

Abstract
Objectives
Multiple sclerosis (MS) is a neurodegenerative disease characterised by inflammation and damage to myelin sheaths. While all current disease‐modifying treatments (DMTs) are very effective at reducing relapses, they do not slow the progression of the disease, and there is little evidence that these treatments are able to repair or remyelinate damaged axons. Recent evidence suggests that activating kappa opioid receptors (KORs) has a beneficial effect on the progression of MS, and this study investigates the effects of KOR agonists treatment in combination with two current DMTs.
Methods
Using the well‐established murine model for immune‐driven demyelination of MS, experimental autoimmune encephalomyelitis, the effect of KOR agonists in combination with DMTs fingolimod or dimethyl fumarate on disease progression, immune cell infiltration and activation as well as myelination were analysed.
Results
Fingolimod in combination with the KOR agonist, nalfurafine, significantly increased each individual beneficial effect as measured by increased recovery of mice and reduced relapses. These beneficial effects correlated with a reduction in immune cell infiltration into the CNS as well as peripheral immune cell alterations including a reduction in autoreactive CD4+ T‐cell cytokine production as well as increased myelination in the spinal cords of co‐treated animals. In contrast, while the use of dimethyl fumarate in combination with nalfurafine did not adversely affect the benefits of nalfurafine, the combination did not significantly enhance those benefits.
Conclusion
This study indicates that KOR agonists can be used in combination with fingolimod and dimethyl fumarate with the nalfurafine–fingolimod combination providing enhanced benefits.
Keywords: experimental autoimmune encephalomyelitis, fingolimod, kappa opioid receptor agonist, nalfurafine, neuroinflammation, remyelination
In this study, we found that the kappa opioid receptor agonist, nalfurafine, significantly enhanced the benefit of fingolimod when used in combination. This co‐treatment effect was demonstrated by increased recovery and reduced relapses in a murine model for immune‐driven demyelination in multiple sclerosis as well as increased remyelination and OPC differentiation.
Introduction
Multiple sclerosis (MS) is a chronic immune‐mediated inflammatory autoimmune disease characterised by infiltration of autoreactive T cells and other inflammatory immune cells from the periphery into the central nervous systems (CNS). The pathology of MS is characterised by damage to the myelin sheaths around nerve axons known as demyelination, which leads to sensory dysfunction and physical and visual impairments. 1 It can be broadly divided into three different subtypes (relapsing–remitting, primary progressive and secondary progressive) with only relapsing–remitting MS (RRMS) having effective treatments available. While over 16 disease‐modifying therapies are FDA approved for RRMS, most of these medicines are immunomodulatory agents that act in the peripheral compartment as a result of the low capacity to cross the intact blood–brain barrier. However, while all these treatments are very effective at reducing relapses, they do not significantly slow disease progression nor is there evidence that these treatments lead to the repair and remyelination of the damaged axons.
We have shown previously that the kappa opioid receptor (KOR) agonist nalfurafine (NalF) reduces disease and promotes remyelination in the well‐established animal model for MS, experimental autoimmune encephalomyelitis (EAE). 2 Using this model, we reported that NalF was effective over a wide range of doses when administered therapeutically in EAE, showing higher recovery and fewer relapses. Strikingly, we demonstrated that NalF was capable of promoting remyelination following immune‐ and non‐immune‐mediated demyelination. 2 To further translate these findings and improve clinical outcomes for people with MS, it is critical to understand whether a combination treatment using NalF along with disease‐modifying treatments (DMTs) would provide superior benefit in reducing relapses and enhancing remyelination compared to either treatment alone.
The currently available DMTs can be classified into three different categories of drugs: generic immune modulators like dimethyl fumarate (DMF) (Tecfidera), S1P receptor modulators like fingolimod (Gilenya) and immune‐targeted monoclonal antibodies. DMF is a methyl ester of the fumaric acid, and in the body, it is rapidly attacked by the detoxification agent, glutathione, leading to the activation of the specific Nrf2 pathway, which reduces inflammation. 3 This effect includes a reduction in inflammatory cytokines and a decreased expression of adhesion molecules on CNS‐infiltrating cells, especially T cells. 4 In addition, DMF is known to shift T helper cells subsets from Th1 (cell‐mediated functions) and Th17 (pro‐inflammatory functions) towards Th2 (i.e. tissue repair). 5 Fingolimod (Gilenya) was approved in 2010 as the first oral DMT for MS. This small molecule drug prevents the egress of T cells from lymph nodes by binding and blocking the sphingosine‐1‐phosphate receptor (S1PR) and thus reducing the circulating pool of T cells that can be recruited into the CNS. Under homeostatic conditions, naïve and memory (both central and effector) T cells circulate through the blood, enter the lymph nodes and egress through the efferent lymphatics in a S1PR‐dependent manner. By binding to S1PR and leading to its degradation, fingolimod causes the retention of T cells in lymph node. 6 , 7 Concurrently, the chemokine receptor, CCR7, is up‐regulated, increasing the effect of fingolimod on T cells sequestration in the lymph node.
In this study, we investigated whether the KOR agonist NalF in combination with different DMTs including fingolimod and DMF resulted in superior therapeutic benefit using the immune‐mediated model of demyelination, EAE.
Results
Co‐treatment of nalfurafine and DMTs on disease outcome
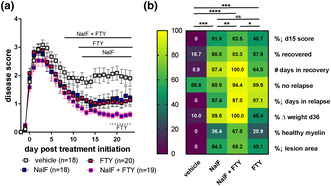
While all DMTs are effective at reducing relapses, they only slow the progression of the disease, and there is little evidence that they are able to repair and remyelinate the damaged axons in vivo. To determine the effect of a dual treatment of NalF with a DMT, we treated animals at the onset of EAE (i.e. score ≥ 1.0) with NalF alone, fingolimod alone or both combined at their optimal dose (0.01 mg kg−1 per day, NalF (based on Denny et al. 2 ; 1.0 mg kg−1 per day fingolimod; Supplementary figure 1a 8 , 9 ). While both NalF and fingolimod alone led to a similar reduction in disease scores, animals co‐treated with NalF and fingolimod had better outcomes overall (Figure 1). In particular, the overall disease score was significantly lower in the co‐treated animals compared to the fingolimod‐treated animals (Figure 1a and e). In addition, the number of animals that recovered and the time spent in recovery increased (Figure 1b and c), and the percentage of mice that relapsed (Figure 1d) was significantly reduced in co‐treated animals compared with the vehicle‐treated animals. Analysis of the spinal cords showed that co‐treatment increased the myelin area more than either NalF or fingolimod alone (Figure 1g). Analysis of historical spinal cord samples further highlights the benefit in myelination by both the co‐treatment and NalF single treatment, compared to vehicle or fingolimod alone (Supplementary figure 1c and d). Additionally, the lesion percentage was significantly lower in all treated groups compared to the vehicle with co‐treatment having the most consistent effect (Figure 1h). While we observed that co‐treatment had a visible effect on multiple different parameters, statistical significance was not reached when looking at each individual parameter alone. Thus, to more holistically compare the type of benefit provided by each therapy, the single outcome parameters were normalised (Table 1) and presented as a heatmap (Figure 1i). Using a mixed effects analysis, the combined treatment showed significantly improved benefit overall compared to all other groups (Figure 1i). While the overall level of benefit was similar in the NalF and fingolimod groups, the specific pattern of benefit was different. These findings indicate that the combination of NalF with fingolimod provides enhanced benefit in this model.
Figure 1.
In vivo treatment of nalfurafine (NalF) in combination with fingolimod (FTY) in experimental autoimmune encephalomyelitis (EAE). (a) Disease scores of vehicle, NalF (0.01 mg kg−1 i.p.), fingolimod (1 mg kg−1 p.o.) or the combination treated animals from disease onset (score ≥ 1) to 23 dpt. Scores aligned to the day of disease onset (day 0 post treatment). Mice were treated daily. Results are combined from six independent experiments (n = 18–30 animals per treatment group, as indicated). Two‐way ANOVA with Tukey's multiple comparisons testing (* comparing treatments to vehicle). (b) Percentage recovery to 23 days post treatment (dpt; recovery = score ≤ 0.5). (c) Number of days in recovery to 23 dpt (% of top recovery). (d) Percentage relapse to 23 dpt (relapse = increase by 1 full point from the lowest score of remission). (e) End score of mice at end of experiment. Non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. (f) Representative images of Black‐Gold II staining in the spinal cord. Myelination (g) and lesion area (h) in the spinal cord quantified by Black‐Gold II staining. (i) Heatmap of all disease parameters analysed by non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Table 1.
Disease parameter calculations
| Heatmap parameters | Parameters | Calculation | Units | Min | Max |
|---|---|---|---|---|---|
| %↓ d15 score | % decrease in score at day 15 post onset | (mean day 15 score vehicle – day 15 score experimental)/(mean day 15 score vehicle) × 100 | % | 0 | 100 |
| % recovered | % of mice that recovered | (# mice with score ≤ 0.5 after a peak of ≥ 2)/(total # of mice) × 100 | % | 0 | 100 |
| # days in recovery | # of days mice were recovered as % of group with top recovery | (mean # of days at a score < 0.5 after a peak of > 2)/(mean # days for top recovery group) × 100 | % | 0 | 100 |
| % no relapse | % of mice that did not relapse | (# mice that had no increase in score ≥ 0.1 after first peak of ≥ 2)/(total # of mice) × 100 | % | 0 | 100 |
| %↓ days in relapse | % reduction in number of days in relapse | (mean # of days vehicle in relapse ‐ # of days in relapse experimental)/(mean # of days vehicle in relapse) × 100 | % | 0 | 100 |
| % ∆ weight d36 | % weight change at 36 days post immunisation | (weight at day 36 post immunisation ‐ weight at baseline)/(weight at baseline) × 100 | % | 0 | 100 |
| % healthy myelin | % healthy myelin area | (% myelin area experimental – mean % myelin area vehicle)/(mean % myelin area healthy – mean % myelin area vehicle) × 100 | % | 0 | 100 |
| %↓ lesion area | % decrease in lesion area | (mean % myelin area vehicle – % myelin area experimental)/(mean % myelin area vehicle) × 100 | % | 0 | 100 |
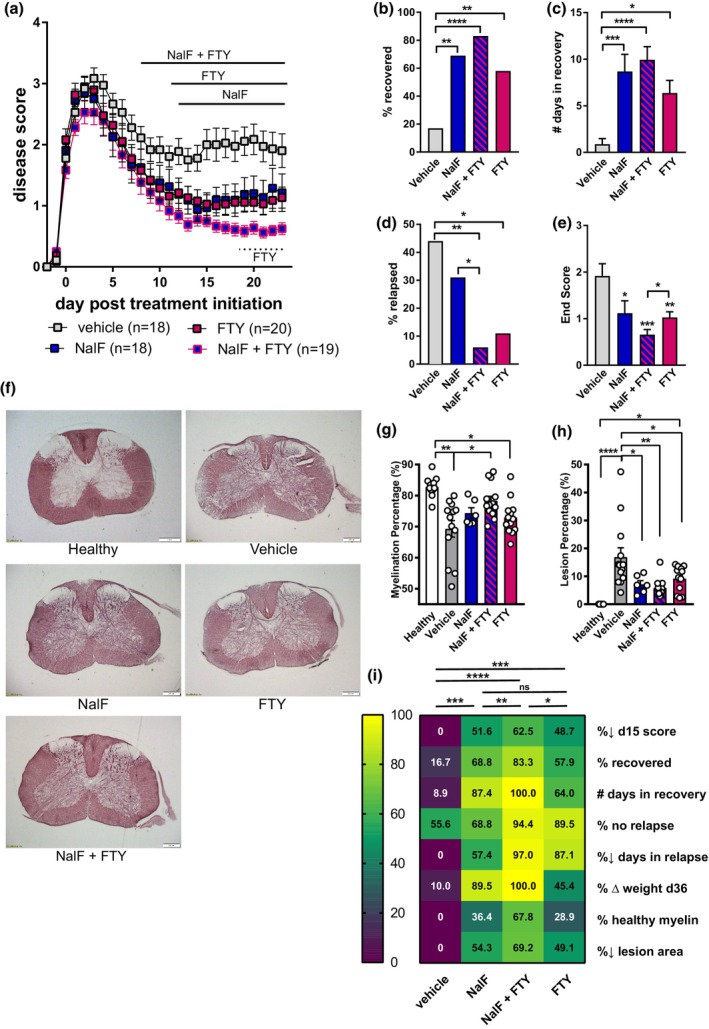
To understand whether enhanced benefit could be achieved using a DMT with a different immune‐modifying mechanism of action, we assessed the combination of DMF and NalF at their optimal doses (0.01 mg kg−1 per day, NalF; 1.0 mg kg−1 per day, DMF). In contrast to the NalF–fingolimod combination, no additional beneficial effect was detected when NalF was combined with DMF in any of the disease parameters including disease score (Figure 2a), recovery (Figure 2b and c) and relapse (Figure 2d and e). Comparing the overall benefit, only NalF was found to provide a significant effect compared to vehicle (Figure 2f). However, positively, no adverse effects of co‐treatment were detected, which is an important finding when considering clinical application.
Figure 2.
In vivo treatment of nalfurafine (NalF) in combination with dimethyl fumarate (DMF). (a) Disease scores of vehicle, NalF (0.01 mg kg−1 i.p.), DMF (1 mg kg−1 p.o.) or the combination treated animals from disease onset (score ≥ 1) to 23 dpt. Scores aligned to the day of disease onset (day 0 post treatment). Mice were treated daily, and results combined and analysed from three independent experiments (n = 9–14 animals per treatment group, as indicated) using two‐way ANOVA comparison against vehicle. (b) Percentage recovery to 23 dpt (recovery = score ≤ 0.5). (c) Number of days in recovery to 23 dpt (% top recovery). (d) Percentage relapse to 23 dpt (relapse = increase by 1 full point from the lowest score of remission). (e) Number of days spent in relapses analysed with non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. (f) Heatmap of all disease parameters analysed with non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Co‐treatment of nalfurafine and fingolimod on immune cells
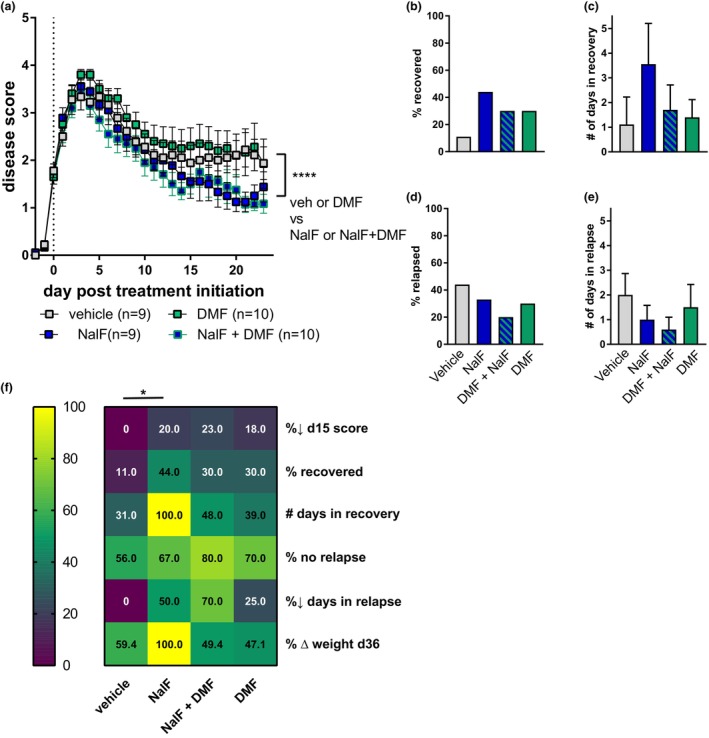
Because the co‐treatment of NalF and fingolimod showed better disease outcomes than either treatment alone, we investigated further how this combination affected the molecular pathways associated with EAE and MS. Since fingolimod is known to target immune cells in the periphery and prevent them from entering the CNS to cause demyelination, we analysed immune cell infiltration into the brain as well as immune cell sub‐populations in the periphery by flow cytometry in EAE animals treated with either NalF, fingolimod or the combination (gating strategies in Supplementary figures 2 and 3a). While many cell types can contribute to EAE and MS pathogenesis, we focused on CD4+ T cells as they are essential to disease development in the EAE model and migrate readily into the brain during EAE (Figure 3a). Other immune cell types and subsets, including CD8+ T cells, B cells and myeloid cells, are depicted in Supplementary figure 4. While NalF and fingolimod treatment alone reduced CD4+ T‐cell infiltration into the brain as shown previously, 2 , 10 , 11 , 12 no synergistic effect of co‐treatment on CD4+ T‐cell infiltration was detected (Figure 3a). Co‐treatment was as effective as NalF or fingolimod single treatment (Figure 3a).
Figure 3.
Analysis of lymphocyte populations from healthy, vehicle, nalfurafine (NalF, 0.01 mg kg−1), fingolimod (FTY, 1 mg kg−1) or the combination in (a) brain tissue, (b) spleen tissue or (c) blood and (d) lymph nodes. (a) All infiltrating immune cells were identified by CD45high expression, and the relative number of cells expressed as a ratio to microglia (CD45intCD11b+). Identification of CNS‐infiltrating lymphocyte immune cell types by a sequential gating strategy (Supplementary figure 2a and b). CD4+ T‐cell lymphocytes and their specific subsets in spleen (b), blood (c) and lymph node (d), gating strategy in Supplementary figures 2c and d and 3a. The results from six independent experiments with 10–18 mice per group for brain and three independent experiments with 8–14 mice per group for spleen, blood and lymph node are shown. Data are mean ± SEM. Non‐parametric one‐way ANOVA with Dunnett's multiple comparisons testing, comparing the mean of each treatment against vehicle within each cell type. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Looking at peripheral immune cell populations, no effect of single or co‐treatment was detected in the splenic CD4+ T‐cell population or its subsets except for a significant decrease in the % of total CD4+ cell in co‐treated animals compared to healthy (Figure 3b, Supplementary figure 1g). In the blood, as expected, CD4+ T cells, and especially naive CD4+ T cells, were significantly reduced by fingolimod alone or in combination with NalF compared to vehicle or NalF alone (for the overall CD4+ T cell compartment), while effector memory (EM) CD4+ T cells were up‐regulated (Figure 3c).
Interestingly, while absolute cell number counts were increased in the lymph node by co‐treatment (Supplementary figure 1f), no effect of any treatments could be seen on the overall CD4+ T cells population in the lymph node (Figure 3d). While a reduction of naïve CD4+ T cells in the co‐treated animals compared to NalF alone was detected (P = 0.0541), co‐treated animals were not significantly different compared to vehicle or fingolimod alone. In addition, EM CD4+ T cells were again increased by fingolimod single treatment or in combination with NalF. In addition, the homing receptor CCR7, known to be down‐regulated on CD4+ T cells by fingolimod, was decreased. While NalF treatment by itself did not seem to effect the effector sub‐population of CD4+ T cells or CCR7+CD4+ T cells, surprisingly, co‐treatment resulted in an intermediate phenotype between single NalF and fingolimod treatment (Figure 3c and d). In summary, NalF and fingolimod alone reduced the infiltration of cells into the CNS with the co‐treatment showing similar beneficial effects. Additionally, these results indicate that treatment with NalF does not impair the fingolimod‐induced alterations in peripheral immune cell migration.
Co‐treatment effect of nalfurafine and fingolimod on T‐cell subsets and cytokine production
While the findings of the peripheral CD4+ T cells were surprising and unexpected, we analysed further what effect NalF, fingolimod and the combination had on T‐cell subsets and inflammatory cytokine production to understand whether the change in the immune subsets was because of systemic changes of the immune environment in vivo or the direct effect of the treatment on the immune cells. For the in vitro subset analysis, we treated healthy splenocytes with NalF, fingolimod or the combination in the absence of stimulation to see what effect the treatments had by themselves, or in the presence of ConA to mimic an inflammatory environment or CD3/CD28 beads to activate and expand T cells specifically (gating strategy in Supplementary figure 3b). In vitro concentrations for NalF, fingolimod and the combination were established by cell viability (MTT, Supplementary figure 5a and b) as well as reduction of inflammatory cytokine production (Supplementary figure 5c and d).
In healthy splenocytes, a significant expansion of CD4+ T cells could be detected when stimulated with CD3/CD28 and treated with fingolimod or NalF and fingolimod in combination compared to unstimulated or NalF single treatment (Figure 4a). In line with the results from EAE mice in vivo, we saw a significant down‐regulation of naïve CD4+ T cells and a significant increase of effector memory T cells in unstimulated splenocytes (Figure 4b and Supplementary figure 6a and b). In addition, CCR7+CD4+ T cells were significantly increased when stimulated with ConA and treated with fingolimod or the combination (Figure 4a). While these results do not reflect the in vivo results, it is important to consider that these are healthy splenocytes and fingolimod had a direct effect on them (Figure 4c).
Figure 4.
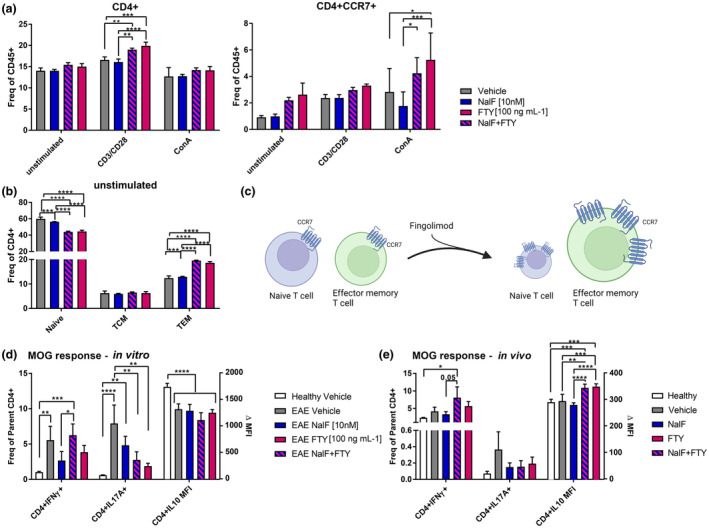
In vitro and in vivo data of nalfurafine (NalF) and fingolimod (FTY) effect of CD4+ T‐cell subsets and cytokine production. (a, b) Healthy splenocytes were either treated with vehicle (grey), NalF (blue, 10 nM), FTY (pink, 100 ng mL−1) or the combination (blue–pink striped) and were left unstimulated or stimulated with ConA or CD3/CD28 for 72 h and the CD4+ T‐cell subsets (naïve; TCM, T‐cell central memory, TEM, T‐cell effector memory) and CD4+CCR7+ T cells were quantified. Data are mean ± SEM from two individual experiments with n = 13 animals per treatment group. Two‐way ANOVA with Tukey's multiple comparisons testing, compare the mean of each stimulation group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (c) Schematic figure of FTY's mode of action created in BioRender. (d) Splenocytes from untreated EAE mice were treated in vitro with vehicle (grey), NalF (blue, 10 nM), FTY (pink, 100 ng mL−1) or the combination (blue–pink striped) and were left unstimulated or stimulated with ConA or MOG for 72 h and intracellular cytokine production in CD4+ T cells were quantified. Data are mean ± SEM from two individual experiments with n = 12 per treatment group. Two‐way ANOVA with Tukey's multiple comparisons testing, compare the mean of each stimulation group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (e) EAE mice were treated in vivo with either vehicle, NalF (0.01 mg kg−1), FTY (1 mg kg−1) or the combination. After splenocyte isolation, cells were left untreated or re‐stimulated with ConA or MOG for 72 h and intracellular cytokine production in CD4+ T cells was quantified. Data are mean ± SEM from two individual experiments with n = 5–8 per treatment group. Two‐way ANOVA with Tukey's multiple comparisons testing comparing the mean of each stimulation group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Even though the abundance of CD4+ T‐cell subsets was unaltered by NalF single treatment, we have observed previously that NalF has immunomodulatory properties by changing the cytokine response. 2 Thus, splenocytes from untreated EAE mice were isolated and cultured in vitro with either vehicle, NalF, fingolimod or the combination and stimulated with myelin oligodendrocyte glycoprotein (MOG35–55) peptide to assess the direct effect of these compounds on antigen‐specific responses using intracellular cytokine analysis of CD4+ T cells (Figure 4d and e and Supplementary figure 6c and d). While single treatment of NalF or fingolimod seemed to significantly reduce IFNγ, co‐treatment did not affect IFNγ production. IL‐17A production was significantly down‐regulated by fingolimod and the combination compared to vehicle treatment with a similar reduction seen after NalF treatment alone (Figure 4d; gating strategy in Supplementary figure 3c).
To determine whether in vivo treatments had similar effects on antigen‐specific recall responses, splenocytes isolated from EAE mice treated in vivo with NalF, fingolimod or the combination were stimulated in vitro with the MOG peptide (Figure 4e). While a similar pattern was observed, the down‐regulation of IL‐17A did not reach significance (Figure 4e) and no effect on IFNγ was observed. However, a significant up‐regulation of IL‐10 was detected in fingolimod single treated and combination treated splenocytes (Figure 4e). This effect was not seen when splenocytes were stimulated in vitro (Figure 4d). In summary, co‐treatment of NalF with fingolimod did not show any significant changes compared to the single treatments with regard to T‐cell subsets and cytokine production. Nonetheless, while there were no clear beneficial effects on cytokine production identified with the co‐treatment, no adverse effects were detected.
Co‐treatment of fingolimod with a different KOR agonist U50,488
To determine whether the co‐treatment effect we observed with fingolimod in combination with the KOR agonist NalF was compound‐ or class specific, we evaluated whether with U50,488, a traditional KOR agonist, 13 in combination with fingolimod would have similar effects. Co‐treatment of U50,488, at the doses tested, and fingolimod improved the disease outcome compared to the single treatments as measured by scores, per cent of mice that recovered and number of days in recovery (Figure 5a and b). While the number of relapses was reduced in the co‐treatment compared to U50,488 alone, the level was similar to vehicle‐treated animals (Figure 5b). Myelination was improved by the co‐treatments as was the reduction in lesion area (Figure 5c), although U50,488 alone did not appear to effectively enhance remyelination in these experiments. Comparing the overall effects of the different treatments on disease outcomes indicates that the co‐treatment provided superior or equal benefits in all parameters, while fingolimod showed equal benefit in relapse reduction (Figure 5d).
Figure 5.
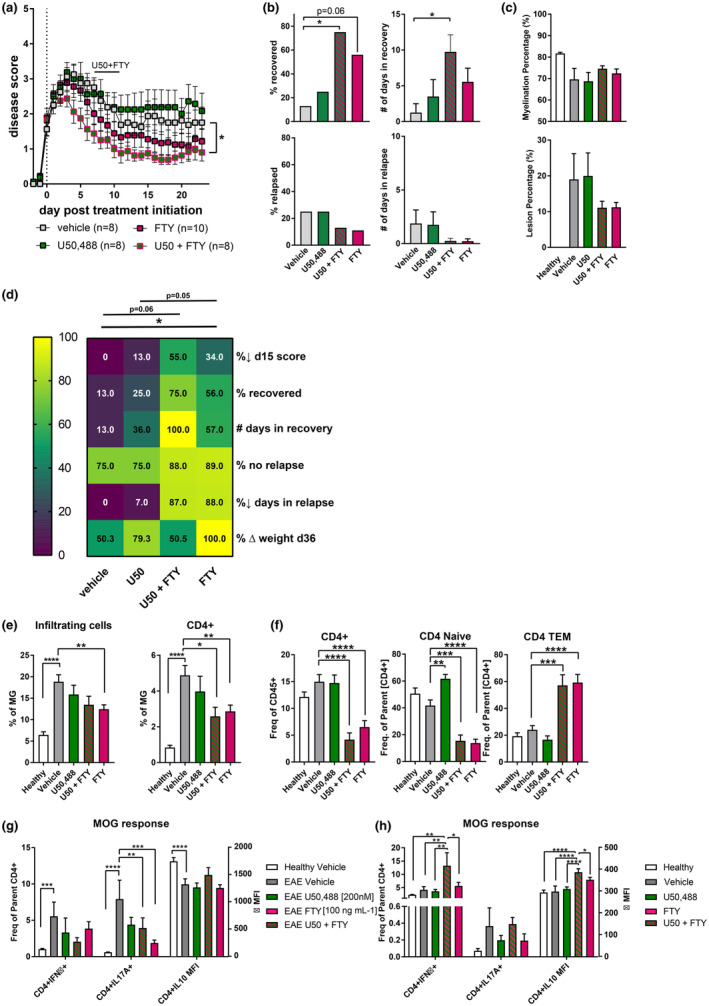
In vivo and in vitro treatment of U50,488 in combination with fingolimod (FTY) in EAE. (a) Disease scores of vehicle, U50,488 (1.6 mg kg−1 i.p.), FTY (1 mg kg−1 p.o.) or the combination treated animals from disease onset (score ≥ 1) to 23 dpt. Scores aligned to the day of disease onset (day 0 post treatment). Mice were treated daily, and results combined and analysed from two independent experiments (n = 8–10 animals per treatment group, as indicated) by two‐way ANOVA with Tukey's multiple comparisons testing and (*) comparing treatments to vehicle. (b) Percentage recovery to 23 dpt (recovery = score ≤ 0.5), number of days in recovery to 23 dpt, percentage relapse to 23 dpt (relapse = increase by 1 full point from the lowest score of remission) and end score of mice at the end of experiment were analysed by non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. (c) Myelination and lesion percentage in the spinal cord quantified by Black‐Gold II staining. (d) Heatmap of all disease parameters analysed by non‐parametric one‐way ANOVA with Tukey's multiple comparisons testing. Analysis of lymphocyte populations in (e) brain tissue and (f) blood. (e) All infiltrating immune cells were identified by CD45high expression, and the relative number of cells is expressed as a ratio to microglia (CD45intCD11b+). (f) CD4+ T‐cell lymphocytes and their specific subsets in blood. Results are combined from two independent experiments. Data are mean ± SEM and analysed by non‐parametric one‐way ANOVA with Dunnett's multiple comparisons testing comparing the mean of each treatment against vehicle within each cell type. (g) Splenocytes from EAE mice were treated ex vivo with vehicle (grey), U50,488 (green, 200 nM), FTY (pink, 100 ng mL−1) or the combination (blue–green striped) and were left unstimulated or stimulated with ConA or MOG for 72 h and intracellular cytokine production in CD4+ T cells was quantified. Data are mean ± SEM from two individual experiments with n = 11 per treatment group and analysed by two‐way ANOVA with Tukey's multiple comparisons testing comparing the mean of each stimulation group. (h) EAE mice were treated in vivo with either vehicle, U50,488 (U50, 1.6 mg kg−1), FTY (1 mg kg−1) or the combination. After splenocyte isolation, cells were left untreated or re‐stimulated with ConA or MOG for 72 h and intracellular cytokine production in CD4+ T cells was quantified. Data are mean ± SEM from one individual experiment with n = 4 or 5 per treatment group and analysed by two‐way ANOVA with Tukey's multiple comparisons testing comparing the mean of each stimulation group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The infiltration of immune cells into the brain was significantly reduced with fingolimod as seen previously, with the co‐treatment and fingolimod alone significantly reducing CD4+ infiltrating T cells (Figure 5e), whereas U50,488 alone did not appear effective in these experiments. In the blood, CD4+ T cells and subsets were not altered by U50,488 treatment, whereas fingolimod and the co‐treatment led to the down‐regulation of CD4+ T cells (especially naïve) and an up‐regulation of effector memory CD4+ T cells (Figure 5f). Cytokine production by splenocytes exposed in vitro or in vivo to the treatments was also assessed. Similar to the findings in Figure 4, IL‐17A production was down‐regulated by fingolimod or the combination, while IFNγ and IL‐10 were not significantly altered by in vitro exposure (Figure 5g). Interestingly, IFNγ and IL‐10 were increased after in vivo exposure to the combination of fingolimod and U50,488 in comparison to either treatment alone or vehicle (Figure 5h).
Taken together, these results are comparable with the combination of fingolimod with NalF; however, when all the experiments are considered together (Supplementary figure 7), the combination of NalF with fingolimod appears to provide superior benefits compared to U50,488 and fingolimod, especially in recovery and relapse reduction as shown by the multi‐parameter analysis (Supplementary figure 7c). Overall, these data suggest that the beneficial effect of co‐treatment with fingolimod and a KOR agonist is a class effect, although superiority of the benefit may be KOR agonist specific.
Co‐treatment effect of fingolimod and KOR agonist on OPC differentiation
To further investigate the mechanism by which NalF in combination with fingolimod increases functional recovery in EAE, primary mouse glial cells containing oligodendrocyte precursor cells (OPCs) were isolated and the ability of NalF, U50,488 and fingolimod alone or in combination to promote differentiation to mature oligodendrocytes (OL) was assessed. Mixed glial cells cultures containing OPCs from postnatal days 5 to 7 C57BL/6J mice were treated with the thyroid hormone 3,3′,5‐Triiodo‐l‐thyronine sodium (T3) as a positive control for differentiation. In parallel, cells were cultured in different concentrations of fingolimod (0.01–100 nM), NalF (20 nM), U50,488 (10 μM) or vehicle (0.1% DMSO/saline) or a combination of fingolimod with NalF or U50,488. After 5 days of drug treatment, cells were fixed and stained for SOX10 (to identify all cells of oligodendrocyte lineage) and MBP (mature OL), and the number of SOX10‐positive cells (expressing MBP) were counted. The number of DAPI+ cells stayed constant over most treatments showing that the cells were similarly viable (Supplementary figure 8a–c). Representative images of all treatments are shown in Figure 6a, c and d.
Figure 6.
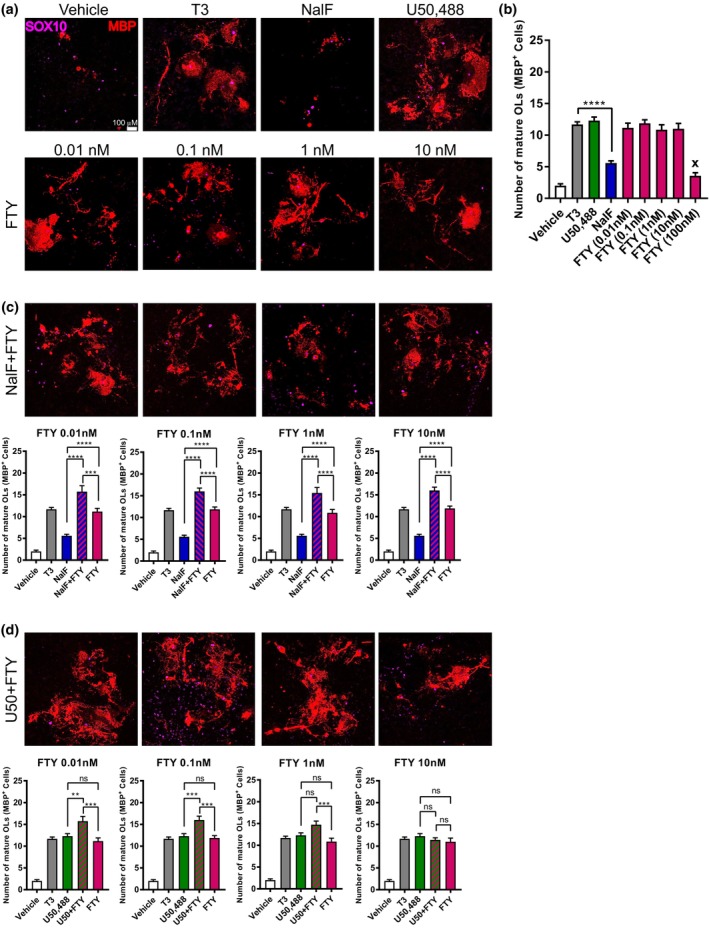
In vitro treatment of nalfurafine (NalF) in combination with fingolimod (FTY) in primary mouse mixed neuronal/glial cultures. (a) Representative images of mouse OPC cultures treated with vehicle (0.1% DMSO), T3 (30 ng mL−1), NalF (20 nM), U50,488 (10 μM), FTY (10 nM, 1 nM, 0.1 nM, 0.01 nM) and a combination of fingolimod with NalF or U50,488. Scale bar = 100 μm. Mixed glial cultures were immunostained for the OL lineage marker SOX10 (purple) and mature OL marker MBP (red). (b) Quantification of number of mature myelinating MBP+ OLs following treatment with U50,488 (10 μM), NalF (20 nM) and FTY (0.01 nM, 0.1 nM, 1 nM, 10 nM, 100 nM). (c) Quantification of number of mature myelinating MBP+ OLs following combination treatment with NalF (20 nM) and FTY (0.01 nM, 0.1 nM, 1 nM, 10 nM). (d) Quantification of number of mature myelinating MBP+ OLs following combined treatment with U50,488 (10 μM) and FTY (0.01 nM, 0.1 nM, 1 nM, 10 nM). Parametric tests were used if data were found to be normally distributed. Data are mean ± SEM, and significance determined by one‐way ANOVA with Bonferroni post‐tests from three individual experiments. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Surprisingly, all concentrations of fingolimod seemed to increase the number of mature OLs (MBP+) to the same level as the positive control T3, except for the highest concentration (100 nM) which appeared to reduce cell viability (Figure 6b). In addition, U50,488 also significantly increased the number of mature OLs, while NalF alone showed a modest but significant increase compared to vehicle (Figure 6b). Interestingly, the combination of NalF with fingolimod significantly increased the number of mature OLs compared to the single treatments (Figure 6c). This enhancement was observed with all tested concentrations of fingolimod. While the same effect was seen with U50,488 in combination with fingolimod at the lower concentrations, the superior effect of the co‐treatment was lost with higher fingolimod concentrations (Figure 6d). Together these data suggest that the increased functional recovery seen in the co‐treatment may be, in part, because of increased differentiation of OPCs into mature OLs leading to repair of the damaged myelin.
Discussion
The aim of this study was to investigate the effect of the co‐treatment of the KOR agonist NalF in combination with different DMTs on the disease course of EAE and elucidate the underlying changes in the immune system and CNS after co‐treatment. On the one hand, we have found that NalF co‐treatment with the DMT fingolimod significantly improved the disease outcome of EAE mice as reflected by increased recovery of these mice and reduced relapses. On the other hand, co‐treatment with the DMT DMF did not further improve NalF's effect on disease recovery but, importantly, also did not cause any adverse events. The latter point is important if considering the use of NalF in the clinical setting to treat MS, since most people with relapsing–remitting MS are on DMTs. Our finding that DMF did not improve EAE recovery has been reported previously by deBruin et al., who found that the tested dose did not improve clinical scores and had no significant effect on the disease course compared to control mice. 14 DMF is currently a first‐line treatment for MS and is still widely used in the clinic because of its good safety profile and tolerability. In contrast to DMF's generic immunomodulatory activity, fingolimod is a highly specific S1PR antagonist which shows high efficacy in EAE at reducing clinical scores and inflammatory cell infiltrates in spinal cord, rescuing neuropathological damage in cerebellum and restoring neuronal function. 15 , 16 , 17 Our studies show that NalF can further improve and complement these effects by promoting functional recovery and remyelination. This study is the first to investigate co‐treatment of a potential remyelinating therapy with two current DMTs and reveals novel effects by which the combination of NalF and fingolimod may be modulating immune cells and glial cells in the CNS.
Immune cell changes
Impaired T‐cell migration to the CNS after treatment with fingolimod, DMF and NalF has been reported previously. 2 , 18 Similarly, in this study, the infiltration of immune cells into the CNS was inhibited by NalF, fingolimod and the combination of both. NalF treatment showed a preferential effect on CD4+ T cells, which is in line with previous published data on CNS infiltration in EAE after NalF treatment, 2 while fingolimod alone reduced all lymphocytes assessed (B and T cells). Fingolimod reduces the entry of lymphocytes into the CNS by binding to the S1PR, which is necessary for lymphocytes to egress from the lymph node. 6 , 7 Thus, a reduction in CNS infiltration after fingolimod treatment is in line with previously published literature. Interestingly, the combination of NalF with fingolimod did not appear to enhance the reduction in CNS infiltration, although it was sustained at similar levels to the single treatments, indicating the co‐treatments did not have antagonistic effects.
Splenic T‐cell populations were unaltered by NalF or fingolimod alone as seen previously. 2 , 19 Interestingly, co‐treatment significantly reduced CD3+, CD4+ and CD8+ T cells in the spleen, suggesting that they were selectively targeted by fingolimod, but leaving the T cells subsets (naïve vs. TEM or TCM) unaltered. In contrast, while blood T cells and their activation subsets were unaltered by vehicle or NalF treatment, they were significantly altered by fingolimod single or co‐treatment as expected. 12 , 20 Interestingly, fingolimod treatment led to a switch in the proportion of CD4+ T‐cell subsets from naïve (down‐regulation) to effector memory (TEM; up‐regulation) in the blood. 21 This, in addition to a reduction in the expression of CCR7, may increase the sequestration of T cells in the lymph node. 22 However, TEM are preferentially kept circulating in the peripheral blood because of their low expression of CCR7. 23 One limitation to note is that the CCR7 staining was performed on ice and not at 37°C, which is optimal; however, reanalysis using this optimal temperature showed similar patterns and did not alter our overall conclusions (Supplementary figure 9). Surprisingly, co‐treatment of fingolimod with NalF shows similar phenotypes as fingolimod single treatment, suggesting that NalF does not interfere with fingolimod's ability to alter T‐cell homing and sequestration.
In the lymph node, NalF or vehicle treatment did not alter the frequency of CD4+ T cells or the subsets. While absolute cell counts were increased in the lymph node as reported in the literature, 22 CD4+ T cells frequency was unaltered. While previous literature has shown that fingolimod's mode of action is impaired by a pertussis‐toxin (PTX)‐sensitive mechanism, 24 the effect in this long‐term mouse model is unclear. However, NalF treatment in combination with fingolimod does not impair fingolimod's mode of action.
To understand whether the change in the immune subsets was as a result of the systemic changes of the immune environment within the mouse or the direct effect of the treatment on the immune cells, isolated immune cells were treated in vitro with NalF, fingolimod or the combination under different stimulation conditions. In line with the in vivo data, naïve CD4+ T cells were down‐regulated with fingolimod single treatment and in combination with NalF, while TEM were up‐regulated. This suggests that the changes in the CD4+ T‐cell subsets are a direct effect of fingolimod on the CD4+ T cells. However, it needs to be further analysed as to whether these changes also occur if other immune cells, like B cells and macrophages, are removed from the cell suspension. Under specific T‐cell‐stimulating conditions with CD3/CD28 activator beads, fingolimod single treatment and in combination with NalF increased the frequency of CD4+ T cells, supporting the idea of the direct effect of these treatments on the CD4+ T cells. The changes seen in the subsets might be as a result of the less stringent requirements for activation of memory T cells compared to naive T cells, including the ability to respond to lower concentrations of antigen than naive T cells as well as memory T cells are less dependent on co‐stimulatory signals than naive T cells, and do not require a long duration of antigenic stimulation. 25 In MS patients on fingolimod treatment, the percentages of T‐cell subsets show significant changes from 2 weeks onwards, where a down‐regulation of naïve CD4+ T cells and an up‐regulation of TEM in PBMCs were reported. 26
As reported previously in Denny et al., 2 one mechanism by which NalF may facilitate its beneficial effect on disease course and recovery is by shaping the cytokine environment and, in particular, reducing pro‐inflammatory cytokines. A reduction of IFNγ‐producing CD4+ T cells as well as IL‐17A‐producing CD4+ T cells was detected after in vitro and in vivo exposure to NalF. Fingolimod alone also had a very similar effect. This finding is in line with previous reports showing reduced IFNγ and IL‐17A in the spinal cord 27 and brain homogenates as well as in the periphery (blood and spleen) 12 , 28 of fingolimod‐treated EAE mice. In addition, Mehling et al. reported reduced levels of IFNγ in the blood of people with MS on fingolimod treatment, 29 while Moreno‐Torres et al. 30 found that changes in IL‐17A before and during treatment were associated with responders and non‐responders to fingolimod treatment where responders showed a down‐regulation of IL‐17A.
Interestingly, in vivo treatment of the combination of fingolimod with either as KOR agonist (NalF or U50,488) seemed to reverse the reduction in IFNγ‐producing CD4+ T cells such that levels were comparable to vehicle‐treated cells. On the one hand, however, fingolimod single or co‐treatment with KOR agonist reduced IL‐10 in CD4+ T cells in vitro, which is in line with Thomas et al. 31 who reported reduced release of pro‐inflammatory cytokines in vitro after fingolimod treatment. Surprisingly, on the other hand, in vivo fingolimod single or co‐treatment with KOR agonist up‐regulate IL‐10 in CD4+ T cells suggesting an increase in Th2 cytokine production and a shift from Th1 to Th2 phenotype. All these changes have been detected in MOG re‐stimulated immune cells, suggesting a specific response of the autoreactive T cells towards the MOG peptide as well as when using the different KOR agonists, suggesting that the effect is not agonist but class specific.
Remyelination and OPC differentiation
As reported previously by electron microscopy and histochemical analysis, NalF treatment increased myelination in the spinal cord and reduced lesion percentage 2 compared to vehicle treatment. In addition, mice treated with fingolimod alone had modestly elevated myelin levels and significantly reduced lesion numbers compared to vehicle‐treated animals. Previous studies on the effect of fingolimod on remyelination are conflicting. Studies using the non‐immune cuprizone model of demyelination have not detected any remyelinating effects with fingolimod treatment. 32 Similarly, other studies using lysolecithin‐induced and viral‐induced models of demyelination did not detect evidence of remyelination, although promising changes to OPC numbers and recruitment were found. 33 , 34 In EAE, many studies are confounded by prophylactic fingolimod treatment preventing demyelination. 35 However, Zhang et al. reported that myelination, measured by luxol fast blue staining in the CNS of the EAE animals, was increased after fingolimod treatment starting on the day of EAE onset. 36 Interestingly, they linked the increase in myelination after fingolimod treatment to an increase in the proliferation and differentiation of OPCs into mature oligodendrocytes in the CNS. While the mechanisms by which fingolimod promoted proliferation and differentiation of OPCs require elucidation, they found that fingolimod activated the Shh/Gli1 pathway, which has been shown to induce oligodendrogenesis. 36 In line with these data, we observed an increase in mature oligodendrocytes using our in vitro OPC cultures exposed to fingolimod over a range of concentrations and to the same level as the positive control T3. As reported previously, U50,488 also significantly increased the number of mature oligodendrocytes to the same level as T3. 37 , 38 NalF promoted a more modest increase in OPC differentiation, suggesting that, although U50,488 and NalF target the same receptor, the downstream signalling may be different. This difference is inversely reflected in the scores and recovery of the EAE mice where NalF performed significantly better than U50,488 in reducing disease severity, increasing recovery and promoting remyelination.
Unexpectedly, the co‐treatment of NalF with fingolimod further increased the myelination and reduced lesion number compared to the single treatments, and this effect is also reflected in the number of mature oligodendrocytes detected after co‐treatment. While this co‐treatment effect is lost when increasing fingolimod doses with U50,488, fingolimod treatment with NalF showed significantly higher numbers of mature oligodendrocytes over all tested concentrations. This increased effect could explain why co‐treatment of fingolimod with NalF was superior to the combination with U50,488 at the dose tested, in terms of reduced disease severity and increased recovery during EAE.
Conclusion
In summary, this study is the first to report that the combined use of the remyelinating agent, NalF, with the DMT, fingolimod, leads to superior benefit in the EAE model of MS. Overall, we have shown that co‐treatment increased the recovery of mice and reduced relapses. This beneficial effect correlated with a reduction in immune cell infiltration into the CNS as well as peripheral immune cells alterations including autoreactive CD4+ T cells and their cytokine profile and increased myelination in the spinal cords of co‐treated animals. Further studies are needed to elucidate the precise mechanism that mediates the observed immune cell changes, increased numbers of mature oligodendrocytes and increased functional recovery. Together this work highlights a novel combination treatment for MS that may not only reduce the symptoms by preventing CNS damage, but also increase CNS repair and recovery.
Methods
Animals
Female C57BL/6J mice (Jackson laboratory; RRID:IMSR_JAX:000664) were purchased from the Biomedical Research Unit of the Malaghan Institute of Medical Research (Wellington, NZ) or bred in‐house and housed in the Victoria University of Wellington PC2 animal facility at a 12‐h dark:12‐h light cycle. Food and water were available ad libitum. Animals were used between 8 and 12 weeks of age for EAE.
Ethics statement
All experiments with animals were carried out in the School of Biological Sciences Animal Facility at the Victoria University of Wellington and were approved by the Victoria University of Wellington Animal Ethics Committee (AEC25295 and AEC29122) and adhere to the ARRIVE reporting guidelines.
Drugs
NalF and U50,488 were synthesised as described previously by the Synthetic Chemical Biology Core Laboratory (University of Kansas, Lawrence, KS, USA). 39 , 40 Fingolimod (hydrochloride) (FTY720) was purchased from Cayman Chemicals (Ann Arbor, MI, USA).
EAE induction and treatments
Mice were immunised subcutaneously (s.c.) in the rear flanks with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (50 μg per mouse; Genescript, Piscataway, NJ, USA) in complete Freund's adjuvant (Sigma‐Aldrich, St. Louis, MO, USA) containing 500 μg per mouse inactivated Mycobacterium tuberculosis H37Ra (Fort Richard, Auckland, New Zealand) and injected intraperitoneally (i.p.) with pertussis toxin (200 ng per mouse; List Biochemicals, Campbell, CA, USA) on days 0 and 2. Mice were weighed and scored daily as follows: 0, normal; 1, partial tail paralysis; 2, full tail paralysis; 3, paralysis in one hind limb; 4, paralysis in both hind limbs; 5, moribund. Experiments were run over 40–55 days and daily treatments were started at disease onset (score ≥ 1; approximately 13–18 days after EAE induction). Mice were sequentially allocated to treatment groups and scored by an investigator blinded to treatment allocations. NalF, U50,488 and vehicle control were administered i.p. in 0.9% saline:DMSO:Tween80 in a ratio of 8:1:1. Fingolimod and its vehicle control were administered p.o. in 0.9% saline.
Primary cell isolation into single‐cell suspension
Following CO2 euthanasia, lymph nodes and spleens were isolated before mice were perfused using PBS. Brains were collected after perfusion, and brains, lymph nodes and spleens were processed into a single‐cell suspension for flow cytometry. Brain was mashed through a 70‐μm cell strainer (Corning, Corning, NY, USA) and centrifuged at 760 g for 5 min. Cell pellets were re‐suspended in 37% Percoll gradient (Sigma‐Aldrich) and centrifuged 30 min at 760 g without brakes. Myelin layer was removed, supernatant discharged and pellet re‐suspended in FACS buffer [2% foetal bovine serum (FBS; Gibco, Billings, MT, USA), 0.1% sodium azide in PBS]. Lymph nodes were mashed through a 70‐μm cell strainer and centrifuged at 760 g for 5 min and cell pellets were re‐suspended for cell counting.
Spleens were mashed through a 70‐μm cell strainer and centrifuged at 760 g for 5 min after which the pellet was loosened and re‐suspended in red blood cell lysis buffer (Sigma‐Aldrich) for 2 min. The cell suspension was incubated with red blood cell lysis buffer for 2 min, wash buffer (DMEM plus 30 mM HEPES and 1% penicillin/streptomycin; all from Gibco) was added and samples were centrifuged at 760 g for 5 min. Blood was collected as terminal bleed via cardiac puncture into tubes containing heparin to prevent coagulation. Red blood cell lysis was performed, and cell pellets were re‐suspended for cell counting using trypan blue.
Analysis of cytokines
Splenocytes were plated in complete T‐cell medium (10% FBS, 2 mM l‐glutamine, 1% penicillin/streptomycin, 10 mM HEPES, 0.1% β‐mercaptoethanol and 0.1% non‐essential amino acids in DMEM; all from Gibco) in a 96‐well plate (Corning) and stimulated with medium, MOG35‐55 peptide (27 μg mL−1), Concanavalin A (ConA; 1 μg mL−1; Sigma‐Aldrich) or CD3/CD28 dynabeads (5 × 104 beads per well; Gibco). NalF (10 nM), U50,488 (200 nM), fingolimod (0.1 μg mL−1) or vehicle (0.01% DMSO) was added before incubation for 72 h at 37°C and 5% CO2. Concentrations were determined by performing a dose–response on the viability of cells (MTT) as a single treatment or as a combination. Extracellular cytokine analysis by ELISA was used to determine the best co‐stimulation effect on the down‐regulation of cytokine. All data are shown in Supplementary figure 5a–c. For intracellular cytokine analysis, splenocyte cultures were stimulated with phorbol 12‐myristate 13‐acetate (PMA; 50 ng mL−1; Sigma‐Aldrich) and ionomycin (500 ng mL−1; Sigma‐Aldrich) in the presence of GolgiStop/Monensin (1 μg per 106 cells; BD Biosciences, Franklin Lakes, NJ, USA) for 4 h at 37°C and 5% CO2 before preparing for flow cytometry.
Flow cytometry
Immunophenotyping was done using CD4‐BV521 (RM4‐5), CD45‐BV510 (30‐F11), CD3‐APC or CD3‐APC‐Cy7 (17.A2), CD25‐PE‐Cy7 (PC61), CD8‐PerCPCy5.5 (53‐6.7), B220‐APC‐Cy7 (RA3‐6B2; BD Biosciences, Franklin Lakes, NJ, USA), CD11b‐PE‐Cy7 (M1/70), Ly6C‐PE (HK1.4), Gr1‐APC‐Cy7 (RB6‐8C5), IA/IE‐BV421 (M5/114.15.2), F4/80‐FITC (BM8), CD11c‐PerCPCy5.5 (N418), CD44‐PE (1M7), CD62L‐FITC (MEL‐14), CCR7‐PE (4B12). All antibodies were from Biolegend (San Diego, CA, USA) unless stated otherwise. Cells were incubated with Fc Block (1 μg per 106 cells; 2.4G2; BD Biosciences) for 15 min prior to staining with fluorescently labelled antibodies for 30 min on ice.
Intracellular cytokines were detected using CD4‐AF488 (RAM4‐5), IFNγ‐BV421 (XMG 1.2), IL‐10‐PE (54 902, BD Bioscience) and IL‐17A‐AF647 (TC11‐18H10). After staining for extracellular proteins, cells were fixed in 4% paraformaldehyde and permeabilised using 0.1% saponin buffer (Sigma‐Aldrich) containing 0.1% bovine serum albumin (Sigma‐Aldrich) and stained with fluorescently labelled antibodies intracellular cytokines overnight on ice.
Flow cytometry was performed on a BD FACS Canto II (BD Biosciences) and analysed using Flowjo software version 10.6 (Treestar, Inc., Ashland, OR, USA).
Tissue processing and Black‐Gold II staining and analysis
Samples were prepared and processed as reported previously. 2 Briefly, following CO2 euthanasia and perfusion with PBS, spinal cords were fixed in 4% paraformaldehyde at 4°C overnight followed by 15% sucrose and 30% sucrose at 4°C overnight or until the tissue sank to the bottom of the tube. The sections were embedded in Cryomatrix embedding resin (Thermo Fisher Scientific, Waltham, MA, USA), snap frozen in isopentane on dry ice and frozen at −80°C for long‐term storage. Transverse 20‐μm sections were cut using a Leica CM3050 S cryostat microtome (Leica Biosystems, Wetzlar, Germany) and mounted on Superfrost plus slides (Thermo Fisher Scientific) and stored at −80°C. Prior to staining, slides were warmed at room temperature for 15–20 min before use, and OCT medium removed by immersing the slides in PBS for 5 min. Black‐Gold II myelin staining (Histo‐Chem, Jefferson, AR, USA) was performed as described by the manufacturer. Quantification was performed as described by Denny et al. 2 The analysis was carried out on five sections per animal, and the sections averaged to generate a mean value for each animal.
Disease parameter calculations
Generation of primary mouse mixed neuronal/glial culture
Primary mouse OPCs were isolated from postnatal days 5 to 7 C57BL/6J mice. Briefly, mice were euthanised by decapitation without perfusion, and brain cortices were dissociated with papain solution (20 U mL−1; Worthington Biochem. Corp., NJ, USA) for 15 min at 37°C. After incubation, cells were centrifuged (100 g for 5 min) and rinsed in 20% FBS and OPC media [DMEM/ F‐12 (Thermo Fischer Scientific), 1× Antibiotic–Antimycotic (Thermo Fischer Scientific), 1× B27 (Gibco), 1× N2 supplement (Thermo Fisher Scientific), 10% FBS, 1× stem pro neural supplement (Thermo Fisher Scientific), Human FGF basic (10 ng mL−1; Miltenyi Biotec, Macquarie Park, NSW, Australia) and PDGF‐AA (30 ng mL−1; Miltenyi Biotec)]. Using a sterile flame‐polished Pasteur pipette, tissue suspension was carefully triturated, increasing speed with increasing tissue dissociation to create a single‐cell suspension. The cell suspension was filtered through a 40‐μm cell strainer and cells were seeded and incubated in poly‐d‐Lysine (100 μg mL−1 Sigma‐Aldrich) coated 96‐well plates (Corning) at a seeding density of 80 000 cells mL−1 and incubated at 37°C in 5% CO2 for 7 days and media changes were performed every other day.
OPC differentiation assay
As a positive control to validate OPC differentiation, cells were cultured in media containing OL differentiation factor thyroid hormone 3,3′,5‐Triiodo‐l‐thyronine sodium (T3; 30 ng mL−1; Sigma‐Aldrich), or treated with the test drugs in different concentrations [fingolimod (100 nM, 10 nM, 1 nM, 0.1 nM, 0.01 nM), NalF (20 nM), U50,488 (10 μM) or vehicle (0.1% DMSO/saline)]. For fingolimod, a concentration range was selected based on previous literature. 41 For the OPC differentiation assay, concentrations of U50,488 and NalF were based on known activity at KOR in potency and activity assays 13 , 38 , 42 , 43 (U50,488 1 nM–100 mM; NalF 0.6–200 nM). From our data, the most effective concentration at promoting OPC differentiation for U50,488 (10 μM) and NalF (20 nM) was utilised for investigating the combination with fingolimod.
After 5 days of drug treatment, cells were fixed in 4% paraformaldehyde in PBS for 20 min. Prior to immunolabelling, cells were blocked in PBS containing 10% donkey serum (Sigma‐Aldrich), 0.1% Triton X‐100 for 10 min at room temperature (RT), followed by incubation with relevant primary antibodies: anti‐SOX10 (Goat, 1:500, R&D Systems Minneapolis, USA, AF2864) and anti‐MBP (Rabbit, 1:200, Abcam, Melbourne, Australia, ab40390). Cells were rinsed with PBS followed by the appropriate fluorophore‐conjugated secondary antibodies [anti‐rabbit AF647 (1:1000; Thermo Fisher Scientific; A21245), anti‐goat AF488 (1:1000; Abcam, Melbourne, Australia; ab150129)] for 60 min at RT. Cells were rinsed twice in PBS before adding 4′,6‐diamidino‐2‐phenylindole (DAPI) solution (300 nM in PBS) (Thermo Fisher Scientific) for 10 min at RT. Imaging was carried out on a confocal microscope (Olympus FV3000, Software number FV31S‐SW, Auckland, NZ), and a high‐throughput confocal microscope (IN Cell Analyzer 6500 HS, Version 7.2, GE Healthcare/Cytiva, WA, USA). The excitation lasers 642 nm (AlexaFlour 647), 561 nm (AlexaFlour 555) and 405 nm (DAPI) were used for capturing 10 images per well using a 20× objective. The quantitative image analysis was carried out using automated unbiased counting methods using Cell profiler 4.1.3 (Broad Institute) 44 and GraphPad Prism V.7 (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical analyses
All graphs and statistical analyses were generated using GraphPad Prism 7 (GraphPad Software, Inc.). Comparisons between two groups were performed using a paired Student's t test. For comparison of more than two groups, one‐way or two‐way analysis of variance (ANOVA) was used with the recommended multiple comparison tests as indicated in the figure legend and as recommended by GraphPad Prism. Differences of P < 0.05 were considered significant.
Conflict of interest
The following authors declare the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: ACL, BMK and TEP are inventors on patent applications that relate to this work and have been licensed to Rekover Therapeutics Ltd. They hold equity in Rekover Therapeutics Ltd. The authors declare no other financial interests.
Author contributions
Katharina Robichon: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft. Rabia Bibi: Data curation; investigation; methodology; writing – review and editing. Mackenzie Kiernan: Data curation; methodology; writing – review and editing. Lisa Denny: Conceptualization; data curation; formal analysis; writing – review and editing. Thomas E Prisinzano: Conceptualization; methodology; resources; writing – review and editing. Bronwyn M Kivell: Conceptualization; formal analysis; funding acquisition; writing – review and editing. Anne Camille La Flamme: Conceptualization; data curation; formal analysis; funding acquisition; supervision; writing – review and editing.
Ethical approval
The Victoria University of Wellington Animal Ethics Committee approved all experimental procedures used in this study (25295 and 29122).
Supporting information
Supplementary figures 1–9
Acknowledgments
The authors thank Sven Sondhauss, Tessa Peck, Afnan Al Abadey and Madeline Griffiths (all from the Victoria University of Wellington) for their help with the experiments. This study was funded by the Ministry of Business, Innovation, and Employment (RTVU1802 to ACL, BMK and TEP), the Neurological Foundation of New Zealand (1639PG to BMK, ACL and TEP), the Health Research Council of New Zealand (18/063 to BMK, ACL and TEP) and the Great New Zealand Trek (to ACL). Open access publishing facilitated by Victoria University of Wellington, as part of the Wiley ‐ Victoria University of Wellington agreement via the Council of Australian University Librarians.
Data availability statement
The datasets used and/or analysed during this study are available from the corresponding author on reasonable request.
References
- 1. Fischer FR, Santambrogio L, Luo Y, Berman MA, Hancock WW, Dorf ME. Modulation of experimental autoimmune encephalomyelitis: effect of altered peptide ligand on chemokine and chemokine receptor expression. J Neuroimmunol 2000; 110: 195–208. [DOI] [PubMed] [Google Scholar]
- 2. Denny L, Al Abadey A, Robichon K et al. Nalfurafine reduces neuroinflammation and drives remyelination in models of CNS demyelinating disease. Clin Transl Immunology 2021; 10: e1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Linker RA, Lee DH, Ryan S et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011; 134: 678–692. [DOI] [PubMed] [Google Scholar]
- 4. Kihara Y, Groves A, Rivera RR, Chun J. Dimethyl fumarate inhibits integrin alpha4 expression in multiple sclerosis models. Ann Clin Transl Neurol 2015; 2: 978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulze‐Topphoff U, Varrin‐Doyer M, Pekarek K et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci USA 2016; 113: 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1‐phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci 2013; 328: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol 2010; 33: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ho H, Cao R, Miao J et al. Fingolimod ameliorates the development of experimental autoimmune encephalomyelitis by inhibiting Akt‐mTOR axis in mice. Int Immunopharmacol 2016; 30: 171–178. [DOI] [PubMed] [Google Scholar]
- 9. Hou H, Cao R, Quan M et al. Rapamycin and fingolimod modulate Treg/Th17 cells in experimental autoimmune encephalomyelitis by regulating the Akt‐mTOR and MAPK/ERK pathways. J Neuroimmunol 2018; 324: 26–34. [DOI] [PubMed] [Google Scholar]
- 10. Salas‐Perdomo A, Miro‐Mur F, Gallizioli M et al. Role of the S1P pathway and inhibition by fingolimod in preventing hemorrhagic transformation after stroke. Sci Rep 2019; 9: 8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fagan SG, Bechet S, Dev KK. Fingolimod rescues memory and improves pathological hallmarks in the 3xTg‐AD model of Alzheimer's disease. Mol Neurobiol 2022; 59: 1882–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rolland WB, Lekic T, Krafft PR et al. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol 2013; 241: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du C, Duan Y, Wei W et al. Kappa opioid receptor activation alleviates experimental autoimmune encephalomyelitis and promotes oligodendrocyte‐mediated remyelination. Nat Commun 2016; 7: 11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Bruin NM, Schmitz K, Schiffmann S et al. Multiple rodent models and behavioral measures reveal unexpected responses to FTY720 and DMF in experimental autoimmune encephalomyelitis. Behav Brain Res 2016; 300: 160–174. [DOI] [PubMed] [Google Scholar]
- 15. Fujino M, Funeshima N, Kitazawa Y et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther 2003; 305: 70–77. [DOI] [PubMed] [Google Scholar]
- 16. Pinschewer DD, Ochsenbein AF, Odermatt B, Brinkmann V, Hengartner H, Zinkernagel RM. FTY720 immunosuppression impairs effector T cell peripheral homing without affecting induction, expansion, and memory. J Immunol 2000; 164: 5761–5770. [DOI] [PubMed] [Google Scholar]
- 17. Balatoni B, Storch MK, Swoboda EM et al. FTY720 sustains and restores neuronal function in the DA rat model of MOG‐induced experimental autoimmune encephalomyelitis. Brain Res Bull 2007; 74: 307–316. [DOI] [PubMed] [Google Scholar]
- 18. Mathias A, Perriot S, Canales M et al. Impaired T‐cell migration to the CNS under fingolimod and dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm 2017; 4: e401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han S, Zhang X, Wang G et al. FTY720 suppresses humoral immunity by inhibiting germinal center reaction. Blood 2004; 104: 4129–4133. [DOI] [PubMed] [Google Scholar]
- 20. Malone K, Diaz Diaz AC, Shearer JA, Moore AC, Waeber C. The effect of fingolimod on regulatory T cells in a mouse model of brain ischaemia. J Neuroinflammation 2021; 18: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Claes N, Dhaeze T, Fraussen J et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12‐month follow‐up study. PloS One 2014; 9: e111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pino M, Paganini S, Deleage C et al. Fingolimod retains cytolytic T cells and limits T follicular helper cell infection in lymphoid sites of SIV persistence. PLoS Pathog 2019; 15: e1008081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friess J, Hecker M, Roch L et al. Fingolimod alters the transcriptome profile of circulating CD4+ cells in multiple sclerosis. Sci Rep 2017; 7: 42087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brinkmann V, Pinschewer D, Chiba K, Feng L. FTY720: a novel transplantation drug that modulates lymphocyte traffic rather than activation. Trends Pharmacol Sci 2000; 21: 49–52. [DOI] [PubMed] [Google Scholar]
- 25. Berard M, Tough DF. Qualitative differences between naive and memory T cells. Immunology 2002; 106: 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song ZY, Yamasaki R, Kawano Y et al. Peripheral blood T cell dynamics predict relapse in multiple sclerosis patients on fingolimod. PloS One 2014; 10: e0124923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kataoka H, Sugahara K, Shimano K et al. FTY720, sphingosine 1‐phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol 2005; 2: 439–448. [PubMed] [Google Scholar]
- 28. Fujii C, Kondo T, Ochi H et al. Altered T cell phenotypes associated with clinical relapse of multiple sclerosis patients receiving fingolimod therapy. Sci Rep 2016; 6: 35314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mehling M, Lindberg R, Raulf F et al. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology 2010; 75: 403–410. [DOI] [PubMed] [Google Scholar]
- 30. Moreno‐Torres I, Gonzalez‐Garcia C, Marconi M et al. Immunophenotype and transcriptome profile of patients with multiple sclerosis treated with Fingolimod: setting up a model for prediction of response in a 2‐year translational study. Front Immunol 2018; 9: 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas K, Proschmann U, Ziemssen T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin Pharmacother 2017; 18: 1649–1660. [DOI] [PubMed] [Google Scholar]
- 32. Alme MN, Nystad AE, Bo L et al. Fingolimod does not enhance cerebellar remyelination in the cuprizone model. J Neuroimmunol 2015; 285: 180–186. [DOI] [PubMed] [Google Scholar]
- 33. Yazdi A, Baharvand H, Javan M. Enhanced remyelination following lysolecithin‐induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 2015; 311: 34–44. [DOI] [PubMed] [Google Scholar]
- 34. Boyd A, Zhang H, Williams A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 2013; 125: 841–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonfiglio T, Olivero G, Merega E et al. Prophylactic versus therapeutic Fingolimod: restoration of presynaptic defects in mice suffering from experimental autoimmune encephalomyelitis. PloS One 2017; 12: e0170825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J, Zhang ZG, Li Y et al. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol Dis 2015; 76: 57–66. [DOI] [PubMed] [Google Scholar]
- 37. Paton KF, Robichon K, Templeton N et al. The Salvinorin analogue, Ethoxymethyl ether Salvinorin B, promotes Remyelination in preclinical models of multiple sclerosis. Front Neurol 2021; 12: 782190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mei F, Mayoral SR, Nobuta H et al. Identification of the kappa‐opioid receptor as a therapeutic target for oligodendrocyte Remyelination. J Neurosci 2016; 36: 7925–7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szmuszkovicz J, Von Voigtlander PF. Benzeneacetamide amines: structurally novel non‐m mu opioids. J Med Chem 1982; 25: 1125–1126. [DOI] [PubMed] [Google Scholar]
- 40. Nagase H, Yamamoto N, Yata M et al. Design and synthesis of potent and highly selective orexin 1 receptor antagonists with a Morphinan skeleton and their pharmacologies. J Med Chem 2017; 60: 1018–1040. [DOI] [PubMed] [Google Scholar]
- 41. Zhang Y, Li X, Ciric B et al. Effect of Fingolimod on neural stem cells: a novel mechanism and broadened application for neural repair. Mol Ther 2017; 25: 401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schattauer SS, Kuhar JR, Song A, Chavkin C. Nalfurafine is a G‐protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal 2017; 32: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Tang K, Inan S et al. Comparison of pharmacological activities of three distinct kappa ligands (Salvinorin a, TRK‐820 and 3FLB) on kappa opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo . J Pharmacol Exp Ther 2005; 312: 220–230. [DOI] [PubMed] [Google Scholar]
- 44. Stirling DR, Swain‐Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 2021; 22: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–9
Data Availability Statement
The datasets used and/or analysed during this study are available from the corresponding author on reasonable request.