

Abstract
Benzimidazole opioids, often referred to as nitazenes, represent a subgroup of new psychoactive substances with a recent increase in fatal overdoses in the USA and Europe. With a variety of analogs emerging on the illicit drug market, forensic laboratories are challenged to identify these potent drugs. We here present a simple quantitative approach for the determination of nine nitazene analogs, namely, clonitazene, etodesnitazene, etonitazene, etonitazepyne, flunitazene, isotonitazene, metodesnitazene, metonitazene and protonitazene in whole blood using liquid-phase microextraction and electromembrane extraction in a 96-well format and liquid chromatography–tandem mass spectrometry. Green and efficient sample preparation was accomplished by liquid-phase microextraction in a 96-well format and resulted in high extraction yields for all analytes (>81%). Here, blood diluted with buffer (1:1, %v) was extracted from a donor compartment across a thin organic liquid membrane and into an aqueous acceptor solution. The acceptor solution was collected and directly injected into the analysis platform. Chromatographic separation was accomplished with a biphenyl column, allowing for a baseline separation of the structural isomers isotonitazene and protonitazene before detection by multiple reaction monitoring. Validation was performed according to Scientific Working Group of Forensic Toxicology guidelines. The calibration range was from 0.5 to 50 nM (except for protonitazene and clonitazene from 0.1 nM) with good linearity and limits of detection down to 0.01 nM. An AGREEprep assessment was performed to evaluate sample preparation greenness, with a final score of 0.71. Nitazenes represent a current threat to public health, and analytical methods that cover a wide range of these analogs are limited. Here, the described method may assist in the detection of nitazenes in whole blood and prevent these substances from being missed in postmortem investigations.
Introduction
New synthetic opioids (NSOs) are becoming an increasing concern to public health due to their high potencies and increased risk of life-threatening side effects. Since 2009, 75 new opioids have been identified in the European drug market and their unregulated nature makes the distribution difficult to monitor (1). Although fentanyls have the highest overdose death rates among synthetic opioids, recent core-structure scheduling of fentanyl-related substances in the USA and China disrupts the supply and encourages clandestine laboratories to synthesize alternative NSOs (2, 3). 2-benzyl benzimidazole opioids, often occurring under the name “nitazenes,” represent such a group of alternative NSOs. These µ-opioid receptor agonists have strong antinociceptive effects similar to morphine and heroin; however, potencies can be up to 1,000 times that of morphine (4–6). The general structure is depicted in Figure 1. Nitazenes contain a benzimidazole ring with an ethylamine in the 1-position and a benzyl group in the 2-position. The synthesis is amenable to incorporating a variety of substituents, producing a large number of analogs.
Figure 1.
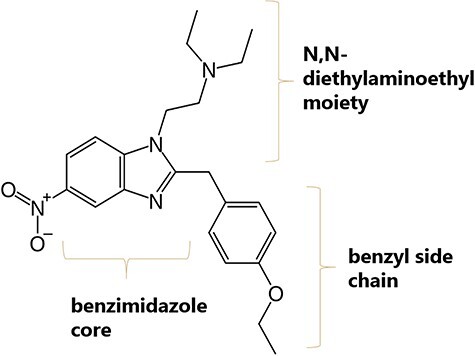
Molecular structure of etonitazene.
Already in the 1950s, nitazenes were investigated for medicinal purposes due to their strong analgesic properties, but were never approved for therapeutic use in humans (7). In 2019, the first findings of the nitazene analog isotonitazene started to appear in the USA (8). According to the National Forensic Laboratory Information System database, 644 reports of isotonitazene have been received as of September 2022 (8). Work by Logan and colleagues (2) confirmed the presence of various nitazene analogs in 288 casework samples between January 2020 and December 2021, demonstrating their popularity among users compared to previous years.
In 2019, the first nitazene was identified in the European drug market, and since then, 10 other analogs have followed. In the most recent European Monitoring Centre for Drugs and Drug Addiction report on new opioids, isotonitazene accounted for 22% of the seized material of new opioids (1). In Norway, three cases of nitazene overdoses have been confirmed in three different locations. Two of these cases had fatal outcomes (9, 10). Norwegian customs have also had four seizures of nitazenes (11).
Nitazenes are often not part of routine toxicology screening and may remain undetected. This is especially relevant when heroin has been fortified with nitazenes, they are constituents in counterfeit medicines or are involved in polydrug abuse. In the USA, most known nitazene analogs are classified as Schedule I–controlled substances under the Controlled Substances Act (12). Eleven nitazene analogs and brorphine will be added to Class A of the Misuse of Drugs Act 1971, and legislation will be brought forward to control these substances (13, 14). The development of analytical methodology for the determination of nitazenes is therefore highly relevant. At present, such validated methods are limited; however, Walton et al. (15) presented a validated quantitative approach for the simultaneous analysis of six nitazene analogs and three associated metabolites alongside the work by Krotulski et al. (16) for the quantification of isotonitazene.
Liquid–liquid extraction, solid-phase extraction and protein precipitation are the commonly encountered sample preparation techniques in forensic analysis. Although robust and simple, these techniques may require large volumes of organic solvents and produce considerable amounts of waste, not aligned with the demands for greener analytical chemistry (17).
Microextraction, in various formats, has proven to be a useful tool to minimize the overall material consumption and has been implemented in numerous analytical applications (18–21). Liquid-phase microextraction is one such technique and is found in three main variations, namely, single-drop microextraction (22), dispersive liquid–liquid microextraction (23) and hollow-fiber liquid-phase microextraction (HF-LPME) (24). With the requirement of a highly robust and reliable sample preparation due to judicial repercussions of analysis outcomes, the lack of commercial equipment for these techniques is a serious shortcoming.
HF-LPME has more recently advanced to a 96-well format (25–28). The 96-well format allows for higher-throughput extractions better suited to forensic analysis. This is a three-phase extraction principle where the analyte is transferred from an aqueous sample solution, across a thin organic layer immobilized in a filter membrane, into an aqueous acceptor solution (Figure 2a). The extraction takes place in a single step, and since extracts are aqueous, they are directly injected into the LC–MS instrument. Extractions can also be enhanced by an electrical field through a technique called electromembrane extraction (EME, Figure 2b).
Figure 2.
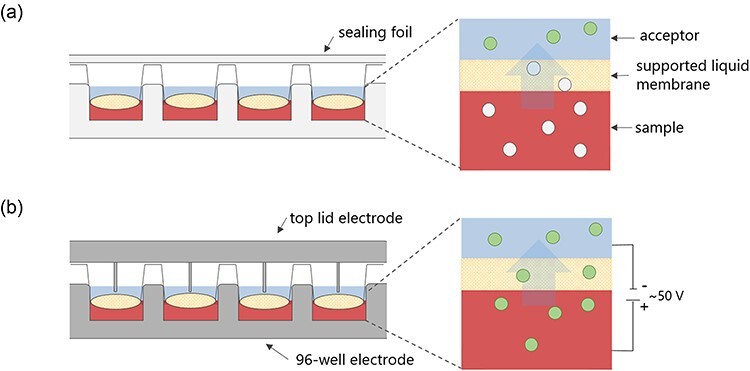
(a) LPME in a 96-well format. Extraction is facilitated by a pH gradient. The uncharged analytes (gray circles in the sample and supported liquid membrane) in an aqueous sample are extracted across an organic liquid membrane and into an aqueous acceptor where the analyte is ionized (green circles in the acceptor). (b) EME in a 96-well format. Extraction of the charged analyte is facilitated by an electric field.
Forensic applications utilizing LPME and EME in a 96-well format have not been extensively explored, but show potential based on existing publications. Vårdal et al. developed a validated method using 96-well LPME for the determination of new psychoactive substances and benzodiazepines in plasma and whole blood (29, 30). In previous work, a method for the determination of tryptamine analogs from whole blood using a 96-well EME was also developed (25).
In the present study, we demonstrate how a 96-well LPME can be employed as a green approach for the simultaneous determination of nine nitazene analogs from whole blood. Both EME and LPME were evaluated for the present application. With the reoccurring incidents of nitazene overdoses, we show how this novel extraction technique is utilized for a highly relevant forensic application.
Experimental procedures
Chemicals and solvents
Metodesnitazene (≥98%), isotonitazene (≥98%), protonitazene (≥98%) and etonitazene-d5 (≥99%) were purchased from Cayman Chemicals (Ann Arbor, Michigan, USA). Metonitazene (≥99%), etonitazepyne (≥99%), etonitazene (≥99%), flunitazene (≥99%), etodesnitazene (≥99%) and clonitazene (≥99%) were purchased from Chiron (Trondheim, Norway). Methanol (Chromasolv LC–MS Ultra) was purchased from Honeywell Research Chemicals (Morris Plains, NJ, USA). Dodecyl acetate (DDA), ammonium formate, 2-nitrophenyl octyl ether (NPOE) and trioctylamine (TOA) were purchased from Sigma-Aldrich (Steinheim, Germany). Dimethyl sulfoxide (DMSO) was purchased from Merck (Darmstadt, Germany). Sodium hydroxide (HPLC grade) and formic acid (FA) (LC–MS grade) were purchased from VWR (Radnor, PA, USA). Methanol (MeOH, Chromasolv™ ≥ 99.9%) was obtained from Honeywell/Riedel-de Haën™ (Seelze, Germany). Water was obtained from a Milli-Q® purification system, Merck Millipore.
Solutions
Human whole blood with sodium fluoride and heparin as additives was supplied by the Blood Bank of Oslo (Oslo University Hospital) and stored in brown glass bottles at −20°C before use. Standard solutions of nitazenes were prepared in 20 mM FA in MeOH:H2O (1:1, v/v) and stored at −20°C. Working solutions of nitazenes were prepared in 50 mM FA and stored at 4°C. A 50 nM nitazene mix was used during optimization experiments. Calibrator and quality control (QC) samples were freshly prepared each day by spiking analyte-free whole blood with respective working solutions (%, v/v) (Supplementary material Table S2).
Liquid-phase microextraction procedure
The equipment has been described previously (31). Extraction was performed with a 96-well polypropylene donor plate with 0.5-mL wells from Agilent (Santa Clara, CA, USA). The acceptor plate was a 96-well MultiScreen-IP filter plate from Merck Millipore (Carrigtwohill, Ireland). The membrane material was polyvinylidene fluoride (PVDF) with a pore size of 0.45 μm. A Platemax Pierceable Aluminum Sealing Film (Axygen, Union City, CA, USA) was used to seal the acceptor plate. Agitation during extraction was accomplished with a Vibramax 100 agitation system from Heidolph (Kellheim, Germany).
Extraction was performed by pipetting 120 µL of analyte-free whole blood, 10 µL of working solution and 120 µL of FA solution spiked with internal standard (IS) into the wells of the donor plate. The filter on the acceptor plate was impregnated with 4 µL of organic solvent, creating the supported liquid membrane. The acceptor and donor plates were clamped together, and the acceptor solution (50 µL) was pipetted into the wells of the acceptor plate before sealing the acceptor plate with aluminum-sealing film. The extraction unit was placed onto the agitation device for extraction for 45 min at 800 rpm. A total of 40 µL of the acceptor solution was transferred to glass autosampler vials. Recovery standards were prepared by spiking 10 µL of working solution to 40 µL of extracted blank.
EME procedure
EME was performed in a laboratory-built 96-well format as previously described (32, 33). Extraction was performed with a laboratory-built donor plate made from steel with 96 wells, serving as the anode. The acceptor plate was a 96-well MultiScreen-IP filter plate from Merck Millipore (Carrigtwohill, Ireland). A laboratory-built steel lid with 96 electrode rods customized for the MultiScreen plate was used to seal the acceptor plate and served as the cathode. Agitation during extraction was accomplished with a Vibramax 100 agitation system from Heidolph (Kellheim, Germany).
The assembly of the extraction device was performed similar to LPME. After the donor and acceptor plates were loaded, the lid was placed on top. The donor plate (anode) and the top lid (cathode) were connected to a power supply (model ES 0300-0.45, Delta Elektronika BV, Zierikzee, the Netherlands) and placed onto the agitation device for the desired time at 700 rpm. The extraction current was recorded using a Fluke287 multimeter at a rate of 8 Hz (Fluke, Everett, WA, USA).
UHPLC–MS-MS
Optimization and validation were performed on separate ultra high performance liquid chromatography–tandem mass spectrometry (UHPLC–MS-MS) platforms. The systems were identical, apart from one system having an injection loop and the other system using flow-through needle injection. Analysis was performed with a UHPLC–MS-MS system comprising an Acquity UPLC system coupled to a Xevo-TQS triple quadrupole by an electrospray ionization interface (Waters, Milford, MA, USA).
During method development, separation was investigated for an Acquity BEH C18 column (2.1 mm I.D. × 100 mm, 1.7 µm) and an Acquity BEH C18 column (2.1 mm I.D. × 50 mm, 1.7 µm) from Waters, an Acquity HSS T3 (2.1 mm I.D. × 100 mm, 1.8 µm), from Waters and a Kinetex biphenyl (2.1 × 100 mm, 1.7 µm) from Phenomenex with a mobile phase consisting of 10 mM ammonium formate pH 3.1 (Solvent A) and MeOH (Solvent B). For the final method, the Kinetex biphenyl column was employed with a flow rate of 0.5 mL/min and separation was performed at 60oC using a 5.5-minute gradient: 0–0.3 min; 10–50% B, 0.3–2 min; 50% B, 2–3.9 min; 50–70% B, 3.9–3.91 min; 50–100% B, 3.91–5 min; 100% B, 5–5.01 min; 100–10% B, 5.01–5.5 min; 10% B. The injection volume was 4 µL. The injection technique was partial loop with overfill and flow-through needle for optimization and validation, respectively.
MS-MS analysis was performed by multiple reaction monitoring (MRM) with positive electrospray ionization. MS-MS parameters were optimized using the built-in source optimization tool by flow injection of 500 nM nitazene mix in MeOH. A capillary voltage of 0.90 kV, an ion source temperature of 150°C, a cone gas flow of 150 L/h and desolvation gas flow of 1000 L/h at 500oC were used. Data acquisition and processing were performed using MassLynx 4.2 software from Waters.
Method validation
The method was validated based on the Scientific Working Group for Forensic Toxicology guidelines (34). Linearity, limit of detection (LOD), limit of quantification (LOQ), inter-day precision and bias, intra-day precision, extraction recovery, matrix effects (ME), autosampler stability, freeze–thaw stability and dilution integrity were studied.
Calibration model
The calibration model was assessed from five assays of six or seven calibrators with one replicate per concentration level with weighted (1/x2) quadratic calibration curves for all nitazene analogs, except 1/x weighting for protonitazene and etonitazene. All data points from the five assays were plotted together and were evaluated using the curve estimation function in IBM SPSS Statistics for Windows version 23.0.
LOD and quantification
LOD was assessed from two replicates for three assays of scalar dilutions (1:4, 1:9, 1:24 and 1:49) of the working solution for the second calibrator (0.5 nM) before being spiked to blood and defined as the lowest concentration with a signal-to-noise ratio (S/N) above three. LOQ was set as the lowest calibrator where inter-day precision and accuracy were ≤±20%.
Precision and accuracy
Precision was determined as the coefficient of variation (%CV). Inter-day precision and accuracy were determined from five assays for four QC sample concentration levels with three replicates per assay. Accuracy was reported as bias (%) calculated as the difference between the true value and the average of all observations at each QC level, and intra-day precision was selected from the assay with the lowest precision. Precision and accuracy were considered acceptable at %CV ≤20% and at bias ≤±20%, respectively.
Recovery and ME
The extraction recovery was calculated for the QC2 and QC4 level with four replicates per level. Recovery was reported as the percent analyte response from pre-spiked extracts relative to the response from post-spiked extracts. Response relates to the analyte area relative to the IS area. The internal standard was spiked post-extraction in both cases. Extraction recoveries above 40% were considered acceptable.
ME was assessed for the QC2 and QC4 level in blood samples from four donors. ME was calculated as the percent analyte peak area from post-spiked extracts relative to the peak area in spiked 50% MeOH in 50 mM FA. IS-corrected ME was calculated from analyte response with IS-spiked post-extraction. MEs between 80% and 120% were considered acceptable.
Autosampler and freeze–thaw stability
Autosampler stability was evaluated by reinjecting both calibrators (one replicate) and QC samples (three replicates) after 1 and 3 days in the autosampler. Autosampler stability was expressed as bias (%) from the nominal QC value. Bias ≤±20% was considered acceptable.
Freeze–thaw stability was evaluated by thawing and refreezing spiked samples at the QC2 and QC3 level with three replicates. The samples were extracted with the fresh calibrators, and stability was reported as bias (%) from the nominal value. Bias ≤±20% was considered acceptable.
Dilution integrity
Dilution was assessed by diluting spiked whole blood (90 nM) 1:1, 1:4 and 1:9 with drug-free whole blood with three replicates for five assays. Precision and accuracy were considered acceptable at a %CV of ≤20% and a bias of ≤±20%, respectively.
Greenness score
Greenness was evaluated based on guidelines proposed by Wojnowski et al. (35). The assessment was based on 10 categories where a score from 0 to 1 was obtained for each category. The sub-scores are weighted based on their importance and recalculated into a final assessment score using open access software obtained from (36). The higher scores represent greener methods.
Results and discussion
Optimization of MRM parameters
The integrated IntelliStart module was used to determine MS tune settings and optimized MRM transitions. The former included ionization mode, capillary voltage, desolvation temperature and desolvation gas flow, for which optimized values are listed in Table I.
Table I.
Optimized MS Tune Settings from IntelliStart
| Ionization mode | Positive electrospray (ES)+ |
|---|---|
| Capillary voltage | 0.90 kV |
| Desolvation temperature | 500℃ |
| Desolvation gas flow | 1000 L/h |
| Cone gas flow | 150 L/h |
The optimized mass spectrometric parameters using Intellistart, including precursor ion, product ion, cone voltage and collision energies, are listed in Table II. The autodwell function in Masslynx was used to generate dwell times.
Table II.
Optimized Mass Spectrometric Parameters and Analyte Retention Time
| Analyte | Retention time (min) | Precursor ion (m/z) | Product ions (m/z)a | Cone (V) | Collision (eV) |
|---|---|---|---|---|---|
| Metodesnitazene | 1.28 | 338.5 | 100.2, 72.2 | 42 | 16, 22 |
| Flunitazene | 1.38 | 371.2 | 100.0, 72.1 | 28 | 22, 30 |
| Metonitazene | 1.49 | 383.2 | 100.1, 72.1 | 24 | 22, 36 |
| Etodesnitazene | 1.56 | 352.3 | 100.1, 72.2 | 42 | 16, 36 |
| Etonitazepyne | 1.65 | 395.5 | 98.1, 107.1 | 82 | 22, 50 |
| Clonitazene | 1.81 | 387.5 | 100.1, 72.1 | 52 | 24, 26 |
| Etonitazene | 1.86 | 397.2 | 72.1, 100.0 | 28 | 38, 22 |
| Isotonitazene | 2.32 | 411.2 | 100.0, 72.1 | 28 | 22, 38 |
| Protonitazene | 2.68 | 411.2 | 100.2, 72.1 | 56 | 42, 22 |
aProduct ions: quantifying ion with corresponding collision energy written in bold, qualifying ion without marking.
Optimization of chromatographic separation
Compound separation was investigated with four columns, namely, Acquity HSS T3 (2.1 × 100 mm, 1.8 µm; Waters), Acquity BEH C18 (2.1 × 100 and 50 mm, 1.7 µm; Waters) and Kinetex Biphenyl (2.1 × 100 mm, 1.7 µm; Phenomenex), and temperatures were tested at 30oC, 45oC and 60oC. With the biphenyl column, additional selectivity was observed and baseline separation of the structural isomers isotonitazene and protonitazene was achieved. Analyte stability was not affected by higher temperatures, and hence, 60oC was selected for separation. The final gradient is found in the UHPLC–MS-MS section. Figure 3 shows the chromatogram from the separation of blood extract at 2 nM.
Figure 3.
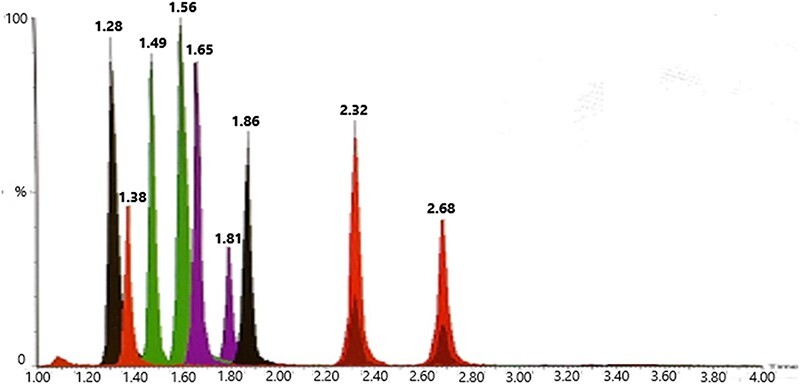
Total ion current chromatogram of whole-blood extract for the calibrator at 2 nM. Peaks are identified as metodesnitazene (1.28), flunitazene (1.38), metonitazene (1.49), etodesnitazene (1.56), etonitazepyne (1.65), clonitazene (1.81), etonitazene (1.86), isotonitazene (2.32) and protonitazene (2.68).
Method development for liquid-phase microextraction
EME and LPME were evaluated for one-step extraction of nitazenes in whole blood, as these techniques are well suited for small sample volumes, offer high-throughput, and are environment-friendly due to low solvent consumption. At this point in method development, only four nitazene analogs were available, namely, metonitazene, flunitazene, etonitazene and isotonitazene. Initial EME and LPME extraction conditions were adapted from the study by Vårdal et al. (29, 30) and previous work (25), respectively, with some modifications as described later.
EME was optimized by altering the acceptor composition, liquid membrane and extraction time. LPME optimization was performed by adding 1.0%, 2.5% and 5.0% TOA to DDA (v/v). This has been shown to be effective to saturate nonspecific binding sites in the PVDF support material used in the currently available 96-well format (30). It should be noted that the use of TOA in EME is not feasible due to the charge interaction of TOA in the electrical field (results not included). Modification of the acceptor solution with 50% DMSO was also tested, as this has been reported to improve analyte partition into the acceptor (30). The average extraction recoveries for the optimization experiments are listed in Table III.
Table III.
Average Extraction Recoveries for Extraction of Nitazenes from Whole Blood with EME and LPME during Method Development, N = 4
| Condition | Technique | Extraction time (min) | Donor composition | Liquid membrane | Acceptor composition | Recovery (%) | %CV |
|---|---|---|---|---|---|---|---|
| 1 | EME | 30 | 50% blood in 100 mM FA | NPOE | 25 mM FA | 56 | <28 |
| 2 | EME | 30 | 50% blood in 100 mM FA | NPOE | 50 mM FA | 61 | <8 |
| 3 | EME | 30 | 50% blood in 100 mM FA | NPOE | 50% DMSO in 100 mM FA | 64 | <22 |
| 4 | EME | 15 | 50% blood in 100 mM FA | NPOE | 50 mM FA | 58 | <58 |
| 1 | LPME | 45 | 50% blood in 100 mM NaOH | DDA | 50 mM FA | 30 | <33 |
| 2 | LPME | 45 | 50% blood in 100 mM NaOH | DDA | 50% DMSO in 100 mM FA | 10 | <33 |
| 3 | LPME | 45 | 50% blood in 100 mM NaOH | 1% TOA in DDA | 50 mM FA | 80 | <13 |
| 4 | LPME | 45 | 50% blood in 100 mM NaOH | 2.5% TOA in DDA | 50 mM FA | 81 | <28 |
| 5 | LPME | 45 | 50% blood in 100 mM NaOH | 5% TOA in DDA | 50 mM FA | 67 | <19 |
Percentages given denotes %v/v.
For EME, the highest average extraction recovery was achieved for Condition 2 at 61% and Condition 3 at 64%, but with Condition 2 being more favorable due to improved robustness (CV < 8%) compared to Condition 3 (CV < 22%). Compared to the previous literature, these relatively low recoveries are unexpected. However, due to the complexity of the sample matrix, such recoveries are not unusual (25, 37). For the LPME extractions, the addition of TOA markedly increased extraction recoveries, with optimal conditions at 1% TOA in DDA, due to favorable robustness with a CV of <13% and a high average extraction recovery at 80%. Based on these results, LPME was found to be more suitable for the extraction of nitazenes from whole blood and is also a more simple extraction system. Although EME is usually the preferred technique due to increased mass transfer efficiency from the electric field, the addition of TOA in LPME outperformed this advantage.
Method validation
Method validation was based on the assessment of linearity, LOD, LOQ, intra-day precision, inter-day precision and bias, extraction recovery, ME, autosampler stability, freeze–thaw stability and dilution integrity.
Table IV includes the calibration model, LOQ and LOD. The calibration range was 0.5–50 nM, except for clonitazene and isotonitazene ranging from 0.1 to 50 nM. The calibration model was quadratic for all nitazenes with weighting 1/x2, except for etodesnitazene and protonitazene with weighting 1/x. LOQ was set as the lowest calibrator, while LOD ranged from 0.01 to 0.1 nM. The correlation coefficient was >0.993 for all analogs, and the regression model was significant by Analysis of Variance (ANOVA).
Table IV.
Calibration Model, LOQ and LOD
| Analyte | Calibration range (nM) | Model | Weighting | LOQ (nM) | LOD (nM) | Correlation (R2) | Origin |
|---|---|---|---|---|---|---|---|
| Metodesnitazene | 0.5–50 | Quadratic | 1/x2 | 0.5 | 0.1 | 0.998 | Excluded |
| Flunitazene | 0.5–50 | Quadratic | 1/x2 | 0.5 | 0.1 | 0.999 | Excluded |
| Metonitazene | 0.5–50 | Quadratic | 1/x2 | 0.5 | 0.05 | 0.998 | Excluded |
| Etodesnitazene | 0.5–50 | Quadratic | 1/x | 0.5 | 0.01 | 0.997 | Excluded |
| Etonitazepyne | 0.5–50 | Quadratic | 1/x2 | 0.5 | 0.01 | 0.999 | Excluded |
| Clonitazene | 0.1–50 | Quadratic | 1/x2 | 0.1 | 0.02 | 0.999 | Excluded |
| Etonitazene | 0.5–50 | Quadratic | 1/x2 | 0.5 | 0.02 | 0.999 | Excluded |
| Isotonitazene | 0.1–50 | Quadratic | 1/x2 | 0.1 | 0.02 | 0.998 | Excluded |
| Protonitazene | 0.5–50 | Quadratic | 1/x | 0.5 | 0.1 | 0.999 | Excluded |
The validation results are presented in Table V. All nitazenes fell within the validation requirements for intra-day precision, inter-day precision and accuracy at all QC levels. The recovery was above 81% with an acceptable CV of ≤16%. ME and IS-corrected ME fell outside requirements for metodesnitazene, etodesnitazene, etonitazepyne, isotonitazene and protonitazene with ion enhancement up to 233%. The observed effects do not, however, impact the assay performance due to matrix-matched calibrators. Additional experiments were conducted to investigate the source of the ME, and it was found that the ME was present independent of the biological matrix, that is, comparing the signal intensity of extracts from spiked blood and spiked buffer (ME < 115%, results not included). Ion enhancement for nitazenes with LC–ESI-MS analysis has also been observed in the literature (15). Freeze–thaw stability was within requirements for all analytes except etodesnitazene (bias 25%) and protonitazene (bias 22%). Repeated freezing of samples with these analytes should be avoided. The nitazenes were generally stable after 1 and 3 days in the autosampler, with some exceptions for metodesnitazene and etodesnitazene.
Table V.
Intra-Day Precision, Inter-Day Precision, Accuracy, Recovery, ME, IS-Corrected ME, Freeze–Thaw Stability and Autosampler Stability
| Analyte, cons. (nM) | Intra-day precision (%CV) | Inter-day precision (%CV) | Accuracy (%bias) | Recovery (%CV) | ME (%CV) | IS-corrected ME (%CV) | Freeze–thaw stability (%bias) | Autosampler stability (%bias) | |
|---|---|---|---|---|---|---|---|---|---|
| 1 day | 3 days | ||||||||
| Metodesnitazene | |||||||||
| 40 | 13 | 9 | 0 | 4 | 22 | ||||
| 8 | 1 | 9 | −1 | 95 (4) | 135 (2) | 151 (10) | 16 | −8 | 0 |
| 0.8 | 10 | 7 | 4 | 91 (5) | 211 (17) | 179 (10) | −2 | 3 | 2 |
| Flunitazene | |||||||||
| 40 | 9 | 5 | −3 | −1 | 4 | ||||
| 8 | 2 | 7 | −1 | 81 (9) | 93 (6) | 101 (5) | 18 | −2 | −2 |
| 0.8 | 5 | 5 | 4 | 86 (8) | 103 (3) | 89 (10) | 5 | −0 | 5 |
| Metonitazene | |||||||||
| 40 | 13 | 10 | 0 | 3 | 19 | ||||
| 8 | 4 | 7 | −3 | 94 (4) | 103 (6) | 111 (9) | 12 | −5 | −2 |
| 0.8 | 6 | 5 | 5 | 95 (9) | 115 (3) | 99 (9) | 2 | 2 | 2 |
| Etodesnitazene | |||||||||
| 40 | 13 | 9 | 1 | 4 | 31 | ||||
| 8 | 5 | 8 | −3 | 97 (5) | 163 (1) | 186 (10) | 25 | −21 | −13 |
| 0.8 | 9 | 6 | 5 | 98 (5) | 233 (17) | 197 (8) | −2 | 6 | 10 |
| Etonitazepyne | |||||||||
| 40 | 8 | 5 | 3 | 4 | 13 | ||||
| 8 | 3 | 5 | 0 | 99 (3) | 119 (8) | 131 (3) | 6 | −6 | 1 |
| 0.8 | 7 | 4 | 3 | 102 (3) | 152 (18) | 130 (11) | 0 | 1 | 3 |
| Clonitazene | |||||||||
| 40 | 3 | 3 | 1 | 1 | 4 | ||||
| 8 | 4 | 3 | −1 | 82 (10) | 100 (13) | 109 (109) | 5 | −4 | −1 |
| 0.8 | 2 | 4 | −2 | 87 (7) | 110 (14) | 95 (9) | −2 | −2 | 4 |
| 0.3 | 3 | 6 | −3 | 11 | 13 | ||||
| Etonitazene | |||||||||
| 40 | 2 | 2 | 1 | 0 | 8 | ||||
| 8 | 6 | 6 | −3 | 93 (8) | 106 (12) | 115 (4) | 5 | −7 | −2 |
| 0.8 | 10 | 7 | 6 | 91 (16) | 118 (12) | 102 (16) | −2 | 3 | 3 |
| Isotonitazene | |||||||||
| 40 | 6 | 5 | 2 | 3 | 16 | ||||
| 8 | 10 | 7 | 0 | 93 (3) | 102 (9) | 110 (2) | 8 | −11 | 3 |
| 0.8 | 12 | 6 | 6 | 91 (13) | 125 (19) | 108 (10) | 13 | 5 | 4 |
| Protonitazene | |||||||||
| 40 | 4 | 6 | 5 | 8 | 2 | ||||
| 8 | 15 | 13 | −3 | 81 (14) | 112 (24) | 122 (16) | 6 | −16 | −10 |
| 0.8 | 10 | 11 | 0 | 94 (6) | 137 (28) | 116 (20) | 22 | −7 | −5 |
| 0.3 | 16 | 13 | 0 | −4 | 3 | ||||
Dilution integrity was evaluated at dilution ratios of 1:1, 1:4 and 1:9 for spiked whole blood. Only the dilution ratio of 1:1 was within validation requirements. For higher dilution factors, concentrations were overestimated by up to 73% and 95% for dilution ratios of 1:4 and 1:9, respectively. Intra-day, inter-day precision and accuracy for the 1:1 dilution ratio are listed in Table VI. From a review by Montanari et al. (38) of eight case reports with 93 fatalities involving nitazene analogs, the highest concentration in postmortem peripheral blood was 33 ng/mL (86 nM) for metonitazene. The method presented here, with a calibration range of up to 50 nM and a dilution ratio of 1:1, is therefore considered acceptable.
Table VI.
Dilution Integrity at a Dilution Ratio of 1:1 for Spiked Whole Blood
| Analyte | Intra-day precision (CV) | Inter-day precision (CV) | Accuracy (bias) |
|---|---|---|---|
| Metodesnitazene | 9 | 9 | 14 |
| Flunitazene | 5 | 5 | 8 |
| Metonitazene | 6 | 7 | 12 |
| Etodesnitazene | 8 | 7 | 14 |
| Etonitazepyne | 8 | 5 | 12 |
| Clonitazene | 6 | 4 | 9 |
| Etonitazene | 6 | 5 | 12 |
| Isotonitazene | 6 | 4 | 10 |
| Protonitazene | 5 | 7 | 12 |
Greenness score
The assignment of a greenness score to new methods can provide valuable insight into analytical method development. It can be used as a tool for routine laboratories implementing new methods or for improving already established methods. We believe that green sample preparation will be the future of analytical laboratories. An array of approaches have been proposed, with varying comprehensiveness and focus on different parts of the analytical workflow (39–41). To solely address the sample preparation, AGREEprep, developed by Wojnowski et al. (35), allows for the evaluation of all aspects of the sample preparation procedure. Figure 4 presents the results from the AGREEprep analysis, and details of the data input for each subcategory are provided in the Supplementary material.
Figure 4.
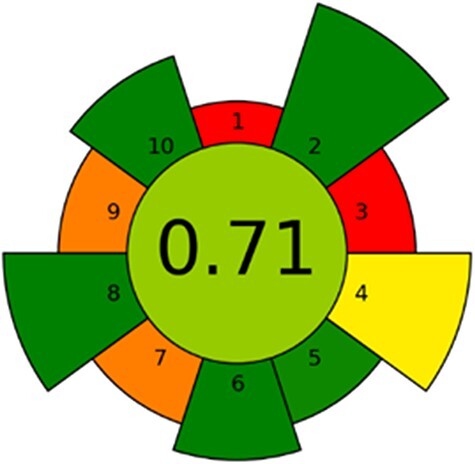
Graphical results from the AGREEprep analysis calculated using the AGREEprep software v.0.9.
The overall score is in the center, while subcriteria (1–10) are located around the inner circle, with the length of each sector representing the weighting of the respective criteria. The overall score of 0.71 is considered very good (35), demonstrating that the 96-well LPME can meet the demands of future analytical laboratories. The lowest scores were received for the sample preparation site and the use of non-reusable equipment. In theory, both these critiques could be addressed; however, on-site sample preparation is not common practice in our laboratory, and reusing plastic equipment is generally not practical in forensic casework.
Conclusion
A validated workflow for the quantification of nine nitazene analogs in whole blood has been developed. Whole blood samples were prepared using 96-well LPME and analyzed using LC–MS-MS. By utilizing a microextraction technique, samples could be prepared with minimal waste and hazardous solvents. Quantification was achieved in the range of 0.5 to 50 nM (except for protonitazene and clonitazene at 0.1 nM), with acceptable linearity, accuracy and precision. The nitazenes included in the assay, namely, clonitazene, etodesnitazene, etonitazene, etonitazepyne, flunitazene, isotonitazene, metodesnitazene, metonitazene and protonitazene, represent not only analogs frequently encountered by authorities but also less known analogs. Therefore, the presented method may be suited for incoming cases of nitazene intoxications.
The presented method aligns with the green analytical chemistry principles and may have the potential to supplement or replace conventional sample preparation techniques. The 96-well LPME is a versatile technique suitable for complex matrixes used in forensic analysis. We hope that the presented results can encourage more researchers to implement greener methodology in their laboratories.
Supplementary Material
Contributor Information
Maria Schüller, Department of Pharmacy, University of Oslo, P.O. Box 1068 Blindern, Oslo 0316, Norway.
Ivana Lucic, Department of Pharmacy, University of Oslo, P.O. Box 1068 Blindern, Oslo 0316, Norway.
Åse Marit Leere Øiestad, Department of Forensic Sciences, Division of Laboratory Medicine, Oslo University Hospital, P.O. Box 4459 Nydalen, Oslo 0424, Norway.
Stig Pedersen-Bjergaard, Department of Pharmacy, University of Oslo, P.O. Box 1068 Blindern, Oslo 0316, Norway; Department of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, Copenhagen 2100, Denmark.
Elisabeth Leere Øiestad, Department of Pharmacy, University of Oslo, P.O. Box 1068 Blindern, Oslo 0316, Norway; Department of Forensic Sciences, Division of Laboratory Medicine, Oslo University Hospital, P.O. Box 4459 Nydalen, Oslo 0424, Norway.
Supplementary data
Supplementary data is available at Journal of Analytical Toxicology online.
Data availability
Data are available in the article and Supplementary Material. Additional data can be provided by the authors upon request.
Funding
No external funding was received for this work.
Author contributions
Maria Schüller (Conceptualization, Methodology, Formal analysis, Investigation, Writing—original draft, Writing—review, and editing, Visualization), Ivana Lucic (Methodology, Formal analysis, Investigation), Åse Marit Leere Øiestad (Conceptualization, Methodology, Formal analysis, Writing—original draft, Writing—review, and editing, Supervision), Stig Pedersen-Bjergaard (Conceptualization, Methodology, Formal analysis, Writing—original draft, Writing—review, and editing), and Elisabeth Leere Øiestad (Conceptualization, Methodology, Formal analysis, Writing—original draft, Writing—review and editing, Supervision).
References
- 1. European Monitoring Centre for Drugs and Drug Addiction . (2023) EU early warning system advisory: overview of the situation with new opioids. Europe, 2021-ongoing.
- 2. Papsun D.M., Krotulski A.J., Logan B.K. (2022) Proliferation of novel synthetic opioids in postmortem investigations after core-structure scheduling for fentanyl-related substances. The American Journal of Forensic Medicine and Pathology, 43, 315–327. 10.1097/PAF.0000000000000787 [DOI] [PubMed] [Google Scholar]
- 3. Rosen L.W., Lawrence S.V., Library of Congress. Congressional Research, S . (2019) China primer: illicit fentanyl and China’s role. IF; 10890, 1 online resource.
- 4. Vandeputte M.M., Krotulski A.J., Walther D., Glatfelter G.C., Papsun D., Walton S.E., et al. (2022) Pharmacological evaluation and forensic case series of N-pyrrolidino etonitazene (etonitazepyne), a newly emerging 2-benzylbenzimidazole ‘nitazene’ synthetic opioid. Archives of Toxicology, 96, 1845–1863. 10.1007/s00204-022-03276-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hunger A., Kebrle J., Rossi A., Hoffmann K. (1960) Benzimidazol-derivate und verwandte hetezrocyclen III. Synthese von 1-Aminoalkyl-2-nenzyl-nitro-benzimidazolen. Helvetica Chimica Acta, 43, 1032–1046. 10.1002/hlca.19600430412 [DOI] [Google Scholar]
- 6. Ujváry I., Christie R., Evans-Brown M., Gallegos A., Jorge R., de Morais J., et al. (2021) DARK classics in chemical neuroscience: etonitazene and related benzimidazoles. ACS Chemical Neuroscience, 12, 1072–1092. 10.1021/acschemneuro.1c00037 [DOI] [PubMed] [Google Scholar]
- 7. Gross F., Turrian H. (1957) Über Benzimidazolderivate mit starker analgetischer Wirkung. Experientia, 13, 401–403. 10.1007/BF02161117 [DOI] [PubMed] [Google Scholar]
- 8. Drug Enforcement Administration; Diversion Control Division; Drug & Chemical Evaluation Section . (2022) Isotonitazene. https://www.deadiversion.usdoj.gov/drug_chem_info/isotonitazene.pdf (accessed Mar 15, 2023).
- 9. TV 2 Nyheter . Frykter stort lager av nytt livsfarlig dop: – Skremmende. https://www.tv2.no/nyheter/innenriks/frykter-stort-lager-av-nytt-livsfarlig-dop-skremmende/15298898/ (accessed Mar 15, 2023).
- 10. NRK . Politiet slår alarm om nytt dødsdop etter dødsfall på Sunnmøre. https://www.nrk.no/mr/politiet-fryktar-fleire-overdosar-etter-funn-av-dodsdopet-etonitazepyn-pa-sunnmore-1.16006580 (accessed March 15, 2023).
- 11. Aftenposten . Ren slump at politiet oppdaget dødsdop på vei inn i Norge. https://www.aftenposten.no/norge/i/2BvGEa/ren-slump-at-politiet-oppdaget-doedsdop-paa-vei-inn-i-norge (accessed Mar 15, 2023).
- 12. United States Drug Enforcement Administration . Alphabetical listing of Controlled Substances. https://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf (accessed Mar 15, 2023).
- 13. Advisory Council on the Misuse of Drugs . ACMD Report – a review of the evidence on the use and harms of 2-benzyl benzimidazole (‘nitazene’) and piperidine benzimidazolone (‘brorphine-like’) opioids. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1091230/Cover_letter_from_ACMD_on_2-benzyl_benzimidazole_and_piperidine_benzimidazolone_opioids.pdf (accessed Jul 18, 2022).
- 14. GOV.UK . Synthetic opioids will be banned as government acts to stop drug deaths. https://www.gov.uk/government/news/synthetic-opioids-will-be-banned-as-government-acts-to-stop-drug-deaths (accessed Mar 15, 2023).
- 15. Walton S.E., Krotulski A.J., Logan B.K. (2022) A forward-thinking approach to addressing the new synthetic opioid 2-benzylbenzimidazole nitazene analogs by Liquid Chromatography–Tandem Quadrupole Mass Spectrometry (LC–QQQ-MS). Journal of Analytical Toxicology, 46, 221–231. 10.1093/jat/bkab117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krotulski A.J., Papsun D.M., Kacinko S.L., Logan B.K. (2020) Isotonitazene quantitation and metabolite discovery in authentic forensic casework. Journal of Analytical Toxicology, 44, 521–530. 10.1093/jat/bkaa016 [DOI] [PubMed] [Google Scholar]
- 17. Armenta S., Garrigues S., de la Guardia M. (2015) The role of green extraction techniques in green analytical chemistry. TrAC Trends in Analytical Chemistry, 71, 2–8. 10.1016/j.trac.2014.12.011 [DOI] [Google Scholar]
- 18. Pego A.M.F., Roveri F.L., Kuninari R.Y., Leyton V., Miziara I.D., Yonamine M. (2017) Determination of cocaine and its derivatives in hair samples by liquid phase microextraction (LPME) and gas chromatography–mass spectrometry (GC–MS). Forensic Science International, 274, 83–90. 10.1016/j.forsciint.2016.12.024 [DOI] [PubMed] [Google Scholar]
- 19. Sharifi V., Abbasi A., Nosrati A. (2016) Application of hollow fiber liquid phase microextraction and dispersive liquid–liquid microextraction techniques in analytical toxicology. Journal of Food and Drug Analysis, 24, 264–276. 10.1016/j.jfda.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manousi N., Samanidou V. (2021) Green sample preparation of alternative biosamples in forensic toxicology. Sustainable Chemistry and Pharmacy, 20, 100388. 10.1016/j.scp.2021.100388 [DOI] [Google Scholar]
- 21. Ares A.M., Fernández P., Regenjo M., Fernández A.M., Carro A.M., Lorenzo R.A. (2017) A fast bioanalytical method based on microextraction by packed sorbent and UPLC–MS/MS for determining new psychoactive substances in oral fluid. Talanta, 174, 454–461. 10.1016/j.talanta.2017.06.022 [DOI] [PubMed] [Google Scholar]
- 22. Liu H., Dasgupta P.K. (1996) Analytical chemistry in a drop. Solvent extraction in a microdrop. Analytical Chemistry, 68, 1817–1821. 10.1021/ac960145h [DOI] [PubMed] [Google Scholar]
- 23. Rezaee M., Assadi Y., Milani Hosseini M.-R., Aghaee E., Ahmadi F., Berijani S. (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. Journal of Chromatography A, 1116, 1–9. 10.1016/j.chroma.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 24. Pedersen-Bjergaard S., Rasmussen K.E. (1999) Liquid-liquid-liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Analytical Chemistry, 71, 2650–2656. 10.1021/ac990055n [DOI] [PubMed] [Google Scholar]
- 25. Schüller M., McQuade T.A.-P., Bergh M.-S.-S., Pedersen-Bjergaard S., Øiestad E.L. (2023) Determination of tryptamine analogs in whole blood by 96-well electromembrane extraction and UHPLC-MS/MS. Talanta Open, 7, 100171. 10.1016/j.talo.2022.100171 [DOI] [Google Scholar]
- 26. Gjelstad A., Rasmussen K., Parmer M., Pedersen-Bjergaard S. (2013) Parallel artificial liquid membrane extraction: micro-scale liquid-liquid-liquid extraction in the 96-well format. Bioanalysis, 5, 1377–1385. 10.4155/bio.13.59 [DOI] [PubMed] [Google Scholar]
- 27. Skaalvik T.G., Øiestad E.L., Trones R., Pedersen-Bjergaard S., Hegstad S. (2021) Determination of psychoactive drugs in serum using conductive vial electromembrane extraction combined with UHPLC-MS/MS. Journal of Chromatography B, 1183, 122926. 10.1016/j.jchromb.2021.122926 [DOI] [PubMed] [Google Scholar]
- 28. Hay A.O., Trones R., Herfindal L., Skrede S., Hansen F.A. (2022) Determination of methotrexate and its metabolites in human plasma by electromembrane extraction in conductive vials followed by LC-MS/MS. Advances in Sample Preparation, 2, 100011. 10.1016/j.sampre.2022.100011 [DOI] [Google Scholar]
- 29. Vårdal L., Askildsen H.-M., Gjelstad A., Øiestad E.L., Edvardsen H.M.E., Pedersen-Bjergaard S. (2017) Parallel artificial liquid membrane extraction of new psychoactive substances in plasma and whole blood. Journal of Chromatography B, 1048, 77–84. 10.1016/j.jchromb.2017.02.010 [DOI] [PubMed] [Google Scholar]
- 30. Vårdal L., Wong G., Øiestad Å.M.L., Pedersen-Bjergaard S., Gjelstad A., Øiestad E.L. (2018) Rapid determination of designer benzodiazepines, benzodiazepines, and Z-hypnotics in whole blood using parallel artificial liquid membrane extraction and UHPLC-MS/MS. Analytical and Bioanalytical Chemistry, 410, 4967–4978. 10.1007/s00216-018-1147-y [DOI] [PubMed] [Google Scholar]
- 31. Ask K.S., Lid M., Øiestad E.L., Pedersen-Bjergaard S., Gjelstad A. (2019) Liquid-phase microextraction in 96-well plates - calibration and accurate quantification of pharmaceuticals in human plasma samples. Journal of Chromatography A, 1602, 117–123. 10.1016/j.chroma.2019.06.013 [DOI] [PubMed] [Google Scholar]
- 32. Skottvoll F.S., Hansen F.A., Harrison S., Boger I.S., Mrsa A., Restan M.S., et al. (2021) Electromembrane extraction and mass spectrometry for liver organoid drug metabolism studies. Analytical Chemistry, 93, 3576–3585. 10.1021/acs.analchem.0c05082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Restan M.S., Pedersen M.E., Jensen H., Pedersen-Bjergaard S. (2019) Electromembrane extraction of unconjugated fluorescein isothiocyanate from solutions of labeled proteins prior to flow induced dispersion analysis. Analytical Chemistry, 91, 6702–6708. 10.1021/acs.analchem.9b00730 [DOI] [PubMed] [Google Scholar]
- 34.Scientific Working Group for Forensic Toxicology. (2013) Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. Journal of Analytical Toxicology, 37, 452–474. 10.1093/jat/bkt054 [DOI] [PubMed] [Google Scholar]
- 35. Wojnowski W., Tobiszewski M., Pena-Pereira F., Psillakis E. (2022) AGREEprep – analytical greenness metric for sample preparation. TrAC Trends in Analytical Chemistry, 149, 116553. 10.1016/j.trac.2022.116553 [DOI] [Google Scholar]
- 36. Wojnowski I.W. (2022) Analytical greenness metric for sample preparation. https://mostwiedzy.pl/pl/wojciech-wojnowski,174235-1/agreeprep (accessed Oct 16, 2022).
- 37. Drouin N., Mandscheff J.-F., Rudaz S., Schappler J. (2017) Development of a new extraction device based on parallel-electromembrane extraction. Analytical Chemistry, 89, 6346–6350. 10.1021/acs.analchem.7b01284 [DOI] [PubMed] [Google Scholar]
- 38. Montanari E., Madeo G., Pichini S., Busardò F.P., Carlier J. (2022) Acute intoxications and fatalities associated with benzimidazole opioid (Nitazene Analog) use: a systematic review. Therapeutic Drug Monitoring, 44, 494–510. 10.1097/FTD.0000000000000970 [DOI] [PubMed] [Google Scholar]
- 39. Gałuszka A., Migaszewski Z.M., Konieczka P., Namieśnik J. (2012) Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends in Analytical Chemistry, 37, 61–72. 10.1016/j.trac.2012.03.013 [DOI] [Google Scholar]
- 40. Płotka-Wasylka J. (2018) A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta, 181, 204–209. 10.1016/j.talanta.2018.01.013 [DOI] [PubMed] [Google Scholar]
- 41. Pena-Pereira F., Wojnowski W., Tobiszewski M. (2020) AGREE—analytical GREEnness metric approach and software. Analytical Chemistry, 92, 10076–10082. 10.1021/acs.analchem.0c01887 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in the article and Supplementary Material. Additional data can be provided by the authors upon request.