

Abstract
The sustained metabolic activation of the brain’s default-mode network is thought to render the system vulnerable to Alzheimer’s disease. Recent results with transgenic mice support this view by linking neuronal activity to interstitial fluid amyloid-β levels and the development of amyloid-β plaques.
As most long-distance runners will tell you, sustained activity comes at a price. The health benefits of regular exercise, with time and age, eventually must compete with side-effects such as tendonitis and aching joints. Is the same true in the nervous system? Specifically, do areas of the brain that are chronically active, areas that foster many of our intellectual and creative gifts, cultivate the seeds of one of our greatest late-life tragedies, Alzheimer’s disease? In this issue of Nature Neuroscience, Bero and colleagues1 present data from transgenic mouse models indicating that this may be so.
A decade ago, Marcus Raichle proposed the concept of a ‘default-mode’ network to describe a collection of interconnected brain regions that maintain robust metabolic activity when the brain is in an otherwise resting state2. The default areas, which consist mainly of certain neocortical regions and the hippocampal formation, seem to subserve such inwardly directed mental activities as introspection, memory retrieval, daydreaming and imagination3. When a person engages in an externally directed task, however, activity in the default-mode network is downregulated and an anti-correlated ‘task-positive network’ is activated3,4. Exceptionally prolonged metabolic activation of the default-mode network is thought to render the system vulnerable to neurological disorders such as Alzheimer’s disease (Fig. 1).
Figure 1: Aβ and senile plaque formation in the default mode network.
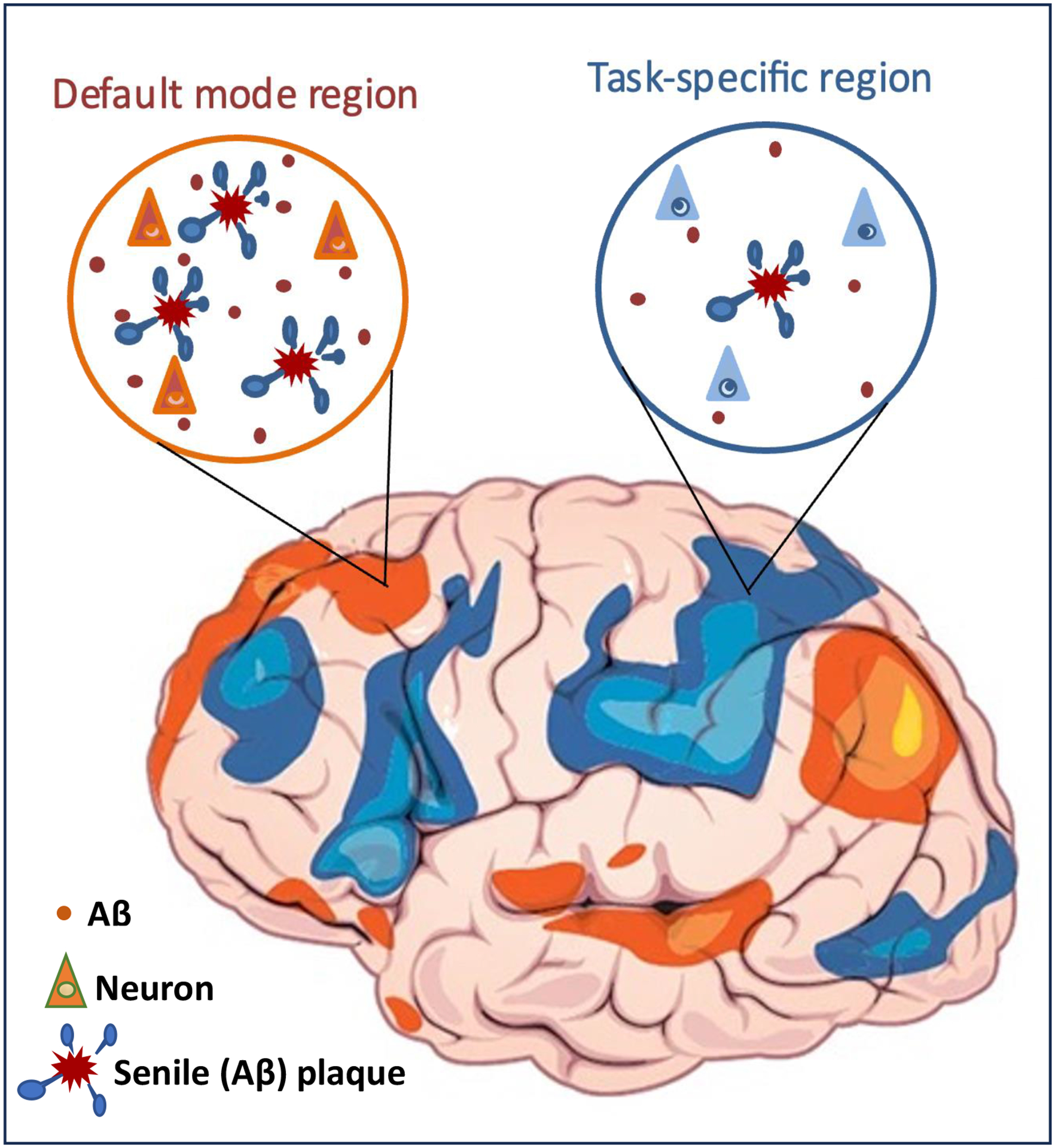
Sustained neuronal activity and elevated aerobic glycolysis in brain areas such as the default-mode network (orange) are thought to be linked to elevated abundance of Aβ and augmented formation of senile (Aβ) plaques compared with those in brain regions with more intermittent, task-directed activation (blue). (The depiction of network activity in the brain is based on reference 3.)
A primary pathogenic occurrence in Alzheimer’s disease is the misfolding and aggregation of the amyloid-β (Aβ) peptide, which activates a cascade of events that culminates in amyloid plaques, neurofibrillary tangles, neuronal degeneration, and dementia5. In vivo visualization of aggregated Aβ in conjunction with functional brain imaging has shown that the default-mode network in people with Alzheimer’s disease is particularly vulnerable to metabolic abnormalities, atrophy and Aβ accumulation6. Although this finding indirectly suggests that the sustained metabolic activation in the default-mode network promotes the aggregation of Aβ, more direct evidence is needed to link neuronal activity to Aβ levels and the development of amyloid plaques.
To address this question, Bero and colleagues1 undertook a series of experiments using transgenic mice that were engineered to develop amyloid plaques consisting of human-sequence Aβ. Using in vivo microdialysis, they found that the amount of soluble Aβ in the interstitial fluid varies markedly among brain areas, and that higher regional levels of soluble Aβ in young mice predict greater Aβ plaque burden in older mice. Hypothesizing that interstitial Aβ levels are governed by local synaptic activity, they then found that the level of lactate, a metabolic byproduct of neuronal metabolism and marker of cerebral aerobic glycolysis, increases with increasing neuronal activity and is correlated with soluble Aβ concentrations. Finally, Bero and colleagues1 demonstrated that changes in neuronal activity affect both the amount of Aβ in the interstitial fluid and the development of amyloid plaques. When neuronal activity is upregulated, soluble Aβ levels rise and plaques increase in size and number; in contrast, when neuronal activity is reduced, Aβ levels decline and the development of senile plaques is abated (Fig. 1).
These findings, although largely correlative, compellingly implicate sustained neuronal activity as a risk factor for the formation of Aβ plaques. On a practical level, this study substantiates the therapeutic strategy of lessening soluble Aβ to diminish the buildup of the protein in the brain and thereby lower the risk of Alzheimer’s disease. Inhibiting the enzymes that generate Aβ (β-secretase and γ-secretase) and anti-Aβ immunization therapy remain enticing, if challenging, therapeutic targets7.
Epidemiological evidence suggests that education may delay the onset of Alzheimer’s disease8. Bero and colleagues1 speculate that education, and presumably the life-long intellectual habits that it promotes, regularly redirects waking brain activity from the default-mode network to the task-specific network, giving the default areas the opportunity to clear the byproducts of chronic activation (such as Aβ). More tentatively, it is also conceivable that certain afflictions accompanying Alzheimer’s disease can accelerate its pathogenesis by increasing interstitial Aβ. Sleep disorders are common in individuals with Alzheimer’s disease9 and sleep in rats is associated with reduced synaptic activity10. A previous study found that sleep diminishes cerebral Aβ and that sleep deprivation increases interstitial Aβ and senile plaque load in APP transgenic mice11. Thus, the coexistence of sleep disorders with Alzheimer’s disease could comprise a vicious cycle in which Alzheimer pathology disrupts sleep, which increases Aβ and thereby accelerates the neurodegenerative process. Epilepsy also frequently occurs with Alzheimer’s disease and may augment regional Aβ levels through the hyperexcitability of certain circuits12. On a speculative note, agents that lower neuronal excitability, even transiently, might have therapeutic value in individuals at risk for Alzheimer’s disease.
Adding to the potential relevance of the findings are the reported sex differences in early Aβ deposition in transgenic mice13 and humans14. It would be interesting to know whether the observation that females are more susceptible than males to β-amyloidosis is related to differences in basal neuronal activity and, if so, why. Another issue is the relationship between activity-induced Aβ increases and neurodegeneration. To what extent and under what conditions does soluble interstitial Aβ convert into injurious oligomers? Is it possible that plaque formation per se serves as a sequestration defense against the toxicity of oligomers? And finally, is the enhanced Aβ aggregation in the default-mode network also a result of the templated aggregation and spreading of Aβ ‘seeds’ along anatomical and functional pathways?15
It is an unfortunate irony that we have learned so much in recent years about the pathogenic role of Aβ in Alzheimer’s disease, but the normal biological function of Aβ, if any, remains enigmatic. Is Aβ simply a detrimental byproduct of cellular metabolism or does it have a useful purpose (useful, at least, to a young brain)? Whether it is beneficial or not, Bero and colleagues’ results1 indicate that reducing the brain’s exposure to Aβ could be helpful, and that it might be possible to achieve this objective in several different ways. Meanwhile, perhaps the best strategy for lessening soluble Aβ in the default mode network may be simply to work diligently, play hard and sleep well.
References
- 1.Bero AW et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci 14, 750–756 (2011). 10.1038/nn.2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raichle ME et al. A default mode of brain function. Proc Natl Acad Sci U S A 98, 676–682 (2001). 10.1073/pnas.98.2.676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckner RL, Andrews-Hanna JR & Schacter DL The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci 1124, 1–38 (2008). 10.1196/annals.1440.011 [DOI] [PubMed] [Google Scholar]
- 4.Broyd SJ et al. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci Biobehav Rev 33, 279–296 (2009). 10.1016/j.neubiorev.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 5.Hardy J & Selkoe DJ The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- 6.Buckner RL et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 25, 7709–7717 (2005). 10.1523/JNEUROSCI.2177-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golde TE, Schneider LS & Koo EH Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 69, 203–213 (2011). 10.1016/j.neuron.2011.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flicker L Modifiable lifestyle risk factors for Alzheimer’s disease. J Alzheimers Dis 20, 803–811 (2010). 10.3233/JAD-2010-091624 [DOI] [PubMed] [Google Scholar]
- 9.Hauw JJ, Hausser-Hauw C, De Girolami U, Hasboun D & Seilhean D Neuropathology of sleep disorders: a review. J Neuropathol Exp Neurol 70, 243–252 (2011). 10.1097/NEN.0b013e318211488e [DOI] [PubMed] [Google Scholar]
- 10.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U & Tononi G Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci 11, 200–208 (2008). 10.1038/nn2035 [DOI] [PubMed] [Google Scholar]
- 11.Kang JE et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007 (2009). 10.1126/science.1180962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noebels J A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia 52 Suppl 1, 39–46 (2011). 10.1111/j.1528-1167.2010.02909.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callahan MJ et al. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol 158, 1173–1177 (2001). 10.1016/s0002-9440(10)64064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corder EH et al. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci 1019, 24–28 (2004). 10.1196/annals.1297.005 [DOI] [PubMed] [Google Scholar]
- 15.Meyer-Luehmann M et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 (2006). 10.1126/science.1131864 [DOI] [PubMed] [Google Scholar]