

Summary
Aneuploidy frequently arises during human meiosis and is the primary cause of early miscarriage and in vitro fertilization (IVF) failure. Individuals undergoing IVF exhibit significant variability in aneuploidy rates, although the exact genetic causes of the variability in aneuploid egg production remain unclear. Preimplantation genetic testing for aneuploidy (PGT-A) using next-generation sequencing is a standard test for identifying and selecting IVF-derived euploid embryos. The wealth of embryo aneuploidy data and ultra-low coverage whole-genome sequencing (ulc-WGS) data from PGT-A have the potential to discover variants in parental genomes that are associated with aneuploidy risk in their embryos. Using ulc-WGS data from ∼10,000 PGT-A biopsies, we imputed genotype likelihoods of genetic variants in embryo genomes. We then used the imputed variants and embryo aneuploidy calls to perform a genome-wide association study of aneuploidy incidence. Finally, we carried out functional evaluation of the identified candidate gene in a mouse oocyte system. We identified one locus on chromosome 3 that is significantly associated with meiotic aneuploidy risk. One candidate gene, CCDC66, encompassed by this locus, is involved in chromosome segregation during meiosis. Using mouse oocytes, we showed that CCDC66 regulates meiotic progression and chromosome segregation fidelity, especially in older mice. Our work extended the research utility of PGT-A ulc-WGS data by allowing robust association testing and improved the understanding of the genetic contribution to maternal meiotic aneuploidy risk. Importantly, we introduce a generalizable method that has potential to be leveraged for similar association studies that use ulc-WGS data.
Keywords: preimplantation genetic testing for aneuploidy, ultra-low-coverage whole-genome sequencing, embryo aneuploidy, genome-wide association study, CCDC66
Graphical abstract
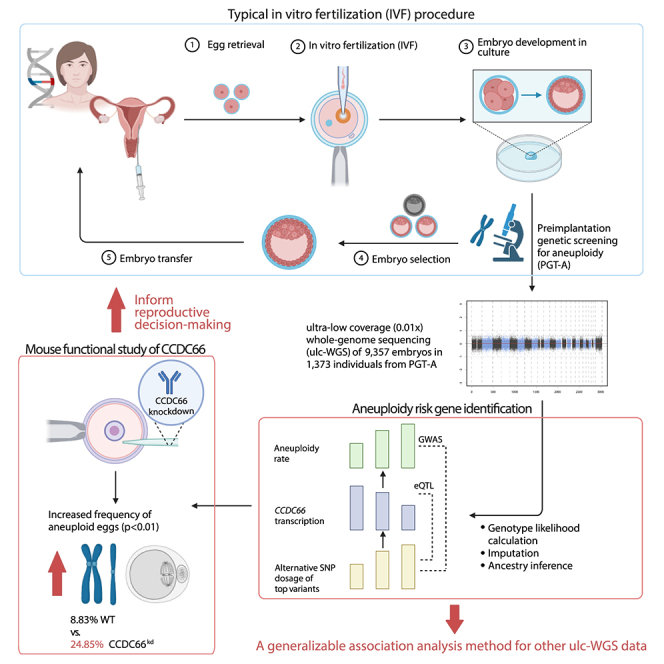
Egg aneuploidy is common in human meiosis and is the primary cause of early miscarriage. Here, we developed a method to analyze ultra-low coverage whole-genome sequencing (ulc-WGS) data from preimplantation genetic testing for aneuploidy (PGT-A) of embryos and identified variants in CCDC66 associated with aneuploid conception risk.
Introduction
Aneuploidy is the most common genetic abnormality in human embryos and the leading genetic cause of miscarriage and in vitro fertilization (IVF) failure.1 Maternal age is well documented as a risk factor for producing aneuploid gametes. However, the propensity to produce aneuploid embryos varies substantially even among mothers of a similar age.1,2,3,4,5 Recently, variants in several genes related to control of chromosome segregation have been implicated in contributing to aneuploidy risk.5,6,7,8 However, many identified variants only contribute to the aneuploidy risk in a small number of individuals, and most of these studies have limited sample sizes. Additional efforts are needed to fully understand the genetic contribution to the aneuploidy risk in populations.
Currently, the most effective treatment of infertility is IVF, where eggs are surgically retrieved after controlled ovarian stimulation and fertilized in a Petri dish with subsequent embryo selection and transfer back to the uterus.9,10 Preimplantation genetic testing for aneuploidy (PGT-A) was developed as an approach to improve IVF outcomes by prioritizing euploid embryos for transfer based on the inferred genetic constitution of an embryo biopsy.11,12,13,14 PGT-A performed on trophectoderm cells isolated from blastocyst-stage embryos has provided a rich resource of aneuploidy measurements. Next-generation sequencing (NGS)-based PGT-A facilitates inference of aneuploidy by comparing the genome sequencing coverage across the chromosomes. For aneuploidy detection, a shallow coverage of the genome (e.g., <0.01× genome coverage per embryo biopsy) is sufficient. However, because of the low sequencing coverage of the NGS-based PGT-A data (referred to as the ultra-low coverage whole-genome sequencing [ulc-WGS] in this study), the genotype information encoded therein is rarely used for genetic studies.12,15
Genome-wide association studies (GWASs) have revolutionized the field of complex disease genetics over the past decade by identifying genotype-phenotype associations based on testing millions of genetic variants across the genomes.16,17 For genetic variants showing strong disease association, further fine-mapping and gene prioritization approaches proceed to identify variants that causally impact the traits.18,19 This approach has identified risk loci for many diseases and traits, such as susceptibility to viral infections and type 2 diabetes.20,21 Applying a GWAS approach to PGT-A data could help identify additional genetic risk factors to embryo aneuploidy.
Here, we describe an integrative approach to identify candidate variants through retrospective analysis of NGS-based PGT-A data. After combining data from sibling embryos and imputing variant dosages, we conducted a GWAS to identify candidate genes. Our analysis identified one genomic region that is associated with embryo aneuploidy risk on chromosome 3. Functional investigation of the variants suggested that the candidate variants are causal expression quantitative trait loci (eQTLs) for coiled-coil domain-containing 66 (CCDC66). Validation experiments in mouse oocytes showed that CCDC66 depletion was associated with higher meiotic aneuploidy rates, likely contributing to elevated risk of aneuploid conception.
Material and methods
Dataset description
PGT-A data were obtained from individuals undergoing IVF between 2017 and 2019 at CCRM Fertility. The samples do not qualify as federally regulated human subjects research, as determined by the Institutional Review Board of Rutgers University (Pro2023001490). One IVF cycle with at least three embryos tested was included for each individual. IVF cycles with maternal age ≥43 years were excluded from the analysis because eggs used in these cycles were from egg donors of unknown age. Embryos underwent trophectoderm biopsy on day 5, 6, or 7 post-fertilization, followed by PGT-A using the Illumina VeriSeq PGS kit and protocol, which entails sequencing on the Illumina MiSeq platform (36-bp single-end reads) (Illumina, USA). Chromosome copy numbers from each embryo biopsy were inferred using the Illumina BlueFuse Multi software suite in accordance with the VeriSeq protocol.22 Each embryo was then noted as “euploid” or “aneuploid” based on the chromosome copy number.
The aneuploidy rate for each IVF cycle was determined with the formula described previously7,8,23:
Sequencing alignment and variant calling
PGT-A sequencing files with <150,000 reads were considered low quality and excluded. After filtering, sequencing files from each IVF cycle were combined into a single file for analysis. The sequencing reads were aligned to the human reference genome (GRCh38) with bwa-mem (version 0.7.17)24 and converted to the binary alignment/map (BAM) format using samtools (v.1.13).25 Ancestry inference was performed using LASER (v.2.0), as previously described.22,26,27 Briefly, principal component (PC) space was defined based on the 1000 Genomes project reference samples. Sequencing samples were then projected onto the space using a Procrustes approach implemented in LASER. Samples were assigned to superpopulations (African [AFR], admixed American [AMR], East Asian [EAS], European [EUR], and South Asian [SAS]) based on genetic similarity to the 1000 Genomes reference panel.
Genotype likelihoods (GLs) were computed with bcftools (v.1.13)28 for each sample at all variable positions of the reference panel (1000 Genomes 30x on GRCh38, https://www.internationalgenome.org/home). Imputation and phasing in the form of GLs were performed using GLIMPSE.29 Specifically, GLIMPSE refines the GLs by iteratively running genotype imputation and haplotype phasing with a Gibbs sampling procedure to produce consensus-based haplotype calls and genotype posteriors at every variant position.29 With imputed data, each variant site was filtered based on the following criteria: imputation score ≥ 0.2, minor allele frequency (MAF) ≥ 5%. After filtering, the imputed genotype dosages of each individual were calculated and used in the association test:
Given the known reference and alternative alleles, of three possible genotypes (i.e., homozygous reference, homozygous alternative, and heterozygous) were multiplied by the number of alternative alleles of each genotype ().
For MAF correlation analysis, the population MAF for each variant was extracted from two reference panels: the 1000 Genomes Projects (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_phased/) and the Genome Aggregation Database (gnomAD) (v.3.1).30 Correlations of MAFs between our imputed data and reference panels were calculated with Pearson correlation coefficient (R).
Association test and eQTL analysis
For the association test, a quasibinomial generalized linear regression model (GLM) was iteratively fit for each variant using the function glm() in R as follows:
glm(data, formula = cbind(aneuploid_embryos_numbers, euploid_embryos_numbers) ∼ age + ancestry_PCs + single_SNP_dosage, family = "quasibinomial")
At each iteration, a single nucleotide polymorphism (SNP) dosage was tested. Maternal age and top four ancestry PCs inferred using LASER were included as covariates. The resulting p values were visualized using a Manhattan plot and checked using a quantile-quantile (Q-Q) plot with the R package GWASTools (v.1.44.0).31 The significant variants were determined using a significance threshold p value ≤ 2e−8 and Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05. Haplotype structure surrounding significant loci was visualized with Locuszoom (http://locuszoom.org/).32
The Genotype-Tissue Expression (GTEx) project includes genotypes, gene expression, and histological and clinical data from 54 non-diseased tissue sites across nearly 1,000 individuals.33 The eQTL information from GTEx (https://www.gtexportal.org/home/eqtlDashboardPage, access date: 06/30/2022) was used to determine the candidate variants’ potential association with expression of nearby (i.e., cis) genes.
Mice and oocyte collection and maturation
C57BL/6 mice (6–10 weeks and 9 months of age) (Jackson Laboratory, USA) were used. Mice were housed with a constant temperature and a standard 12-h light/12-h dark cycle in the animal facility at Rutgers University (NJ, USA). All animal experiments performed in this study were approved by the Rutgers IACUC (protocol #201702497) and followed guidelines set by the National Institutes of Health. For oocyte collection, mice were primed with pregnant mare serum gonadotropin (PMSG; Lee Biosolutions, #493-10) two days before collection. Prophase I-arrested oocytes were collected as described before34 in minimum essential medium (MEM) (Sigma, #M0268) with 2.5 μM milrinone (Sigma, #M4659) to prevent spontaneous meiotic resumption. The oocytes were then incubated in Chatot, Ziomek, and Bavister (CZB) media without milrinone in 5% CO2 at 37°C for the desired time of maturation depending on the meiotic stages to be evaluated (0 h for prophase I, 5 h for pro-metaphase I, 7 h for metaphase I, and 16 h for metaphase II).
Knockdown of CCDC66 in mouse oocytes
To deplete CCDC66, we used the Trim-away strategy.7,35,36 Rabbit anti-CCDC66 antibody (Bethyl Laboratories, #A303-339A) and control immunoglobulin G (IgG) antibody (Merck Millipore, #12–370) were purified using Amicon Ultra 0.5-mL Centrifugal Filter (Merk Millipore, #UFC5003096). pGEMHE-Cherry-TRIM21 (Addgene, #105522) or pGEMHE-mEGFP-mTrim21 (Addgene, #105519) were linearized with Asc I (New England Biolabs, #R0558S) and in vitro transcribed using a T7 mMessage mMachine Kit (Ambion, #AM1340). Prophase I-arrested oocytes were co-microinjected with the fluorescently tagged Trim21 cRNA and with either rabbit anti-CCDC66 antibody (0.5 mg/mL) or IgG antibody (0.5 mg/mL) in the control group. Injections were performed using a Xenoworks digital microinjector (Sutter Instruments) in MEM supplemented with 2.5 μM milrinone. The oocytes were incubated in milrinione-containing CZB media for at least 3 h in 5% CO2 at 37°C before starting meiotic maturation by washing out the milrinone and culturing in CZB medium. Oocytes were fixed at metaphase I stage (7 h post-milrinone washout) and immunostained to evaluate CCDC66 knockdown efficiency.
Antibodies and immunofluorescence
The following antibodies were used: rabbit anti-CCDC66 antibody (1:50, Bethyl Laboratories, A303-339A), mouse anti-α-tubulin ((B-5-1-2) Alexa Fluor 488) (1:100, Invitrogen, 322588), and human anti-centromeric antigen (ACA) (1:30, Antibodies Incorporated, 15–234). These secondary antibodies (1:200) were used: donkey-anti-rabbit Alexa Fluor 568 (Life Technologies, A10042) and goat-anti-human Alexa Fluor 633 (Life Technologies, A21091).
Immunofluorescence was performed as previously described.37 Oocytes were fixed with 2% paraformaldehyde (PFA) (Sigma-Aldrich, P6148) in phosphate-buffered saline (PBS) at room temperature for 20 min. The fixative was then washed out by incubating the oocytes in blocking buffer (0.3% BSA containing 0.01% Tween 20 in PBS) three times for 10 min. Oocytes were then permeabilized in PBS containing 0.2% Triton X-100 for 20 min and blocked in blocking buffer for 10 min. Primary antibody incubation was performed by incubating the oocytes overnight at 4°C (CCDC66) or 1 h at room temperature (ACA) in a dark, humidified chamber followed by three washes of 10 min each in blocking solution. Then oocytes were incubated in secondary antibody for 1 h in a dark, humidified chamber followed by three washes of 10 min each in blocking buffer. Finally, oocytes were mounted in 10 μL of Vectashield containing 4, 6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Life Technologies, D1306).
In situ chromosome counting
As described previously,38,39 the microinjected prophase I-arrested oocytes from young and old mice were matured in CZB media without milrinone in a humified incubator (5% CO2, 37°C) for 16 h until they completed meiosis I and arrested at metaphase of meiosis II. Then, eggs were cultured for at least 2 h in 100 μM Monastrol (Sigma #M8515) to collapse the spindle and facilitate the separation of the chromosomes. The eggs were fixed with 2% PFA in PBS for 20 min and permeabilized in PBS containing 0.2% Triton X-100 for 20 min. Eggs were stained with ACA antibody to detect centromeres and DAPI to detect DNA. A normally developing mouse egg at metaphase II has 20 sister chromatids; any deviation of this number was considered an aneuploid egg. Chromosome counting was performed with ImageJ software (NIH, https://imagej.net/ij/index.html) using cell counter plugins.
Imaging
Images were acquired with Leica SP8 confocal microscopes equipped with a 40×, 1.30 NA oil-immersion objective or a 63×, 1.40 NA oil-immersion objective. For each image, optical z sections were obtained using 0.5-μm step with zoom of 4.5. For comparison of pixel intensities, the laser power was kept constant for each oocyte in an experiment. All oocytes in the same experiment were processed at the same time.
Results
Project overview, sample cohort, variant calling, and ancestry inference
To identify genomic loci associated with aneuploidy in the embryos of individuals who underwent IVF, we analyzed embryo biopsy sequences collected from the PGT-A procedure (Figure 1). The dataset included 10,011 embryo biopsies from 1,467 IVF cycles. After removing data from egg donors and low quality (<150,000 reads), 9,357 embryo biopsies from 1,373 cycles remained, with maternal age ranging from 23 to 42 years (median = 35) (Figure 2A). To improve the coverage for analysis, we pooled all sequenced embryos from each IVF cycle. Because embryos in a cycle are equivalent of full siblings, this combined file captured both maternal and paternal genomes. After pooling, the median coverage of each sample was 0.056× (Figure S1A). As expected, the mean coverage per sample was linearly associated with the number of sequenced embryo biopsies (Figure S1B).
Figure 1.

Overall strategy for PGT-A data analysis
For each IVF cycle, the number of aneuploid and euploid embryo biopsies were determined using PGT-A. ulc-WGS data from PGT-A for each IVF cycle were combined for analysis. Genotype likelihood and dosage were imputed for each variant, and ancestry of the samples was inferred using the genotype likelihood. Association tests were performed between the imputed genetic variants and the aneuploidy rate. eQTL analysis was used to determine the candidate gene associated with the top variants from the association test. CCDC66 was selected for functional studies using a mouse oocyte model.
Figure 2.

Phenotypic characterization and ancestry inference of the individuals
(A) Aneuploidy rate versus age. x axis: maternal age in years; y axis: blastocyst aneuploidy rate. The whiskers in the box-whisker plot span a range that is 1.5× the interquartile range (IQR).
(B and C) Ancestry inference using the PGT-A data. Principal component axes (PC1 and 2 in B, PC2 and 3 in C) were defined based on analysis of 1000 Genomes reference samples and colored according to superpopulation annotations (African [AFR], admixed American [AMR], East Asian [EAS], European [EUR], South Asian [SAS]). PGT-A samples were then projected onto these axes using a Procrustes approach with LASER.26,27
We next performed ancestry inference based on the sequence data using the program LASER. Our analysis revealed a diverse cohort, consistent with the demographic composition of the local population (Figures 2B and 2C). Specifically, according to the superpopulation reference panel defined by the 1000 Genomes Project,40 788 samples (57.4%) have genetic similarity with EUR reference samples, 223 (16.2%) with AMR reference samples, 168 (12.2%) with AFR reference samples, 143 (10.4%) with SAS reference samples, and 52 (3.8%) with EAS reference samples.
Using the program GLIMPSE, we identified variants and performed GL imputation across the sample cohort (see material and methods for details). A total of 10,740,080 variants were imputed, among which 4,353,993 variants had INFO scores ≥0.2 (Figure S2A). After selecting variants with ≥5% MAF, 2,549,983 variants remained (Figure S2B). After imputation, MAFs of imputed variants in our sample were highly correlated with large population databases: the 1000 Genomes (R = 0.95, p < 2.2e−16, Figure S3A) and the gnomAD (R = 0.97, p < 2.2e−16, Figure S3B).
Genome-wide association analysis for aneuploidy
To identify aneuploidy risk loci, we next investigated the association between aneuploidy rate and genotype dosage for each variant using a GLM, incorporating four ancestry PCs and the maternal age as covariates (see material and methods for details).
Three SNPs on chromosome 3 reached genome-wide significance for association with aneuploidy at the level of p ≤ 2e−8 and FDR ≤ 0.05 (Figure 3A; Tables 1 and S1). The Q-Q plot did not show strong inflation of the test statistics (Figure 3B), suggesting that confounding factors, such as population structure, were generally controlled. The three significant SNPs were located in ELKS/RAB6-interacting/CAST family member 2 (ERC2), which has not been reported as associated with maternally derived aneuploidy in OMIM (Figure 3C). Within the locus, the three significant SNPs are in strong linkage disequilibrium with each other (Table 1). The top SNP, rs12495172 (chr3:55959628G>A [GRCh38]), is located in intron 12–13 of ERC2. The mean depth of coverage of the 1-Mbp window covering the significant variants had a median of 0.066 among all samples, comparable to 0.055 for the entire chromosome (Figure S3C; Table 1). As indicated by the positive beta values (e.g., 0.079 for the rs12495172), the alternative allele of each significant variant in ERC2 is positively associated with aneuploidy rate.
Figure 3.

Association test for aneuploidy
(A) Manhattan plot depicts −log10(p value) of association tests of each SNP versus the aneuploid embryo count. The rs numbers of the three significant SNPs on chromosome 3 are labeled. The dashed line indicates the genome-wide significance threshold of p value < 5 × 10−8.
(B) Q-Q plot describes the distribution of observed −log10(p value) versus those expected under the null hypothesis.
(C) LocusZoom plot shows lead SNP (rs12495172) and SNPs in the region around ERC2. The lead SNP is shown in purple diamond and the heatmap shows the linkage disequilibrium (LD) between the lead and nearby SNPs. Recombination rates are plotted as blue lines.
Table 1.
SNPs associated with embryo aneuploidy
| Chr | Position | Ref/Alt | INFO | AF | AF gnomAD | AF 1KG | p | FDR | beta | LD | Distance | rsID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 55959628 | G/A | 0.237 | 33.8% | 39.4% | 40.8% | 5.48E−09 | 8.67E−03 | 0.079 | – | – | rs12495172 |
| 3 | 55952031 | C/T | 0.244 | 32.8% | 38.9% | 39.4% | 6.80E−09 | 8.67E−03 | 0.084 | 0.563 | 7597 | rs11130489 |
| 3 | 55959515 | A/G | 0.234 | 35.0% | 42.3% | 44.1% | 1.41E−08 | 1.20E−02 | 0.085 | 0.567 | 113 | rs897966 |
| 3 | 55967692 | G/C | 0.242 | 34.8% | 41.6% | 43.0% | 6.23E−08 | 3.10E−02 | 0.082 | 0.559 | 8064 | rs9881130 |
| 3 | 55962557 | C/T | 0.245 | 34.2% | 41.8% | 43.1% | 6.30E−08 | 3.10E−02 | 0.078 | 0.613 | 2929 | rs6797130 |
| 3 | 55955490 | C/G | 0.239 | 35.0% | 42.8% | 44.4% | 7.29E−08 | 3.10E−02 | 0.079 | 0.621 | 4138 | rs9311590 |
| 3 | 55965325 | A/G | 0.247 | 32.8% | 38.1% | 38.8% | 9.96E−08 | 3.63E−02 | 0.080 | 0.559 | 5697 | rs6770904 |
| 3 | 55962257 | T/C | 0.235 | 34.7% | 39.8% | 41.0% | 1.20E−07 | 3.83E−02 | 0.083 | 0.618 | 2629 | rs6763168 |
Position: chromosomal position based on the human reference genome (GRCh38).
INFO: IMPUTE info quality score.
AF: alternative allele frequency.
AF gnomAD: alternative allele frequency in the gnomAD project.
AF 1KG: alternative allele frequency in the 1000 Genomes project.
beta: regression coefficient.
LD: linkage disequilibrium (r2) between the SNP and the top SNP (chr3:55959628G>A).
Distance: the distance (bps) between the SNP and the top SNP (chr3:55959628G>A).
Next, we aimed to identify the candidate genes associated with the top variants. A previous study showed that variants discovered by GWASs are more likely to affect the expression of nearby genes (i.e., as eQTLs), and the altered expression can ultimately influence the phenotypic trait.41 Therefore, integrating GWASs with gene expression data can facilitate candidate gene prioritization.19 To determine the effect of the top SNPs on nearby gene expression, we examined eQTL signals using data from the GTEx project (GTEx analysis release v.8, dbGaP: phs000424.v.8.p2). The GTEx data suggested that alternative alleles of the top variants were associated with reduced expression of a nearby gene, CCDC66, in two tissues (thyroid and tibial nerve, see Figure 4A as one example). CCDC66 has a wide expression profile, and ovary showed the second highest expression in females (Figure S4A), suggesting its potential function in female reproduction. Next, we determined the expression of CCDC66 in ovaries from different female age groups (20–29, 30–39, and 40–49). Possibly because of the small sample size and the large variation among samples, CCDC66 expression is not significantly different among the three age groups (Figure S4B). Given that there was no eQTL signal for other genes, including ERC2, we selected CCDC66 as the candidate aneuploidy risk gene whose reduction in expression may be associated with increased aneuploidy rate.
Figure 4.

Gene prioritization for casual variants and genes
(A) GTEx eQTLs of the lead significant variants rs12495172 (chr3:55959628G>A) on CCDC66 expression. The decreased expression of CCDC66 correlates with alternative alleles of rs12495172 (number of individuals is shown below each genotype).
(B) The genotype dosages of the alternative allele of rs12495172. The alternative allele genotype dosages in samples were divided into 3 bins of roughly equal size. The aneuploidy rates among the samples were positively correlated with the alterative allele genotype dosages. The whiskers in the box-whisker plot span a range that is 1.5× the interquartile range (IQR).
CCDC66 regulates meiotic progression and chromosome segregation fidelity
CCDC66 encodes a microtubule-associated protein that regulates microtubule nucleation and organization during cell division.42,43 In mitosis, CCDC66 regulates centrosome maturation via recruitment of core pericentriolar material (PCM) proteins and microtubule organization via its cross-linking activity.42
Because the vast majority of aneuploidies have a maternal origin (i.e., from oocytes),44,45 we focused our experimental analysis on oocyte meiosis. Human oocytes are challenging to obtain in significant numbers. We therefore elected to determine the role of CCDC66 in meiosis using mouse oocytes, a robust meiotic experimental system. First, we evaluated localization of the protein during meiotic maturation via immunostaining of oocytes fixed at different meiotic stages (Figure 5A). We detected CCDC66 in prophase I-staged oocytes with slight enrichment in the nucleus. In pro-metaphase I and metaphase I oocytes and in metaphase II eggs, CCDC66 was enriched around the spindle (Figure 5A). This localization pattern suggested a requirement of CCDC66 during mouse oocyte meiotic maturation.
Figure 5.

CCDC66 is important for meiotic progression and production of euploid eggs
(A) Localization of CCDC66 in different stages of mouse oocyte meiotic maturation. Oocytes were stained with anti-CCDC66 antibody (gray) at (0, 5, 7, and 16) hours of maturation corresponding to prophase I, pro-metaphase I, metaphase I, and metaphase II, respectively. Tubulin and DAPI were used to label the spindle and DNA (green and blue, respectively). Scale bars: 15 and 4 μm (insets).
(B) Prophase-I arrested oocytes were co-microinjected with Trim21 cRNA and either CCDC66 antibody or IgG. Oocytes were fixed at metaphase I and stained to detect CCDC66 (gray). Tubulin (green) and DAPI (blue) were used to label the spindle and DNA.
(C) Relative CCDC66 intensity from (B), two-tailed unpaired Student’s t test (∗∗∗p < 0.001).
(D) Quantification of percentage of polar body extrusion (PBE).
(E) Percentage of aneuploid metaphase II eggs after knockdown of CCDC66 in young and old mouse oocytes (one-way ANOVA, ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001). Number of oocytes examined: young IgG, 34; young CCDC66, 37; old IgG, 29; old CCDC66, 31. These experiments were repeated 3 times.
To evaluate a requirement for CCDC66 in oocyte meiotic maturation, we depleted the protein using the Trim-Away strategy35 and confirmed ∼95% depletion by subsequent immunocytochemistry (Figures 5B and 5C). To determine the effect of CCDC66 depletion on meiotic progression and meiosis I chromosome segregation, we calculated the percentage of oocytes that extruded polar bodies (PBEs) and percentage of aneuploid metaphase II eggs, respectively. In reproductively young mice (6–10 weeks of age, equivalent to ∼20 years of human age46), 73.18% of control-injected oocytes extruded a polar body. This rate decreased significantly to 66.16% in the CCDC66 depletion group (p < 0.05) (Figure 5D). In oocytes from young mice, the average rate of aneuploidy in metaphase II eggs was 2.56% in the control group and increased significantly to 13.24% in the depletion group (p < 0.05). Therefore, decreased levels of CCDC66 increase the chances of chromosome segregation errors during meiosis I in oocytes from reproductively young mice.
Elevated egg aneuploidy is associated with advanced maternal age (>35 years), but some women experience higher egg aneuploidy rate at younger-than-average ages. To evaluate the interplay between genetics and maternal age, we also conducted the PBE and aneuploidy rate assessment experiments in reproductively older mice (9 months, equivalent to ∼38 years in humans46). Control-injected oocytes from older mice had a reduced PBE rate (66.59%) compared with oocytes in the young control-injected group (73.18%). Furthermore, depletion of CCDC66 in older oocytes also significantly reduced PBE compared with older oocyte controls (57.07% versus 66.59%, respectively; p < 0.05) (Figure 5D). In accordance with having an age-related reduction in PBE rate, control-injected oocytes from reproductively old mice had an elevated aneuploidy incidence (8.83%). Depletion of CCDC66 in oocytes from old mice had a more severe phenotype with a higher incidence of aneuploidy compared with controls (24.85%, p < 0.01). Furthermore, oocytes from 9-month-old mice were significantly more likely to be aneuploid when CCDC66 was depleted than oocytes from young mice. Taken together, these data demonstrate that decreased levels of CCDC66 are associated with increased egg aneuploidy rates, a phenotype which becomes more severe with reproductive aging.
Discussion
The key to reproductive success lies in faithful chromosome segregation in meiosis to create a euploid zygote upon fertilization.1,47 The error-prone nature of meiosis often results in low-quality gametes, leading to spontaneous abortion of aneuploid embryos.3,4,48 Recent studies suggest oocyte meiotic maturation is susceptible to dysregulation by maternal genetic variants that contribute to aneuploid concepti, such as variants in CEP120 and AURKB (reviewed in Biswas et al., Capalbo et al., and Volozonoka et al.47,49,50). These maternal genetic variants are strong candidates for clinical validation as predictive biomarkers of IVF outcomes. Identifying and validating additional genetic variants will contribute to a complete panel of infertility biomarkers. This can be used to complement existing clinical approaches to infertility, and genetic evaluations as the prognostic indicator of conception success could substantially improve pregnancy outcomes.
A major hurdle in identifying aneuploidy biomarkers is the lack of individual samples with both egg aneuploidy phenotypes and genome sequencing information. To overcome this limitation, we developed an integrated method for analyzing PGT-A data and illustrated the utility of these data for understanding risk factors of embryo aneuploidy. Unlike most discoveries focused on maternal genomes for aneuploidy risk variants,47 our method has the potential to identify risk factors of both maternal and paternal origins. Here, we provide one example of how by leveraging the power of imputation and GLs, even ulc-WGS data are sufficient to identify common variant associations with aneuploidy risk, especially when aggregating sibling embryo sequences from the same individual. We discovered one locus associated with aneuploidy on chromosome 3. Further eQTL analysis suggests that CCDC66 is the candidate gene for embryo aneuploidy risk.
Through functional studies, we found that CCDC66 is important for the completion of meiotic progression and the production of euploid eggs. In mouse oocytes, the gene is expressed at all meiotic stages, and we observed a significant reduction of PBE in young and old mice after depleting endogenous CCDC66. Depletion of the protein in eggs also increased the incidence of aneuploidy, a phenotype that is exaggerated in aged mice. When the age and aneuploidy rate interaction was included as a co-variate in our association analysis, it did not show significant association with the aneuploidy rate variation. However, our limited sample size might have contributed to the result. In mitotic cells, CCDC66 function indicates that it is a microtubule-associated protein that localizes to centrosomes, centriolar satellites, and the primary cilium throughout the cell cycle.42,43 More importantly, in our OMIM search, we did not find other studies focusing on the function of CCDC66 in meiosis. Additional studies are needed to better understand its function in both mitosis and meiosis.
Our current study has a few limitations. First, in addition to errors of meiotic origin, aneuploidy detected by PGT-A could also arise from chromosome mis-segregation during early embryonic mitotic divisions. These mitotic errors could cause mosaicism in the embryos and potentially confound the meiotic aneuploidy phenotype of interest.51,52,53,54 To circumvent this limitation, we recently developed a haplotype-based approach to isolate the subset of aneuploidies with characteristic signatures of meiotic error.22 In the future when the sample size is sufficiently large, we can apply this method to disentangle the genetic underpinnings of mitotic versus meiotic errors. Analysis of these sub-phenotypes will allow us to evaluate whether certain alleles predispose to meiotic errors, mitotic errors, or both. Second, to increase the sequencing coverage, we combined embryo biopsy sequences from the same IVF cycle. Genetically, these embryos are equivalent of full siblings, and the combined sequences contain genomic variation from both maternal and paternal genomes. Therefore, some parts of the genome could be tetraploid rather than diploid. However, given the low coverage in the combined samples (median coverage 0.056×), we expect most of the sites are not affected, and our analyses based on the diploid assumption are still valid. Third, our functional analysis is based on a mouse oocyte system. More functional studies in model organisms, such as knock-in mutations to mimic the human genetic condition, would help elucidate the role of the variants in candidate genes in relation to their parental origin.
Conclusion
Sufficient large sample size is fundamentally important in addressing biological questions in population and medical genetics. Large low-coverage sequencing datasets have become more accessible for analyses as costs of sequencing continue to plummet. Given the same sequencing depth, low-coverage sequencing of many individuals tends to be more powerful than deep sequencing of fewer individuals.55,56 Recent studies have demonstrated the application of low-coverage sequencing data in GWASs,20,57 polygenic risk score calculation,58 and population genomics.59,60 In addition, computational tools that are specialized for low-coverage sequencing data are also being actively developed.29,61,62 These developments allow for future applications of low-coverage sequencing data.
Recently, a large number of ulc-WGS data have been generated from different sources, such as PGT-A,63 cell-free DNA (cfDNA)64 including non-invasive prenatal testing (NIPT),20 and off-target sequencing reads from targeted sequencing experiments.65 These sequences have not been fully investigated due to the difficulties in interpreting the sparse genotype observations. Our results show that when applied to large datasets, global patterns emerge even at the very low depth of coverage and can provide insight into the biological origins of aneuploidy. Once fully developed, we believe that our method, with the consideration of genotype uncertainty in a probabilistic framework, would be applicable to other ulc-WGS datasets and could help improve the overall utility of the ulc-WGS data in the genetics field.
Data and code availability
The summary statistics of the GWAS variants with p < 1e−3 are available in Table S1, and the summary statistics of all variants have been submitted to the GWAS catalog (https://www.ebi.ac.uk/gwas/) (GWAS Catalog: GCST90292548). The data used for the analyses described in Figures 4 and S4 were obtained from the GTEx Portal on 09/21/23 and GTEx Analysis Release v.8 (dbGaP: phs000424.v.8.p2) on 09/21/23. The analysis codes are available at https://github.com/JXing-Lab/PGTA_aneuploidy.
Acknowledgments
This work is partly supported by the NIH/NICHD grant R01-HD091331 to K.S. and J.X. R.C.M. is supported by grant R35GM133747 from the NIH/NIGMS. We thank the GTEx project for providing the eQTL data in the GTEx Portal. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Author contributions
Conceptualization: R.C.M., J.X.; investigation: S.S., M.A., D.A., M.E.H., C.R.W., M.D., A.B., M.V., M.K.-J., R.C.M., K.S., J.X.; data curation: D.A., M.E.H., C.R.W., M.V., M.K.-J.; formal analysis: S.S., M.A., D.A., R.C.M., K.S., J.X.; initial draft: S.S., M.A., J.X.; supervision: R.C.M., K.S., J.X. All authors reviewed the manuscript and agreed to the published version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: November 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.11.002.
Supplemental information
References
- 1.Hassold T., Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 2.Franasiak J.M., Forman E.J., Hong K.H., Werner M.D., Upham K.M., Treff N.R., Scott R.T., Jr. The nature of aneuploidy with increasing age of the female partner: a review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil. Steril. 2014;101:656–663.e1. doi: 10.1016/j.fertnstert.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 3.Kubicek D., Hornak M., Horak J., Navratil R., Tauwinklova G., Rubes J., Vesela K. Incidence and origin of meiotic whole and segmental chromosomal aneuploidies detected by karyomapping. Reprod. Biomed. Online. 2019;38:330–339. doi: 10.1016/j.rbmo.2018.11.023. [DOI] [PubMed] [Google Scholar]
- 4.Kuliev A., Zlatopolsky Z., Kirillova I., Spivakova J., Cieslak Janzen J. Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod. Biomed. Online. 2011;22:2–8. doi: 10.1016/j.rbmo.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 5.McCoy R.C., Demko Z., Ryan A., Banjevic M., Hill M., Sigurjonsson S., Rabinowitz M., Fraser H.B., Petrov D.A. Common variants spanning PLK4 are associated with mitotic-origin aneuploidy in human embryos. Science. 2015;348:235–238. doi: 10.1126/science.aaa3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen A.L., Marin D., Zhou A., Gentilello A.S., Smoak E.M., Cao Z., Fedick A., Wang Y., Taylor D., Scott R.T., Jr., et al. Identification and characterization of Aurora kinase B and C variants associated with maternal aneuploidy. Mol. Hum. Reprod. 2017;23:406–416. doi: 10.1093/molehr/gax018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyc K.M., El Yakoubi W., Bag A., Landis J., Zhan Y., Treff N.R., Scott R.T., Tao X., Schindler K., Xing J. Exome sequencing links CEP120 mutation to maternally derived aneuploid conception risk. Hum. Reprod. 2020;35:2134–2148. doi: 10.1093/humrep/deaa148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun S., Miller M., Wang Y., Tyc K.M., Cao X., Scott R.T., Jr., Tao X., Bromberg Y., Schindler K., Xing J. Predicting embryonic aneuploidy rate in IVF patients using whole-exome sequencing. Hum. Genet. 2022;141:1615–1627. doi: 10.1007/s00439-022-02450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe J., Shanks A.L. StatPearls. Treasure Island (FL); 2023. In Vitro Fertilization. [Google Scholar]
- 10.Sunderam S., Kissin D.M., Crawford S.B., Folger S.G., Boulet S.L., Warner L., Barfield W.D. Assisted Reproductive Technology Surveillance - United States, 2015. MMWR. Surveill. Summ. 2018;67:1–28. doi: 10.15585/mmwr.ss6703a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermeesch J.R., Voet T., Devriendt K. Prenatal and pre-implantation genetic diagnosis. Nat. Rev. Genet. 2016;17:643–656. doi: 10.1038/nrg.2016.97. [DOI] [PubMed] [Google Scholar]
- 12.Viotti M. Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements. Genes. 2020;11:602. doi: 10.3390/genes11060602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neal S.A., Morin S.J., Franasiak J.M., Goodman L.R., Juneau C.R., Forman E.J., Werner M.D., Scott R.T., Jr. Preimplantation genetic testing for aneuploidy is cost-effective, shortens treatment time, and reduces the risk of failed embryo transfer and clinical miscarriage. Fertil. Steril. 2018;110:896–904. doi: 10.1016/j.fertnstert.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 14.Rubio C., Bellver J., Rodrigo L., Castillón G., Guillén A., Vidal C., Giles J., Ferrando M., Cabanillas S., Remohí J., et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: a randomized, controlled study. Fertil. Steril. 2017;107:1122–1129. doi: 10.1016/j.fertnstert.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Li S., Yan B., Li T.K.T., Lu J., Gu Y., Tan Y., Gong F., Lam T.W., Xie P., Wang Y., et al. Ultra-low-coverage genome-wide association study-insights into gestational age using 17,844 embryo samples with preimplantation genetic testing. Genome Med. 2023;15:10. doi: 10.1186/s13073-023-01158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tam V., Patel N., Turcotte M., Bossé Y., Paré G., Meyre D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019;20:467–484. doi: 10.1038/s41576-019-0127-1. [DOI] [PubMed] [Google Scholar]
- 17.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broekema R.V., Bakker O.B., Jonkers I.H. A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol. 2020;10 doi: 10.1098/rsob.190221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaid D.J., Chen W., Larson N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018;19:491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S., Huang S., Chen F., Zhao L., Yuan Y., Francis S.S., Fang L., Li Z., Lin L., Liu R., et al. Genomic Analyses from Non-invasive Prenatal Testing Reveal Genetic Associations, Patterns of Viral Infections, and Chinese Population History. Cell. 2018;175:347–359.e14. doi: 10.1016/j.cell.2018.08.016. [DOI] [PubMed] [Google Scholar]
- 21.Xue A., Wu Y., Zhu Z., Zhang F., Kemper K.E., Zheng Z., Yengo L., Lloyd-Jones L.R., Sidorenko J., Wu Y., et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018;9:2941. doi: 10.1038/s41467-018-04951-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ariad D., Yan S.M., Victor A.R., Barnes F.L., Zouves C.G., Viotti M., McCoy R.C. Haplotype-aware inference of human chromosome abnormalities. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2109307118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyc K.M., Wong A., Scott R.T., Jr., Tao X., Schindler K., Xing J. Analysis of DNA variants in miRNAs and miRNA 3'UTR binding sites in female infertility patients. Lab. Invest. 2021;101:503–512. doi: 10.1038/s41374-020-00498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C., Zhan X., Liang L., Abecasis G.R., Lin X. Improved ancestry estimation for both genotyping and sequencing data using projection procrustes analysis and genotype imputation. Am. J. Hum. Genet. 2015;96:926–937. doi: 10.1016/j.ajhg.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C., Zhan X., Bragg-Gresham J., Kang H.M., Stambolian D., Chew E.Y., Branham K.E., Heckenlively J., FUSION Study. Fulton R., et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet. 2014;46:409–415. doi: 10.1038/ng.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubinacci S., Ribeiro D.M., Hofmeister R.J., Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat. Genet. 2021;53:120–126. doi: 10.1038/s41588-020-00756-0. [DOI] [PubMed] [Google Scholar]
- 30.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gogarten S.M., Bhangale T., Conomos M.P., Laurie C.A., McHugh C.P., Painter I., Zheng X., Crosslin D.R., Levine D., Lumley T., et al. GWASTools: an R/Bioconductor package for quality control and analysis of genome-wide association studies. Bioinformatics. 2012;28:3329–3331. doi: 10.1093/bioinformatics/bts610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boughton A.P., Welch R.P., Flickinger M., VandeHaar P., Taliun D., Abecasis G.R., Boehnke M. LocusZoom.js: Interactive and embeddable visualization of genetic association study results. Bioinformatics. 2021;37:3017–3018. doi: 10.1093/bioinformatics/btab186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguet F., Anand S., Ardlie K.G., Gabriel S., Getz G.A., Graubert A., Hadley K., Handsaker R.E., Huang K.H., Kashin S., et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aboelenain M., Schindler K. Aurora kinase B inhibits aurora kinase A to control maternal mRNA translation in mouse oocytes. Development. 2021;148:dev199560. doi: 10.1242/dev.199560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clift D., So C., McEwan W.A., James L.C., Schuh M. Acute and rapid degradation of endogenous proteins by Trim-Away. Nat. Protoc. 2018;13:2149–2175. doi: 10.1038/s41596-018-0028-3. [DOI] [PubMed] [Google Scholar]
- 36.So C., Menelaou K., Uraji J., Harasimov K., Steyer A.M., Seres K.B., Bucevičius J., Lukinavičius G., Möbius W., Sibold C., et al. Mechanism of spindle pole organization and instability in human oocytes. Science. 2022;375 doi: 10.1126/science.abj3944. [DOI] [PubMed] [Google Scholar]
- 37.Blengini C.S., Schindler K. Immunofluorescence technique to detect subcellular structures critical to oocyte maturation. Methods Mol. Biol. 2018;1818:67–76. doi: 10.1007/978-1-4939-8603-3_8. [DOI] [PubMed] [Google Scholar]
- 38.Blengini C.S., Nguyen A.L., Aboelenain M., Schindler K. Age-dependent integrity of the meiotic spindle assembly checkpoint in females requires Aurora kinase B. Aging Cell. 2021;20 doi: 10.1111/acel.13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein P., Schindler K. Mouse oocyte microinjection, maturation and ploidy assessment. J. Vis. Exp. 2011 doi: 10.3791/2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fairley S., Lowy-Gallego E., Perry E., Flicek P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2020;48:D941–D947. doi: 10.1093/nar/gkz836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batman U., Deretic J., Firat-Karalar E.N. The ciliopathy protein CCDC66 controls mitotic progression and cytokinesis by promoting microtubule nucleation and organization. PLoS Biol. 2022;20 doi: 10.1371/journal.pbio.3001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Odabasi E., Conkar D., Deretic J., Batman U., Frikstad K.A.M., Patzke S., Firat-Karalar E.N. CCDC66 regulates primary cilium length and signaling via interactions with transition zone and axonemal proteins. J. Cell Sci. 2023;136:jcs260327. doi: 10.1242/jcs.260327. [DOI] [PubMed] [Google Scholar]
- 44.Rabinowitz M., Ryan A., Gemelos G., Hill M., Baner J., Cinnioglu C., Banjevic M., Potter D., Petrov D.A., Demko Z. Origins and rates of aneuploidy in human blastomeres. Fertil. Steril. 2012;97:395–401. doi: 10.1016/j.fertnstert.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 45.McCoy R.C., Demko Z.P., Ryan A., Banjevic M., Hill M., Sigurjonsson S., Rabinowitz M., Petrov D.A. Evidence of Selection against Complex Mitotic-Origin Aneuploidy during Preimplantation Development. PLoS Genet. 2015;11 doi: 10.1371/journal.pgen.1005601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fox J.G. Elsevier; 2007. The Mouse in Biomedical Research. [Google Scholar]
- 47.Biswas L., Tyc K., El Yakoubi W., Morgan K., Xing J., Schindler K. Meiosis interrupted: the genetics of female infertility via meiotic failure. Reproduction. 2021;161:R13–R35. doi: 10.1530/REP-20-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carp H., Toder V., Aviram A., Daniely M., Mashiach S., Barkai G. Karyotype of the abortus in recurrent miscarriage. Fertil. Steril. 2001;75:678–682. doi: 10.1016/s0015-0282(00)01801-x. [DOI] [PubMed] [Google Scholar]
- 49.Capalbo A., Poli M., Riera-Escamilla A., Shukla V., Kudo Høffding M., Krausz C., Hoffmann E.R., Simon C. Preconception genome medicine: current state and future perspectives to improve infertility diagnosis and reproductive and health outcomes based on individual genomic data. Hum. Reprod. Update. 2021;27:254–279. doi: 10.1093/humupd/dmaa044. [DOI] [PubMed] [Google Scholar]
- 50.Volozonoka L., Miskova A., Kornejeva L., Kempa I., Bargatina V., Gailite L. A systematic review and standardized clinical validity assessment of genes involved in female reproductive failure. Reproduction. 2022;163:351–363. doi: 10.1530/REP-21-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fragouli E., Wells D. Aneuploidy in the human blastocyst. Cytogenet. Genome Res. 2011;133:149–159. doi: 10.1159/000323500. [DOI] [PubMed] [Google Scholar]
- 52.Northrop L.E., Treff N.R., Levy B., Scott R.T., Jr. SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocysts. Mol. Hum. Reprod. 2010;16:590–600. doi: 10.1093/molehr/gaq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Capalbo A., Bono S., Spizzichino L., Biricik A., Baldi M., Colamaria S., Ubaldi F.M., Rienzi L., Fiorentino F. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and trophoblast: insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum. Reprod. 2013;28:509–518. doi: 10.1093/humrep/des394. [DOI] [PubMed] [Google Scholar]
- 54.Johnson D.S., Cinnioglu C., Ross R., Filby A., Gemelos G., Hill M., Ryan A., Smotrich D., Rabinowitz M., Murray M.J. Comprehensive analysis of karyotypic mosaicism between trophectoderm and inner cell mass. Mol. Hum. Reprod. 2010;16:944–949. doi: 10.1093/molehr/gaq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y., Sidore C., Kang H.M., Boehnke M., Abecasis G.R. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res. 2011;21:940–951. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fumagalli M. Assessing the Effect of Sequencing Depth and Sample Size in Population Genetics Inferences. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasaniuc B., Rohland N., McLaren P.J., Garimella K., Zaitlen N., Li H., Gupta N., Neale B.M., Daly M.J., Sklar P., et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat. Genet. 2012;44:631–635. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Homburger J.R., Neben C.L., Mishne G., Zhou A.Y., Kathiresan S., Khera A.V. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome-wide polygenic scores. Genome Med. 2019;11:74. doi: 10.1186/s13073-019-0682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rustagi N., Zhou A., Watkins W.S., Gedvilaite E., Wang S., Ramesh N., Muzny D., Gibbs R.A., Jorde L.B., Yu F., Xing J. Extremely low-coverage whole genome sequencing in South Asians captures population genomics information. BMC Genom. 2017;18:396. doi: 10.1186/s12864-017-3767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lou R.N., Jacobs A., Wilder A.P., Therkildsen N.O. A beginner's guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. 2021;30:5966–5993. doi: 10.1111/mec.16077. [DOI] [PubMed] [Google Scholar]
- 61.Davies R.W., Flint J., Myers S., Mott R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 2016;48:965–969. doi: 10.1038/ng.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korneliussen T.S., Albrechtsen A., Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinf. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ariad D., Madjunkova S., Madjunkov M., Chen S., Abramov R., Librach C., McCoy R.C. Aberrant landscapes of maternal meiotic crossovers contribute to aneuploidies in human embryos. bioRxiv. 2023 doi: 10.1101/gr.278168.123. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wan J.C.M., Stephens D., Luo L., White J.R., Stewart C.M., Rousseau B., Tsui D.W.Y., Diaz L.A., Jr. Genome-wide mutational signatures in low-coverage whole genome sequencing of cell-free DNA. Nat. Commun. 2022;13:4953. doi: 10.1038/s41467-022-32598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gusev A., Groha S., Taraszka K., Semenov Y.R., Zaitlen N. Constructing germline research cohorts from the discarded reads of clinical tumor sequences. Genome Med. 2021;13:179. doi: 10.1186/s13073-021-00999-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary statistics of the GWAS variants with p < 1e−3 are available in Table S1, and the summary statistics of all variants have been submitted to the GWAS catalog (https://www.ebi.ac.uk/gwas/) (GWAS Catalog: GCST90292548). The data used for the analyses described in Figures 4 and S4 were obtained from the GTEx Portal on 09/21/23 and GTEx Analysis Release v.8 (dbGaP: phs000424.v.8.p2) on 09/21/23. The analysis codes are available at https://github.com/JXing-Lab/PGTA_aneuploidy.