

Abstract
Background
The clinical diagnosis of manifest Huntington's disease (HD) relies on a high level of clinical confidence (99% confidence) of HD‐consistent motor signs. Longitudinal data have reliably identified cognitive and behavioral dysfunction predating clinical motor diagnosis by up to 15 years. Reliance on motor signs to establish a diagnosis of HD increases risk of early misdiagnosis or delayed diagnosis. Clinical neuropsychologists are uniquely positioned to advise on the clinical application of the Movement Disorder Society Task Force's recently proposed non‐motor diagnostic criteria for HD.
Objectives
To provide (1) a recommended clinical approach toward non‐motor diagnostic criteria in persons with HD and facilitation of accurate diagnosis; (2) recommended practices for medical treatment providers to screen and longitudinally monitor non‐motor signs of HD.
Methods
The Huntington Study Group re‐established the Neuropsychology Working Group, then recruited a multi‐disciplinary group of neuropsychologists, neurologists, and psychiatrists to conduct an unstructured literature review and discuss expert opinions on practice, to facilitate an informal consensus opinion to accomplish the objectives.
Results
The opinion and an example protocol for medical treatment providers to screen, monitor, and triage non‐motor signs and symptoms of Huntington's disease is provided.
Conclusions
Clinical diagnosis of non‐motor HD is empirically justified and clinically important. Screening and triage by non‐neuropsychologist clinicians can aid in detecting and monitoring non‐motor Huntington's disease manifestation. The Neuropsychology Working Group consensus advances good clinical practice, clinical research, and quality of life. A companion position paper presenting the details of our consensus opinion regarding evidence‐based guidelines for neuropsychological practice is forthcoming.
Keywords: Huntington's disease, neuropsychology, non‐motor, assessment, diagnosis
Huntington's disease (HD) is an inherited neurodegenerative condition characterized by progressive movement, cognitive, and psychiatric dysfunction. Standard clinical practice for diagnosis of manifest HD relies on motor criteria, whereby a clinician has 99% confidence that extrapyramidal movement signs are consistent with HD criteria in a person with a family history of HD or genetic testing indicating that the person is an HD gene expansion carrier. However, longitudinal data indicate that predominantly cognitive and behavioral/psychiatric phenotypes are common, even a plurality, in persons with HD. 1 , 2 Non‐motor signs of sufficient severity to cause functional impairment often emerge a decade or more before clinical motor decline. 3 These cognitive and behavioral changes, while often overlooked, can profoundly impact a person with HD's life, affecting their ability to work, maintain relationships, and participate in family life. 4 While these changes encompass a broad spectrum in terms of severity and manifestations, they can, in a minority of cases, also result in unlawful behaviors. 5 , 6 The burden of these non‐motor symptoms often surpasses that of the motor symptoms, underscoring the necessity of their recognition and management.
People who exhibit cognitive and neuropsychiatric signs and symptoms may be diagnosed with symptomatic/syndrome‐based disorders rather than HD (eg, early psychiatric symptoms may be diagnosed as bipolar disorder), frequently in cases of non‐motor symptom predominant prodromes and/or when specialist care is difficult to access. 7 These instances delay or mislead clinical intervention, prognosis, and functional planning. Eligibility for clinical trials and studies is also affected by exclusive reliance on motor signs for a diagnosis of HD; persons with predominantly non‐motor manifestations of HD pathology are often missed in recruitment efforts for large‐scale longitudinal studies, perpetuating a cycle of defining and confirming motor signs as the only valid method of disease diagnosis for HD. 8 While large‐scale registries have measured and documented subtle cognitive impairment and associated functional decline years prior to clinical motor diagnosis (eg, TRACK‐HD, 9 PREDICT‐HD, 10 PHAROS 11 ), the patient populations for most therapeutic trials have usually been stage I/II (early clinical diagnosis) per motor and functional scores of the Unified Huntington's Disease Rating Scale (UHDRS), 12 based in part on the motor outcome measures being amenable to detecting treatment‐effect. A by‐product of the motor‐contingent diagnostic criteria and use as clinical‐trial end‐point is a relatively lesser attention for development and analysis of novel disease‐modifying therapies for HD pathology in persons manifesting predominantly non‐motor symptoms. The HD‐ISS staging system is a recently developed research diagnostic criteria that includes a cognitive impairment component, though the factor is assessed via a single measure and the ISS is not designed or intended for clinical application. 13
The authors determined that pursuing a “white paper” type report to the field would help address these issues; specifically, providing (1) a recommended clinical approach toward non‐motor diagnostic criteria in persons with HD and facilitation of accurate diagnosis; (2) recommended practices for medical treatment providers to screen and longitudinally monitor non‐motor signs of HD.
Methods
To begin to address these issues, the movement disorder society (MDS) commissioned a Task Force on HD Diagnostic Categories, to discuss and produce a set of recommendations for classification of HD that considers cognitive signs. Briefly, the Task Force highlighted the importance of cognition and recommended the use of the Diagnostic and Statistical Manual, Fifth Edition (DSM‐5) 14 for diagnostic purposes. However, consensus was reached to retain motor criteria for diagnosis of HD. The aim of this paper is to further the conversation regarding the inclusion of cognitive signs in the diagnosis of HD.
Neuropsychologists specialize in assessing brain‐behavior relationships in the clinical setting via integration of clinical history with empirically validated examination of cognitive performance and behavioral/emotional symptoms. Neuropsychological evaluation informs differential diagnoses, improves sensitivity in monitoring of clinical status, and guides the multidisciplinary treatment plan for persons with neurobehavioral disorders. Thus, neuropsychologists with expertise in HD are well‐positioned to offer opinions on how to approach non‐motor diagnostic criteria in HD and protocols for medical treatment providers to screen and longitudinally monitor non‐motor signs of HD.
The Huntington Study Group (HSG) re‐established its dormant Neuropsychology Working Group (NPWG) in 2019 (Co‐Chairs: CMC, MAR). The NPWG recruited a group of neuropsychologists (CMC, MAR, VDB, KA, SAA, ASC, ALN‐S, AP, JCS) with a combination of clinical, research, and HD‐specific leadership exceeding 10 decades in combination. To ensure multi‐disciplinary input, the group recruited two physicians with a combined five decades of experience in clinical and research related to neurological and psychiatric aspects of HD, along with well‐defined HD‐specific leadership roles (ALT, MCE). The group met monthly and engaged in continual email communications to discuss these objectives over a 6 month period in 2021–2022. This position paper summarizes the consensus opinion of the group regarding (1) recommended clinical approach toward non‐motor diagnostic criteria in persons with HD and facilitation of accurate diagnosis; (2) recommended practices for medical treatment providers to screen and longitudinally monitor non‐motor signs of HD. The HSG Research Advisory Council (RAC) reviewed this opinion and comments were considered and integrated. This content is intended for consideration across all levels and domains of clinical providers who work with HD populations. A companion position paper by the same group outlines the consensus opinion of the NPWG regarding best clinical neuropsychological practice guidelines for working with persons with HD.
Results
Objective 1: Non‐Motor Clinical Diagnostic Criteria in HD
Historical Perspective
Initial investigations regarding cognitive deficits preceding motor manifestation of HD began more than 40 years ago. Prior to the availability of genetic testing, studies from the 1970s and early 1980s showed that subtle cognitive deficits could be detected several years before clinical motor diagnosis of HD, suggesting that cognitive changes could be a potential predictor of HD. 15 In the late 1980s and early 1990s, studies with persons who were at high risk of developing HD as determined via linkage analysis showed mixed findings, with some noting the presence of cognitive impairments in executive functions, visuospatial functions, and memory, 16 and some not detecting group differences in cognition between high‐ and low‐risk individuals. 17 After analysis to identify mutation carriers became available in 1993, 18 several more studies of cognitive impairment in pre‐diagnosis individuals were published. Thereafter, attempts to address methodological limitations in the previous investigations produced an equivocal landscape, with some data demonstrating significant cognitive impairment in at‐risk individuals 19 , 20 and some failing to detect a difference compared to non‐HD CAG expansion carriers. 15 , 21 , 22
In the last decade, there has been increased advocacy for integrating non‐motor signs into the HD diagnostic schema. 23 Paulsen argued that cognitive signs should carry more weight in diagnostic criteria given their significant impact on functional decline and family burden, as well as the protracted period of cognitive deficit that can precede motor manifestation of HD. 24 A large‐scale, international study of prodromal cognitive impairment, Predict‐HD, solidified the robustness of cognitive impairment as a clinical indicator of manifest HD. 25 Duff and colleagues showed that nearly 40% of pre‐diagnosed HD CAG expansion carriers in the Predict‐HD study met formal criteria for Mild Cognitive Impairment when using comparative normative data from at‐risk gene‐negative participants. 26 However, a specific diagnostic approach incorporating non‐motor signs of HD was not introduced until recently by Ross and colleagues 27 following the first explicit discussion of using cognitive exam findings in the diagnostic context. 28
Movement Disorder Task Force Diagnostic Approach: Neurocognitive Disorder Due to Huntington's Disease
The NPWG reached a consensus supporting the approach outlined by the MDS Task Force. 27 The MDS Task Force reviewed clinical and research definitions of HD in 2017 to offer updated nosology incorporating non‐motor signs into the diagnostic process. First, the Task Force proposed a new lexicon separating HD into Premanifest and Manifest stages. The Premanifest stage was further subdivided into a pre‐symptomatic stage, for those with confirmed HD CAG expansion who exhibited no motor signs; and a prodromal stage, for those who exhibited detectable motor signs that were not yet sufficient to warrant a diagnosis of HD (see Fig. 1). With regard to integrating non‐motor signs into the manifest diagnosis, the Task Force noted that (1) longitudinal studies have thus far not supported the presence of significant cognitive impairment without at least the presence of detectable motor signs, and (2) the most common psychiatric symptoms of HD (eg, apathy, irritability, depressive suicidality, anxiety) are non‐specific to HD CAG expansion carriers. Thus, the group advised combining the Diagnostic Confidence Level (DCL) template used for motor‐based diagnosis of HD (see Table 1) with the DSM‐5 approach toward diagnostic characterization of Mild and Major Neurocognitive Disorder (NCD; see Table S1). The specific Mild and Major NCD due to Huntington's Disease diagnostic criteria in the DSM‐5 specifies that beyond the general criteria, there should be evidence of insidious onset and gradual progression, clinical (motor) manifest HD or risk due to family history or genetic testing, and the absence of a better explainable medical or mental condition. Whereas a motor‐manifest diagnosis requires a DCL of 4, the task force proposed that a DCL of 3 combined with a Mild or Major NCD would represent manifest HD, and a DCL of 2 combined with either a Mild or Major NCD would represent prodromal HD (see Table 2).
Figure 1.
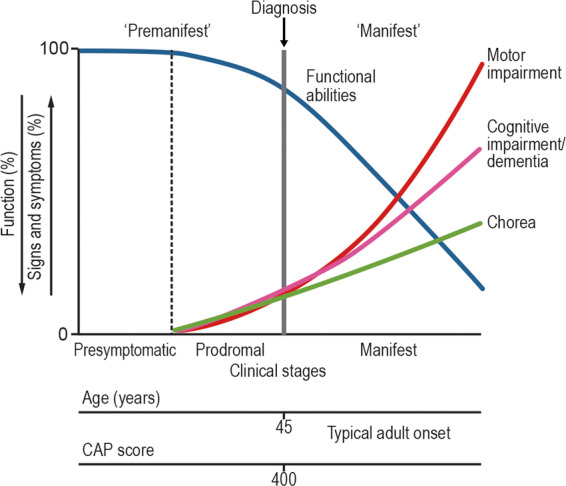
Clinical stages of Huntington's disease. Image is used under open‐source agreement of the associated publication, with acknowledgement to Ross and colleagues. 18
TABLE 1.
HD diagnostic confidence levels
| 1. | Symptoms mild, < 50% confidence |
| 2. | Symptoms possibly related to HD (50–89% confidence) |
| 3. | Symptoms probably related to HD (90–98% confidence) |
| 4. | Definitive HD motor signs (>99% confidence); Severity per Total Motor Score 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 |
Abbreviation: HD, Huntington's disease.
TABLE 2.
Criteria for diagnosis in individuals with CAG‐repeat expansion in Huntington's disease
| Diagnosis | Motor | Cognitive | Potential treatment |
|---|---|---|---|
| 1. Presymptomatic HD | Dx conf 0–2 | Normal | Disease modifying |
| 2. Prodromal HD (either A or B) | A) Dx conf 2 | A) + Minor or Major cognitive changes | Symptomatic or disease modifying |
| B) Dx conf 3 | B) With normal/unchanged cognition | ||
| 3. Manifest HD (either A or B) | A) Dx conf 3 | A) +Minor or Major cognitive changes | Symptomatic or disease modifying |
| B) Dx conf 4 | B) With normal/unchanged cognition |
Note: Adapted from Ross et al. 27 Potential treatments apply to each of the 3 diagnoses regardless of the criteria for meeting the diagnosis. It is expected that the ability to define signs and symptoms would be enhanced by longitudinal follow‐up and assessments. Recommend that neuropsychological testing be used to make determination of cognitive impairment; if necessary, an alternative quantified clinical assessment may be used to either document substantial impairment or document decline over time.
Abbreviations: Dx conf, diagnostic confidence; HD, Huntington's disease.
Behavioral/Psychiatric Manifestations and their Impact on Cognition
Behavioral disorders and psychiatric phenomena are among the earliest and most functionally impairing signs of HD 29 , 30 , 31 and can include anxiety, apathy, depression, irritability, perseveration, obsessions, and psychosis. However, the heterogeneity with which these symptoms are expressed in HD complicates their use as diagnostic criteria. For example, these symptoms tend to fluctuate non‐linearly, and are subject to environmental and treatment factors. 32 , 33 It is also debatable whether psychiatric and behavioral syndromes in HD can be separated into pathology‐related syndromes (eg, apathy, irritability) and non‐specific syndromes associated with HD (eg, PTSD, depressive syndromes). 34 , 35 , 36 That said, recent updates to diagnostic criteria, reflected in the DSM‐5‐TR, do provide specific codes for the presence/absence of “behavioral disturbance” even in the Mild NCD diagnosis; this represents a potential pathway of integrating neuropsychiatric symptoms into the overall formulation of non‐motor symptoms into manifest HD diagnosis.
For these reasons, the NPWG reached the following consensus on the operationalization of MDS Task Force positions on behavioral/psychiatric manifestations in relation to cognitive signs in HD:
Although HD diagnostic criteria and staging systems (ie, the ISS, most recently) have traditionally excluded behavioral and psychiatric manifestations from their framework, 27 the presence of these symptoms in a confirmed HD CAG expansion carrier warrants close monitoring of cognitive and motor status.
The NPWG acknowledges that behavioral disorders are intrinsically and reciprocally linked to cognitive function. 37 For example, behavioral disorders (eg, anxiety, apathy) negatively affect cognitive functioning and influence the results of cognitive assessments. Further, common cognitive deficits in HD, such as early declines in social cognition, also have important implications for behavioral functioning.
The MDS Task Force did not address the DSM‐5 approach toward capturing the presence of neuropsychiatric signs presumed related to the neuropathology producing the Neurocognitive Disorders (NCD), that is, “with or without behavioral disturbance.” The NPWG recommends using the “behavioral disturbance” modifier to incorporate documentation of behavioral/psychiatric manifestations of HD into a unitary NCD diagnosis, when the behavioral/psychiatric feature is favored to represent a neuropsychiatric syndrome associated with the HD pathology implicated in the NCD diagnosis itself. When this etiological connection is unclear, a separate primary psychiatric disorder diagnosis should be independently assigned.
The NPWG expects future research to delineate the presentation and course of neuropsychiatric signs in HD, including their association with other symptomatic dimensions and functioning, possibly leading to recognition of a predominantly behavioral HD phenotype.
Special Considerations in Clinical Application
The DSM‐5 criteria for supportive evidence warranting a NCD is not absolute; the guidelines require evidence of significant or modest cognitive decline supported by patient/informant concern and impairment in cognitive performance. We strongly recommend that neuropsychological testing be used to make this determination, or if necessary, an alternative quantified clinical assessment should document decline over time. Depending on clinical setting, the upcoming ICD‐11 terminology of Mild Neurocognitive Disorder and Dementia and associated criteria could easily be used with our recommendations. As applied to the HD population, there may be circumstances in which the patient is not concerned due to anosognosia; thus, an emphasis on the report of informant/collateral is important. Additionally, given the implications of assigning a manifest HD diagnosis, the field may wish to consider whether (a) recommendations for stricter severity of performance deficits (eg, >2 SD below normative expectations, rather than 1 or 1.5 SD) and (b) evidence of decline between two assessment points should be applied in this patient population to meet the gradual progression criteria (rather than report sufficing). Relatedly, discussion is warranted on if/how to operationalize the clinical confidence in the absence of better explaining medical or mental conditions criterion.
Finally, future discussion and empirical research is required to assess whether requiring a minimum DCL of 3 with NCD is useful or too stringent for a non‐motor HD manifest diagnosis. In fact, it is the opinion of the authors that future consideration should be given to shifting the overall approach to diagnosis of HD to include: (1) a genetic diagnosis step, following confirmation of CAG expansion, and (2) assigning a symptomatic HD diagnosis in those genetic diagnosis HD patients who are judged to be presenting neurobehavioral symptomatology (ie, motor and/or non‐motor features), which when taken as a whole and in the context of any clinically indicated additional diagnostics, is deemed probably attributable to HD pathology. Such a shift in diagnostic criteria would intentionally allow a diagnosis of symptomatic HD, in a confirmed genetic carrier, even in the absence of motor signs. Finally, the referring provider and clinical neuropsychologist should both obtain informed consent from a motor non‐manifest, at‐risk carrier, that undergoing neuropsychological evaluation may result in diagnostic evidence of manifest HD.
Objective 2: Protocols for Medical Treatment Providers to Screen and Longitudinally Monitor Non‐Motor Symptoms of HD
With the goal of detecting possible non‐motor manifestations of HD as early as possible, it is important that medical providers screen and monitor those at high risk of HD and those with concerning subclinical features of non‐motor manifest HD. Following a positive screening, we recommend triage to appropriate specialists (eg, behavioral neurologist, neuropsychiatrist, movement disorders specialist, neuropsychologist, occupational therapist, genetic counselor) to aid in evaluating for a non‐motor manifest HD diagnosis and to prevent false positive diagnosis. As previously mentioned, informed consent about the potential impacts of this non‐motor workup should be obtained, given the implications of a possible manifest HD diagnosis being assigned.
Recommended Screening, Triage, and Monitoring of Cognitive, Psychiatric, and Functional Changes
Cognitive Screening Tests
We acknowledge that neuropsychological evaluation is not always feasible. In such circumstances, we recommend documentation of cognitive signs and symptoms, including by patients and caregivers when possible, and cognitive screening 38 with the caveat that cognitive screening is not diagnostic or equivalent to neuropsychological evaluation. 39 , 40 Collateral informant reporting is important given the frequency of both denial and anosognosia in people with HD. Anosognosia can fluctuate based on disease stage and predisposing psychological factors. 41 , 42 , 43 Additionally, language and cultural diversity factors should be considered to avoid false positives or negatives, both of which are possible.
If cognitive screening measures are the only means available, we recommend using a screening tool that has empirically demonstrated sensitivity and specificity to clinically significant cognitive impairment [ie, set at or near prodromal or mild cognitive impairment (MCI) cut‐offs]. Similarly, the tool must have sensitivity across a range of cognitive severity such that it can detect cognitive signs across the range from pre‐symptomatic to early‐manifest disease stage. Furthermore, the measure should ideally have been characterized with respect to determining its Reliable Change Index, 44 so that if repeated screening is conducted, the clinician has the statistical basis to judge if there has been a significant change (ie, decline) in performance. The screening tool needs to assess multiple cognitive domains relevant to HD (attention/executive, memory, visuospatial, language) rather than focusing on a single aspect of cognition, such as memory. Moreover, the cognitive screening test should have demographically adjusted norms and be developed from a study with a robust sample size that can provide the basis for judging the patient's performance. Formally translated screeners and suitable normative data are crucial when evaluating patients from various linguistic and cultural backgrounds to avoid pathologizing exposure‐based factors (eg, education, occupation, culture) and incorrectly conceptualizing the individual as exhibiting cognitive impairment when none exists. 45 , 46 , 47
In 2017, MDS commissioned a subcommittee tasked with completing a systematic review of the professional literature to identify cognitive screening measures to evaluate “their context of use and validation in HD.” 48 Following an in‐depth review of cognitive screening measures, no measures were designated as “recommended.” In their review, MDS designated the Montreal Cognitive Assessment (MoCA) as “recommended with caveats” for assessing the severity of cognition in HD and “suggested” for its ability to detect cognitive impairment. 48 While noting that the MoCA assesses a broad range of cognitive domains, the authors also identified that the utility of the MoCA is limited because it includes too few items to enable assessment of individual domains of cognition. Eight other cognitive screening measures met the “suggested” designation, a notch below the recommended category, including the Mini Mental Status Exam (MMSE), which is the most commonly used screening test for providing an overall measure of cognitive impairment in clinical and research settings. 49 , 50 Primary critiques of the MMSE included limitations in assessing executive functions and insensitivity to assessing cognitive change over time. The Unified Huntington's Disease Rating Scale (UHDRS)‐Cognitive Assessment also received “suggested” status, but it was judged to be constrained by its limited breadth with regard to cognitive constructs affected in HD and the lack of validation of the sum score.
In consideration of the MDS Task Force 2017 review, the NPWG recommends the MoCA for cognitive screening purposes when clinically indicated. Alternative screening measures can be found in Table 3. The MoCA meets many of the requirements previously outlined, including being more sensitive to cognitive dysfunction in HD when compared to the MMSE. 38 , 51 Additionally, the MoCA's total score is corrected for education, 52 providing some level of sensitivity for the potential confounding effects of limited demographic variables. However, we urge caution against over‐reliance on, or over‐exposure to, a single test such as the MoCA, particularly in the setting of clinical trials involving HD patients. Multiple versions of the MoCA are available and their use can help mitigate practice effects that may occur due to frequent exposure to the same test. Furthermore, we advocate for diversifying cognitive assessments, integrating other validated tests such as the UHDRS‐Cognitive Assessment when appropriate. Though studies examining the MoCA's efficacy in longitudinally tracking cognition in HD populations are lacking, one study demonstrated the UHDRS‐Cognitive Assessment's superior validity in monitoring cognitive changes in HD patients after 12 months, compared to the MoCA and MMSE. 44 As such, it may serve as a viable alternative in this context. Otherwise, we recommend the MoCA with the Chelune formula to assess for treatment response or progression of cognitive decline. 53 When an assessment is necessary for mid‐stage HD patients, the UHDRS‐FAP is a consideration but is limited by its need for further study in HD populations. 48 In addition to cognitive screening tests, the NPWG also recommends screening for behavioral and psychiatric signs and symptoms using well‐established but brief symptom‐reports (per patient and informant/s), discussed further below.
TABLE 3.
Example of screening measures for non‐motor HD signs
| Domain | Measure | Clinical criteria | Alternatives |
|---|---|---|---|
| Cognition | MoCA | Total score ≤26 | MMSE, Kokmen, UHDRS‐Cog |
| SLUMS | |||
| Functional Status | WHODAS 2.0 (36 item) | None: 0–0.49 | UHDRS‐FAS/TFC/IS, Lawton‐Brody iADL, FAQ |
| Mild: 0.5–1.49 | |||
| Moderate: 1.5–2.49 | |||
| Severe: 2.5–3.49 | |||
| Extreme: 3.5–5 | |||
| Mood | PHQ‐9 | Total score ≥5 | HADS |
| GAD‐7 | |||
| Sleep–wake | ISI | Total score ≥8 | PROMIS‐SD‐short & SRI‐short, ESS, PSQI |
| Apathy | b‐DAS | Executive subscale ≥4 | AES |
| Emotional subscale ≥5 | |||
| Initiation subscale ≥6 | |||
| Dangerous Behaviors | C‐SSRS‐Screen | C‐SSRS‐s Ideation ≥3 | PHQ‐9 item #9 |
| BVC‐short (last month) | C‐SSRS‐s Behavior = 2 | ||
| BVC‐s ≥3 | |||
| Collateral Observations | NPI‐Q | Clinical judgment per item | Clinical interview |
Abbreviations: MoCA, Montreal Cognitive Assessment 52 ; MMSE‐2, MiniMental Status Exam, Second Edition 54 ; Kokmen STMS, Kokmen Short Test ofMental Status 55 ; SLUMS, Saint Louis University Mental Status Examination 56 ; UHDRS‐Cog, Unified Huntington’s Disease Rating Scale‐Cognitive Function 57 ; UHDRS‐TFC, Unified Huntington’s Disease Rating Scale‐Total Function Capacity 57 ; WHODAS 2.0, World Health Organization Disability Assessment Schedule 2.0 58 ; Lawton‐Brody iADL, Instrumental Activities of Daily Living 59 ; FAQ, Functional Activities Questionnaire 60 ; PHQ‐9, Patient Health Questionnaire 61 ; GAD‐7, 7‐item Generalized Anxiety Disorder Scale 62 ; HADS, The Hospital Anxiety and Depression Scale 63 ; ISI, Insomnia Severity Index 64 ; PROMIS, Patient‐Reported Outcomes Measurement Information System; SD,Sleep Disturbance; SRI, Sleep‐related Impairment 65 ; ESS, Epworth Sleepiness Scale 66 ; PSQI, Pittsburgh Sleep Quality Index 67 ; b‐DAS, brief Dimensional Apathy Scale 68 ; AES, Apathy Evaluation Scale 69 ; C‐SSRS‐Screen, Columbia Suicide Severity Rating Scale Screen Version 70 ; BVC, The Brøset Violence Checklist 71 ; NPI‐Q, Neuropsychiatric Inventory Questionnaire. 72
We recognize that the MoCA is globally acknowledged and frequently used among health professionals, and thus, we endorse its use for cognitive screening when clinically relevant. One aspect of this determination should be the frequency of, and interval between, administration of a cognitive screen. Practice‐effects can impact diagnostic sensitivity and therefore delay detection of relevant symptomatology and/or impact clinical trial eligibility. The MoCA does have alternate forms, but it would be advisable to reserve use for annual visits or when clinical‐functional status decline is reported. Furthermore, it is crucial to consider that other recommended screening tools may not be as universally recognized or accessible, which could hinder their application in some circumstances. We advise considering this factor as a potential constraint when adapting our recommendations.
Functional Screening Measures
Functional status is important in determining the risk of early signs of non‐motor manifestation of HD and important in forming recommendations. We suggest documenting functional status, ideally with comments (based on clinical judgment) as to whether the dysfunction is primarily related to non‐motor or motor signs. We suggest screening for functional impairment, ideally using both a patient and a collateral informant questionnaire. The WHO Disability Assessment Schedule 2.0 (WHODAS 2.0) is an open‐source, self‐report measure with an available proxy questionnaire that has a 36‐item short‐form (with 12‐item brief version available), which covers basic activities of daily living (ADLs); instrumental ADLs; and high‐complexity domains such as mobility, life activities, vocation, and interpersonal/social activity. 73 The inclusion of more complex functional domains increases sensitivity in patients with subtle symptoms impacting independence; additionally, both versions have been psychometrically validated across HD stages. 74 , 75 However, we should also acknowledge the more disease‐specific and frequently used functional screens used in clinical‐research with HD, namely the UHDRS sub‐scales related with function: the 5‐item clinician‐patient/informant interview score for Total Functional Capacity, the expanded 25‐item Functional Assessment Scale, and the single clinician‐rated Independence Scale score. We suggest these as acceptable for substitution, though point out that each of psychometric weaknesses (namely, incomplete reliability testing) and low “ceilings” (reducing sensitivity in early stage/subtle symptom cases). 76 A new measure that bridges the advantages of the WHODAS and UHDRS functional sub‐scales is the Functional Rating Scale 2.0, which may offer another option when available with validity data and cut‐offs. 77 Please see Table 3 for additional recommendations and alternatives for functional assessment screening measures.
Behavioral/Psychiatric Screening Measures
Behavioral and psychiatric symptoms fluctuate, and therefore need to be considered frequently; these symptoms can impact the interpretation of cognitive and functional screening tools. We advise documenting the presence and severity of these symptoms per patient and collateral informant report. 78 Short‐forms of questionnaires with empirically based cut‐offs for clinically significant symptoms should be considered for depression, anxiety, apathy, and sleep–wake disturbance (Table 3). Screening for safety‐related behavioral symptoms (ie, suicidality, aggression/violence toward others) should also be incorporated into the assessment due to the elevated incidence in HD and profound morbidity/mortality risk associated with the symptoms; a secondary benefit of including screening steps is ensuring the domain is not overlooked or forgotten during clinical contacts. 79 , 80 , 81 , 82 Psychotic symptoms typically require clinical judgment based on comparing patient reports to observations and collateral reports, as well as monitoring for atypical behavioral manifestations in clinic. Finally, behavioral/psychiatric symptoms should also be noted independent from performance/report on screening measures. Both patient and collateral informant perspectives should be considered given the high incidence of anosognosia among HD patients.
Triage Response
For determining next steps, a triage approach is applied to the information gathered from a clinical interview in addition to the cognitive, functional, and behavioral/psychiatric screeners. This triage response will ensure that patients are referred to appropriate providers for follow‐up, which will aid in supporting a non‐motor manifest HD diagnosis if appropriate and facilitate treatment planning. An example is outlined in Table 4, which is intended to apply to new patients or those previously seen without elevated concern for clinically significant non‐motor HD signs. In brief, we suggest that the screening results be organized into High, Moderate, Low, or Lowest risk of non‐motor manifest HD. These categories are not substitutes for a neurocognitive or psychiatric diagnoses, nor do they represent a diagnostic impression on whether HD pathology is the likeliest etiology for the symptoms. However, they do suggest reasonable next steps in the diagnostic workup.
TABLE 4.
Example of screening categories & triage for patients at risk for non‐motor HD signs
| Category | Cognitive or psychiatric screens (consider patient & informant) | Motor symptoms | Functional Status (associated with non‐motor symptoms) | Triage Response |
|---|---|---|---|---|
| Lowest risk | Asymptomatic, normal findings on cognitive and psychiatric screens | Asymptomatic | Intact |
|
| Low risk | Subtle symptoms, normal findings on cognitive and psychiatric screens | Asymptomatic | Intact |
|
| Modest risk | Atypical findings on cognitive and/or psychiatric screens | Asymptomatic or symptomatic | Intact |
|
| High risk | Atypical findings on cognitive and/or psychiatric screens | Asymptomatic or symptomatic | Impaired |
|
| Any critical item/screen is positive | Asymptomatic or symptomatic | Impaired or not impaired |
|
Abbreviations: OT, occupational therapy; PT, physical therapy; SLT, speech‐language therapy.
Monitoring over Time
In the setting of the proposed triage model, we recommend follow‐up monitoring for all HD patients. For those in the Lowest and Low categories, clinical judgment, established cut‐scores, and RCI on the cognitive screen can be used to document the stability, or degree of progression, of the symptoms. Discussing the concept of baseline neuropsychological evaluation can have a role in the Lowest category and can be encouraged after appropriate management of modifiable comorbid confounds in the Low category. The triage categories and responses remain relevant in follow‐up; however, the NPWG has identified several common referrals that warrant referral for full neuropsychological evaluation:
Is there evidence of marked cognitive and behavioral decline from baseline/prior assessment in a patient without an NCD or with Mild NCD due to HD?
Are cognitive and behavioral symptoms linked to functional decline or functional safety considerations (eg, access to firearms/weapons, heavy machinery, driving, independent management of medications, financial capacity, decision‐making capacity)?
Is there a comorbid condition exacerbating HD dysfunction?
Is the patient applying for, or are they suited to disability, vocational rehabilitation services, or in‐home care?
These are important clinical considerations regarding diagnosis (ie, transition to Major NCD) as well as available resources and treatment planning. Follow‐up monitoring can also help track changes associated with treatment decisions, such as response or side‐effects to HD‐related medications and the management of comorbid conditions. Repeat assessments should be performed at the request of the patient or family. If an HD patient is high functioning and has a job that has significant financial and legal responsibilities (ie, judge, CEO, financial advisor), or safety concerns (eg, police, pilot, nurse, physician/surgeon), then baseline and repeat neuropsychological evaluations are essential. As we have described, a full neuropsychological evaluation is more sensitive than cognitive screeners for detecting cognitive impairment in high functioning individuals, such as physicians. 83 , 84
Some circumstances have a significant likelihood of not indicating follow‐up for neuropsychological re‐evaluation, and thus we advise consultation with the neuropsychologist prior to making a formal referral when possible. These circumstances include:
Neuropsychological decline consistent with the HD disease course in a patient already diagnosed with Major Neurocognitive disorder due to HD
Clinically unstable psychiatric presentation (active psychosis, hypo/mania, intoxication, substance abuse not yet in early remission)
Sensorimotor and/or non‐verbal status precludes significant engagement in cognitive testing
Strong patient resistance or refusal (consider counseling)
Of note, without a diagnostic confidence level characterization of motor symptoms, an HD‐related NCD diagnosis is not possible using our proposed framework; thus, we would recommend a neurological exam prior to triage to neuropsychological evaluation, if possible. If such an examination is not immediately possible, a neuropsychologist may nonetheless diagnose a Major or Mild NCD, but the etiology would be unclear.
Conclusions
Clinical diagnosis of non‐motor HD is empirically justified and clinically important. While a diagnostic approach differs from establishing research criteria, clinical practice and research are reciprocal and inextricably linked toward the pursuit of developing disease‐modifying therapies for HD. Establishing a common lexicon and diagnostic approach for non‐motor manifest HD signs and symptoms serves to increase our understanding of the implications of early detection and intervention on clinical outcomes across the spectrum of HD phenotypes. As the NPWG diagnostic approach has outlined in detail above, appropriate screening and triage by non‐neuropsychologist clinicians can aid in detecting non‐motor HD manifestation and effectively monitoring the complete clinical course of HD. Further expert discussion and empirical research is required to address the potential implications of applying this clinical approach, including (i) importance of collateral/informant concerns given anosognosia in the HD population, (ii) operationalization of impaired cognitive performance and insidious/progressive course, (iii) operationalization of clinical confidence that the supportive findings are HD related rather than alternative medical or mental condition, and (iv) informed consent clarifying the potential result of evaluation being a manifest HD diagnosis. Ultimately, accurately diagnosing these symptoms is not only good clinical practice, but also advances clinical research and lays the foundation for optimal quality of life for patients living with HD and their families. A companion position paper by the NPWG with a consensus opinion regarding evidence‐based guidelines for neuropsychological practice is forthcoming.
Author Roles
(1) Research review project: A. Conception, B. Organization, C. Execution; (2) Qualitative Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
C.M.C.: 1ABC, 2ABC, 3AB
M.A.R.: 1BC, 2ABC, 3AB
V.A.D.B.: 1BC, 2ABC, 3AB
K.A.: 1BC, 2ABC, 3AB
S.A.A.: 1C, 2ABC, 3AB
A.S.C.: 1C, 2ABC, 3AB
M.C.E.: 1C, 2ABC, 3AB
A.L.N.S.: 1C, 2ABC, 3AB
A.P.: 1C, 2ABC, 3AB
A.T.: 1C, 2ABC, 3AB
J.C.S.: 1C, 2ABC, 3B
Ethical Compliance Statement
All procedures involved in this review were consistent with the regulations established by the Helsinki Declaration of the World Medical Association. Informed patient consent was not necessary for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Disclosures
Funding Sources and Conflicts of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no financial disclosures that are applicable to the research or would qualify as a conflict of interest.
Supporting information
Table S1. DSM‐5, Diagnostic and statistical manual of mental disorders, fifth edition, text revision. HD, Huntington disease.
M. Agustina Rossetti, Victor A. Del Bene, Kendra Anderson, Sharlet A. Anderson, Andrea S. Celka, Mary C. Edmondson, Amelia L. Nelson‐Sheese, Adam Piccolino and Antonio L. Teixeira are equally contributed to this work.
References
- 1. Biglan KM, Zhang Y, Long J, Geschwind M, Kang G, Killoran A, et al. Refining the diagnosis of Huntington disease: the PREDICT‐HD study. Front Aging Neurosci 2013;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vinther‐Jensen T, Larsen IU, Hjermind LE, et al. A clinical classification acknowledging neuropsychiatric and cognitive impairment in Huntington's disease. Orphanet J Rare Dis 2014;9(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaganti SS, McCusker EA, Loy CT. What do we know about late onset Huntington's disease? J Huntingtons Dis 2017;6(2):95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Exuzides A, Matos JE, Patel AM, Martin AA, Ricker B, Bega D. Understanding the burdens associated with Huntington's disease in manifest patients and care partners–comparing to Parkinson's disease and the general population. Brain Sci 2022;12(2):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McDonell KE, Brown BK, Hale L, et al. Medicolegal aspects of Huntington disease. J Am Acad Psychiatry Law 2021;49(4):565–571. [DOI] [PubMed] [Google Scholar]
- 6. Brown JM, Weinkauf E. Forensic mental health: a source guide for professionals. Createspace Independent Publishing Platform. 2018. [Google Scholar]
- 7. Mustafa FA. Misdiagnosis of Huntington's disease. Lancet Psychiatry 2017;4(1):21. [DOI] [PubMed] [Google Scholar]
- 8. McAllister B, Gusella JF, Landwehrmeyer GB, Lee JM, MacDonald ME, Orth M, et al. Timing and impact of psychiatric, cognitive, and motor abnormalities in Huntington disease. Neurology 2021;96:e2395–e2406. 10.1212/WNL.0000000000011893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: analysis of 36‐month observational data. Lancet Neurol 2013;12(7):637–649. [DOI] [PubMed] [Google Scholar]
- 10. Paulsen JS, Long JD, Ross CA, et al. Prediction of manifest Huntington's disease with clinical and imaging measures: a prospective observational study. Lancet Neurol 2014;13(12):1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biglan KM, Shoulson I, Kieburtz K, et al. Clinical‐genetic associations in the prospective Huntington at risk observational study (PHAROS): implications for clinical trials. JAMA Neurol 2016;73(1):102–110. [DOI] [PubMed] [Google Scholar]
- 12. Huntington Study Group . Unified Huntington's disease rating scale: reliability and consistency. Mov Disord 1996;11(2):136–142. [DOI] [PubMed] [Google Scholar]
- 13. Tabrizi SJ, Schobel S, Gantman EC, et al. A biological classification of Huntington's disease: the integrated staging system. Lancet Neurol 2022;21(7):632–644. [DOI] [PubMed] [Google Scholar]
- 14. DSM‐5 Task Force . Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013. [Google Scholar]
- 15. Blackmore L, Simpson SA, Crawford JR. Cognitive performance in UK sample of presymptomatic people carrying the gene for Huntington's disease. J Med Genet 1995;32(5):358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jason GW, Pajurkova EM, Suchowersky O, Hewitt J, Hilbert C, Reed J, Hayden MR. Presymptomatic neuropsychological impairment in Huntington's disease. Arch Neurol 1988;45(7):769–773. [DOI] [PubMed] [Google Scholar]
- 17. Strauss ME, Brandt J. Are there neuropsychologic manifestations of the gene for Huntington's disease in asymptomatic, at‐risk individuals? Arch Neurol 1990;47(8):905–908. [DOI] [PubMed] [Google Scholar]
- 18. Baig SS, Strong M, Rosser E, Taverner NV, Glew R, Miedzybrodzka Z, et al. 22 years of predictive testing for Huntington's disease: the experience of the UK Huntington's prediction consortium. Eur J Hum Genet 2016;24(10):1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foroud T, Siemers E, Kleindorfer D, et al. Cognitive scores in carriers of Huntington's disease gene compared to noncarriers. Annals Neurol 1995;37(5):657–664. [DOI] [PubMed] [Google Scholar]
- 20. Kirkwood SC, Siemers E, Stout JC, Hodes ME, Conneally PM, Christian JC, Foroud T. Longitudinal cognitive and motor changes among presymptomatic Huntington disease gene carriers. Arch Neurol 1999;56(5):563–568. [DOI] [PubMed] [Google Scholar]
- 21. Giordani B, Berent S, Boivin MJ, et al. Longitudinal neuropsychological and genetic linkage analysis of persons at risk for Huntington's disease. Arch Neurol 1995;52(1):59–64. [DOI] [PubMed] [Google Scholar]
- 22. de Boo GM, Tibben A, Lanser JBK, Jennekens‐Schinkel A, Hermans J, Maat‐Kievit A, Roos RAC. Early cognitive and motor symptoms in identified carriers of the gene for Huntington disease. Arch Neurol 1997;54(11):1353–1363. [DOI] [PubMed] [Google Scholar]
- 23. Ross CA, Pantelyat A, Kogan J, Brandt J. Determinants of functional disability in Huntington's disease: role of cognitive and motor dysfunction. Mov Disord 2014;29(11):1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paulsen JS. Cognitive impairment in Huntington disease: diagnosis and treatment. Curr Neurol Neurosci Rep 2011;11(5):474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stout JC, Paulsen JS, Queller S, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology 2011;25(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duff K, Paulsen J, Mills J, et al. Mild cognitive impairment in prediagnosed Huntington disease. Neurology 2010. Aug 10;75(6):500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ross CA, Reilmann R, Cardoso F, McCusker EA, Testa CM, Stout JC, et al. Movement disorder society task force viewpoint: Huntington's disease diagnostic categories. Mov Disord Clin Practice 2019;6(7):541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington's disease based on natural history. Mov Disord 2014;29(11):1335–1341. [DOI] [PubMed] [Google Scholar]
- 29. Van Duijn E, Craufurd D, Hubers AA, Giltay EJ, Bonelli R, Rickards H, et al. European Huntington's disease network Behavioural phenotype working group. Neuropsychiatric symptoms in a European Huntington's disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry 2014;85(12):1411–1418. [DOI] [PubMed] [Google Scholar]
- 30. Epping EA, Kim JI, Craufurd D, et al. PREDICT‐HD investigators and coordinators of the Huntington study group longitudinal psychiatric symptoms in prodromal Huntington's disease: a decade of data. Am J Psychiatry 2016;173(2):184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sellers J, Ridner SH, Claassen DO. A systematic review of neuropsychiatric symptoms and functional capacity in Huntington's disease. J Neuropsychiatry Clin Neurosci 2020;32(2):109–124. [DOI] [PubMed] [Google Scholar]
- 32. Anderson KE, Van Duijn E, Craufurd D, Drazinic C, Edmondson M, Goodman N, et al. Clinical management of neuropsychiatric symptoms of Huntington disease: expert‐based consensus guidelines on agitation, anxiety, apathy, psychosis and sleep disorders. J Huntingtons Dis 2018;7(4):355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teixeira AL, De SLC, Rocha NP, Furr‐Stimming E, Lauterbach EC. Revisiting the neuropsychiatry of Huntington's disease. Dementia Neuropsychol 2016;10:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dale M, van Duijn E. Anxiety in Huntington's disease. J Neuropsychiatry Clin Neurosci 2015;27(4):262–271. [DOI] [PubMed] [Google Scholar]
- 35. Oosterloo M, Craufurd D, Nijsten H, van Duijn E. Obsessive‐compulsive and perseverative behaviors in Huntington's disease. J Huntingtons Dis 2019;8(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andrews SC, Langbehn DR, Craufurd D, et al. Apathy predicts rate of cognitive decline over 24 months in premanifest Huntington's disease. Psychol Med 2020;1–7:1338–1344. [DOI] [PubMed] [Google Scholar]
- 37. Poudel GR, Driscoll S, Domínguez DJF, Stout JC, Churchyard A, Chua P, et al. Functional brain correlates of neuropsychiatric symptoms in presymptomatic Huntington's disease: the IMAGE‐HD study. J Huntingtons Dis 2015;4(4):325–332. [DOI] [PubMed] [Google Scholar]
- 38. Ringkøbing SP, Larsen IU, Jørgensen K, Vinther‐Jensen T, Vogel A. Cognitive screening tests in Huntington gene mutation carriers: examining the validity of the mini‐mental state examination and the Montreal cognitive assessment. J Huntingtons Dis 2020;9(1):59–68. [DOI] [PubMed] [Google Scholar]
- 39. Block CK, Johnson‐Greene D, Pliskin N, Boake C. Discriminating cognitive screening and cognitive testing from neuropsychological assessment: implications for professional practice. Clin Neuropsychol 2017. Apr;31(3):487–500. [DOI] [PubMed] [Google Scholar]
- 40. Roebuck‐Spencer TM, Glen T, Puente AE, Denney RL, Ruff RM, Hostetter G, Bianchini KJ. Cognitive screening tests versus comprehensive neuropsychological test batteries: a national academy of neuropsychology education paper. Arch Clin Neuropsychol 2017;32(4):491–498. [DOI] [PubMed] [Google Scholar]
- 41. Isaacs D, Gibson JS, Stovall J, Claassen DO. The impact of anosognosia on clinical and patient‐reported assessments of psychiatric symptoms in Huntington's disease. J Huntingtons Dis 2020;9:1–12. [DOI] [PubMed] [Google Scholar]
- 42. Sitek EJ, Thompson JC, Craufurd D, Snowden JS. Unawareness of deficits in Huntington's disease. J Huntingtons Dis 2014;3(2):125–135. [DOI] [PubMed] [Google Scholar]
- 43. Wibawa P, Zombor R, Dragovic M, et al. Anosognosia is associated with greater caregiver burden and poorer executive function in Huntington disease. J Geriatr Psychiatry Neurol 2020;33(1):52–58. [DOI] [PubMed] [Google Scholar]
- 44. Toh EA, MacAskill MR, Dalrymple‐Alford JC, Myall DJ, Livingston L, Macleod SAD, et al. Comparison of cognitive and UHDRS measures in monitoring disease progression in Huntington's disease: a 12‐month longitudinal study. Transl Neurodegenerat 2014;3(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heaton RK, Ryan L, Grant I. Demographic Influences and Use of Demographically Corrected Norms in Neuropsychological Assessment; 2009.
- 46. Rivera Mindt M, Byrd D, Saez P, Manly J. Increasing culturally competent neuropsychological services for ethnic minority populations: a call to action. Clin Neuropsychol 2010;24(3):429–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Romero HR, Lageman SK, Kamath V, Irani F, Sim A, Suarez P, et al. Challenges in the neuropsychological assessment of ethnic minorities: summit proceedings. Clin Neuropsychol 2009;23(5):761–779. [DOI] [PubMed] [Google Scholar]
- 48. Mestre TA, Bachoud‐Lévi A, Marinus J, Stout JC, Paulsen JS, Como P, et al. Rating scales for cognition in Huntington's disease: critique and recommendations. Mov Disord 2018;33(2):187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 50. Hunt HA, Van Kampen S, Takwoingi Y, Llewellyn DJ, Pearson M, Hyde CJ. The comparative diagnostic accuracy of the mini mental state examination (MMSE) and the general practitioner assessment of cognition (GPCOG) for identifying dementia in primary care: a systematic review protocol. Diagn Prognostic Res 2017;1(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mickes L, Jacobson M, Peavy G, Wixted JT, Lessig S, Goldstein JL, Corey‐Bloom J. A comparison of two brief screening measures of cognitive impairment in Huntington's disease. Mov Disord 2010;25(13):2229–2233. [DOI] [PubMed] [Google Scholar]
- 52. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53(4):695–699. [DOI] [PubMed] [Google Scholar]
- 53. Chelune GJ, Naugle RI, Lüders H, Sedlak J, Awad IA. Individual change after epilepsy surgery: practice effects and base‐rate information. Neuropsychology 1993;7(1):41–52. [Google Scholar]
- 54. Folstein MF, Folstein SE, White T, Messer MA. Mini‐mental state examination. 2nd ed. Lutz, FL: Psychological Assessment Resources; 2010. [Google Scholar]
- 55. Kokmen E, Smith GE, Petersen RC, Tangalos E, Ivnik RC. The short test of mental status. Correlations with standardized psychometric testing. Arch Neurol 1991. Jul;48(7):725–728. [DOI] [PubMed] [Google Scholar]
- 56. Morley T. Saint Louis university mental status examination (SLUMS); Aging Successfully 2002.
- 57. Health Organization W . World Health Organization disabilty assessment schedule: WHODAS II. Phase 2 field trials. Health Services Research [Internet]; 2000. Available from: https://apps.who.int/iris/bitstream/handle/10665/68350/a80933.pdf. [Google Scholar]
- 58. Lawton MP, Brody EM. Instrumental activities of daily living (Iadl) scale‐self‐rated version. Psychopharmacol Bull 1988;24(4):789–791. [PubMed] [Google Scholar]
- 59. Pfeffer RI, Kurosaki TT, Harrah CH Jr, Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol 1982. May;37(3):323–329. [DOI] [PubMed] [Google Scholar]
- 60. Kroenke K, Spitzer RL, Williams JBW. The PHQ‐9. J Gen Intern Med 2001. Sep;16(9):606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Williams N. The GAD‐7 questionnaire. Occup Med 2014;64(3):224. [Google Scholar]
- 62. Snaith RP. The hospital anxiety and depression scale. Health Qual Life Outcomes 2003;1(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morin CM. Insomnia: psychological assessment and management. New York: Guilford Press; 1993. [Google Scholar]
- 64. Yu L, Buysse DJ, Germain A, et al. Development of short forms from the PROMIS™ sleep disturbance and sleep‐related impairment item banks. Behav Sleep Med 2011;10(1):6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991;14(6):540–545. [DOI] [PubMed] [Google Scholar]
- 66. Buysse DJ, Reynolds CF 3rd, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh sleep quality index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28(2):193–213. [DOI] [PubMed] [Google Scholar]
- 67. Radakovic R, McGrory S, Chandran S, et al. The brief dimensional apathy scale: a short clinical assessment of apathy. Clin Neuropsychol 2020;34(2):423–435. [DOI] [PubMed] [Google Scholar]
- 68. Marin RS, Biedrzycki RC, Firinciogullari S. Reliability and validity of the apathy evaluation scale. Psychiatry Res 1991;38(2):143–162. [DOI] [PubMed] [Google Scholar]
- 69. Bjureberg J, Dahlin M, Carlborg A, Edberg H, Haglund A, Runeson B. Columbia‐suicide severity rating scale screen version: initial screening for suicide risk in a psychiatric emergency department. Psychol Med 2021;26:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Woods P, Almvik R. The Brøset violence checklist (BVC). Acta Psychiatr Scand Suppl 2002;412:103–105. [DOI] [PubMed] [Google Scholar]
- 71. Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI‐Q, a brief clinical form of the neuropsychiatric inventory. J Neuropsychiatry Clin Neurosci 2000;12(2):233–239. [DOI] [PubMed] [Google Scholar]
- 72. Kremer Study Group . Unified Huntingtons disease rating scale: reliability and consistency. Mov Disord [Internet]. 1996; Available from: https://www.narcis.nl/publication/RecordID/oai:repository.ubn.ru.nl:2066%2F23289 [DOI] [PubMed]
- 73. Üstün TB, Kostanjsek N, Chatterji S, Rehm J. Measuring Health and Disability: Manual for WHO Disability Assessment Schedule WHODAS 2.0. World Health Organization; 2010. [Google Scholar]
- 74. Kim JI, Long JD, Mills JA, Downing N, Williams JK, Paulsen JS. Performance of the 12‐item WHODAS 2.0 in prodromal Huntington disease. Eur J Hum Genet 2015;23(11):1584–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Downing NR, Kim JI, Williams JK, Long JD, Mills JA, Paulsen JS. WHODAS 2.0 in prodromal Huntington disease: measures of functioning in neuropsychiatric disease. Eur J Hum Genet 2014;22(8):958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mestre TA, Busse M, Davis AM, et al. Rating scales and performance‐based measures for assessment of functional ability in Huntington's disease: critique and recommendations. Mov Disord Clin Practice 2018;5(4):361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Roché M, Feigenbam P, Fuller R, et al. F25 The functional rating scale 2.0 (FuRST 2.0): from focus groups to focus‐HD. J Neurol, Neuros Psychiatry 2022; 93:A45. [Google Scholar]
- 78. Chatterjee A, Anderson KE, Moskowitz CB, Hauser WA, Marder KS. A comparison of self‐report and caregiver assessment of depression, apathy, and irritability in Huntington's disease. J Neuropsychiatry Clin Neurosci 2005;17(3):378–383. [DOI] [PubMed] [Google Scholar]
- 79. Estalagem A, Bastos H, Cruz MDC. Huntington's disease‐a case of early psychiatric symptoms and suicide. Eur Psychiatry 2021;64(S1):S645. [Google Scholar]
- 80. Kachian ZR, Cohen‐Zimerman S, Bega D, Gordon B, Grafman J. Suicidal ideation and behavior in Huntington's disease: systematic review and recommendations. J Affect Disord 2019. May;1(250):319–329. [DOI] [PubMed] [Google Scholar]
- 81. Paulsen JS, Hoth KF, Nehl C, Stierman L, Group Huntington Study . Critical periods of suicide risk in Huntington's disease. Am J Psychiatry 2005;162(4):725–731. [DOI] [PubMed] [Google Scholar]
- 82. Fisher CA, Sewell K, Brown A, Churchyard A. Aggression in Huntington's disease: a systematic review of rates of aggression and treatment methods. J Huntingtons Dis 2014;3(4):319–332. [DOI] [PubMed] [Google Scholar]
- 83. Del Bene VA, Brandt J. Identifying neuropsychologically impaired physicians. Clin Neuropsychol 2020;34(2):318–331. [DOI] [PubMed] [Google Scholar]
- 84. Gaudet CE, Del Bene VA. Neuropsychological assessment of the aging physician: a Review & Commentary. J Geriatr Psychiatry Neurol 2021;35:279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. DSM‐5, Diagnostic and statistical manual of mental disorders, fifth edition, text revision. HD, Huntington disease.