

Abstract
The field of protein engineering has seen tremendous expansion in the last decade, with researchers developing novel proteins with specialised functionalities for a range of uses, from drug discovery to industrial biotechnology. The emergence of computational tools and high‐throughput screening technology has substantially sped up the process of protein engineering. However, much of the expertise required to engage in such projects is still concentrated in the hands of a few specialised individuals, including computational biologists and structural biochemists. The international Genetically Engineered Machine (iGEM) competition represents a platform for undergraduate students to innovate in synthetic biology. Yet, due to their complexity, arduous protein engineering projects are hindered by the resources available and strict timelines of the competition. The authors highlight how the 2022 iGEM Team, ‘Sporadicate’, set out to develop InFinity 1.0, a computational framework for increased accessibility to effective protein engineering, hoping to increase awareness and accessibility to novel in silico tools.
Keywords: biochemical engineering, synthetic biology
Protein engineering has seen a large growth over the past decade fuelled by innovation in AI and machine learning tools. However, these often remain inaccessible to the average researcher. The authors present their experience as the ‘Sporadicate iGEM Team’ in the development of an in silico framework to increase awareness and accessibility to novel in silico tools.
1. PROTEIN ENGINEERING: AN OVERVIEW
Protein engineering is a rapidly evolving field in which efforts can be broadly categorised into two classes: the re‐engineering of natural proteins to either fine‐tune (substrate specificity and affinity, catalytic properties, stability) or introduce new functionality; or de novo protein design (referred to as design rather than engineering) where proteins are artificially built from first principles and engineered to perform a desired function [1] (Figure 1).
FIGURE 1.
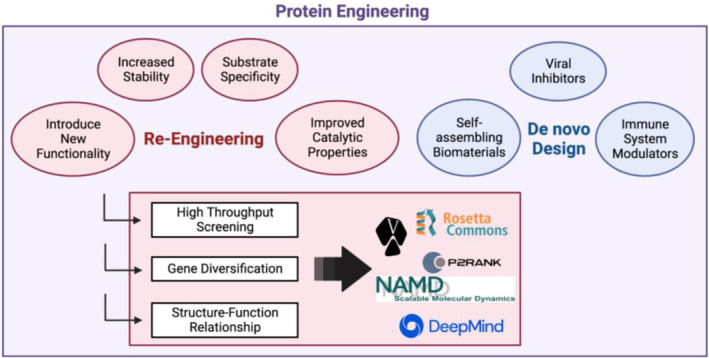
Areas of research in protein engineering.
Within protein re‐engineering, approaches range from the highly rational to solely combinatorial. Historically, researchers have relied on rational engineering approaches to introduce changes in natural protein structures [2]. Albeit requiring prior knowledge of the protein's folding and functional characteristics, these techniques rely on a small library size to screen for the desired behaviour, with many successful examples documented extensively in the literature [3].
When structural and functional information is not easily accessible, directed evolution strategies employ random mutagenesis such as error‐prone PCR (epPCR) to generate a large protein library to screen for functionality. Existing directed evolution techniques, however, are limited in scalability due to the laborious process of in vitro high throughput screening and subsequent selection [4]. Additionally, directed evolution relies on manual staging of each cycle. This limits its ability to explore the functional sequence space deeply and extensively [5].
At the intersection between directed evolution and rational design lie combinatorial approaches, which combine stochastic elements and rational information to generate small yet effective protein pools, able to constrain the protein folding space and optimise the chances of success [6].
1.1. Computational tools for addressing challenges in protein engineering
Two key challenges in protein engineering are: (1) deciphering the sequence‐structure‐function relationship in proteins and (2) effectively navigating the protein design space defined as 20 n , where n is the number of amino acids [7].
Computational tools addressing both these issues have been developed extensively in the last decade, with in silico strategies for predicting protein structure (AlphaFold, RoseTTAFold), their interactions with other ligands (AutoDock Vina, UCSF Dock, P2Rank), as well as dynamic system simulations (molecular dynamics software, such as GROMACS, NAMD, and AMBER) finding regular applications in the cutting edge.
In particular, AI‐based methods have revolutionised the field of rational design through use of the ever increasing amount of data provided by ‐omics projects to investigate rules for engineering functional biomolecules [8]. Machine learning approaches have been applied to a range of processes that span all areas of protein design, including protein secondary structure prediction, fold recognition, contact matrix calculations, genomics, proteomics, and systems biology [8]. The development of deep learning approaches in particular, driven by the increasing availability of computer power and the development of GPU‐based calculations has resulted in the release of several landmark rational design papers within the last decade [9, 10].
Adoption of these tools unlocks the possibility of semi‐rational engineering approaches, characterised by smaller and enriched variant protein libraries. This not only mitigates the limitation of screening numerous variants, a time and resource‐intensive step in wet lab settings, but increases the likelihood of engineering a functional protein.
As highlighted by Walker, Yallapragada and Tangney [11], one of the main limitations concerning protein engineering and its widespread adoption is the lack of intuitive workflows for in silico tools. In recent years, there have been efforts to tackle this issue (Table 1), an in depth discussion of tools considered approachable to non‐specialists can be found in recent reviews [24, 25]. Yet, none of these address the specificity and affinity engineering problem in a manner that is one‐stop and intuitive to non‐specialists, whilst being open‐source. Indeed, to exploit the capabilities of ever‐evolving algorithms, expertise in both computational biology and structural biochemistry is required, resulting in a high barrier to entry. A great case study of the phenomenon is iGEM—(International Genetically Engineered Machine competition) an annual synthetic biology competition, wherein multidisciplinary student teams pursue a project of their own design.
TABLE 1.
Recent computational tools considered approachable to non‐specialist protein engineers.
| Tool | Functionality | Features and limitations | Reference |
|---|---|---|---|
| AlphaFold 2.0 | Computational tool designed for predicting three‐dimensional protein structure using a deep learning system trained on structures in the PDB. |
|
[12] |
| ROSIE | The gold‐standard, a web platform hosting dozens of tools from the Rosetta suite of programs that enable modelling and protein design, including: Molecular docking, prediction and design for protein stability and solubility. |
|
[14] |
| HotSpot Wizard 3.0 | Web application that allows identification of hot spots for mutagenesis. |
|
[15] |
| FuncLib | To redesign an active site and create multiple‐point designs. Based on conservation analysis and energy calculations. |
|
[16] |
| CaverWeb 1.2 | To calculate trajectory and interaction energy profiles of a ligand travelling through a protein tunnel. |
|
[17] |
| DockingApp (Autodock Vina) | Platform‐independent application for setting up, performing and analysing results from AutoDock Vina. |
|
[18] |
| LoopGrafter | For transplanting loops between two structurally related proteins, with a focus on the analysis of dynamic properties of the selected loops to transplant. |
|
[19] |
| Protein Repair One‐Stop‐Shop (PROSS) | Automated web platform aimed at improving protein thermostability and functional yield. |
|
[21] |
| SoluProt | Predicts the solubility of a protein specified by input sequence, in Escherichia coli. |
|
[22] |
| DeepSoluE | Predicts the solubility of a protein specified by input sequence, in Escherichia coli. |
|
[23] |
In 2022, we set out to compete in the iGEM competition with Sporadicate, a novel, broadspectrum, and timely crop biofungicide system [26]. In order to target pathogens over both space and time, we designed a Bacillus subtilis spore‐based system that would trigger the release of our biocontrol agent—vegetative Bacillus subtilis cells—in response to a common biomarker of pathogenic fungi (N‐acetylglucosamine—i.e. chitin monomer). Our sensing system required transduction of our desired input signal into germination, the process by which dormant spores awaken into cells. Natively, protein receptors in the coat of spores known as germinant receptors perform this task, sensing for nutrients such as glucose as well as amino acids [27]. We planned to modify native germinant receptor GerA (Figure 2), a sensing protein detecting L‐alanine [28], with enhanced specificity towards N‐acetylglucosamine.
FIGURE 2.
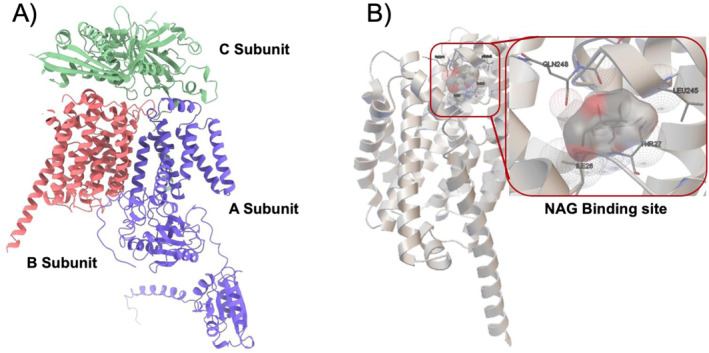
Binding site identification. (a) GerA complex obtained through AlphaFold. The protein consists of three smaller subunits, AA, AB and AC. The B subunit is thought to sense and bind with the germinant L‐alanine, whereas the A subunit is responsible for transducing the signal from B [28] upon activation. The function of the C subunit is unclear, but it is not necessary for germinant receptor activation [29]. (b) NAG Binding site identified in GerAB through Autodock. NAG, n‐acetylglucosamine.
Given the limited available information on the protein structure and how this relates to function—we needed to adopt a combinatorial strategy. However, as GerA comprises 1220 residues, a protein design space of 201220 would have been impossible to explore in practice. Hence, we devised a potential workflow integrating novel computational tools that would enable us to design a smaller, richer protein library. This not only would have maximised our chances of success, but could lay out the foundations of accessible protein engineering research, in iGEM and beyond.
2. DESIGN
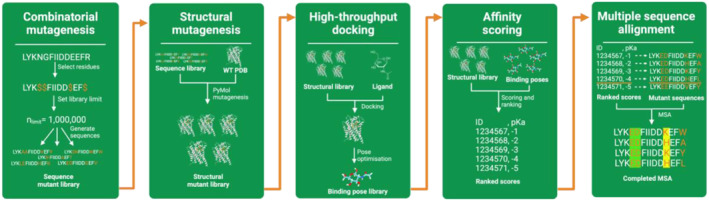
With the advent of computational tools that can accurately predict protein structures and significant progress in the field of molecular docking, we pose the question if said developments could be incorporated into a framework for streamlined protein engineering [12, 30, 31]. We propose InFinity 1.0, an open‐source computational framework, mimicking in vivo techniques (Figure 3). Specifically, this consists of mutant library generation, positive and negative selection finally followed by analysis of commonly occurring motifs. Here, we present how such a framework could be constructed (Figure 3).
FIGURE 3.
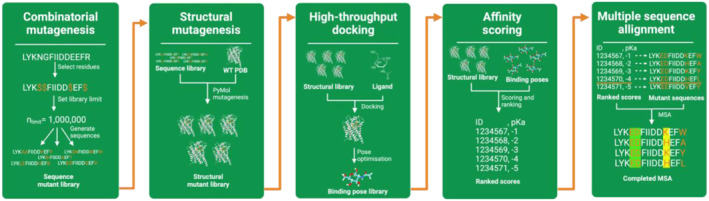
Overview of proposed framework for computational protein engineering. The framework will benefit from advances in structural modelling and molecular docking. Adapting these for use in computational protein engineering could allow for high‐throughput screening of mutants, aiding in design and testing to be carried out in the lab.
First, taking either a predicted (e.g. Alphafold) or experimentally derived protein structure, mutations should combinatorically be introduced within the targeted ligand binding pocket. Sequence‐wise this can be done quickly and efficiently with simple combinatorics of a supplied FASTA sequence. Accurately modelling the mutations structurally is perhaps the greatest challenge, and a compromise has to be met between computational efficiency and accuracy. For example, PyMol's mutagenesis tool can efficiently model changes to side‐chains, avoiding steric clashes but does not account for changes to the scaffold and effect on local and global energetics as molecular dynamic simulations do. Nonetheless, at this step, the desired input is simply an initial protein structure and residue positions targeted for mutagenesis, while the output is a library of mutant structures.
For positive selection, this can be performed with relatively computationally efficient docking and scoring. In recent years, we have seen advances in the use of machine‐learning based docking and scoring functions for computational drug discovery, and some have shown significant improvements in the predictive accuracy of ligand‐binding affinities; for example, DiffDock, GNINA and scoring functions adapted with an XGB model [30, 32, 33]. Taking the mutant library previously derived, the desired ligand could be docked and affinities are calculated using a concoction of the leading docking tools and scoring functions. The final output would then be a ranked list of which mutants' structures showed the greatest binding affinities.
The use of negative selection will be desirable especially in cases where computational efficiency has been prioritised over accuracy in earlier steps. Here, we aim to investigate top scoring mutants from positive selection, specifically screening how mutants could have had detrimental effects on overall protein folding. Missense3D [34] is one example of a computationally efficient way of achieving this while Molecular Dynamics simulations could be used to gain more granularity.
The final step would then be ranking and consensus evaluation of common motifs. For use in iGEM and other protein engineering projects, a selection of the top motifs could then potentially be tested in vivo. The specific benefit of this framework over current implementations is its use of open source tools and modularity, opening the door for the integration of novel tools that could improve the performance of the overall pipeline.
3. INFINITY 1.0 PIPELINE
Throughout the iGEM competition period, we produced an initial proof of concept prototype for the InFinity 1.0 pipeline (Figure 4). Our prototype links several established open‐source tools, which allows inexperienced users to achieve high‐throughput and accurate mutant screening for protein design. Users can input sequence information which is then used to generate a mutant library, this is then sampled for high‐throughput ligand docking which produces binding affinity information. Binding affinities are scored and ranked, and finally multiple sequence alignment is applied to the highest‐ranked mutants to derive information about common motifs which is passed back to the user. Our pipeline along with a user guide can be found on our team's GitHub Page (See Code Availability below).
FIGURE 4.
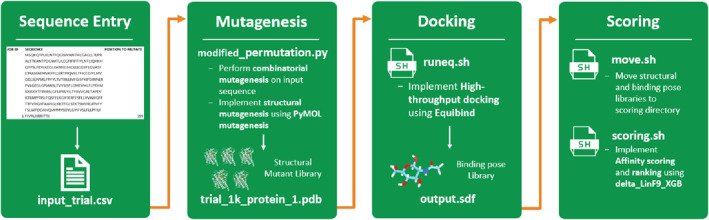
Overview of implemented Infinity 1.0 pipeline. Process begins with a csv file in which users can input the sequence and mutations of their choice, and ends with the corresponding ranked affinity scores with the ligand of choice. For each stage, a script has been created to interface between the user and incorporated tool. Aside from the initial sequence information and computational resource specification, the pipeline requires minimal user input to run.
3.1. Creating a library of mutants
Our approach to combinatorial mutagenesis was highly customisable, allowing the user to select which residues to mutate and wrote a script that generated a random subset of mutants from the associated combinatorial space. To make our software accessible to iGEM users and individuals without a background in structural biology or protein engineering, we added a simple .csv file that can be easily filled and is seamlessly integrated into the software. Users can select their desired sequence and specify which amino acids they want to mutate, as well as the positions of these amino acids.
We used PyMol's mutagenesis tool [35] in our proof of concept to introduce the generated mutant sequences into the wildtype protein structure. The tool substitutes residue sidechains and adjusts using the most optimal rotamer for each, and as such, is best suited for fewer and less structurally significant mutations, such as those required to alter protein specificity or affinity slightly which would be accounted for in a negative selection step.
As our implementation of the pipeline was more of an exploratory proof of concept within the limited iGEM time frame, we did not perform the negative selection step of the design, which would involve modelling the relaxed structure for each mutant and screen for large structural disruptions. We instead relied on the less computationally expensive positive selection and affinity score ranking processes to select our final mutants of interest.
3.2. Positive selection
After obtaining the structural library, high‐throughput rigid docking was performed using an adapted version of EquiBind [36], a recently released deep learning‐based docking tool. EquiBind's use of a SE(3)‐equivariant geometric deep learning model significantly improves the computational efficiency of docking and has higher accuracy of binding poses compared to comparable baselines. Using GPU acceleration, a single GPU can evaluate approximately 200,000 mutants per day. This step can also be parallelised using multiple GPUs by splitting the mutant dataset.
Affinity scoring was then applied to the high‐throughput docking results using the ΔLin_F9XGB scoring function [33], an improved version of the linear empirical scoring function Lin_F9, utilising extreme gradient boosting and Δ‐machine learning. ΔLin_F9XGB is open‐source and has been shown to be on par or superior to some of the leading scoring functions when tested against the CASF‐2016 benchmark [37]. The scoring function takes the .mol2 binding poses generated using equibind and can be massively parallelised according to the maximum number of CPU processing cores available to the user.
3.3. Ranking
Finally, alignment and consensus screening were performed on the top‐ranking mutants to identify trends in the specific types of residue substitutions. Through deriving consensus motifs, desirable properties of the binding pocket can be derived, and in turn aid in the selection of mutants to be tested experimentally.
3.4. Validation
To test the protocol's ability to enrich for mutants with altered binding affinity/specificity, the protocol must be run to completion and a subset of top mutants would have to be synthesised and tested in vitro. Owing to the time and resource constraints of iGEM, we sought to instead investigate possible approaches to testing the protocol in silico using test sets from literature. The closest alternative to in vitro testing would in this case be evaluating the protocol's accuracy of affinity predictions following in silico mutagenesis. We attempted this on a test set of 26 ABL kinase mutants (PDB ID: 4WA9) with associated binding affinities from a study by Hauser et. al. [38]. First, taking the wild type ABL kinase structure, in‐silico structural mutations were introduced to match those from the test‐set. Affinity prediction was then carried out on these in silico mutants and changes in affinity between the wild type and mutants were compared to those same differences, determined experimentally. With this limited test‐set, we were unable to show a significant correlation between predicted and literature affinities. However, this was an expected result as a much greater test‐set would be required to detect meaningful relationships owing to the inherent low accuracy of current scoring functions. Yet, datasets of mutants and associated binding affinities are much sparser in literature as compared to wild‐type datasets, and the largest mutant databases with experimental affinities have become outdated or are no longer publicly available [39, 40] To aid in the development of future protein engineering tools aimed at altering binding affinity we therefore call for the development and curation of new and comprehensive databases of mutants akin to PDBBind [41] in scope. However, even with such a database, evaluation would be restricted to affinity prediction and fail to address the specific aim of enriching for mutants with improved ligand binding affinity/specificity and as such in vitro evaluation is still necessary, and future work should attempt this.
In terms of computational efficiency we were able to screen 20,000 mutants/day with a quad‐core intel‐based system. With the structural mutagenesis step being rate limiting, we expect significant improvements could be made by better parallelising this step or utilising different mutagenesis tools.
4. CASE STUDY: INFINITY 1.0 COMPARISON TO COMMERCIAL TOOLS
Tools similar to those integrated into the InFinity 1.0 pipeline, such as those for docking and scoring, are often commercially applied within the context of drug discovery services. One such company offering this service is MedChemExpress (MCE), which offers precise virtual screening (VS) as an efficient alternative to high‐throughput screening of ligands in early‐stage drug discovery. MCE's VS uses structure‐based virtual screening (SBVS) to dock and rank molecules using 3D target structures.
In a 2022 study, Kong et al. used MCE for molecular coupling, a tool to align substrates to protein binding sites with the aim of identifying potential binding pockets on amyrin synthase, amyrin synthase enzyme (CrMAS), for the substrate 2,3‐oxidosqualene. The method revealed five binding pockets on CrMAS (Figure S1), with Site 1 showing the highest affinity to 2,3‐oxidosqualene.
Our case study aimed to compare these findings with affinity predictions on the same sites using InFinity 1.0. The outcome of the scoring function delta_LinF9_XGB, used following docking performed by Equibind, was converted from its native units to Gibbs Free energy (ΔG) using a rearranged formula from the paper by Yang and Zhang [33].
Figure 5 and Table 2 show that the results from Autodock Vina, Kong et al. [42] and Equibind exhibit a similar trend in binding affinity, a property that is inversely proportional to ΔG value. Additionally, our tool effectively differentiated between sites of high (Sites 1, 2, and 5) and low binding affinity (Sites 3 and 4), mirroring the findings of Kong et al. [42].
FIGURE 5.
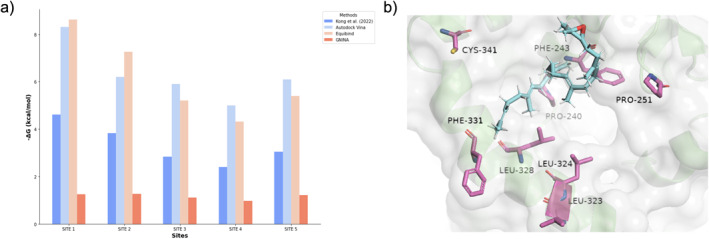
Comparison of CrMAS Binding Sites between MedChemExpress and InFinity 1.0 with 2,3‐oxidosqualene as the Ligand. (a) Comparative analysis of various scoring functions—EquiBind, Autodock Vina, and GNINA—against the findings presented in Kong et al. [42] following the docking process. (b) PyMOL visualisation of the binding site at Site 1 with 2,3‐oxidosqualene inside. CrMAS, amyrin synthase enzyme.
TABLE 2.
Gibbs energy scores derived from various scoring functions for different sites during the docking of 2,3‐oxidosqualene to CrMAS.
| Sites | −ΔG (kcal/mol) | |||
|---|---|---|---|---|
| Kong et al. [42] | Autodock Vina | Equibind (docking) and delta_LinF9_XGB (scoring) | GNINA | |
| Site 1 | 4.62 | 8.3 | 8.62 | 1.25 |
| Site 2 | 3.83 | 6.2 | 7.27 | 1.26 |
| Site 3 | 2.84 | 5.9 | 5.21 | 1.12 |
| Site 4 | 2.41 | 5.0 | 4.32 | 0.97 |
| Site 5 | 3.05 | 6.1 | 5.4 | 1.22 |
Abbrebiation: CrMAS, amyrin synthase enzyme.
To demonstrate the modularity of Infinity 1.0, we incorporated another scoring function, GNINA. GNINA, a fork of smina [43] and Autodock Vina [30], integrates convolutional neural networks (CNNs) for advanced scoring refinement. This integration of GNINA resulted in the identification of the top three highest (Sites 1, 2, and 5) and lowest (Sites 3 and 4) affinity sites, as shown in Table 2. These findings align with the results documented by Kong et al. [42]. The observed low binding affinity at Site 4, relative to the other sites, may be attributed to steric hindrance, as illustrated in Figure S1. Steric hindrance arises when atoms or groups within a molecule obstruct each other, thereby inhibiting optimal intermolecular interactions (e.g., hydrogen bonds, van der Waals forces, or ionic bonds) and preventing the establishment of efficient binding [44].
Our study demonstrates that InFinity 1.0 identifies sites exhibiting both high and low binding affinity to a given ligand, as shown when compared to the findings of Kong et al. [42]. InFinity 1.0 yielded more positive ΔG values for Site 1 and Site 2, indicating a heightened affinity of CrMAS for 2,3‐oxidosqualene at these sites relative to Sites 3 and 4, which aligns with Kong et al. [42] finding. Furthermore, InFinity 1.0's rapid docking and scoring capabilities surpass those of traditional methods, such as Autodock Vina, allowing quick screening within proteins to pinpoint sites for further investigation. This accelerated screening process not only reduces the time required but also minimises the financial expenditure for iGEM teams. Furthermore, as our program is open‐source, it provides iGEM competitors with free access for use in their projects, allowing them not only to use but also to expand upon the software without incurring costs.
5. DISCUSSION
Our proposed implementation serves the intended purpose of demonstrating how a pipeline could conceivably be constructed, repurposing breakthrough tools utilised for structural prediction and computational drug discovery. As more effective tools are developed, they can replace those in our current implementation, with the aim to suggest a small subset of potential mutants to test in vitro. This future improved implementation would then open the door for researchers with time and funding‐constrained projects such as those in iGEM to perform specificity and affinity engineering. The main aspect that calls for improvements is accuracy in modelling the structural impact of combinatorial mutants. In our study, we have employed PyMOL mutagenesis to introduce novel mutations into the 3D structure, due to its open‐source nature and quick integration into our pipeline. However, this approach has its limitations as it does not alter the underlying scaffold and does not consider steric clashes. To overcome these limitations, an alternative software tool, Rosetta CoupledMoves could be used [45]. This tool couples protein side‐chain, backbone flexibility, as well as ligand degrees of freedom for the improved redesign of viable mutants. The adoption of this alternative tool promises to enhance the accuracy of our structural modelling efforts, enabling us to more effectively investigate protein function and interaction. We have included command‐line instructions for its use within the InFinity 1.0 pipeline context in our Github repository (see Code Availability).
Finally, seeing inherent flexibility and potential inaccuracies in structural prediction, we propose docking programs should utilise flexible docking. One interesting avenue would be utilising the open‐source GNINA [46] platform which builds on AutoDock Vina [30], by using ML‐based scoring functions.
As opposed to the currently available dominant in silico‐based protein engineering tools that focus on enzyme engineering, InFinity 1.0 aims to alter binding affinity and specificity. This has applications namely for biosensor design, with use‐cases readily seen in iGEM, and can include sensors for pollutant detection [47], medical diagnostic tests [48], agricultural use and microbiology research with the possibility of monitoring metabolites in real time [49]. Additionally it can be applied to alter cellular functions, by changing natural‐sense and response systems. Its applicability could perhaps also translate to drug design, investigating mutants' effect on drug binding‐specificity, which could prove useful in for example, antiviral drug development [50].
6. CONCLUSION
In silico tools have developed exponentially in the last 5 years, with innovation in the sector set to revolutionise the current biotech landscape. In this study, we proposed a framework to increase accessibility and effectiveness of protein engineering techniques, stemming from our individual experience in the iGEM landscape. Focusing on altering ligand binding specificity, we enable users to generate a pool of combinatorial protein variants, which are then screened and ranked based on binding affinity predictions and common motifs, allowing for an output consisting of a library of 20,000 variants.
Seeing that our developed framework takes care of combinatorial mutagenesis, future work could adapt the software pipeline, simply changing the tools used for structural mutagenesis and docking/scoring without too extensive modification.
AUTHOR CONTRIBUTIONS
Shirin Bamezai: Conceptualisation; funding acquisition; investigation; methodology; writing – original draft. Giovanni Maresca di Serracapriola: Conceptualisation; funding acquisition; investigation; methodology; writing – original draft. Freya Morris: Conceptualisation; investigation; methodology; software; writing – original draft. Rasmus Hildebrandt: Conceptualisation; investigation; methodology; software; writing – original draft. Marc Augustine Sojerido Amil: Conceptualisation; investigation; methodology; software; writing – original draft. Sporadicate iGEM Team: Conceptualisation. Rodrigo Ledesma‐Amaro: Funding acquisition; project administration; resources; supervision; writing – review and editing.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
Supporting information
Figure S1
ACKNOWLEDGEMENTS
This study was funded thanks to the contributions of: iGEM Impact Grant, Eurekare, EraCoBioTECH, Faculty of Life Sciences Imperial, SynbiCITE, SynBioUK, Hummingbird, Gandhi Centre for Inclusive Innovation, Imperial College Business School, Potter Clarkson.
Bamezai, S. , et al.: Protein engineering in the computational age: an open source framework for exploring mutational landscapes in silico . Eng. Biol. 7(1-4), 29–38 (2023). 10.1049/enb2.12028
The below authors were also part of the Sporadicate iGEM Team: Giovanni Maresca di Serracapriola, Shirin Bamezai, Freya Morris, Rasmus Hildebrandt, Marc Amil, Rodrigo Ledesma Amaro.
Shirin Bamezai, Giovanni Maresca di Serracapriola, Freya Morris, Rasmus Hildebrandt and Marc Augustine Sojerido Amil have contributed equally to this work.
Contributor Information
Rodrigo Ledesma‐Amaro, Email: r.ledesma-amaro@imperial.ac.uk.
Sporadicate iGEM Team:
Haoran Xin, Jay Montogomery‐Johnson, Youzi Bi, Yuancheng Ding, Fontaine Gibbs, Alessandro Serafini, Emese Klug, Anvita Srinath, Tairan Wang, Allegra Maresca di Serracapriola, Mohamed Almarei, and Joaquin Caro Astorga
DATA AVAILABILITY STATEMENT
Access at: https://github.com/MarcAmil30/InFinity1.0.
REFERENCES
- 1. Lutz, S. , Iamurri, S.M. : Protein engineering: past, present, and future. In: Bornscheuer, U.T. , Höhne, M. (eds.) Protein Engineering, pp. 1–12. Springer New York (Methods in Molecular Biology), New York: (2018). 10.1007/978-1-4939-7366-8_1 [DOI] [PubMed] [Google Scholar]
- 2. Korendovych, I.V. : Rational and semirational protein design. In: Bornscheuer, U.T. , Höhne, M. (eds.) Protein Engineering, pp. 15–23. Springer New York (Methods in Molecular Biology), New York: (2018). 10.1007/978-1-4939-7366-8_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang, H. , et al.: Microbial production and molecular engineering of industrial enzymes. In: Biotechnology of Microbial Enzymes, pp. 151–165. Elsevier; (2017). 10.1016/B978-0-12-803725-6.00006-6 [DOI] [Google Scholar]
- 4. Vidal, L.S. , et al.: A primer to directed evolution: current methodologies and future directions. RSC Chemical Biology 4(4), 271–291 (2023). 10.1039/D2CB00231K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ravikumar, A. , et al.: Scalable, continuous evolution of genes at mutation rates above genomic error thresholds. Cell 175(7), 1946–1957.e13 (2018). 10.1016/j.cell.2018.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lutz, S. : Beyond directed evolution ‐ semi‐rational protein engineering and design. Curr. Opin. Biotechnol. 21(6), 734–743 (2010). 10.1016/j.copbio.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Setiawan, D. , Brender, J. , Zhang, Y. : Recent advances in automated protein design and its future challenges. Expet Opin. Drug Discov. 13(7), 587–604 (2018). 10.1080/17460441.2018.1465922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paladino, A. , et al.: Protein design: from computer models to artificial intelligence. WIREs Computational Molecular Science 7(5), e1318 (2017). 10.1002/wcms.1318 [DOI] [Google Scholar]
- 9. Yeh, A.H.‐W. , et al.: De novo design of luciferases using deep learning. Nature 614(7949), 774–780 (2023). 10.1038/s41586-023-05696-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu, H. , Chen, Q. : Computational protein design with data‐driven approaches: recent developments and perspectives. WIREs Computational Molecular Science n/a(n/a), e1646 (2022). 10.1002/wcms.1646 [DOI] [Google Scholar]
- 11. Walker, S.P. , Yallapragada, V.V.B. , Tangney, M. : Arming yourself for the in silico protein design revolution. Trends Biotechnol. 39(7), 651–664 (2021). 10.1016/j.tibtech.2020.10.003 [DOI] [PubMed] [Google Scholar]
- 12. Jumper, J. , et al.: Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589 (2021). 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leman, J.K. , et al.: Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17(7), 665–680 (2020). 10.1038/s41592-020-0848-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moretti, R. , et al.: Web‐accessible molecular modeling with Rosetta: the Rosetta online server that includes everyone (ROSIE). Protein Sci. 27(1), 259–268 (2018). 10.1002/pro.3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sumbalova, L. , et al.: HotSpot Wizard 3.0: web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 46(W1), W356–W362 (2018). 10.1093/nar/gky417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khersonsky, O. , et al.: Automated design of efficient and functionally diverse enzyme repertoires. Mol. Cell 72(1), 178–186.e5 (2018). 10.1016/j.molcel.2018.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Musil, M. , et al.: Fully automated virtual screening pipeline of FDA‐approved drugs using Caver Web. Comput. Struct. Biotechnol. J. 20, 6512–6518 (2022). 10.1016/j.csbj.2022.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Muzio, E. , Toti, D. , Polticelli, F. : DockingApp: a user friendly interface for facilitated docking simulations with AutoDock Vina. J. Comput. Aided Mol. Des. 31(2), 213–218 (2017). 10.1007/s10822-016-0006-1 [DOI] [PubMed] [Google Scholar]
- 19. Planas‐Iglesias, J. , et al.: LoopGrafter: a web tool for transplanting dynamical loops for protein engineering. Nucleic Acids Res. 50(W1), W465–W473 (2022). 10.1093/nar/gkac249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peleg, Y. , et al.: Community‐wide experimental evaluation of the PROSS stability‐design method. J. Mol. Biol. 433(13), 166964 (2021). 10.1016/j.jmb.2021.166964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldenzweig, A. , et al.: Automated structure‐ and sequence‐based design of proteins for high bacterial expression and stability. Mol. Cell 63(2), 337–346 (2016). 10.1016/j.molcel.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hon, J. , et al.: SoluProt: prediction of soluble protein expression in Escherichia coli . Bioinformatics 37(1), 23–28 (2021). 10.1093/bioinformatics/btaa1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, C. , Zou, Q. : Prediction of protein solubility based on sequence physicochemical patterns and distributed representation information with DeepSoluE. BMC Biol. 21(1), 12 (2023). 10.1186/s12915-023-01510-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vasina, M. , et al.: Tools for computational design and high‐throughput screening of therapeutic enzymes. Adv. Drug Deliv. Rev. 183, 114143 (2022). 10.1016/j.addr.2022.114143 [DOI] [PubMed] [Google Scholar]
- 25. Sequeiros‐Borja, C.E. , Surpeta, B. , Brezovsky, J. : Recent advances in user‐friendly computational tools to engineer protein function. Briefings Bioinf. 22(3), bbaa150 (2021). 10.1093/bib/bbaa150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. ICL iGEM Team : Sporadicate ‐ iGEM Wiki (2022). https://2022.igem.wiki/imperial-college-london/. Accessed: 1 May 2023
- 27. Ross, C. , Abel‐Santos, E. : The Ger receptor family from sporulating bacteria. Curr. Issues Mol. Biol. 12(3), 147–158 (2010) [PMC free article] [PubMed] [Google Scholar]
- 28. Blinker, S. , et al.: Predicting the structure and dynamics of membrane protein GerAB from Bacillus subtilis. Int. J. Mol. Sci. 22(7), 3793 (2021). 10.3390/ijms22073793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li, Y. , et al.: Structure‐based functional studies of the effects of amino acid substitutions in GerBC, the C subunit of the Bacillus subtilis GerB spore germinant receptor. J. Bacteriol. 193(16), 4143–4152 (2011). 10.1128/JB.05247-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trott, O. , Olson, A.J. : AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31(2), 455–461 (2010). 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eberhardt, J. , et al.: AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 61(8), 3891–3898 (2021). 10.1021/acs.jcim.1c00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corso, G. , et al.: DiffDock: diffusion steps, twists, and turns for molecular docking. arXiv (2022). 10.48550/arXiv.2210.01776 [DOI] [Google Scholar]
- 33. Yang, C. , Zhang, Y. : Delta machine learning to improve scoring‐ranking‐screening performances of protein–ligand scoring functions. J. Chem. Inf. Model. 62(11), 2696–2712 (2022). 10.1021/acs.jcim.2c00485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khanna, T. , et al.: Missense3D‐DB web catalogue: an atom‐based analysis and repository of 4M human protein‐coding genetic variants. Hum. Genet. 140(5), 805–812 (2021). 10.1007/s00439-020-02246-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schodinger, L. , DeLano, W. : PyMOL. http://www.pymol.org/pymol (2020)
- 36. Stärk, H. , et al.: EquiBind: geometric deep learning for drug binding structure prediction. arXiv (2022). 10.48550/arXiv.2202.05146 [DOI] [Google Scholar]
- 37. Su, M. , et al.: Comparative assessment of scoring functions: the CASF‐2016 update. J. Chem. Inf. Model. 59(2), 895–913 (2019). 10.1021/acs.jcim.8b00545 [DOI] [PubMed] [Google Scholar]
- 38. Hauser, K. , et al.: Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free‐energy calculations. Commun. Biol. 1(1), 1–14 (2018). 10.1038/s42003-018-0075-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pires, D.E.V. , Blundell, T.L. , Ascher, D.B. : Platinum: a database of experimentally measured effects of mutations on structurally defined protein–ligand complexes. Nucleic Acids Res. 43(D1), D387–D391 (2015). 10.1093/nar/gku966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hurst, J.M. , et al.: The SAAPdb web resource: a large‐scale structural analysis of mutant proteins. Hum. Mutat. 30(4), 616–624 (2009). 10.1002/humu.20898 [DOI] [PubMed] [Google Scholar]
- 41. Wang, R. , et al.: The PDBbind database: methodologies and updates. J. Med. Chem. 48(12), 4111–4119 (2005). 10.1021/jm048957q [DOI] [PubMed] [Google Scholar]
- 42. Kong, J. , et al.: Enhanced production of amyrin in Yarrowia lipolytica using a combinatorial protein and metabolic engineering approach. Microb. Cell Factories 21(1), 186 (2022). 10.1186/s12934-022-01915-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koes, D.R. , Baumgartner, M.P. , Camacho, C.J. : Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 53(8), 1893–1904 (2013). 10.1021/ci300604z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hlavacek, W.S. , Posner, R.G. , Perelson, A.S. : Steric effects on multivalent ligand‐receptor binding: exclusion of ligand sites by bound cell surface receptors. Biophys. J. 76(6), 3031–3043 (1999). 10.1016/S0006-3495(99)77456-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ollikainen, N. , Jong, R.M.de , Kortemme, T. : Coupling protein side‐chain and backbone flexibility improves the re‐design of protein‐ligand specificity. PLoS Comput. Biol. 11(9), e1004335 (2015). 10.1371/journal.pcbi.1004335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McNutt, A.T. , et al.: GNINA 1.0: molecular docking with deep learning. J. Cheminf. 13(1), 43 (2021). 10.1186/s13321-021-00522-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ray, S. , Panjikar, S. , Anand, R. : Design of protein‐based biosensors for selective detection of benzene groups of pollutants. ACS Sens. 3(9), 1632–1638 (2018). 10.1021/acssensors.8b00190 [DOI] [PubMed] [Google Scholar]
- 48. Veetil, J.V. , Jin, S. , Ye, K. : A glucose sensor protein for continuous glucose monitoring. Biosens. Bioelectron. 26(4), 1650–1655 (2010). 10.1016/j.bios.2010.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teng, Y. , et al.: Biosensor‐enabled pathway optimization in metabolic engineering. Curr. Opin. Biotechnol. 75, 102696 (2022). 10.1016/j.copbio.2022.102696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leonard, A.C. , Whitehead, T.A. : Design and engineering of genetically encoded protein biosensors for small molecules. Curr. Opin. Biotechnol. 78, 102787 (2022). 10.1016/j.copbio.2022.102787 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
Access at: https://github.com/MarcAmil30/InFinity1.0.