sewastianik M.; Barrasa M. I.; Bartel D. P.; Fink G. R.. New CRISPR Mutagenesis Strategies Reveal Variation in Repair Mechanisms among Fungi. mSphere 2018, 3 ( (2), ). 10.1128/mSphere.00154-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G. D.; Denning D. W.; Gow N. A.; Levitz S. M.; Netea M. G.; White T. C. Hidden killers: human fungal infections. Sci. Transl Med. 2012, 4 (165), 165rv113. 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- Bongomin F.; Gago S.; Oladele R. O.; Denning D. W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi (Basel) 2017, 3 (4), 57. 10.3390/jof3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nnadi N. E.; Carter D. A. Climate change and the emergence of fungal pathogens. PLoS Pathog 2021, 17 (4), e1009503 10.1371/journal.ppat.1009503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin D. S.; Rautemaa-Richardson R.; Alastruey-Izquierdo A. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis 2017, 17 (12), e383–e392. 10.1016/S1473-3099(17)30316-X. [DOI] [PubMed] [Google Scholar]
- Schwartz I. S.; Patterson T. F. The Emerging Threat of Antifungal Resistance in Transplant Infectious Diseases. Curr. Infect Dis Rep 2018, 20 (3), 2. 10.1007/s11908-018-0608-y. [DOI] [PubMed] [Google Scholar]; Hendrickson J. A.; Hu C.; Aitken S. L.; Beyda N. Antifungal Resistance: a Concerning Trend for the Present and Future. Curr. Infect Dis Rep 2019, 21 (12), 47. 10.1007/s11908-019-0702-9. [DOI] [PubMed] [Google Scholar]
- Satoh K.; Makimura K.; Hasumi Y.; Nishiyama Y.; Uchida K.; Yamaguchi H. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol. Immunol. 2009, 53 (1), 41–44. 10.1111/j.1348-0421.2008.00083.x. [DOI] [PubMed] [Google Scholar]; Hanson B. M.; Dinh A. Q.; Tran T. T.; Arenas S.; Pronty D.; Gershengorn H. B.; Ferreira T.; Arias C. A.; Shukla B. S. Candida auris Invasive Infections during a COVID-19 Case Surge. Antimicrob. Agents Chemother. 2021, 65 (10), e0114621 10.1128/AAC.01146-21. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lyman M.; Forsberg K.; Sexton D. J.; Chow N. A.; Lockhart S. R.; Jackson B. R.; Chiller T. Worsening Spread of Candida auris in the United States, 2019 to 2021. Ann. Int. Med. 2023, 176 (4), 489–495. 10.7326/M22-3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citorik R. J.; Mimee M.; Lu T. K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32 (11), 1141–1145. 10.1038/nbt.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bikard D.; Euler C. W.; Jiang W.; Nussenzweig P. M.; Goldberg G. W.; Duportet X.; Fischetti V. A.; Marraffini L. A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32 (11), 1146–1150. 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gomaa A. A.; Klumpe H. E.; Luo M. L.; Selle K.; Barrangou R.; Beisel C. L. Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. mBio 2014, 5 (1), e00928–00913. 10.1128/mBio.00928-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mayorga-Ramos A.; Zuniga-Miranda J.; Carrera-Pacheco S. E.; Barba-Ostria C.; Guaman L. P. CRISPR-Cas-Based Antimicrobials: Design, Challenges, and Bacterial Mechanisms of Resistance. ACS Infect Dis 2023, 9 (7), 1283–1302. 10.1021/acsinfecdis.2c00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P. D.; Lander E. S.; Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157 (6), 1262–1278. 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Bikard D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016, 44 (9), 4243–4251. 10.1093/nar/gkw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. M.; Hong E. L.; Amundsen C.; Balakrishnan R.; Binkley G.; Chan E. T.; Christie K. R.; Costanzo M. C.; Dwight S. S.; Engel S. R.; et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–705. 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Lin Y.; Liu T.; Lai F. L.; Zhang C. T.; Gao F.; Zhang R. DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Res. 2021, 49 (D1), D677–D686. 10.1093/nar/gkaa917. [DOI] [PMC free article] [PubMed] [Google Scholar]; Motaung T. E.; Ells R.; Pohl C. H.; Albertyn J.; Tsilo T. J. Genome-wide functional analysis in Candida albicans. Virulence 2017, 8 (8), 1563–1579. 10.1080/21505594.2017.1292198. [DOI] [PMC free article] [PubMed] [Google Scholar]; Roemer T.; Jiang B.; Davison J.; Ketela T.; Veillette K.; Breton A.; Tandia F.; Linteau A.; Sillaots S.; Marta C.; et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2003, 50 (1), 167–181. 10.1046/j.1365-2958.2003.03697.x. [DOI] [PubMed] [Google Scholar]
- Strope P. K.; Skelly D. A.; Kozmin S. G.; Mahadevan G.; Stone E. A.; Magwene P. M.; Dietrich F. S.; McCusker J. H. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 2015, 25 (5), 762–774. 10.1101/gr.185538.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh B. O.; Symington L. S. Recombination proteins in yeast. Annu. Rev. Genet 2004, 38, 233–271. 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Gao S.; Honey S.; Futcher B.; Grollman A. P. The non-homologous end-joining pathway of S. cerevisiae works effectively in G1-phase cells, and religates cognate ends correctly and non-randomly. DNA Repair (Amst) 2016, 42, 1–10. 10.1016/j.dnarep.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Pan Q.; Gendron P.; Zhu W.; Guo F.; Cen S.; Wainberg M. A.; Liang C. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell reports 2016, 15 (3), 481–489. 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]; Yoder K. E.; Bundschuh R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/Cas9. Sci. Rep. 2016, 6 (1), 1–12. 10.1038/srep29530. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang G.; Zhao N.; Berkhout B.; Das A. T. CRISPR-Cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Molecular Therapy 2016, 24 (3), 522–526. 10.1038/mt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deveau H.; Barrangou R.; Garneau J. E.; Labonté J.; Fremaux C.; Boyaval P.; Romero D. A.; Horvath P.; Moineau S. Phage Response to CRISPR-Encoded Resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190 (4), 1390–1400. 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo J. E.; Norville J. E.; Mali P.; Rios X.; Aach J.; Church G. M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic acids research 2013, 41 (7), 4336–4343. 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza B. J.; Trinh C. T. Enhanced guide-RNA design and targeting analysis for precise CRISPR genome editing of single and consortia of industrially relevant and non-model organisms. Bioinformatics 2018, 34 (1), 16–23. 10.1093/bioinformatics/btx564. [DOI] [PubMed] [Google Scholar]; Mendoza B.; Fry T.; Dooley D.; Herman J.; Trinh C. T. CASPER: An Integrated Software Platform for Rapid Development of CRISPR Tools. CRISPR Journal 2022, 5 (4), 609–617. 10.1089/crispr.2022.0025. [DOI] [PubMed] [Google Scholar]
- Gibson D.; Young L.; Chuang R.; Venter J.; Hutchison C.; Smith H. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data of this study are included in the article and SI Appendix including Figures S1–S4 and Tables S1–S4.