

Summary
The c.1222C>T (p.Arg408Trp) variant in the phenylalanine hydroxylase gene (PAH) is the most frequent cause of phenylketonuria (PKU), the most common inborn error of metabolism. This autosomal-recessive disorder is characterized by accumulation of blood phenylalanine (Phe) to neurotoxic levels. Using real-world data, we observed that despite dietary and medical interventions, most PKU individuals harboring at least one c.1222C>T variant experience chronic, severe Phe elevations and do not comply with Phe monitoring guidelines. Motivated by these findings, we generated an edited c.1222C>T hepatocyte cell line and humanized c.1222C>T mouse models, with which we demonstrated efficient in vitro and in vivo correction of the variant with prime editing. Delivery via adeno-associated viral (AAV) vectors reproducibly achieved complete normalization of blood Phe levels in PKU mice, with up to 52% whole-liver corrective PAH editing. These studies validate a strategy involving prime editing as a potential treatment for a large proportion of individuals with PKU.
Keywords: CRISPR, gene editing, genome editing, inborn error of metabolism, metabolic disease, phenylketonuria, prime editing, rare disease
Graphical abstract
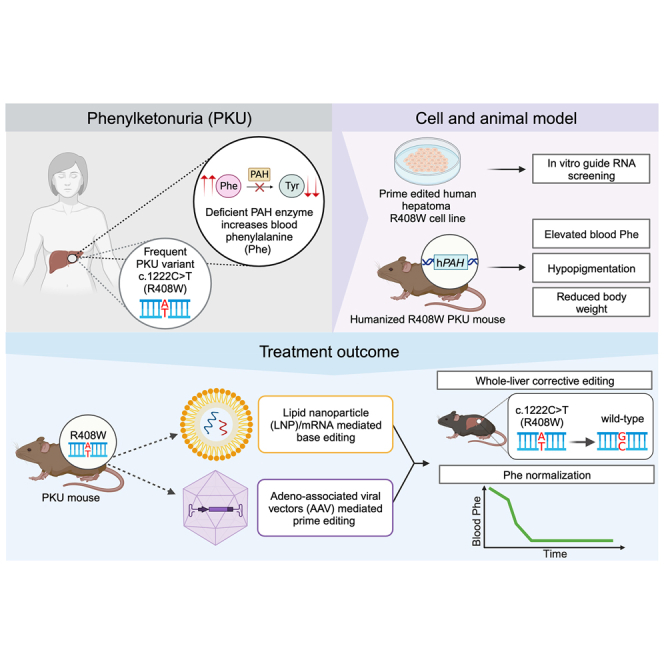
The PAH c.1222C>T variant is the most frequent variant reported in phenylketonuria (PKU) individuals. Here, the authors show these individuals have poor metabolic control, and they use prime editing, delivered by adeno-associated viral vectors, to directly correct the variant in the liver in humanized PKU mice and definitively treat the disease.
Introduction
Phenylketonuria (PKU [MIM: 261600]) is an autosomal-recessive disorder caused by mutations in the gene encoding phenylalanine hydroxylase (PAH), resulting in the accumulation of phenylalanine (Phe) to neurotoxic levels because of the inability to convert Phe into tyrosine. Untreated PKU can manifest in high blood Phe levels of >1200 μmol/L, whereas current treatment guidelines state that blood Phe levels should ideally be maintained in the range of 120–360 μmol/L.1 The existing treatment options range from a strict low-Phe diet to an oral medication (sapropterin, a cofactor of PAH) to an injectable enzyme-substitution therapy (with pegvaliase). All are chronic, everyday interventions to which affected individuals must adhere for their lifetimes if they are to prevent cognitive impairment and a range of neuropsychiatric complications. The burden of intensive Phe control and monitoring faced by individuals with PKU and their families is profound, especially with treatments that can be unpalatable and cost-prohibitive. Lack of adherence to the guidelines on the part of many individuals with PKU is not only a serious risk, but also to be expected. Studies have shown that more than 70% of adults with PKU are noncompliant with therapy.2 As such, durable and, ideally, curative therapies would be needed to optimally address the medical needs of individuals with PKU.
More than 1,000 PAH variants have been cataloged in PKU individuals.3 These vary in their consequences for PAH activity, from having little or no effect to eliminating PAH activity completely. The most frequently occurring pathogenic PAH variant worldwide is the c.1222C>T (p.Arg408Trp) variant (GenBank: NM_000277.3) (MIM: 612349), which is most prevalent in European populations (e.g., present in 98.9% of PKU individuals in Estonia, 89.2% in Poland, 75.7% in Russia, 43.6% in Sweden, and 35.7% in Germany), Australia (34.7%), and the United States (32.9%).3 Individuals homozygous for this variant have the most severe form of PKU and do not respond to sapropterin,4 limiting their treatment options. Individuals with the c.1222C>T variant do respond to pegvaliase, a once-daily injected bacterial-derived enzyme that directly catabolizes Phe, albeit with a substantial risk of anaphylaxis. However, in clinical trials, pegvaliase had only a mean Phe reduction of 51% at one year after initiation (from 1,233 μmol/L to 565 μmol/L), meaning that a large proportion of the individuals did not achieve the guideline-directed Phe goal of <360 μmol/L.5
A safe one-time therapy that permanently normalizes blood Phe levels would be transformative for the care of individuals with PKU. Previous studies have suggested that reverting only ≈10% of the PAH alleles to wild-type in the liver, where PAH is largely expressed, would achieve this goal.6,7,8,9 In vivo CRISPR editing is an emerging therapeutic approach that makes changes in DNA sequence in organs such as the liver in an individual’s body. Nuclease editors have been demonstrated to be effective in non-human primates and in human individuals, but their use is limited to disruption or deletion of genes or regulatory elements in the genome.10 Base editors have the ability to directly and precisely correct disease-causing variants, but their utility is highly dependent on target-sequence context and is generally restricted to single-nucleotide transitions (C→T, A→G).11,12 Prime editors have the precision of base editing, with the advantage that they can make all possible single-nucleotide changes, can correct indels, and have greater target-site flexibility.13
A prime editor comprises a Cas9 variant that nicks one DNA strand at the target site, a reverse transcriptase enzyme fused to Cas9, and a guide RNA (called prime-editing guide RNA, or pegRNA) that in complex with Cas9 specifies the target site (via a matching protospacer sequence at the DNA target site, just upstream of the Cas9 protein’s preferred protospacer-adjacent motif [PAM], typically an NGG sequence) and has a 3′ extension with (1) a primer-binding site (PBS) that hybridizes with the nicked DNA strand upstream of the nick and (2) a reverse transcriptase template (RTT) that serves as a template for the reverse transcriptase to build a DNA flap anchored to the nicked DNA strand at the nick site.13 The desired edit is encoded within the RTT sequence and is thus incorporated into the synthesized DNA flap. The DNA flap competes with the original sequence on the nicked DNA strand downstream of the nick for incorporation into the final repaired double-strand DNA product. Mismatch repair (MMR) favors the original sequence by acting against the desired prime-editing outcome.14 Successful prime editing is facilitated by: (1) using an optimized prime-editor protein (e.g., PEmax); (2) using an engineered pegRNA with a stabilizing structured RNA motif at its 3′ end; (3) inhibiting MMR, e.g., by adding extra mismatches into the RTT, namely synonymous edits that will not compromise the correction of a disease-causing variant; (4) using a nicking guide RNA (ngRNA) that directs Cas9 to nick the DNA strand opposite the DNA flap, instigating a nick repair process that favors incorporation of the DNA flap into the final product; and (5) using an ngRNA whose protospacer sequence overlaps with the site of the edit(s).13,14,15
In this work, we sought to define the degree of adherence to Phe treatment and monitoring goals by PKU individuals with the PAH c.1222C>T variant. We used real-world data from a PKU cohort managed at a metabolic specialty clinic at an academic medical center. Upon establishing a high degree of unmet medical need among these individuals, we created an in vitro cellular model and in vivo humanized PKU mouse models in order to develop and validate a corrective prime-editing therapeutic for the PAH c.1222C>T variant. Using adeno-associated viral (AAV) vectors for delivery of a lead prime-editing candidate, we demonstrated efficient corrective editing and normalization of blood Phe levels in homozygous and compound-heterozygous mice.
Subjects and methods
Enrollment
PKU-affected individuals born prior to January 1, 2021, and followed in the Children’s Hospital of Philadelphia (CHOP) Metabolism Clinic were considered for inclusion in the study. All available genotype information was reviewed, and subjects who were either homozygous or compound-heterozygous for the PAH c.1222C>T variant were included in the final study cohort.
This study was reviewed by the CHOP institutional review board (IRB 23–021115) and found to meet exemption criteria for human subjects research per 45 CFR 46.104(d) 4(iii). In addition, a waiver of HIPAA authorization was granted per 45 CFR 164.512(i)(2)(ii) for accessing subjects’ PAH genotypes and longitudinal Phe levels.
Vectors
The following prime-editor-expressing plasmids were obtained from Addgene as gifts from Dr. David Liu: pCMV-PEmax-P2A-hMLH1dn (Addgene plasmid #174828; http://n2t.net/addgene:174828; RRID: Addgene_174828), pU6-tevopreq1-GG-acceptor (Addgene plasmid #174038; http://n2t.net/addgene:174038; http://n2t.net/addgene:174038; RRID: Addgene_174038), v3em-Nterm-PE2max (Addgene plasmid #198734; http://n2t.net/addgene:198734; RRID: Addgene_198734), and v3em-Cterm-PE2max-ΔRNaseH-dualU6 (Addgene plasmid #198735; http://n2t.net/addgene:198735; RRID: Addgene_ 198735).14,15,16 Each pegRNA was expressed from pU6-tevopreq1-GG-acceptor, with the spacer sequence (plus an additional 5′ G to facilitate U6 expression) and PBS and RTT sequences indicated in Table S1 cloned into the plasmid. Each ngRNA was expressed from pGuide (Addgene plasmid #64711; http://n2t.net/addgene:64711; RRID: Addgene_64711); the spacer sequence (plus an additional 5′ G to facilitate U6 expression) indicated in Table S2 was cloned into the plasmid. For the lead pegRNA/ngRNA combination (P56/N19), the sequences were cloned into the v3em-Cterm-PE2max-ΔRNaseH-dualU6 plasmid (Table S3). This last vector and the v3em-Nterm-PE2max vector were transferred to PackGene Biotech to generate AAV vectors with the AAV8 serotype on a fee-for-service basis. Any of the newly generated plasmids is readily available from the authors via a “transfer of research material” agreement with the University of Pennsylvania.
Culture and transfection of HuH-7 cells
The methods were similar to those previously described.9 HuH-7 cells were maintained in Dulbecco’s modified Eagle’s medium (containing 4 mM L-glutamine and 1 g/L glucose) with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C with 5% CO2. HuH-7 cells were seeded on 6-well plates (Corning) at 3.5 × 105 cells per well. At 16–24 h after seeding, cells were transfected at approximately 80%–90% confluency with 9 μL TransIT-LT1 Transfection Reagent (MIR2300, Mirus), 1,500 ng prime-editor plasmid (pCMV-PEmax-P2A-hMLH1dn), 750 ng pegRNA plasmid, and 750 ng ngRNA plasmid per well according to the manufacturer’s instructions. Cells were cultured for 72 h after transfection, and then media were removed, cells were washed with 1× DPBS (Corning), and genomic DNA was isolated with the DNeasy Blood and Tissue Kit (QIAGEN) according to the manufacturer’s instructions.
Generation of PAH c.1222C>T homozygous HuH-7 cells
The methods were similar to those previously described.9 HuH-7 cells in a well of a 6-well plate were transfected with 9 μL TransIT-LT1 Transfection Reagent, 1,500 ng prime editor plasmid (pCMV-PEmax-P2A-hMLH1dn), 750 ng pegRNA plasmid, and 750 ng ngRNA plasmid. Cells were dissociated with trypsin 48 h after transfection and replated onto 10 cm plates (5,000 cells/plate) with conditioned medium to facilitate recovery, and genomic DNA was isolated from the remainder of the cells as a pool for PCR and Sanger sequencing of the PAH c.1222C>T site. Single cells were permitted to expand for 7–14 days so that clonal populations could be established. Colonies were manually picked and replated into individual wells of a 96-well plate. Genomic DNA was isolated from individual clones, and PCR and Sanger sequencing were performed for identification of c.1222C>T homozygous HuH-7 clones. One representative clone was expanded for use in subsequent studies. This c.1222C>T homozygous HuH-7 cell line is readily available from the authors via a “transfer of research material” agreement with the University of Pennsylvania.
Generation of humanized PKU mouse models
Humanized mice with either the PAH c.1222C>T (p.Arg408Trp) variant (hereafter referred to as R408W mice) or the PAH c.1066−11G>A variant (hereafter referred to as 1066 mice) were generated by Ozgene on a fee-for-service basis. The schemes are outlined in Figures S1, S2, and S3. In brief, the humanized c.1222C>T allele was created by knocking in human exon 12 and approximately 500 bp of genomic DNA both 5′ and 3′ of the knockin exon in place of the corresponding genomic region of murine Pah via homologous recombination in C57BL/6 mouse embryonic stem cells. The knockin exon included the c.1222C>T variant, introduced by alteration of the arginine codon CGG to the tryptophan codon TGG. The neo antibiotic selection cassette used for facilitating knockin was subsequently removed by Cre recombinase. The humanized c.1066−11G>A allele was created by knockin of genomic DNA of human PAH from approximately 500 bp upstream of exon 10 through approximately 600 bp downstream of exon 12 in place of the corresponding genomic region of murine Pah via homologous recombination in C57BL/6 mouse embryonic stem cells. The knockin human region included the c.1066−11G>A variant, introduced by alteration of the corresponding position in intron 11 from a G to an A. The neo antibiotic selection cassette used for facilitating knockin was subsequently removed by Cre recombinase. After the generation of chimeric mice by blastocyst injection, the humanized alleles were bred to heterozygosity, homozygosity, or compound heterozygosity through crosses of the founder mice and subsequent generations with wild-type mice or with each other. Each of the humanized mouse models is readily available from the authors via a “transfer of research material” agreement with the University of Pennsylvania.
Mouse studies
The methods were similar to those previously described.9 All procedures used in mouse studies were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania (protocol #805887), where the studies were performed, and were consistent with local, state, and federal regulations, including the National Institutes of Health Guide for the Care and Use of Laboratory Animals, as applicable. Mice were maintained on a 12 h light/12 h dark cycle in a temperature range of 65°F–75°F and a humidity range of 40%–60% and were fed ad libitum with a chow diet (LabDiet, Laboratory Autoclavable Rodent Diet 5010). Homozygous and compound-heterozygous humanized PKU mice, as well as heterozygous humanized non-PKU mice, were generated as littermates or colony mates via timed breeding; in some cases wild-type C57BL/6J mice (stock no. 000664; RRID:IMSR_JAX:000664) from The Jackson Laboratory were used. Genotyping was performed via PCR amplification from genomic DNA samples (prepared from clipped tails and ears) followed by Sanger sequencing or by next-generation sequencing. Age-matched female and male colony mates were used for experiments at 6 weeks of age or 10 weeks of age; animals were randomly assigned to various experimental groups when applicable, and collection and analysis of data were performed in a blinded fashion when possible. AAV vectors were generated from the v3em-Nterm-PE2max and v3em-Cterm-PE2max-ΔRNaseH-dualU6 plamids, as described above. The AAV vectors were administered as 1:1 mixes to the mice at total doses of 4 × 1011 vg, 8 × 1011 vg, or 1 × 1012 vg via retro-orbital injection under anesthesia with 1%–2% inhaled isoflurane. The AAV dose range was chosen on the basis of a prior study with similar vectors;16 the exact doses were determined by the sizes of aliquots provided by PackGene. Mice were euthanized at about 6 weeks after AAV treatment. Euthanasia in all instances was achieved via terminal inhalation of carbon dioxide followed by secondary euthanasia through cervical dislocation or decapitation, consistent with the 2020 American Veterinary Medical Association Guidelines on Euthanasia. For all treated mice, eight liver samples (two from each lobe) were obtained on necropsy and processed with the DNeasy Blood and Tissue Kit (QIAGEN) as per the manufacturer’s instructions for isolation of genomic DNA. Next-generation sequencing results from the liver samples were averaged to provide quantification of whole-liver editing. Blood samples were collected via the tail tip at various timepoints (pre-treatment to establish the baseline level, week 1, week 2, week 3, week 4, and week 5) in the early afternoon to account for diurnal variation in blood phenylalanine levels.
Measurement of blood analytes
The methods were similar to those previously described.9 The blood phenylalanine levels were measured by an enzymatic method with the Phenylalanine Assay Kit (MAK005, Millipore Sigma) according to the manufacturers’ instructions. Plasma samples were deproteinized with a 10 kDa MWCO spin filter (CLS431478-25EA, Millipore Sigma) and pre-treated with 5 μL of tyrosinase for 10 min at room temperature prior to the start of the assay. Infrequently, a sample was lost during preparation, and the value for that sample was omitted from the final dataset for that reason. Reaction mixes were made according to the manufacturers’ instructions, and the fluorescence intensity of each sample was measured (λex = 535/λem = 587 nm). Aspartate aminotransferase (AST) (MAK055-1KT, Millipore Sigma) and alanine aminotransferase (ALT) (MAK052-1KT, Millipore Sigma) activities were measured according to the manufacturers’ instructions.
RNA production
For ONE-seq, a 120-mer pegRNA was chemically synthesized under solid-phase synthesis by a commercial supplier (Integrated DNA Technologies) with end modifications: 5′-mA∗mC∗mU∗UUGCUGCCACAAUACCUGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCGAGAAGGGACGUGGUAUUGUG∗mG∗mC∗mA-3′, where “m” and ∗ indicate 2′-O-methylation and phosphorothioate linkage, respectively.
ONE-seq and in silico nomination of candidate off-target sites
The methods were similar to those previously described.9,17 The human ONE-seq library for the protospacer sequence of the lead pegRNA (5′-ACTTTGCTGCCACAATACCT-3′) was designed with the GRCh38 Ensembl v98 reference genome. In comparison to the on-target site, sites with up to four mismatches; up to three mismatches plus up to one DNA or RNA bulge; and up to two mismatches plus up to two DNA or RNA bulges were identified with Cas-Designer v1.2.18 The final oligonucleotide sequences were generated with a script,17 and the oligonucleotide library was synthesized by Twist Biosciences. The library was PCR-amplified and subjected to 0.9× AMPure XP (Beckman Coulter) bead purification. After incubation at 25°C for 10 min in NEBuffer r3.1 (New England Biolabs), ribonucleoproteins from 17 nM recombinant Cas9 protein (New England Biolabs) and 34 nM pegRNA (Integrated DNA Technologies) were mixed with 30 ng of the purified library and incubated at 37°C for 2 h. Proteinase K was added to quench the reaction at 37°C for 45 min, followed by 1.2× AMPure XP bead purification. The reaction was then incubated with Phusion High-Fidelity Polymerase (New England Biolabs) at 72°C for 10 min, followed by 1.2× AMPure XP bead purification. The reaction was ligated with an annealed adaptor oligonucleotide duplex at 25°C for 10 min to facilitate PCR amplification of the cleaved library products, and 2× AMPure XP bead purification followed. Size selection of the ligated reaction was performed on a BluePippin system (Sage Science) so that DNA of 150–200 bp would be isolated on a 3% agarose gel cassette; two subsequent rounds of PCR amplification generated a barcoded library, which underwent paired-end sequencing on an Illumina MiSeq System as described below. The analysis pipeline17 used for processing the data assigned a score quantifying the cleavage efficiency for each potential off-target site, relative to the on-target site. Sites were ranked on the basis of this ONE-seq score, and the mean ONE-seq score between duplicate experiments was used for site prioritization. The top 24 ONE-seq sites had scores ranging from 1.0 (on-target site) to below a threshold of 0.01 (1/100th of the cleavage efficiency observed with the on-target site). In addition to the top 24 ONE-seq sites, we selected all remaining genomic sites (55 in total) with up to three mismatches, up to two mismatches plus up to one DNA or RNA bulge, and up to one mismatch plus up to two DNA or RNA bulges, in comparison to the on-target site, as identified in silico with Cas-Designer v1.2. The results of next-generation sequencing (see below) for these sites are listed in Table S4.
Next-generation sequencing
The methods were similar to those previously described.9 PCR reactions were performed with NEBNext Polymerase (NEB) and the primer sets listed in Table S5; these were designed with Primer3 v4.1.0 (https://primer3.ut.ee/). The following program was used for all genomic DNA PCRs: 98°C for 20 s, 35 × (98°C for 20 s, 57°C for 30 s, 72°C for 10 s), and 72°C for 2 min. PCR products were visualized via capillary electrophoresis (QIAxcel, QIAGEN) and then purified and normalized via an NGS Normalization 96-Well Kit (Norgen Biotek Corporation). A secondary barcoding PCR added Illumina barcodes (Nextera XT Index Kit V2 Set A and/or Nextera XT Index Kit V2 Set D) through the use of ∼15 ng of first-round PCR product as a template, followed by purification and normalization. Final pooled libraries were quantified on a Qubit 3.0 Fluorometer (Thermo Fisher Scientific); then, after denaturation, dilution to 10 pM, and supplementation with 15% PhiX, libraries underwent single-end or paired-end sequencing on an Illumina MiSeq System. We analyzed the amplicon sequencing data with CRISPResso2 v219 and scripts to quantify editing. For on-target editing, corrective T-to-C editing was quantified at the site of the c.1222C>T variant. For candidate off-target sites, we considered all aligned reads in which there was any mismatch to the reference sequence in any position within a window spanning from the position 5′ of the non-target-strand nick site to the 3′ end of the portion of the non-target strand overlapping with the RTT, 14 positions downstream of the nick site—thus accounting for single-nucleotide substitutions and indels within the window—to be potentially edited. In some cases, PCR amplicons were subjected to Sanger sequencing, performed by GENEWIZ, instead of next-generation sequencing.
Data analysis
Sequencing data were analyzed as described above. Other data were collected and analyzed with GraphPad Prism v10.0.2.
Results
PKU individuals with PAH c.1222C>T variants demonstrate very poor metabolic control
Among 129 PKU-affected individuals followed by the Children’s Hospital of Philadelphia (CHOP) Metabolic Clinic, 32 (24.8%) were found to be compound heterozygous for the PAH c.1222C>T allele, and four (3.1%) were homozygous for the variant. This might be an underestimate of true c.1222C>T prevalence because genotype information was not available for some older individuals with PKU.
Consensus management guidelines from the American College of Medical Genetics recommend individuals with PKU maintain Phe levels in the 120–360 μmol/L range.1 Levels above 600 μmol/L can be neurotoxic and are associated with poor psychiatric and neurocognitive outcomes.20,21,22 In our study cohort, 33 of 36 (91.6%) treated individuals had at least a single Phe level above 360 μmol/L, and 25 of 36 (69.4%) had at least a single level above 600 μmol/L (Figure 1). Furthermore, 10 of 36 (27.7%) had lifetime average Phe levels above 360 μmol/L. The four individuals homozygous for the c.1222C>T allele demonstrated especially poor metabolic control.
Figure 1.

Lifetime recorded measurements of blood phenylalanine concentrations in PKU individuals with PAH c.1222C>T variants
All available Phe concentrations from 32 PKU individuals harboring the c.1222C>T variant in trans with a second pathogenic allele are shown on the left, where all available levels from four individuals homozygous for c.1222C>T are shown on the right. Most individuals had several recorded concentrations above 360 μmol/L, the maximum recommended concentration per PKU guidelines, as indicated by the dotted line.
In addition to high Phe levels, individuals with c.1222C>T variants demonstrated poor adherence to Phe monitoring schedules. Although exact recommended intervals can vary by clinical scenario, general guidelines include weekly Phe monitoring prior to 1 year of age, every-2-week monitoring for ages 1–12 years, and every-2–4 weeks monitoring in adolescents and adults (Figure 2, gray box).1 We calculated the interval since the last recorded value for every Phe measurement in our cohort (Figure 2, black dots). The interval between monitoring labs was longer than recommended for 1,556 of 2,535 (61.3%) of the Phe values measured. Collectively, these data demonstrate that most individuals with c.1222C>T variants have poor metabolic control and are at risk for chronic neurologic damage.
Figure 2.

Intervals between measurements of blood phenylalanine concentrations in PKU individuals with PAH c.1222C>T variants
The durations of intervals between Phe-monitoring checks are plotted by age. Recommended guidelines for how often Phe should be checked varies with age and is indicated in gray. Most individuals did not check Phe concentrations frequently enough, as evidenced by the fact that most monitoring intervals fell outside the recommended range.
Screening and optimization of prime editing in an edited c.1222C>T hepatocyte cell line
Motivated by the severe unmet medical need in PKU individuals with PAH c.1222C>T variants, we set out to determine whether prime editing could productively correct the variant in cellular and animal models and thus potentially serve as the basis of a future therapeutic intervention for these individuals. Lacking access to c.1222C>T-individual-derived hepatocytes, we used prime editing to introduce the c.1222C>T variant into HuH-7 human hepatoma cells, a commonly used hepatocyte-like substitute for primary human hepatocytes. Four pegRNA protospacer sequences have NGG PAMs with positioning near the site of the c.1222C>T variant and amenable to prime editing of the variant (Figures 3 and 4). We found that our initial attempt at the use of PEmax with an engineered pegRNA (designated P1)—harboring one of the four protospacer sequences, with a PBS length of 12 nt and an RTT length of 12 nt (lengths consistent with recommendations by previous publications on prime editing)—and either of two ngRNAs, designated N1 and N2, could achieve 34% and 25% insertion of the c.1222C>T variant, respectively, into bulk HuH-7 cells when delivered via plasmid transfection (Figures 3 and 4, insertion set). We used single-cell cloning to expand edited HuH-7 cell lines and identified lines that were homozygous for the c.1222C>T variant.
Figure 3.

Prime editing to introduce PAH c.1222C>T variant in human hepatocytes in vitro
(A) Schematic depiction of the genomic site of the PAH c.1222C>T variant, adapted from the UCSC Genome Browser (GRCh38/hg38). The vertical blue bar outlined by the red box indicates the G altered to A (in red) by the variant on the antisense strand. The four horizontal lines near the site of the variant indicate four potential NGG PAM sequences for use with pegRNAs or ngRNAs; the horizontal line to the far right of the variant indicates a potential NGG PAM for use with an ngRNA only. The labeled horizontal lines (PAM #1, PAM #2) correspond to the PAMs used in the schematic shown in (D).
(B) Sanger sequencing chromatogram of the genomic site of the PAH c.1222C>T variant in wild-type HuH-7 cells. The boxed nucleotide is at the site of the variant.
(C) Chromatogram of the site in PAH c.1222C>T homozygous HuH-7 cell line. The boxed nucleotide is at the site of the edited variant, in the position indicated by the red arrow.
(D) Schematic depiction of prime editing with the P1/N1 pegRNA/ngRNA combination to insert the c.1222C>T variant into the PAH locus in the human genome. Boxes or underlines indicate nucleotides at the site of the variant.
Figure 4.

Prime editing to correct the PAH c.1222C>T variant in human hepatocytes in vitro
Efficiencies of PAH c.1222C>T editing by various pegRNA/ngRNA combinations (Px/Nx, refer to Tables S1 and S2 for sequences); the first two combinations in the table/graph were for insertion of the c.1222C>T variant in wild-type HuH-7 cells (used to generate the PAH c.1222C>T homozygous HuH-7 cell line in Figure 3), and the remainder for correction of the variant in homozygous HuH-7 cells. For each letter sequence, blue indicates the PBS of the pegRNA, and black indicates the RTT of the pegRNA. Red lowercase letters indicate edits at the site of the variant; magenta lowercase letters indicate additional synonymous edits. The short lines at the top of the table indicate NGG PAM sequences used for pegRNAs and/or ngRNAs. Each long line immediately below a pegRNA PBS/RTT sequence in the table indicates the span of the protospacer sequence of the ngRNA used with that pegRNA. n = 1 biological replicate for each pegRNA/ngRNA combination except for the last set, for which n = 2 biological replicates. Lines in the graph = mean values.
The c.1222C>T variant is the result of a C→T change that could in principle be reversed with adenine base editing on the antisense strand. However, the lack of an appropriately positioned NGG PAM relative to the variant, as well as the possibility of unwanted bystander editing, militated against the use of a base editor. We therefore pursued prime editing as a therapeutic strategy. With a c.1222C>T homozygous HuH-7 cell line, we screened a variety of pegRNA and ngRNA candidates for corrective activity with iterative experiments. We initially focused on the four candidate pegRNA protospacer sequences by using a PBS length of 12 nt and an RTT length adjusted on the basis of the distance of the PAM from the site of the c.1222C>T variant, paired with ngRNAs (Figure 4, correction set #1). For the pegRNAs with the two protospacer sequences with PAMs closest to the site of the c.1222C>T variant, we also assessed the effect of introducing one or more synonymous variants (limited to transition variants) to inhibit mismatch repair, in the hope of enhancing the prime editing efficiency. We found that three of the protospacer sequences supported corrective editing activity with double-digit percentages; the best result of 24% resulted from the use of the same protospacer with which the c.1222C>T homozygous HuH-7 cell lines were generated, along with two synonymous variants closely flanking the site of the c.1222C>T variant. We selected this protospacer, along with an RTT harboring the two synonymous variants (pegRNA P4) and a matching ngRNA (N5), for further optimization.
Our next step was to vary the PBS and RTT lengths, ranging from 10 to 14 nt each (Figure 4, correction set #2A). Among these PBS/RTT combinations, we found that a PBS length of 10 nt and RTT length of 14 nt (pegRNA/ngRNA combination P31/N5) performed best, with corrective editing activity of 47%, although several other combinations also had activity in the 40%–50% range, suggesting that there would be no single optimal PBS/RTT configuration that would substantially outperform all others. We extended this work by testing a PBS length of 10 nt with a wider range of RTT lengths (as high as 20 nt) and an RTT length of 14 nt with a wider range of PBS lengths (as low as 9 nt and as high as 16 nt) (Figure 4, correction set #2B). None of these additional combinations exceeded the corrective editing activity of the P31/N5 combination. Taking advantage of the longer-length RTTs in some pegRNAs, which had RTTs of 17–20 nt, we also tested the addition of a third synonymous variant and found that it improved the corrective editing activity beyond that achieved by two synonymous variants (Figure 4, correction set #2B); a PBS length of 10 nt and RTT length of 19 nt (P47/N7 combination) had 48% activity.
We undertook further optimization of the P31/N5 combination by testing all of the nine possible sets of substitutions (whether transition or transversion) in the positions of the two synonymous variants, along with matching ngRNAs (Figure 4, correction set #3). We found that the identity of the substitutions had a meaningful effect on corrective editing activity and that distinct trends were apparent in the two positions. The activity ranged from 37% to 55%. We selected the pegRNA/ngRNA combination (P56/N19) that yielded 55% corrective editing activity in HuH-7 cells for testing in vivo.
Generation of humanized PKU mouse models
To generate humanized PKU mice with the PAH c.1222C>T variant (R408W mice), we undertook homologous recombination in C57BL/6 embryonic stem cells to knockin the human PAH exon 12 sequence harboring the c.1222C>T variant, as well as ∼500 bp of flanking genomic sequence both 5′ of the exon (intron 11) and 3′ of the exon (intron 12) and thereby replaced the orthologous portion of the endogenous mouse Pah (Figure S1). Homozygous R408W mice were generated from the recombinant embryonic stem cells and confirmed to have elevated blood Phe levels, as well as hypopigmentation and reduced weight, consistent with the mice’s having PKU (Figures 5A, 5B, and S2).
Figure 5.

Prime editing to correct PAH c.1222C>T variant in humanized mice
(A) Changes in blood phenylalanine levels in homozygous PKU mice after treatment with 8 × 1011 vg AAV dose (n = 4 animals) or with 4 × 1011 vg AAV dose (n = 3 animals); concentrations at various timepoints up to 5 weeks after treatment were compared to concentrations in untreated homozygous PKU control (n = 3 animals) and untreated heterozygous non-PKU control (n = 3 animals) age-matched (6 weeks of age) colony mates (one blood sample per timepoint).
(B) Changes in blood phenylalanine concentrations in homozygous PKU mice after treatment with 8 × 1011 vg AAV dose (n = 3 animals). Concentrations at various timepoints up to 5 weeks after treatment to were compared to concentrations in untreated heterozygous non-PKU control (n = 2 animals) age-matched (10 weeks of age) colony mates (one blood sample per timepoint).
(C) Changes in blood phenylalanine concentrations in compound-heterozygous PKU mice after treatment with 1 × 1012 vg AAV dose (n = 3 animals). Concentrations at various timepoints up to 5 weeks after treatment were compared to concentrations in untreated heterozygous non-PKU control (n = 2 animals) age-matched (6 weeks of age) colony mates (one blood sample per timepoint).
(D) Corrective PAH c.1222C>T editing (determined from genomic DNA) in the whole liver (mean value from eight liver samples) in each of the treated groups of mice. For compound-heterozygous mice, each displayed number is the estimated percentage of edited c.1222C>T alleles (editable alleles).
(E) Standard CRISPResso output for a liver sample from the treated homozygous PKU mouse with the highest level of editing. Lines in graphs = mean values.
We sought to assess therapeutic prime editing not only in homozygous R408W mice—which would model human PKU individuals homozygous for the c.1222C>T allele—but also in mice harboring one c.1222C>T allele and another loss-of-function Pah allele—which would model human PKU individuals compound heterozygous for the c.1222C>T allele; such individuals far outnumber homozygous individuals (Figure 1). We reasoned that using an existing loss-of-function mouse Pah allele, such as the allele in the commonly used Pahenu2 PKU mouse model, would not be optimal; this allele would not have the wild-type human sequence at the site of the c.1222C>T variant in the endogenous Pah exon 12, and interactions with the prime editor would not be reflective of the interactions of a wild-type PAH exon 12 sequence in human cells. Accordingly, we also generated humanized PKU mice with a different severe, frequent PAH allele, the c.1066−11G>A variant (1066 mice).
We undertook homologous recombination in C57BL/6 embryonic stem cells to knock in the human PAH exon 11 sequence harboring the c.1066-11G>A variant, as well as the entirety of the flanking introns (10 and 11), the flanking exons (10 and 12), and ≈500–600 base pairs (bp) of flanking genomic sequence both 5′ of exon 10 (intron 9) and 3′ of exon 12 (intron 12), replacing the orthologous portion of the endogenous mouse Pah (Figure S3). Mice bearing the humanized c.1066−11G>A allele were generated from the recombinant embryonic stem cells and then crossed with humanized R408W mice, resulting in compound-heterozygous R408W/1066 offspring that were confirmed to have elevated blood Phe levels consistent with the mice’s having PKU (Figure 5C).
In vivo corrective editing of the c.1222C>T variant in humanized PKU mice
To deliver the prime editor into the mouse liver, we used a recently reported dual-AAV system: one vector encodes the N-terminal part of the PEmax protein fused to a C-terminal split-intein; the other vector encodes the C-terminal part of the PEmax protein (lacking the RNase H domain) fused to the N-terminal split-intein, an expression cassette for the pegRNA, and an expression cassette for the ngRNA (Figure S4).16 We produced AAV8 vectors for delivery of the optimized pegRNA/ngRNA combination described above (P56/N19) by mixing the two vectors in equal proportion for intravenous administration to mice.
In our initial in vivo study, we treated four age-matched (6 weeks of age) homozygous R408W (PKU) mice each with a combined 8 × 1011 viral genomes (vg) dose and three age-matched homozygous R408W (PKU) mice with a combined 4 × 1011 vg dose; three untreated homozygous R408W (PKU) colony mates and three untreated heterozygous R408W (non-PKU) colony mates served as controls (Figure 5A). In aggregate, the PKU mice had a mean baseline blood Phe level of 1,376 μmol/L; the non-PKU mice, 93 μmol/L. All of the treated PKU mice displayed substantially decreased Phe levels by 2 weeks after treatment and were well below the threshold of 360 μmol/L by 5 weeks; the higher-dosed mice were at 109 μmol/L, and the lower-dosed mice at 165 μmol/L. Upon necropsy around 6 weeks, next-generation sequencing of genomic DNA from whole-liver samples to determine corrective editing activity showed a range from 31% to 52% (mean 41%) in the higher-dosed mice and from 17% to 35% (mean 26%) in the lower-dosed mice (Figure 5D).
In a second study with older mice, we treated three age-matched (10 weeks of age) homozygous R408W (PKU) mice each with a combined 8 × 1011 vg dose (Figure 5B). Starting with a mean baseline blood Phe level of 1,948 μmol/L, once again all of the treated PKU mice displayed substantially decreased Phe levels by 2 weeks after treatment and, at 151 μmol/L, were well below the threshold of 360 μmol/L by 5 weeks. Whole-liver editing (genomic DNA) ranged from 17% to 52% (mean 33%) in the treated mice (Figures 5D and 5E).
In a third study, we treated three age-matched (6 weeks of age) compound-heterozygous R408W/1066 mice with a combined 1 × 1012 vg dose (Figure 5C). Starting with a mean baseline blood Phe level of 1,867 μmol/L, yet again all of the treated PKU mice displayed substantially decreased Phe levels by 2 weeks after treatment and, at 154 μmol/L, were well below the threshold of 360 μmol/L by 5 weeks. Because the pegRNA/ngRNA combination had the potential to edit wild-type alleles (i.e., introduction of two synonymous variants only), we were only able to estimate the corrective editing (genomic DNA) of the c.1222C>T alleles in the livers of the compound-heterozygous mice; estimates ranged from 22% to 56% (mean 40%) (Figure 5D).
In all three studies, blood aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels in the treated mice remained normal throughout the study period (Figure S5).
Off-target assessment
To evaluate off-target editing of our optimized pegRNA/ngRNA combination (P56/N19), we performed ONE-seq17 with a synthetic human genomic library selected by homology to the pegRNA protospacer sequence, treated with recombinant Cas9 protein and the appropriate pegRNA. We reasoned that off-target prime editing activity would be unlikely to occur at a genomic site if Cas9 nuclease activity was not supported at that site. We assessed the top 24 ONE-seq-nominated sites (including the on-target PAH site, which was the top-ranked site) with next-generation sequencing of targeted PCR amplicons from pegRNA/ngRNA-transfected versus control-transfected c.1222C>T homozygous HuH-7 genomic DNA samples (Figure 6). To supplement the off-target analysis, we assessed an additional 55 candidate genomic sites nominated by in silico prediction based on sequence similarity to the on-target PAH site (Figure 6). We observed no evidence of off-target sequence alteration, either single-nucleotide substitutions or indels, at any of the interrogated sites.
Figure 6.

Assessment of off-target editing
On-target or off-target editing at top ONE-seq-nominated or in-silico-nominated candidate sites calculated as the proportion of aligned sequencing reads with alteration of ≥1 base pair within the target sequence (ranging from the position proximal to the pegRNA nick site to the distal position spanned by the RTT sequence, capturing both single-nucleotide substitutions and indels) in PAH c.1222C>T homozygous HuH-7 cells that underwent plasmid transfection with the lead pegRNA/ngRNA (P56/N19) combination (n = 3 treated and 3 untreated biological replicates; y axis is log10 scale). Sites with unsuccessful sequencing are omitted. Refer to Table S4 for candidate site sequences and numerical values.
Discussion
We and others have reported the use of base editing to correct PKU pathogenic variants in vivo, either the non-human variant present in the homozygous Pahenu2 PKU mouse model6,7 or the human c.842C>T (p.Pro281Leu) variant (one of the most frequent PAH variants worldwide, albeit much less frequent that the c.1222C>T variant) in a humanized PKU mouse model.9 In our work on the c.842C>T variant, we found delivery of an adenine base editor mRNA and a guide RNA via lipid nanoparticles (LNPs) achieved efficient corrective editing and the complete normalization of blood Phe levels in humanized PKU mice within a few days of treatment.9 In a previous effort to use a prime editor delivered either via dual AAV vectors or a single adenoviral vector to correct the non-human variant present in the homozygous Pahenu2 PKU mouse model, AAV treatment did not significantly modify blood Phe levels (minimal corrective editing), and adenoviral vector treatment moderately reduced blood Phe levels but generally not below the 360 μmol/L threshold (<10% corrective editing).8 In this current study, using an optimized prime editor configuration for a highly clinically relevant human PAH variant, we achieved reduction of blood Phe levels to well below the 360 μmol/L threshold in every single treated PKU mouse with the variant, even in compound-heterozygous mice with only half the editable alleles of homozygous mice.
We acknowledge the limitations of this study. AAV delivery has disadvantages in comparison to LNP delivery when applied for clinical use. The use of AAV vectors entails (1) the prolonged expression of the editor, which could elicit cytolytic immune responses and exacerbate off-target editing, (2) the risk of vector sequence integration, especially at any double-strand-break sites that should be induced by the editor or should spontaneously occur, and (3) the inability to re-dose the therapy if an initial treatment does not achieve the ∼10% corrective editing threshold. LNP delivery shares none of these detrimental aspects of AAV therapy. Current obstacles to LNP delivery of a prime editor include (1) synthesis and encapsulation of the substantially longer mRNA encoding the prime editor protein, in comparison to an mRNA encoding Cas9 nuclease or a base editor, (2) the challenges imposed with regard to scale and purity by the chemical synthesis of a pegRNA that is substantially longer than a standard guide RNA, and (3) the need to co-deliver two guide RNAs rather than one guide RNA. As described in a parallel manuscript,23 our initial attempt to deploy the P56/N19 prime editing configuration via LNP delivery in humanized R408W PKU mice was unsuccessful, whereas deployment of a base editing configuration to correct the c.1222C>T variant via LNP delivery achieved substantial whole-liver editing and normalized Phe levels in humanized R408W PKU mice.
Another limitation is uncertainty as to the optimal approach to off-target assessment for prime editors. Most prior studies have used Cas9 nuclease-oriented techniques as the means to predict sites of off-target prime editing, as we have done in this study, although very new approaches have been designed to assess the reverse transcriptase activity inherent to prime editors.24,25 We did not perform multiple replicates for each pegRNA/ngRNA combination when screening for corrective editing activity in c.1222C>T homozygous HuH-7 cells because our objective was not accuracy with respect to absolute values of editing activity but rather efficiency in iteratively arriving at a highly active prime-editing configuration that could be tested in vivo. We were only able to assess a small subset of all possible pegRNA/ngRNA combinations, and it is possible that combinations with more optimal editing activity than P56/N19 exist. Finally, we did not include vehicle-treated control mice or control mice treated with AAVs encoding an unrelated non-PAH-targeting pegRNA/ngRNA in our experiments; we reasoned that saline vehicle or AAV vectors would not intrinsically cause substantial reductions in blood Phe levels or achieve corrective editing.
Our analysis of real-world clinical data from a group of individuals with PAH c.1222C>T variants unequivocally demonstrate the shortcomings of the current treatment regime with respect to metabolic control and highlight the high unmet medical need of these individuals. On the basis of the results reported here, we envision a prime-editing therapy that uses the same or a similar pegRNA/ngRNA combination as the one optimized in this study and that is demonstrated to achieve as high as >50% whole-liver corrective editing of the c.1222C>T variant. (That high level of editing, when adjusted for a liver hepatocyte fraction of ∼70%, signifies corrective editing of >70% of the c.1222C>T alleles in the hepatocytes; this percentage is well over the threshold needed to address any recessive inborn error of metabolism). Such a therapy, deployed as a treatment to normalize blood Phe levels durably and even permanently, could go a long way toward definitively addressing the medical needs of a large plurality of individuals with PKU. We also anticipate that the workflow demonstrated in this study can be productively applied to the development of prime-editing therapeutics for other PKU variants and for pathogenic variants causing other inborn errors of metabolism.
Data and code availability
The accession number for the next-generation sequencing data reported in this paper is Sequence Read Archive: PRJNA1015257. Other data are available in the Supplemental Data. The GRCh38 Ensembl v98 reference genome annotation was used.
Acknowledgments
This work was supported by grants R35-HL145203, R01-HL148769, and U19-NS132301 from the U.S. National Institutes of Health (K.M.); a career development award from the American Heart Association (X.W.); a graduate research fellowship from the U.S. National Science Foundation (M.N.W.); the Children’s Hospital of Philadelphia Research Institute Frontier Program (R.C.A.-N.); and the Winkelman Family Fund in Cardiovascular Innovation. The graphical abstract was created with BioRender.com.
Author contributions
K.M., R.C.A.-N., and X.W. supervised the work. D.L.B., M.N.W., P.Q., K.M., and X.W. contributed to wet laboratory experiments. R.C.A.-N. performed clinical data analyses. K.M. performed bioinformatic analyses. D.L.B., K.M., R.C.A.-N., and X.W. drafted the manuscript, and all authors contributed to the editing of the manuscript.
Declaration of interests
K.M. is an advisor to and holds equity in Verve Therapeutics and Variant Bio and is an advisor to LEXEO Therapeutics. R.C.A.-N. is an advisor to Latus Bio. All other authors have no financial conflicts of interests to disclose.
Published: November 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.10.005.
Supplemental information
References
- 1.Vockley J., Andersson H.C., Antshel K.M., Braverman N.E., Burton B.K., Frazier D.M., Mitchell J., Smith W.E., Thompson B.H., Berry S.A., American College of Medical Genetics and Genomics Therapeutics Committee Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med. 2014;16:188–200. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 2.Jurecki E.R., Cederbaum S., Kopesky J., Perry K., Rohr F., Sanchez-Valle A., Viau K.S., Sheinin M.Y., Cohen-Pfeffer J.L. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet. Metabol. 2017;120:190–197. doi: 10.1016/j.ymgme.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Hillert A., Anikster Y., Belanger-Quintana A., Burlina A., Burton B.K., Carducci C., Chiesa A.E., Christodoulou J., Đorđević M., Desviat L.R., et al. The genetic landscape and epidemiology of phenylketonuria. Am. J. Hum. Genet. 2020;107:234–250. doi: 10.1016/j.ajhg.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leuders S., Wolfgart E., Ott T., du Moulin M., van Teeffelen-Heithoff A., Vogelpohl L., Och U., Marquardt T., Weglage J., Feldmann R., Rutsch F. Influence of PAH genotype on sapropterin response in PKU: results of a single-center cohort study. JIMD Rep. 2014;13:101–109. doi: 10.1007/8904_2013_263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burton B.K., Longo N., Vockley J., Grange D.K., Harding C.O., Decker C., Li M., Lau K., Rosen O., Larimore K., et al. Pegvaliase for the treatment of phenylketonuria: results of the phase 2 dose-finding studies with long-term follow-up. Mol. Genet. Metabol. 2020;130:239–246. doi: 10.1016/j.ymgme.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D., et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 7.Villiger L., Rothgangl T., Witzigmann D., Oka R., Lin P.J.C., Qi W., Janjuha S., Berk C., Ringnalda F., Beattie M.B., et al. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng. 2021;5:179–189. doi: 10.1038/s41551-020-00671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böck D., Rothgangl T., Villiger L., Schmidheini L., Matsushita M., Mathis N., Ioannidi E., Rimann N., Grisch-Chan H.M., Kreutzer S., et al. In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abl9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks D.L., Carrasco M.J., Qu P., Peranteau W.H., Ahrens-Nicklas R.C., Musunuru K., Alameh M.-G., Wang X. Rapid and definitive treatment of phenylketonuria in variant-humanized mice with corrective editing. Nat. Commun. 2023;14:3451. doi: 10.1038/s41467-023-39246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O’Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 11.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.F., Chen C., Nelson J.W., Newby G.A., Sahin M., et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652.e29. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A., Liu D.R. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022;40:402–410. doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis J.R., Banskota S., Levy J.M., Newby G.A., Wang X., Anzalone A.V., Nelson A.T., Chen P.J., Hennes A.D., An M., et al. Efficient prime editing in mouse brain, liver and heart with dual AAVs. Nat. Biotechnol. 2023:1–12. doi: 10.1038/s41587-023-01758-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petri K., Kim D.Y., Sasaki K.E., Canver M.C., Wang X., Shah H., Lee H., Horng J.E., Clement K., Iyer S., et al. Global-scale CRISPR gene editor specificity profiling by ONE-seq identifies population-specific, variant off-target effects. bioRxiv. 2021 doi: 10.1101/2021.04.05.438458v1. Preprint at. [DOI] [Google Scholar]
- 18.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romani C., Manti F., Nardecchia F., Valentini F., Fallarino N., Carducci C., De Leo S., MacDonald A., Palermo L., Leuzzi V. Adult cognitive outcomes in phenylketonuria: explaining causes of variability beyond average Phe levels. Orphanet J. Rare Dis. 2019;14:273. doi: 10.1186/s13023-019-1225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashe K., Kelso W., Farrand S., Panetta J., Fazio T., De Jong G., Walterfang M. Psychiatric and cognitive aspects of phenylketonuria: the limitations of diet and promise of new treatments. Front. Psychiatr. 2019;10:561. doi: 10.3389/fpsyt.2019.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas L., Olson A., Romani C. The impact of metabolic control on cognition, neurophysiology, and well-being in PKU: a systematic review and meta-analysis of the within-participant literature. Mol. Genet. Metabol. 2023;138 doi: 10.1016/j.ymgme.2022.106969. [DOI] [PubMed] [Google Scholar]
- 23.Brooks D.L., Whittaker M.N., Said H., Dwivedi G., Qu P., Musunuru K., Ahrens-Nicklas R.C., Alameh M.-G., Wang X. A base editing strategy using mRNA-LNPs to correct the most frequent phenylketonuria variant in vivo. HGG Adv. 2023;2023:2003–2014. doi: 10.1016/j.xhgg.2023.100253. [DOI] [PubMed] [Google Scholar]
- 24.Kwon J., Kim M., Bae S., Jo A., Kim Y., Lee J.K. TAPE-seq is a cell-based method for predicting genome-wide off-target effects of prime editor. Nat. Commun. 2022;13:7975. doi: 10.1038/s41467-022-35743-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang S.Q., Liu P., Ponnienselvan K., Suresh S., Chen Z., Kramme C., Chatterjee P., Zhu L.J., Sontheimer E.J., Xue W., Wolfe S.A. Genome-wide profiling of prime editor off-target sites in vitro and in vivo using PE-tag. Nat. Methods. 2023;20:898–907. doi: 10.1038/s41592-023-01859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the next-generation sequencing data reported in this paper is Sequence Read Archive: PRJNA1015257. Other data are available in the Supplemental Data. The GRCh38 Ensembl v98 reference genome annotation was used.