

Abstract
Methylmalonic acidemia (MMA) is a severe inborn error of metabolism that is characterized by pleiotropic metabolic perturbations and multiorgan pathology. Treatment options are limited and non-curative as the underlying causative molecular mechanisms remain unknown. While earlier studies have focused on the potential direct toxicity of metabolites such as methylmalonic and propionic acid as a mechanism to explain disease pathophysiology, new observations have revealed that aberrant acylation, specifically methylmalonylation, is a characteristic feature of MMA. The mitochondrial sirtuin enzyme SIRT5 is capable of recognizing and removing this PTM, however, reduced protein levels of SIRT5 along with other mitochondrial SIRTs 3 and 4 in MMA and potentially reduced function of all three indicates aberrant acylation may require clinical intervention. Therefore, targeting posttranslational modifications may represent a new therapeutic approach to treat MMA and related organic acidemias.
Keywords: methylmalonyl-CoA mutase, MMA, organic acidemia, PTM, sirtuin
1 |. THE COMPLEX PATHOPHYSIOLOGY OF MMA: HISTORICAL OVERVIEW
Methylmalonic aciduria (MMA) was first identified as an inborn error of metabolism in 1967.1,2 Several years prior, an association between increased methylmalonic acid levels and pernicious anemia had been noted but a direct connection of a B12-dependent enzyme underlying a human metabolic disorder remained suspected, but unknown until Oberholtzer and Stokke published definitive papers on the clinical and biochemical features of the disorder subsequently called MMA.1,2 Soon thereafter, a cobalamin responsive form of the disorder was recognized, firmly establishing a role for vitamin B12 in human intermediary metabolism.3,4 While the original reports documented key features of the disorder, including neonatal lethality, propensity toward infections, kidney disease, poor growth, and recurrent metabolic instability. Concurrently (1961–1969), another related inborn error of metabolism was characterized first as ketotic hyperglycinemia and later as propionic acidemia (PA).5–7 PA presents with many characteristics of MMA, including neonatal lethality,8 and the clinical, biochemical, and molecular features of these inborn errors of metabolism (IEMs) have continued to expand over the decades.
Methylmalonic acidemia is now recognized as genetically heterogeneous inborn error of metabolism, primarily resulting from deficiencies of methylmalonyl-CoA mutase (MMUT) or the enzymes that transport and metabolize vitamin B12 into the active cofactor, 5′-deoxyadenosylcobalamin.9–11 The enzyme MMUT is responsible for the conversion of methylmalonyl-CoA to succinyl-CoA, a TCA cycle intermediate, during the oxidative metabolism of valine, isoleucine, methionine, threonine, cholesterol, and odd-chain fatty acids.9–11 Upstream of MMUT is the propionyl-CoA carboxylase (PCC) complex which converts propionyl-CoA to methylmalonyl-CoA.8 PA is the result of deficiencies in PCC components propionyl-CoA carboxylase alpha chain (PCCA) or propionyl-CoA carboxylase beta chain (PCCB).8 Newborn screening has been implemented in the United States and many but not all European countries to detect elevated levels of propionylcarnitine (C3) in the blood to allow for the early diagnosis and treatment of MMA and PA, which is currently focused on restriction of propiogenic amino acids via a dietary protein restriction.5,12–14 Carnitine supplementation is common15,16 and may operate by reducing the accretion of propionyl-CoA and methylmalonyl-CoA, via carnitine acylation and excretion.14,16 The wide range of organ system involvement in MMA patients (Figure 1) is well appreciated clinically,10 but remains mysterious in terms of molecular mechanisms leading to the disease pathophysiology.
FIGURE 1.
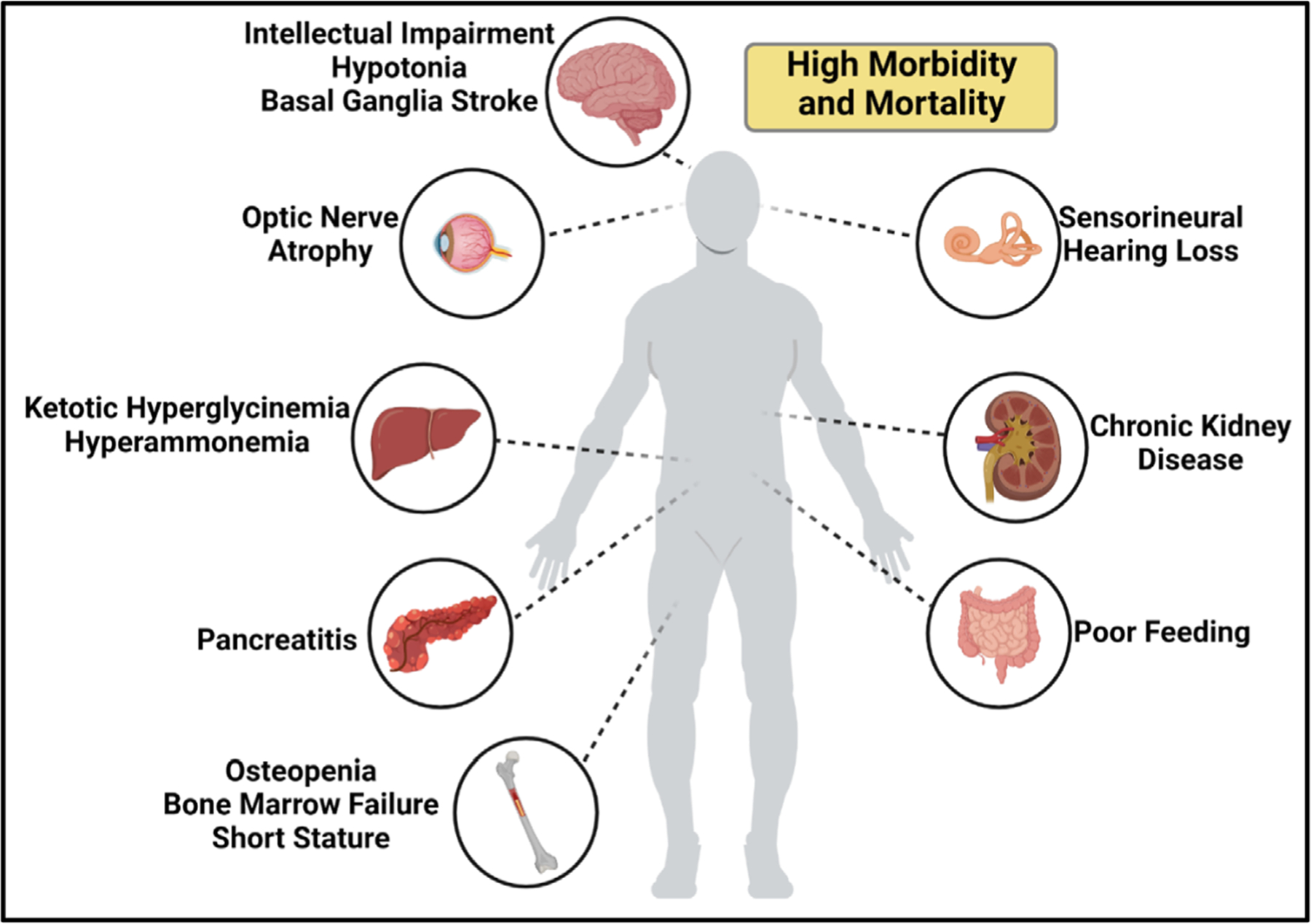
Manifestations of methylmalonic acidemia. (A) pictorial representation of the pathophysiology of MMA and the organ/organ systems most affected.
Patients with severe forms of MMA present early in life, usually within the first few days after birth, with symptoms such as vomiting, lethargy, muscle weakness, seizures, and coma.14 Multiple metabolic perturbations are frequently noted, including hypoglycemia, ketonuria, and hyperammonemia.14 After an affected infant is stabilized, the main management approach is to restrict the intake of branched-chain amino acids, provide aggressive management of symptoms, monitor for long term complications, and in very severe patients, consider an elective liver transplantation to provide metabolic stability. After a liver transplant (LT) or liver and kidney transplant (LKT), risk of metabolic decompensations is reduced,17,18 and there is increased chance of preservation and even improvement in neurocognition.19,20 However, despite successful LT/LKT, patients still manifest biochemical abnormalities such as high methylmalonic acid levels in the blood and cerebral spinal fluid and in those receiving LT only, there is still an significant risk of developing renal insufficiency, basal ganglia injury, and optic nerve disease.19,21,22 In addition, transplantation is accompanied by the risk of organ rejection, and the need for long-term immunosuppression, which can be toxic and increases the risk of malignancy.19,22
2 |. BIOCHEMICAL PATHOLOGY OF MMA
The loss of conversion of methylmalonyl-CoA to succinyl-CoA by MMUT leads to the accumulation of methylmalonyl-CoA, propionyl-CoA and propionyl-CoA derived metabolites such as 2-methylcitrate, which are subsequently converted to their respective acids, especially methylmalonic and propionic acids, which massively accumulate in the tissues and bodily fluids of affected individuals. Initial theories centered on linking the toxicity of CoA and accumulated acids species on metabolic processes, including the postulation that anaplerotic mechanisms might be operative.1,23 In one of the earliest studies, Utter et al demonstrated that methylmalonyl-CoA could compete with acetyl-CoA for the allosteric activating site of pyruvate carboxylase (PC) and inhibit its activity converting pyruvate to oxaloacetate.23 Indeed, Oberholzer et al proposed that in MMA, the metabolic block in the conversion of methylmalonyl-CoA to succinyl-CoA, in concert with methylmalonyl-CoA inhibition of PC production of oxaloacetate from pyruvate, would not only decrease TCA cycle function but also reduce hepatic gluconeogenesis, perhaps explaining the propensity for hypoglycemia development in MMA.1 Following these discoveries, numerous studies shifted to examining the toxicity of accumulated acids.
2.1 |. Direct toxicity of MMA
Early investigations focused on the effects of methylmalonate on metabolite transport in the liver and brain.24–41 Through studies using liver mitochondria isolated from rats and hepatocytes from guinea pigs, methylmalonate was proposed to inhibit malate oxidation as well as PC and downstream gluconeogenesis by blocking malate transport.24,25 Other experiments carried out in rat brains indicate methylmalonate incubation via subcutaneous injection at day 15 of life increased brain glucose uptake but decreased uptake of β-hydroxybutyrate and thus led to indirect inhibition of β-hydroxybutyrate dehydrogenase (HBDH).26 In addition to metabolite transport, methylmalonate was postulated to have direct toxic effects on neural tissue through overproduction of reactive oxidative species (ROS) and dysregulation of cytoskeletal elements.
Propionate and methylmalonate incubation in the cerebral cortexes of 21-day-old rats demonstrated increased free radical generation27 and mice injected with methylmalonate into their cerebral cortexes presented with increased neural inflammation, glial dysfunction, and oxidative damage.28 Increased inflammation and oxidative damage were postulated to be driven by methylmalonate, and was therefore suggested to drive cognitive impairment, memory defects and cerebral dysfunction observed in treated mice.28 This finding was advanced as an explanation for related pathophysiologies in human MMA patients.28 Additionally, methylmalonate and propionate injection into rat cerebral cortexes led to decreased phosphorylation of cytoskeletal proteins which was hypothesized to further contribute to altered brain cytoskeletal metabolism resulting in altered structures in the central nervous system (CNS).29 More recent studies in which ammonia and/or methylmalonate was administered as an intracerebroventricular injection into newborn mice (12 h of age), suggest these metabolites can reduce working memory,30 and activate glial cells resulting in increased cytokine production which could ultimately adversely affect brain development.30 In addition to oxidative damage, cytoskeletal arrangement alteration, and glial cell activation, methylmalonate was tested for direct inhibition of mitochondrial enzymatic functions resulting in energy production deficiencies seen in MMA.
In a study where methylmalonate was incubated on cortex slices from 1-week-old rats, 3-HBDH activity was lower via measurement of 3-hydroybutyrate conversion to acetoacetate and propionate inhibited cerebral lipid synthesis.31 In studies carried out in both rat liver and brain tissues, methylmalonate addition led to inhibition of succinate dehydrogenase (SDH) as well as Complex I and Complex II + III, and therefore became the attributed cause of reduced mitochondrial respiration.32–38 Decreased respiration in the brain was hypothesized to lead to increased lactate production and glucose uptake and perhaps contributed to the convulsions seen in rodents that were administered methylmalonate directly into the brain.34–37 Effects of methylmalonate on ATP depletion, creatine kinase (CK), and phosphocreatine production were also noted.36,39 As a result, creatine supplementation was suggested as a therapy for MMA.36,40 Later studies using rat liver and brain homogenates suggested increased lactate was also the result of methylmalonate inhibition of lactate dehydrogenase (LDH) preventing the conversion of lactate to pyruvate.41
2.2 |. Indirect toxicity and secondary effects: Ammonia metabolism and altered anaplerosis
Hyperammonemia can be a deadly complication of MMA and the related disorder, PA, with secondary disturbance of ammonia metabolism postulated to occur through three major mechanisms. The first is propionyl-CoA competition, the second is the reduction of ammonia scavengers glutamine, alanine, and asparagine, and the third is propionic acid inhibition of TCA cycle anaplerosis. In MMA and PA, increased concentrations of propionyl-CoA can outcompete acetyl-CoA for the active pocket of N-acetylglutamate synthase (NAGS), which normally generates N-acetylglutamate (NAG) from glutamate and acetyl-CoA. NAG is the essential co-factor necessary for carbamoyl phosphate synthetase 1 (CPS1) which initiates the first step in the urea cycle, consuming ATP in the condensation reaction between ammonia and bicarbonate to generate carbamoyl phosphate. If propionyl-CoA outcompetes acetyl-CoA due to differences in abundance, propionyl-glutamate is produced instead and is a much weaker activator of CPS1 leading to increased ammonia in rat liver mitochondria.42 As a result, N-carbamylglutamate has been advocated as a treatment for hyperammonemia seen in MMA and PA to activate CPS1 by circumventing the need for NAG.43,44
It has also been proposed that decreased levels of nitrogen amino acid scavengers contributes to hyperammonemia in MMA and PA. Normally, plasma glutamine, alanine, and asparagine serve as a temporary sink for nitrogen when there is a deficiency in the urea cycle or buildup of ammonia.45 Glutamine is normally generated from glutamate and ammonia in the liver and muscle by glutamine synthetase (GS), alanine is generated via transamination of pyruvate by alanine transaminase in muscle, and asparagine is generated through the transamidation of glutamine by asparagine synthetase.45 PA patients were shown to have significantly reduced levels of plasma glutamine, alanine, and asparagine compared to patients with other forms of urea cycle disorders and it has been postulated that GS is inhibited in PA.45 Furthermore, experiments in 3D rat and mouse MMUT knockout brain cell aggregates indicated methylcitrate may inhibit GS leading to increased ammonia production in the brain.46,47 Methylcitrate is formed from the condensation of propionic acid with oxaloacetate and methylcitrate is elevated in plasma of MMA and PA patients.48 In addition to increased methylcitrate production, excess propionic acid would deplete mitochondrial oxaloacetate and inhibit production of citrate, reducing the amount available to drive the TCA cycle.48 Ultimately, the reduction of oxaloacetate would lead to reduction in α-ketoglutarate, which would drive reverse reactions by glutamate dehydrogenase (GDH) to produce α-ketoglutarate and ammonia from glutamate thus contributing to hyperammonemia.48
In addition to reduced oxaloacetate and α-ketoglutarate, studies performed on human cell lines and tissue extracts from MMA patients demonstrated reduction of succinate and malate.49–52 This is likely due to decreased succinyl-CoA levels as well as observed decreased expression of succinate dehydrogenase flavoprotein subunit A.50 Therefore, anaplerotic therapies such as dimethyl-oxoglutarate have been suggested as a treatment.48,53 Since most theories behind the pathophysiology of MMA centered on the accumulation of toxic acylic acid metabolites, therapies have been targeted toward reducing metabolite load.
3 |. LIMITATIONS OF THE TOXIC METABOLITE THEORY AS AN EXPLANATION FOR MMA PATHOPHYSIOLOGY
Many of the previous studies were carried out using wildtype animals or tissue homogenates derived from normal rats and mice. In other experiments, cell lines were used to carry out testing in the presence of increased metabolites in the cell culture medium. Therefore, these experimental systems may not accurately reflect the disease state, where the metabolites are concentrated in the mitochondria and the affected cells, which are deficient in the MMUT enzyme, appear to have a tissue-specific propensity toward the development of mitochondriopathy that characterizes this disorder.
Both animal and cellular studies using MMUT deficient models illustrate that metabolites alone are insufficient to generate disease pathophysiology.54,55 Mice engineered to express Mmut in the hepatocytes from a germline transgene (Mmut −/− ;TgIns-Alb-Mut) are fully rescued from the lethality of the full knock out state but manifest significant methylmalonic acidemia, similar to MMA patients who have received an LT.54 When control (Mmut +/−;TgIns-Alb-Mut) and affected mice (Mmut−/−; TgIns-Alb-Mut) were placed on high protein diet over the course of 6 months, plasma methylmalonic acid levels in affected Mmut−/−;TgIns-Alb-Mut mice reached approximately 2 mM, mirroring what is seen in MMA patients when renal dysfunction is present.54 Affected mice developed renal pathologies following protein stress including megamitochondria with loss of cristae, however, liver cells which expressed very low levels of MMUT were resistant and displayed totally normal ultrastructure of the mitochondria.54 The accumulation of methylmalonic acid over the course of several months, to massive levels, had no untoward effects on Mmut−/−;TgIns-Alb-Mut hepatocytes, demonstrating that the acylic acid, itself, is not primarily responsible for the secondary metabolic perturbations in MMA, and illustrating that the pathophysiology of MMA exhibits conditional autonomy, with very low levels of MMUT fully protective against the secondary mitochondriopathy that is characteristic of MMA.54 The results in the Mmut−/−;TgIns-Alb-Mut mice suggest that the formation of megamitochondria is a hallmark of MMA pathology and should be further studied in studies using wildtype animals and cell lines incubated with methylmalonic acid.56,57
The role of direct methylmalonic acid toxicity has been further studied in a cellular model. Ramon et al treated human BJ5ta-MMUT fibroblasts with a variety of stressors attempting to induce a phenotype. One regimen studied was the addition of methylmalonic acid at concentrations of 100 mM in the culture medium and like the studies in the MMA mice, no phenotype was observed.55 Additionally, experiments of toxic metabolite addition have not consistently yielded the same results. Several studies were unable to confirm direct inhibition of cytochrome oxidase in rat liver mitochondrial following incubation with methylmalonic, tigilic, or propionic acid, inhibition of dicarboxylic carrier protein, or inhibition of electron transport chain complexes.33,58–60 In aggregate, the animal model and cellular studies strengthen the hypothesis that other mechanisms may be major drivers of disease pathophysiology in MMA, including impaired anaplerosis53 and aberrant posttranslational modifications (PTMs),61 the focus of this review.
4 |. ACYLATIONS, THEIR REGULATION, AND METABOLIC DISEASE
Protein PTMs are chemical moieties covalently installed onto the side chains of amino acids.62 In most instances, placement and removal of PTMs are enzymatically driven62 and PTMs provide a layer of protein regulation as they can increase or decrease protein enzymatic activity or stability, or alter protein localization, charge, or affinity for substrate, cofactor, or other protein binding partner.63 Therefore, a single PTM has the potential to alter an entire pathway or cellular process. Therefore, it is important to evaluate how PTMs are affected in various forms of human disease especially in instances where they can occur nonenzymatically.64 Specifically, forms of acylation such as acetylation, malonylation, glutrylation, propionylation, butyrylation, succinylation, and methylmalonylation in the alkaline environment of the mitochondria move onto protein lysine residues from CoA carriers without enzymatic assistance61,65 (Figure 2). Furthermore, the imbalances of acyl-CoA groups in metabolic disease and disorders further drive nonenzymatic acylation when specific pools of acyl-CoAs become overabundant leading to metabolic dysfunction.66
FIGURE 2.
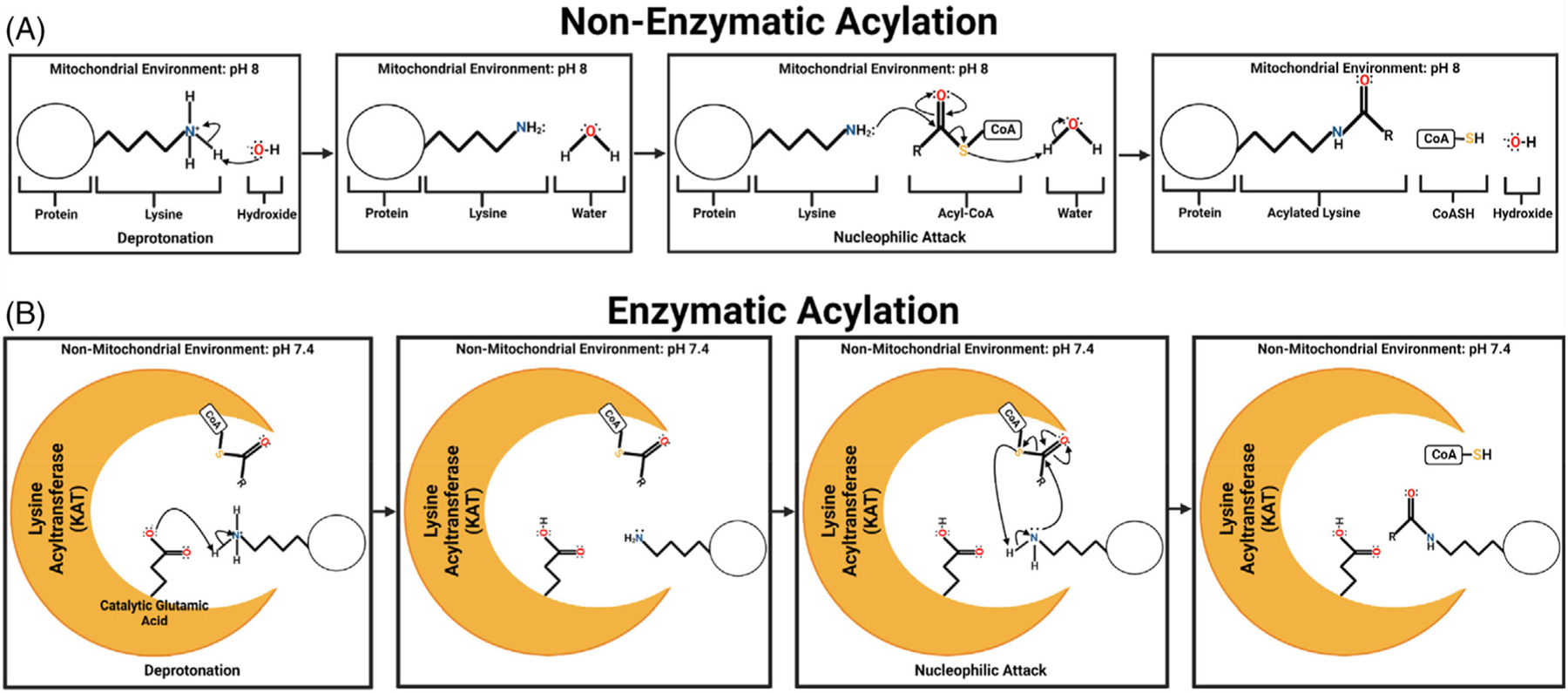
Nonenzymatic and Enzymatic Acylation. (A) Nonenzymatic acylation of lysine substrate. In the alkaline environment of the mitochondria, free hydroxide can serve as the proton acceptor to initiate nucleophilic attack of the Nε amino group of lysine on the reactive thioester of acyl-CoAs. The results are an acylated protein lysine residue, free CoASH, and reformation of hydroxide. (B) Enzymatic acylation of lysine substrate. Rather than free hydroxide, enzymatic acylation is initiated in the active pocket of a lysine acyltransferase or KAT through a catalytic amino acid (in this example it is glutamic acid) that initiates deprotonation. Deprotonation allows for nucleophilic attack of the Nε amino group of lysine on the reactive thioester of acyl-CoA which is held in proximity via binding to the KAT.
Many of the characterized mitochondrial acylation events inhibit the modified protein’s function, which is intuitive given many of these enzymes interact with negatively charged substrates such as amino and carboxylic acids.67 In these cases the positive charge of lysine residues can increase affinity—and therefore activity—toward negatively charged substrates that is negated by acylation.67 Due to the regulatory potential of lysine acylation, mammals have evolved deacylase enzymes responsible for ensuring these modifications are well-regulated. Multiple mechanistically distinct classes of deacylases have been characterized. For the purposes of this discussion, we focus on the class III NAD+ dependent sirtuins (SIRT) due to their central role in the control of metabolism.
The sirtuin protein family consists of seven members that are distinguishable based on their cell compartmentalization, substrate preference or both68–73 (Figure 3A) but share a common mechanism of substrate deacylation74 (Figure 4). Three of these SIRTs (SIRT3–5) localize to the mitochondria to regulate nonenzymatic acylation and thereby maintain homeostasis of key metabolic pathways.67 Loss of these mitochondrial SIRTs, especially in the presence of induced hyperacylation by diet in mice, leads to metabolic dysfunction and demonstrates increased acylation’s role in the pathophysiology of metabolic disease.
FIGURE 3.
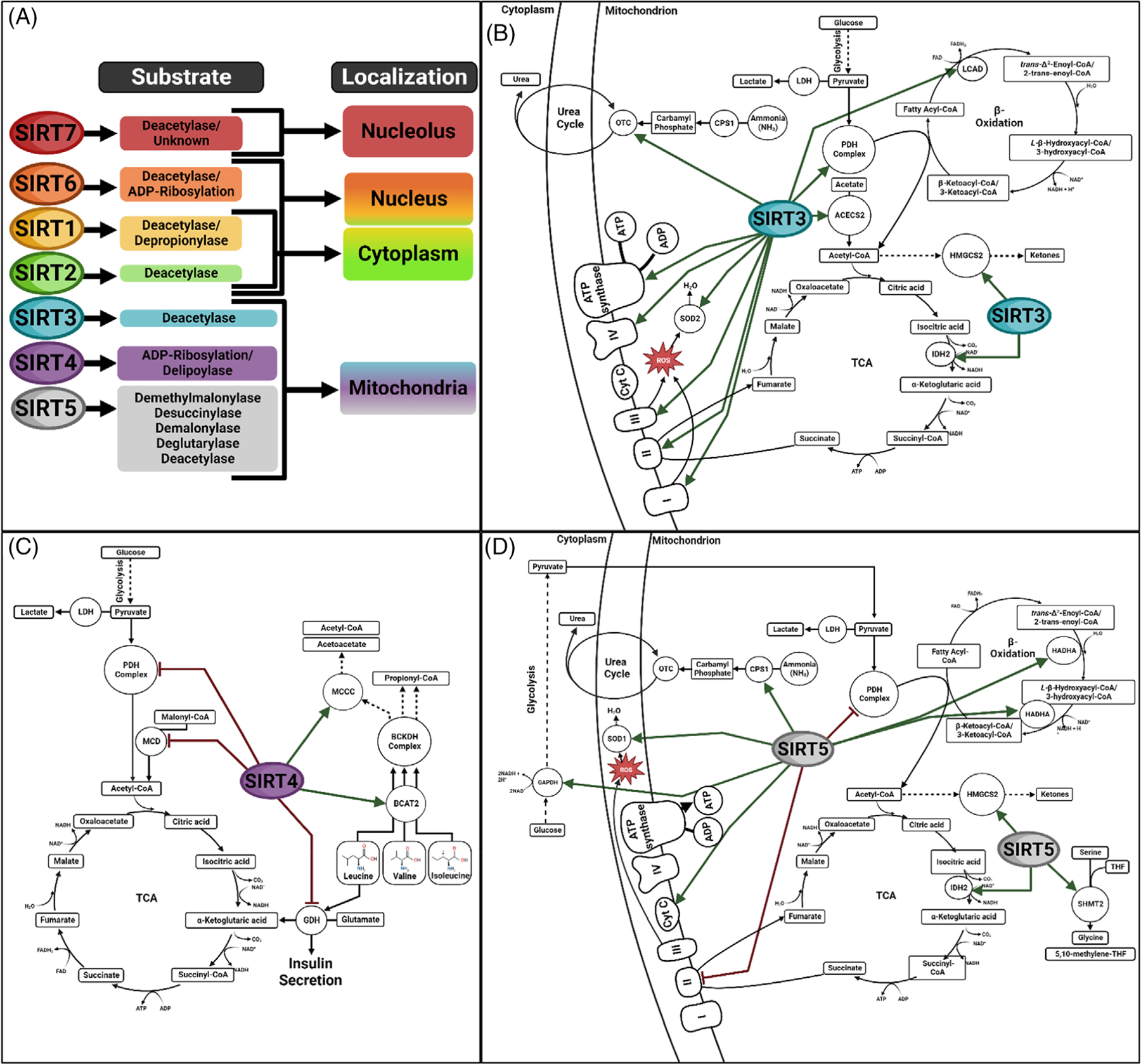
SIRTIUN enzymes: compartmentalization and function. (A) Diagram demonstrating substrate preference and cellular compartment localization of each SIRT family member as well as established function of the mitochondrial SIRTs necessary for preservation of metabolic function.(B) SIRT3 regulated metabolic pathways. Green arrows indicate increased protein activity following modification by SIRT3. (C) SIRT4 regulated metabolic pathways. Green arrows indicate increased protein activity and red bars indicate reduced protein activity following modification by SIRT4. (D) SIRT5 regulated metabolic pathways. Green arrows indicate increased protein activity and red bars indicate reduced protein activity following modification by SIRT5.
FIGURE 4.
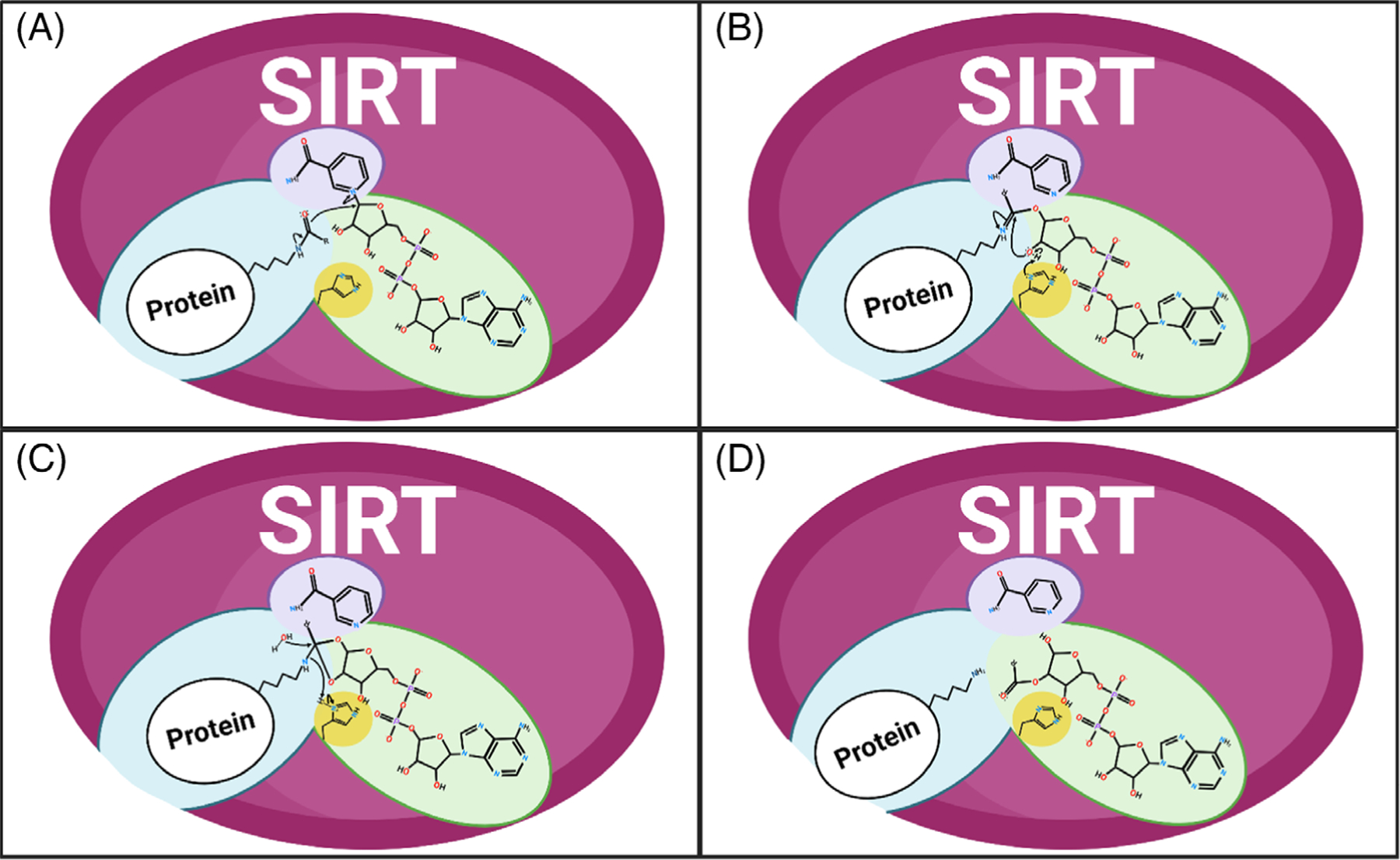
Sirtuin enzymatic deacylation of substrate. (A) Nicotinamide cleavage. Nicotinamide adenine dinucleotide (NAD+) is shown in the purple and green pockets of the representative SIRT enzyme. Acylated lysine protein substrate is highlighted in the blue pocket. (B,C) Formation of the alpha-1’-O-alkylamidate intermediate. Nicotinamide is shown in the purple pocket, the alpha-1’-O-alkylamidate intermediate in the blue and green pockets, and the SIRT catalytic histidine in the yellow pocket. (D) Deacylation of lysine. The reaction yields deacylated lysine (blue pocket), nicotinamide (purple pocket), and 2’-O-acetyl-adenosine diphosphate ribose (green pocket).
A life-threatening complication of methylmalonic acidemia is acute metabolic decompensation or AMD which can present with hyperammonemia, hypoglycemia, metabolic acidosis, ketonuria, hyperglycinemia, and lactic acidosis.4,75,76 Investigations also demonstrate increased oxidative stress and deficiencies in the electron transport chain, the TCA cycle, and TCA anaplerotic pathways in MMA murine and human tissues.53,57,61,77 Similar metabolic dysfunction is seen with the knockout of mitochondrial sirtuins which lead to the hyperacylation of specific metabolic pathways or as a result of hyperacylation. SIRT3 is a mitochondrial deacetylase whose protein targets reside in a variety of metabolic pathways including the electron transport chain, the pyruvate dehydrogenase (PDH) complex, ketogenesis, fatty-acid oxidation, regulation of reactive oxidative species (ROS) levels, and the urea cycle (Figure 3B).67,78–84 Thus when SIRT3 activity is lost or reduced, hyperacetylation occurs in these pathways leading metabolic dysfunction including a significant reduction in ATP production.85–87 SIRT3 is reduced in MMA patient liver tissues which could contribute to metabolic dysregulation.61 One of the primary functions of SIRT3 is to increase energy production through upregulation of the PDH complex, TCA cycle, and electron transport chain while simultaneously minimizing oxidative stress induced by the increase in ROS generation through upregulation of superoxide dismutase 2 (SOD2).67,80 SIRT3 also deacetylates and upregulates isocitrate dehydrogenase (IDH2) in order to increase NADPH which is in turn used to reduce oxidized glutathione (GSSG) to GSH for ROS detoxification.88 In addition, SIRT3 switches to upregulate β-oxidation, ketogenesis through deacetylation and activation of hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), and conversion of acetate to acetyl-CoA by acetyl-CoA synthetase 2 (AceCS2) in times of caloric restriction to fulfill energy demands.78 Simultaneously, SIRT3 increases ornithine transcarbamoylase (OTC) activity to upregulate the urea cycle to offset increased ammonia production from nitrogen catabolic pathways.78,84 In MMA patient livers, SIRT3 protein levels are reduced61 which could contribute to poor cellular energy production regulation. The activities of SIRT4 and SIRT5 or lack thereof are also associated with metabolic perturbations due to altered PTM landscapes.
SIRT4 is a unique SIRT in that it has multiple characterized activities, including the ability to remove acylation and lipoylation marks and catalyze ADP-ribosylation PTMs (Figure 3C). One of the most potent effects of SIRT4 activity is the suppression of insulin secretion in response to glucose.89,90 SIRT4 deacylates and activates enzymes of the methylcrotonyl-CoA carboxylase complex (MCCC), reducing leucine levels necessary for activation of GDH while simultaneously ADP-ribosylating and inactivating GDH directly to reduce insulin secretion in the pancreas.91 Through mouse tissue studies, it has been further determined that SIRT4 KO mice islets of Langerhans cells in the pancreas have higher leucine-stimulated as well as glucose-stimulated insulin secretion as a result of reduced suppression of GDH and reduced activation of MCCC.91 In addition to upregulation of MCCC, studies in human Hek293T and pancreatic cancer cell lines indicated SIRT4 likely deacylates and stabilizes branched-chain amino acid transaminase 2 (BCAT2).92 Increased stabilization leads to increased catabolism of isoleucine and valine in addition to leucine.92
Additionally, SIRT4 suppresses PDH complex activity through delipoylation of the E2 component dihydrolipoyllysine acetyltransferase (DLAT),91 and suppresses the activity of malonyl-CoA decarboxylase (MCD).93 Down-regulation of PDH and MCD would hinder acetyl-CoA production for the TCA cycle. Overall, many of the functions of SIRT4 could have a negative impact in MMA tissues. Perhaps this is why SIRT4 protein levels are reduced in mouse and human MMA liver tissues61 as its depletion would drive reduced catabolism of isoleucine and valine, increased insulin secretion, and upregulation of MCD and the PDH complex. Unlike SIRT3 and SIRT4, SIRT5 targets negatively charged acylations including malonylation, succinylation, glutarylation, and more recently methylmalonylation and has weak activity against neutral acyl groups.61,72,73,94–98
Within the catalytic domain of SIRT5, there are two important amino acid residues arginine 105 and tyrosine 102 which interact with negatively charged acyl substrates.72 Studies have identified succinylated, glutarylated, and malonylated SIRT5 substrates in amino acid degradation, the TCA cycle, ketogenesis, β-oxidation, ketogenesis, fatty acid metabolism, PDH complex, SDH complex, glycolysis, and the urea cycle (Figure 3D).73,94–104 In opposition to SIRT3 function, SIRT5 inhibits the PDH complex and SDH through desuccinylation.95 It is possible that in part, SIRT5 normally inhibits these complexes in response to caloric restriction in order to help shift energy production through β-oxidation, ketogenesis, and fatty acid metabolism. It has also been postulated that reduction of SDH complex activity is a mechanism of ROS reduction/control101 as SIRT5 also upregulates the electron transport chain via upregulation of cytochrome C. This is backed by SIRT5 desuccinylation and activation of superoxide dismutase 1 (SOD1),100 as well as decsuccinylation and activation of IDH2 and demalonylaiton of glucose-6-phosphate dehydrogenase (GAPDH) to produce NADPH.99 This upregulation of GAPDH also serves to increase glycolysis.97 Similar to SIRT3, SIRT5 also upregulates ketogenesis and β-oxidation through desuccinylation of HMGCS2 and hydroxyacyl-CoA dehydrogenase (HADHA), respectively96,102 while protecting against ammonia accumulation through deacylation and activation of CPS1 in the urea cycle.73,94,98,103 SIRT5 can also upregulate amino acid metabolism through desuccinylation of serine hydroxymethyltransferase (SHMT2).104 Just as with SIRT4, SIRT5 protein levels are reduced in the liver of MMA mice and patients61 further reducing the cells ability to reduce harmful hyperacylation and subsequent metabolic dysregulation which likely to drives pathophysiology both in MMMA and potentially other inherited metabolic disease.
5 |. HYPERACYLATION IN MMA AND THE ROLE OF SIRT5
Much like increased global glutarylation that was noted in a mouse model of glutaric acidemia type I (glutaryl-CoA dehydrogenase KO mice),73 there are significant increases in malonyl-, propionyl-, and methylmalonyl-CoA pools in MMA that drive hypermalonylation, hyperpropionylation and hypermethylmalonylation of mitochondrial enzymes.61 Mass spectrometry analysis revealed hyperacylation of metabolic pathways including the TCA cycle, β-oxidation, amino acid metabolism, the urea cycle, glutathione metabolism, PPAR signaling, oxidative stress, mitochondrial DNA replication and stability, carbon metabolism, fatty acid metabolism, glycolysis and gluconeogenesis in MMA patient and mouse tissues as compared to control tissue.61 Further in-depth study confirmed methymalonylation in MMA mouse tissues on components of the urea cycle including CPS1, enzymes in the TCA cycle, and components of the mitochondrial DNA replication and stability complex.61 Overlap with previously identified sites of acylation-dependent enzyme function have suggested a model where in MMA, PTMs drive pathophysiology associated with reduced TCA enzyme function,61,99 hyperammonemia,61,73,94,98 increased activity of GDH161,105 and reduced mitochondrial DNA copy number106 (Figure 5). Furthermore, hyperacylation of glycine cleavage system H protein (GCSH) was discovered using a candidate pathway approach and has provided the foundation for a mechanistic explanation of the hyperglycinemia that historically characterized the disorder61 (Figure 5).
FIGURE 5.
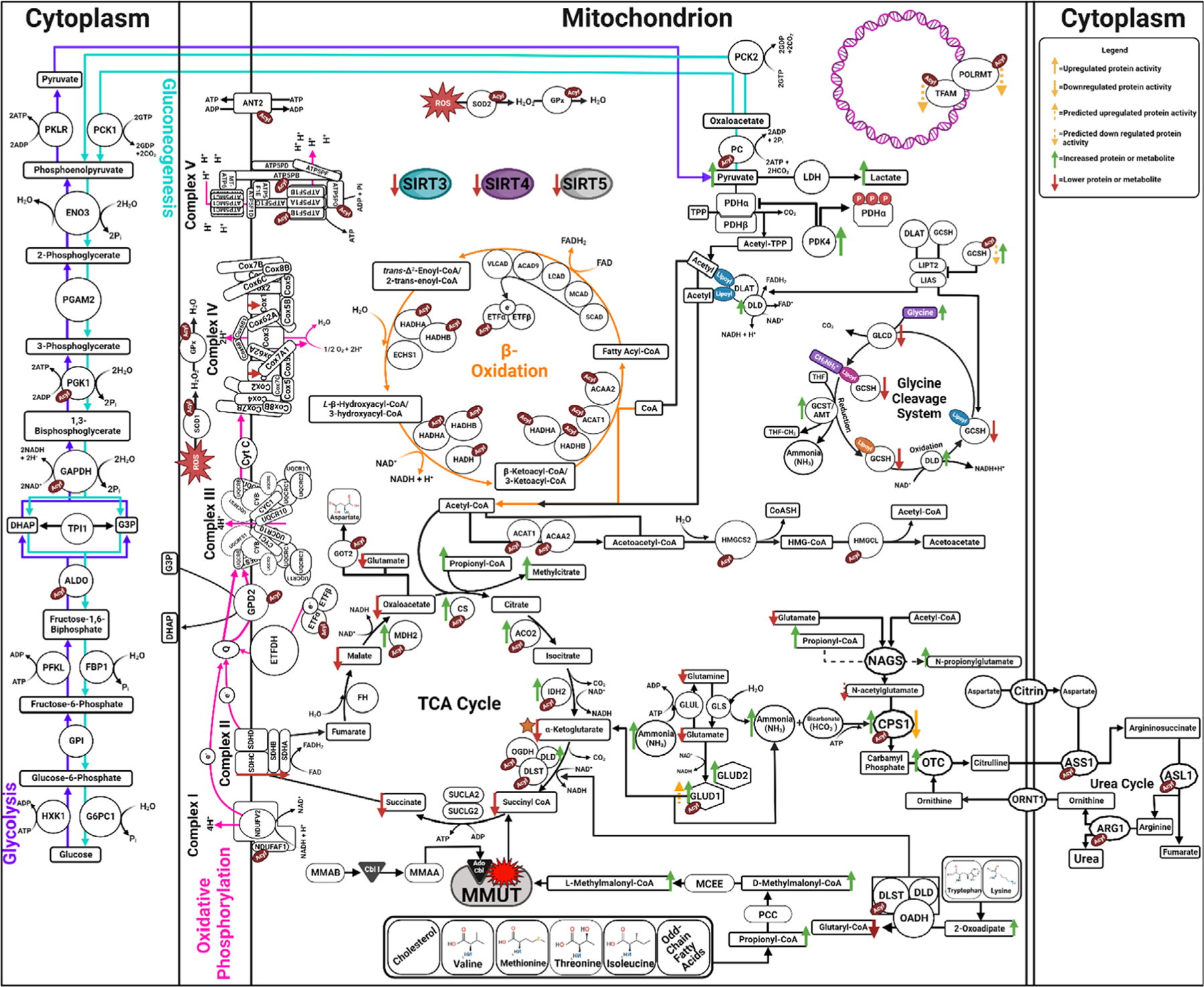
Altered pathway functions in MMA. A summary of alterations seen in the TCA cycle, the urea cycle, and the glycine cleavage cycle in the context of MMA. The red symbol over MMUT is representative of loss of protein function in MMA. Green arrows indicate noted increase in protein abundance or increased metabolite level in MMA. Red arrows indicate decreased protein abundance or metabolite level in MMA. Yellow arrows indicate increased protein function when in the upward direction or decreased protein function when in the downward direction. Dashed line arrows indicate predicted change. Red acyl tags indicate the marked protein exhibits aberrant methylmalonylation, malonylation or both in MMA as found by mass spectrometry analysis in Head et al.61 Blue lipoyl tags indicate the marked protein exhibits lipoylation PTMs. The red phospho tags (P) indicate phosphorylation events. The orange star indicates the point in TCA cycle where synthetic anaplerotic supplementation could be beneficial to increasing TCA function in MMA.
In addition, genetic studies further support the importance of SIRT5 and PTM metabolism as a contributor to disease severity in MMA. SIRT5 KO mice, when crossed to a mildly affected MMA mouse (MMUTG715V/G715V), resulted in increased runting, increased global methylmalonylation in liver extracts especially of CPS1, and increased blood ammonia levels in the knock-out transgenic progeny compared to parental strains or controls, further demonstrating the role of SIRT5 in methylmalonyl-lysine metabolism and as a mediator of disease severity such as hyperammonemia.61 Furthermore, MMA mouse and patient liver tissues exhibited significant reduction in SIRT5 protein levels even SIRT5 mRNA levels were not reduced in MMA mice.56,61 MMA pathophysiology is exacerbated with SIRT5 loss and reduced when SIRT5 is overexpressed.61
The hypermethylmalonylation of the hepatic MMA proteome, reduction of endogenous SIRT5, and in vitro activity of SIRT5 against methylmalonyl-lysine led to a proof-of-concept study to determine whether addition of a modified SIRT5 enzyme resistant to inactivation by aberrant acylation (superSIRT5) was effective as a therapeutic independent of MMUT activity.61 Indeed, when an AAV8 vector configured to express superSIRT5 in the liver was delivered to Mmut—/—;TgINS-MCK-Mmut mice (MMA mice with limited tissue specific expression of MMUT in skeletal muscle), MMA mice demonstrated increased weight gain, reduced global methylmalonylation, and reduced ammonia levels as compared to MMA mice treated with a vector similarly configured but designed to express eGFP.61 Whether such an approach may benefit renal or CNS manifestations is unknown and under further study. These findings suggest a new treatment approach for MMA, and potentially other organic acidemias, based on increased deacylase activity. At this time, we are unaware of specific SIRT5 agonists, and while pan-SIRT activators could have unsuitable off target effects, they may represent an interesting new avenue for small molecule modulation of deleterious PTMs in MMA and related disorders.
6 |. FUTURE DIRECTIONS: DEFINING AND MANIPULATING THE MMA-ASSOCIATED ACYLOME
The identification of sirtuin-regulated lysine methylmalonylation has expanded the mechanistic landscape of MMA. However, the profiling and functional characterization of this PTM remains in its infancy. Expanding our knowledge of hyperacylated proteins–not only in the mitochondria, but in all cellular compartments across multiple cell and tissue types–in MMA and other IEMs will be necessary to better understand the contributions of this phenomenon to the molecular underpinnings of disease pathophysiology. In addition to traditional genetic and biological techniques, going forward we envision chemical biology could play a critical role in defining and manipulating aberrant acylation in MMA and related disorders. Unlike canonical enzyme-catalyzed histone acetylation, substantial evidence indicates that SIRT5-regulated protein malonylation, succinylation, and glutarylation is driven nonenzymatically by the reactivity of their acyl-CoA precursors.65,107–110 In particular, the high reactivity of malonyl-CoA has been attributed to the electron-withdrawing effect of the β-carboxy group, an attribute that should also extend to methylmalonyl-CoA.111,112 In the future we envision that chemoproteomic studies of thioester reactivity may be helpful in identifying new protein targets of aberrant hyperacylation.111 Continued development of small molecule modulators of SIRT5-regulated acylation, including nonenzymatic “acylators”111 and drug-like inhibitors of this enzyme,113 will enable the determination of how hyperacylation contributes to MMA pathology without the need for genetic manipulation of SIRT5. Recent studies have indicated the presence of substantial propionyl-CoA in the nucleus capable of fueling histone modifications.114 It may be intuited this pool is enlarged by propionate accumulation in MMA. We anticipate that newly developed drug-like inhibitors of known histone propionyltransferases like EP300/CREBBP and KAT6A/KAT6B,115,116 as well as biorthogonal metabolites capable of identifying new propionylation substrates,117 may help elucidate how MMA alters these nuclear signaling processes and inform novel therapeutic avenues. Beyond acylation, numerous functions have been attributed to the reversible interaction of MMA metabolites such as methylmalonyl-CoA, methylmalonic acid, and propionate with enzyme targets. Here again, applying the arsenal of methods commonly applied in drug target identification (e.g., affinity-based chemoproteomics, cellular thermal shift assay) to identify new targets of these metabolites will likely yield new insights into their function and selectivity.118 In addition to identifying new targets of aberrant acylation and metabolite-regulated signaling, strategies aimed at intervening in these processes will also be a critical priority. Approaches to reduce deleterious PTMs, such as attempting to increase NAD+ levels by supplementing with nicotinamide riboside or competitively consume CoA,119 could provide a foundation for new therapies designed to mitigate the chemical pathology, and hopefully clinical symptoms, seen in a variety of IEMs.120
FUNDING INFORMATION
PamelaSara E. Head was supported by a Postdoctoral Research Associate (PRAT) fellowship from the National Institute of General Medical Sciences (NIGMS), award number 1FI2GM137781-01. Charles P. Venditti was supported by the Intramural Research Program of the NHGRI through 1ZIAHG200318-16. Jordan L. Meier was supported by the Intramural Research Program of the NCI through 1TZIABC011488-09.
National Cancer Institute, Grant/Award Number: 1TZIABC011488-09; National Human Genome Research Institute, Grant/Award Number: 1ZIAHG200318-16; National Institute of General Medical Sciences, Grant/Award Number: 1FI2GM137781-01
Footnotes
CONFLICT OF INTEREST STATEMENT
Venditti reports previous nonfinancial support and other from Moderna Therapeutics, nonfinancial support and other from LogicBio Therapeutics, nonfinancial support and other from Translate Bio, nonfinancial support and other from AskBio, all outside the submitted work. Venditti currently receives nonfinancial support and other from Selecta Biosciences. In addition, on the behalf of Venditti and Head, the NIH has filed of number of patents related to medical devices, gene therapy, and biomarkers. Meier reports no conflicts.
ETHICS STATEMENT
Authors declare human ethics approval was not needed for this publication.
PATIENT CONSENT STATEMENT
This article does not contain any studies with human subjects performed by any of the authors.
ANIMAL RIGHTS STATEMENT
This article does not contain any studies with animals.
DATA AVAILABILITY STATEMENT
Supporting data in this review has been published and cited in the article.
REFERENCES
- 1.Oberholzer VG, Levin B, Burgess EA, Young WF. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis. Arch Dis Child 1967;42:492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stokke O, Eldjarn L, Norum KR, Steenjoh J, Halvorsen S. Methylmalonic acidemia—a new inborn error of metabolism which may cause fatal acidosis in neonatal period. Scand J Clin Lab Inv 1967;20:313–328. [Google Scholar]
- 3.Rosenberg LE, Lilljeqvist A, Hsia YE. Methylmalonic aciduria: metabolic block localization and vitamin B 12 dependency. Science 1968;162:805–807. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg LE, Lilljeqvist AC, Hsia YE. Methylmalonic aciduria—an inborn error leading to metabolic acidosis, long-chain ketonuria and intermittent hyperglycinemia. N Engl J Med 1968;278:1319–1322. [DOI] [PubMed] [Google Scholar]
- 5.Childs B, Nyhan WL, Borden M, Bard L, Cooke RE. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I Pediatrics 1961;27:522–538. [PubMed] [Google Scholar]
- 6.Nyhan WL, Borden M, Childs B. Idiopathic hyperglycinemia: a new disorder of amino acid metabolism. II. The concentrations of other amino acids in the plasma and their modification by the administration of leucine. Pediatrics 1961;27:539–550. [PubMed] [Google Scholar]
- 7.Hsia YE, Scully KJ, Rosenberg LE. Defective propionate carboxylation in ketotic hyperglycinaemia. Lancet 1969;1: 757–758. [DOI] [PubMed] [Google Scholar]
- 8.Shchelochkov OA, Carrillo N, Venditti C. In: Adam MP et al. , eds. GeneReviews((R)) Univeristy of Washington; 1993. [Google Scholar]
- 9.Fenton WA, Gravel RA, Rosenblatt DS. Disorders of propionate and methylmalonate metabolism. The Metabolic and Molecular Bases of Inherited Disease McGraw Hill Medical; 2001:2165–2193. [Google Scholar]
- 10.Manoli I, Sloan JL, Venditti CP. In: Adam MP et al. , eds. GeneReviews((R)) Univeristy of Washington; 1993. [Google Scholar]
- 11.Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr 2016; 28:682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumgartner MR, Hörster F, Dionisi-Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014; 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manoli I, Myles JG, Sloan JL, Shchelochkov OA, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet Med 2016;18: 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manoli I, Venditti CP. Disorders of branched chain amino acid metabolism. Transl Sci Rare Dis 2016;1:91–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brass EP, Fennessey PV, Miller LV. Inhibition of oxidative metabolism by propionic acid and its reversal by carnitine in isolated rat hepatocytes. Biochem J 1986;236:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brass EP, Stabler SP. Carnitine metabolism in the vitamin B-12-deficient rat. Biochem J 1988;255:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forny P, Hörster F, Ballhausen D, et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: first revision. J Inherit Metab Dis 2021; 44:566–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niemi AK, Kim IK, Krueger CE, et al. Treatment of methylmalonic acidemia by liver or combined liver-kidney transplantation. J Pediatr 2015;166:1455–1461 e1451. [DOI] [PubMed] [Google Scholar]
- 19.Sloan JL, Manoli I, Venditti CP. Liver or combined liver-kidney transplantation for patients with isolated methylmalonic acidemia: who and when? J Pediatr 2015;166:1346–1350. [DOI] [PubMed] [Google Scholar]
- 20.Martinelli D, Catesini G, Greco B, et al. Neurologic outcome following liver transplantation for methylmalonic aciduria. J Inherit Metab Dis 2023;46:450–465. [DOI] [PubMed] [Google Scholar]
- 21.Chakrapani A, Sivakumar P, McKiernan PJ, Leonard JV. Metabolic stroke in methylmalonic acidemia five years after liver transplantation. J Pediatr-us 2002;140:261–263. [DOI] [PubMed] [Google Scholar]
- 22.Brassier A, Krug P, Lacaille F, et al. Long-term outcome of methylmalonic aciduria after kidney, liver, or combined liver-kidney transplantation: the French experience. J Inherit Metab Dis 2020;43:234–243. [DOI] [PubMed] [Google Scholar]
- 23.Utter MF, Keech DB, Scrutton MC. A possible role for acetyl coa in the control of gluconeogenesis. Adv Enzyme Regul 1964;2:49–68. [DOI] [PubMed] [Google Scholar]
- 24.Halperin ML, Schiller CM, Fritz IB. The inhibition by methylmalonic acid of malate transport by the dicarboxylate carrier in rat liver mitochondria. A possible explantation for hypoglycemia in methylmalonic aciduria. J Clin Invest 1971;50:2276–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arinze IJ, Waters D, Donaldson MK. Effect of methylamalonic acid on gluconeogenesis in isolated rat and Guinea-pig hepatocytes. Biochem J 1979;184:717–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutra JC, Wajner M, Wannmacher CF, Dutra-Filho CS, Wannmacher CMD. Effects of methylmalonate and propionate on uptake of glucose and ketone bodies in vitro by brain of developing rats. Biochem Med Metab Biol 1991;45:56–64. [DOI] [PubMed] [Google Scholar]
- 27.Fontella FU, Pulrolnik V, Gassen E, et al. Propionic and L-methylmalonic acids induce oxidative stress in brain of young rats. Neuroreport 2000;11:541–544. [DOI] [PubMed] [Google Scholar]
- 28.Gabbi P, Ribeiro LR, Jessié Martins G, et al. Methylmalonate induces inflammatory and apoptotic potential: a link to glial activation and neurological dysfunction. J Neuropathol Exp Neurol 2017;76:160–178. [DOI] [PubMed] [Google Scholar]
- 29.De Mattos-Dutra A, De Freitas MS, Lisboa CSF, Pessoa-Pureur R, Wajner M. Effects of acute and chronic administration of methylmalonic and propionic acids on the in vitro incorporation of 32P into cytoskeletal proteins from cerebral cortex of young rats. Neurochem Int 1998;33:75–82. [DOI] [PubMed] [Google Scholar]
- 30.Gabbi P, Nogueira V, Haupental F, et al. Ammonia role in glial dysfunction in methylmalonic acidemia. Toxicol Lett 2018;295:237–248. [DOI] [PubMed] [Google Scholar]
- 31.Patel MS, Owen OE, Raefsky C. Effect of methylmalonate on ketone body metabolism in developing rat brain. Life Sci 1976;19:41–47. [DOI] [PubMed] [Google Scholar]
- 32.Dutra JC, Dutra-Filho CS, Cardozo SEC, Wannmacher CMD, Sarkis JJF, Wajner M. Inhibition of succinate dehydrogenase and β-hydroxybutyrate dehydrogenase activities by methylmalonate in brain and liver of developing rats. J Inherit Metab Dis 1993;16:147–153. [DOI] [PubMed] [Google Scholar]
- 33.Toyoshima S, Watanabe F, Saido H, Miyatake K, Nakano Y. Methylmalonic acid inhibits respiration in rat liver mitochondria. Journal of Nutrition 1995;125:2846–2850. [DOI] [PubMed] [Google Scholar]
- 34.De Mello CF, Begnini J, Jiménez-Bernal RE, et al. Intrastriatal methylmalonic acid administration induces rotational behavior and convulsions through glutamatergic mechanisms. Brain Res 1996;721:120–125. [DOI] [PubMed] [Google Scholar]
- 35.Brusque AM, Borba Rosa R, Schuck PF, et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem Int 2002; 40:593–601. [DOI] [PubMed] [Google Scholar]
- 36.Royes LFF, Fighera MR, Furian AF, et al. Creatine protects against the convulsive behavior and lactate production elicited by the intrastriatal injection of methylmalonate. Neuroscience 2003;118:1079–1090. [DOI] [PubMed] [Google Scholar]
- 37.Marisco PD, Ribeiro MCP, Bonini JS, et al. Ammonia potentiates methylmalonic acid-induced convulsions and TBARS production. Exp Neurol 2003;182:455–460. [DOI] [PubMed] [Google Scholar]
- 38.Narasimhan P, Sklar R, Murrell M, Swanson RA, Sharp FR. Methylmalonyl-CoA mutase induction by cerebral ischemia and neurotoxicity of the mitochondrial toxin methylmalonic acid. J Neurosci 1996;16:7336–7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuck PF, Rosa RB, Pettenuzzo LF, et al. Inhibition of mitochondrial creatine kinase activity from rat cerebral cortex by methylmalonic acid. Neurochem Int 2004;45:661–667. [DOI] [PubMed] [Google Scholar]
- 40.Royes LF, Fighera MR, Furian AF, et al. Effectiveness of creatine monohydrate on seizures and oxidative damage induced by methylmalonate. Pharmacol Biochem Behav 2006;83:136–144. [DOI] [PubMed] [Google Scholar]
- 41.Saad LO, Mirandola SR, Maciel EN, Castilho RF. Lactate dehydrogenase activity is inhibited by methylmalonate in vitro. Neurochem Res 2006;31:541–548. [DOI] [PubMed] [Google Scholar]
- 42.Coude FX, Sweetman L, Nyhan WL. Inhibition by propionylcoenzyme a of N-acetylglutamate synthetase in rat liver mitochondria. A possible explanation for hyperammonemia in propionic and methylmalonic acidemia. J Clin Invest 1979;64: 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics 2010;126:e208–e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yap S, Leong HY, Abdul Aziz F, et al. N-Carbamylglutamate is an effective treatment for acute neonatal hyperammonaemia in a patient with methylmalonic aciduria. Neonatology 2016;109:303–307. [DOI] [PubMed] [Google Scholar]
- 45.Tuchman M, Yudkoff M. Blood levels of ammonia and nitrogen scavenging amino acids in patients with inherited hyperammonemia. Mol Genet Metab 1999;66:10–15. [DOI] [PubMed] [Google Scholar]
- 46.Cudre-Cung HP, Zavadakova P, do Vale-Pereira S, et al. Ammonium accumulation is a primary effect of 2-methylcitrate exposure in an in vitro model for brain damage in methylmalonic aciduria. Mol Genet Metab 2016;119: 57–67. [DOI] [PubMed] [Google Scholar]
- 47.Remacle N, Forny P, Cudré-Cung HP, et al. New in vitro model derived from brain-specific Mut−/− mice confirms cerebral ammonium accumulation in methylmalonic aciduria. Mol Genet Metab 2018;124:266–277. [DOI] [PubMed] [Google Scholar]
- 48.Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, Longo N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol Genet Metab 2006;88: 123–130. [DOI] [PubMed] [Google Scholar]
- 49.Knerr I, Weinhold N, Vockley J, Gibson KM. Advances and challenges in the treatment of branched-chain amino/keto acid metabolic defects. J Inherit Metab Dis 2012;35:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caterino M, Chandler RJ, Sloan JL, et al. The proteome of methylmalonic acidemia (MMA): the elucidation of altered pathways in patient livers. Mol Biosyst 2016;12:566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Dirbashi O, Alfadhel M, Al-Thihli K, et al. Assessment of methylcitrate and methylcitrate to citrate ratio in dried blood spots as biomarkers for inborn errors of propionate metabolism. Sci Rep 2019;9:12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collado MS, Armstrong AJ, Olson M, et al. Biochemical and anaplerotic applications of in vitro models of propionic acidemia and methylmalonic acidemia using patient-derived primary hepatocytes. Mol Genet Metab 2020;130:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forny P, Bonilla X, Lamparter D, et al. Integrated multi-omics reveals anaplerotic rewiring in methylmalonyl-CoA mutase deficiency. Nat Metab 2023;5:80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manoli I, Sysol JR, Li L, et al. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc Natl Acad Sci U S A 2013;110: 13552–13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramon C, Traversi F, Burer C, Froese DS, Stelling J. Cellular and computational models reveal environmental and metabolic interactions in MMUT-type methylmalonic aciduria. J Inherit Metab Dis 2022:1–15. [DOI] [PubMed] [Google Scholar]
- 56.Manoli I, Sysol JR, Epping MW, et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight 2018;3:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chandler RJ, Zerfas PM, Shanske S, et al. Mitochondrial dysfunction in Mut methylmalonic acidemia. FASEB Journal 2009;23:1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayasaka K, Metoki K, Satoh T, et al. Comparison of cytosolic and mitochondrial enzyme alterations in the livers of propionic or Methylmalonic acidemia: a reduction of cytochrome oxidase activity. Tohoku J Exp Med 1982;137:329–334. [DOI] [PubMed] [Google Scholar]
- 59.Kölker S, Schwab M, Hörster F, et al. Methylmalonic acid, a biochemical Hallmark of Methylmalonic acidurias but No inhibitor of mitochondrial respiratory chain. J Biol Chem 2003;278:47388–47393. [DOI] [PubMed] [Google Scholar]
- 60.Kölker S, Okun JG. Methylmalonic acid—an endogenous toxin? Cell Mol Life Sci 2005;62:621–624. [DOI] [PubMed] [Google Scholar]
- 61.Head PE, Myung S, Chen Y, et al. Aberrant methylmalonylation underlies methylmalonic acidemia and is attenuated by an engineered sirtuin. Sci Transl Med 2022;14:eabn4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zee BM, Garcia BA. Discovery of lysine post-translational modifications through mass spectrometric detection. Essays Biochem 2012;52:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parekh RB, Rohlff C. Post-translational modification of proteins and the discovery of new medicine. Curr Opin Biotechnol 1997;8:718–723. [DOI] [PubMed] [Google Scholar]
- 64.Baker PR 2nd, Friederich MW, Swanson MA, et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2014;137:366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem 2013;288:29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pougovkina O, Te Brinke H, Wanders RJ, Houten SM, de Boer VC. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. J Inherit Metab Dis 2014;37:709–714. [DOI] [PubMed] [Google Scholar]
- 67.Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial Acylome emerges: proteomics, regulation by Sirtuins, and metabolic and disease implications. Cell Metab 2018;27:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flick F, Luscher B. Regulation of sirtuin function by post-translational modifications. Front Pharmacol 2012;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol 2012;4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi JE, Mostoslavsky R. Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev 2014;26:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldman JL, Dittenhafer-Reed KE, Kudo N, et al. Kinetic and structural basis for acyl-group selectivity and NAD(+) dependence in Sirtuin-catalyzed deacylation. Biochemistry 2015;54:3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du J, Zhou Y, Su X, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011;334: 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan M, Peng C, Anderson KA, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 2014;19:605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawse WF, Hoff KG, Fatkins DG, et al. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure 2008;16:1368–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henriquez H, el Din A, Ozand PT, Subramanyam SB, al Gain SI. Emergency presentations of patients with methylmalonic acidemia, propionic acidemia and branched chain amino acidemia (MSUD). Brain Dev 1994;16:86–93. [DOI] [PubMed] [Google Scholar]
- 76.Haijes HA, Jans JJM, van der Ham M, van Hasselt PM, Verhoeven-Duif NM. Understanding acute metabolic decompensation in propionic and methylmalonic acidemias: a deep metabolic phenotyping approach. Orphanet J Rare Dis 2020;15:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Keyzer Y, Valayannopoulos V, Benoist JF, et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr Res 2009;66:91–95. [DOI] [PubMed] [Google Scholar]
- 78.Hirschey MD, Shimazu T, Capra JA, Pollard KS, Verdin E. SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging (Albany NY) 2011;3:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol 2011;76:267–277. [DOI] [PubMed] [Google Scholar]
- 80.Jing E, O’Neill BT, Rardin MJ, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013;62:3404–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fan J, Shan C, Kang HB, et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell 2014;53:534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hirschey MD, Shimazu T, Goetzman E, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Y, Zhang J, Lin Y, et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011;12:534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hallows WC, Yu W, Smith BC, et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell 2011;41:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahn BH, Kim HS, Song S, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 2008;105:14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010;49:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kendrick AA, Choudhury M, Rahman SM, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 2011;433:505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Someya S, Yu W, Hallows WC, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010;143:802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haigis MC, Mostoslavsky R, Haigis KM, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006;126:941–954. [DOI] [PubMed] [Google Scholar]
- 90.Ahuja N, Schwer B, Carobbio S, et al. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem 2007;282:33583–33592. [DOI] [PubMed] [Google Scholar]
- 91.Anderson KA, Huynh FK, Fisher-Wellman K, et al. SIRT4 is a lysine Deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017;25:838–855 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lei MZ, Li XX, Zhang Y, et al. Acetylation promotes BCAT2 degradation to suppress BCAA catabolism and pancreatic cancer growth. Signal Transduct Target Ther 2020;5:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laurent G, German NJ, Saha AK, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol Cell 2013;50:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakagawa T, Guarente L. Urea cycle regulation by mitochondrial sirtuin, SIRT5. Aging (Albany NY) 2009;1:578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Park J, Chen Y, Tishkoff DX, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 2013;50:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rardin MJ, He W, Nishida Y, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 2013;18:920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nishida Y, Rardin MJ, Carrico C, et al. SIRT5 regulates both cytosolic and mitochondrial protein Malonylation with glycolysis as a major target. Mol Cell 2015;59:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009;137:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou L, Wang F, Sun R, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep 2016;17:811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin ZF, Xu HB, Wang JY, et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun 2013;441:191–195. [DOI] [PubMed] [Google Scholar]
- 101.Tang X, Chen XF, Chen HZ, Liu DP. Mitochondrial Sirtuins in cardiometabolic diseases. Clin Sci (Lond) 2017;131:2063–2078. [DOI] [PubMed] [Google Scholar]
- 102.Sadhukhan S, Liu X, Ryu D, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A 2016;113: 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ogura M, Nakamura Y, Tanaka D, et al. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem Biophys Res Commun 2010;393:73–78. [DOI] [PubMed] [Google Scholar]
- 104.Yang X, Wang Z, Li X, et al. SHMT2 Desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res 2018;78: 372–386. [DOI] [PubMed] [Google Scholar]
- 105.Aleshin VA, Mkrtchyan GV, Kaehne T, Graf AV, Maslova MV, Bunik VI. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J Neurochem 2020; 153:80–102. [DOI] [PubMed] [Google Scholar]
- 106.King GA, Hashemi Shabestari M, Taris KKH, et al. Acetylation and phosphorylation of human TFAM regulate TFAM-DNA interactions via contrasting mechanisms. Nucleic Acids Res 2018;46:3633–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wagner GR, Bhatt DP, O’Connell TM, et al. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab 2017;25:823–837 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baeza J, Smallegan MJ, Denu JM. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol 2015;10: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.James AM, Smith AC, Ding S, et al. Nucleotide-binding sites can enhance N-acylation of nearby protein lysine residues. Sci Rep 2020;10:20254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anmangandla A, Ren Y, Fu Q, Zhang S, Lin H. The acyl-CoA specificity of human lysine acetyltransferase KAT2A. Biochemistry 2022;61:1874–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kulkarni RA, Worth AJ, Zengeya TT, et al. Discovering targets of non-enzymatic acylation by thioester reactivity profiling. Cell Chem Biol 2017;24:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Walsh CT Jr, Hildebrand JG, Spector LB. Succinyl phosphate. Its nonenzymatic hydrolysis and reaction with coenzyme a. J Biol Chem 1970;245:5699–5708. [PubMed] [Google Scholar]
- 113.Yan D, Franzini A, Pomicter AD, et al. Sirt5 is a druggable metabolic vulnerability in acute myeloid leukemia. Blood Cancer Discov 2021;2:266–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Trefely S, Huber K, Liu J, et al. Quantitative subcellular acyl-CoA analysis reveals distinct nuclear metabolism and isoleucine-dependent histone propionylation. Mol Cell 2022; 82:447–462 e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baell JB, Leaver DJ, Hermans SJ, et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 2018;560:253–257. [DOI] [PubMed] [Google Scholar]
- 117.Sinclair WR, Shrimp JH, Zengeya TT, et al. Bioorthogonal pro-metabolites for profiling short chain fatty acylation. Chem Sci 2018;9:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kulkarni RA, Montgomery DC, Meier JL. Epigenetic regulation by endogenous metabolite pharmacology. Curr Opin Chem Biol 2019;51:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Armstrong AJ, Henke BR, Collado MS, et al. Identification of 2,2-Dimethylbutanoic acid (HST5040), a clinical development candidate for the treatment of propionic acidemia and Methylmalonic acidemia. J Med Chem 2021;64:5037–5048. [DOI] [PubMed] [Google Scholar]
- 120.Canto C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 2012;15:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supporting data in this review has been published and cited in the article.