

Abstract
The objective of this study was to investigate the late cardioprotective effect of exercise preconditioning (EP) on isoproterenol (ISO)-induced myocardial injury in rats and the role of protein kinase C (PKC) in EP. Rats were injected with ISO 24 h after running on a treadmill for four periods of 10 min each at 28–30 m/min with intervening periods of rest of 10 min. Nonselective PKC inhibitor chelerythrine (CHE) was injected before EP. The myocardial injury was evaluated quantitatively in terms of the serum cardiac troponin I (cTnI) levels, the myocardial ischemia/hypoxia area, and the integral optical density (IOD) of haematoxylin–basic fuchsin–picric acid (HBFP) staining, and qualitatively in terms of the myocardial ultrastructure. EP markedly attenuated the ISO-induced myocardial ischemia/hypoxia and ultrastructural damage with lower serum cTnI levels. CHE injection before EP did not block the protective effect of EP, displaying a mild myocardial ischemia/hypoxia and well-preserved ultrastructure with even lower serum cTnI levels. The results indicate that EP can exert a late cardioprotection against ISO-induced myocardial injury, and that an injection of the nonselective PKC inhibitor CHE before EP may have a different effect on ISO-induced myocardial injury. Further investigation needs to be conducted to define the role of different PKC isozymes in EP by using isozyme-selective inhibitors.
Keywords: Exercise preconditioning, Isoproterenol, Haematoxylin–basic fuchsin–picric acid staining, Ultrastructure, Cardiac troponin I, Protein kinase C
Introduction
Ischemic preconditioning (IPC), a brief episode of repetitive cardiac ischemia/reperfusion (I/R), exerts a cardioprotective effect against subsequent lethal periods of ischemia and is a powerful endogenous form of cardioprotection against ischemic injury [1]. Similar to IPC, a single bout of exercise has been associated with improved functional recovery, enhanced contractility, and reduced myocardial infarction during subsequent lethal ischemic injury in a biphasic manner [2–6], with the early phase occurring immediately after the exercise [5] and the late phase developing 24 h post-exercise [2–4, 6]. These protective effects, known as exercise preconditioning (EP), cannot be explained by changes in hemodynamic variables, collateral flow, and ischemia [2]. One possible mechanism may be related to the cellular and molecular adaptation induced by exercise, which shares common cellular transduction pathways with IPC [2, 3].
As a cellular mediator, protein kinase C (PKC) has been proposed to play a vital role in IPC by translocating to the particulate fraction, whereby protein phosphorylation is likely to be achieved [7, 8]. Evidence in rat hearts has also shown that PKC inhibitor chelerythrine (CHE) attenuated the late cardioprotection of EP against I/R injury [4, 6]. However, because the experimental models for the induction of acute myocardial injury used in previous EP studies were based on mechanical occlusion of the coronary artery in vivo or in the isolated Langendorff system in vitro, and they require open chest surgery and complicated instrumentation, it seems to be important to validate the results with experimental models that mimic the ischemic situation more closely. Isoproterenol (ISO) has been used as a model compound in various animal species to induce ischemia and infarct-like lesions comparable to those taking place in human myocardial infarction [9, 10]. The haematoxylin–basic fuchsin–picric acid (HBFP) staining technique has proved useful in histopathological investigations of myocardial ischemia [11, 12] and provides a clear and striking demonstration of early myocardial ischemia with the ischemic fibers staining a vivid crimson color in contrast to the light brown color of nonischemic tissue.
In the present work, we investigated the late cardioprotective effect of EP on ISO-induced myocardial injury in rats for the first time as well as the role of PKC as a mediator of signal transduction in the molecular mechanisms of EP. We hypothesized that a single bout of interval exercise would attenuate the ISO-induced myocardial injury at 24 h post-exercise, and PKC inhibitor CHE may abolish the cardioprotective effect of EP. To test this hypothesis, we measured the serum cTnI levels, and the area and integral optical density (IOD) of myocardial ischemia/hypoxia with HBFP staining as quantitative evaluation of the injury, and observed the alteration of the myocardial ultrastructure qualitatively in ISO-injected rats pretreated by EP or EP with CHE.
Materials and methods
Animals
Adult (12-week-old) male Sprague Dawley (220–300 g) rats obtained from Chinese Academy of Sciences (Shanghai, China) were housed in standard rat cages maintained at constant temperature and humidity with a 12:12 h light–dark cycle and were fed and watered ad libitum. All animal care and experimental procedures were conducted in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences and approved by the Ethics Committee for Science Research of the Shanghai University of Sport.
Experimental protocol
All animals underwent a light exercise familiarization on the treadmill for 6 consecutive days. The velocity on the treadmill was 15 m/min, and the exercise duration was 10–20 min/day. Two days later, animals were randomly assigned to five experimental groups: Group C was placed on the treadmill without belt movement. Group ISO was injected with ISO (4 mg/kg, intraperitoneally). Group EP was subjected to a single bout of interval exercise. Group EP + ISO was injected with ISO (4 mg/kg, intraperitoneally) 24 h after a single bout of interval exercise. Group CHE + EP + ISO was treated similarly to group EP + ISO, but CHE (5 mg/kg, Sigma, USA) was injected intraperitoneally 10 min before exercise.
The EP protocol was designed with minor modifications of the protocol described by Domenech et al. [2] and Lennon et al. [3]. Rats were allowed to run on a treadmill for four periods of 10 min each at 28–30 m/min with intervening periods of rest of 10 min at 0% grade. Exercise began and ended with 5 min “warm up” and “cool down” periods at 15 m/min and 0% grade. The work rate running at 28–30 m/min represents an estimated 75% of maximum oxygen consumption [13].
Rats in groups C and EP were sacrificed 24 h after the last exercise session. At the same time, rats in groups ISO, EP + ISO, and CHE + EP + ISO were injected with ISO and sacrificed 2 h later. Animals were anesthetized with trichloroacetaldehyde monohydrate (400 mg/kg, intraperitoneally), the blood was drawn from the inferior caval vein, and the heart was prepared for HBFP staining and transmission electron microscopy analysis.
Immunoassay for cTnI
Blood samples were centrifuged immediately after collection, and the sera were frozen at −80°C until assayed. Serum cTnI levels were measured by automated immunochemiluminescence on Access 2 immunoassay system (Beckman Coulter, USA). This assay is based on a single-step sandwich principle with paramagnetic particles coated as the solid phase and two monoclonal anti-human cTnI antibodies, which are effective at detecting cardiac injury in rats [14]. The sensitivity threshold for cTnI was 0.01 μg/L.
HBFP staining and image analysis
After rats were anesthetized, the blood was drawn from the inferior caval vein, and the heart was exposed for perfusion fixation as described previously [15], then the heart was post-fixed, dehydrated, hyalinized, macerated, and embedded in paraffin, sectioned, and mounted. Sections were stained by the HBFP technique [11, 16]. The result was observed with a microscope (Olympus, Tokyo, Japan). Five visual fields from each section, with five sections per group, totaling 25 visual fields, were randomly taken for morphometric analysis using the Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) [15]. The positive area (crimson red) and the IOD of HBFP staining were calculated. The contrast and magnification of all the images were identical.
Transmission electron microscopy analysis
Hearts were excised rapidly after the blood was drawn. The samples for the transmission electron microscopy analysis were taken from the tissue beneath the endocardial surface of the left ventricular anterior free wall at the level of the near apex. The tissue was quickly cut into 1 mm3 pieces on a dry ice-cooled sample plate, then transferred to vials containing 3% glutaraldehyde, fixed for at least 4 h, and then postosmicated in 1% osmium tetroxide. After osmium fixation, the tissue was dehydrated in a graded ethanol series, rinsed in propylene oxide, embedded in Poly/Bed 812, and stained with 0.5% methylene blue. Ultrathin sections were cut with a Reichert-Jung (Vienna, Austria) ultrathin microtome into 50–60 nm thickness and stained with uranium acetate–lead citrate. Samples were examined with a transmission electron microscope (model H-7000, Hitachi, Tokyo, Japan), and representative areas were photographed.
Statistical analysis
Results are expressed as mean ± SD, and values were compared using a one-way analysis of variance (SPSS 10.0; SPSS, Chicago, IL). Upon confirmation of a significant main effect, individual differences were determined with post-hoc analysis. A value of P < 0.05 was considered significant.
Results
Serum levels of cTnI
Serum cTnI levels measured by immunochemiluminescence are shown in Fig 1. Serum cTnI levels were found to be close to the lowest limit of quantitation in group C (n = 14) and EP (n = 15) rats (0.04 ± 0.02 and 0.05 ± 0.03 μg/L, respectively), while they were significantly elevated in group ISO (n = 14) and EP + ISO (n = 15; P < 0.05). EP attenuated the myocardial injury, resulting in significantly decreased serum cTnI levels in group EP + ISO when compared with that in group ISO (23.38 ± 17.77 vs. 35.75 ± 26.50 μg/L, P < 0.05). However, in contradiction with our initial hypothesis, serum cTnI levels in group CHE + EP + ISO (n = 12) were significantly lower than those in group ISO (8.37 ± 4.11 vs. 35.75 ± 26.50 μg/L, P < 0.05), and even significantly lower than those in group EP + ISO (8.37 ± 4.11 vs. 23.38 ± 17.77 μg/L, P < 0.05).
Fig. 1.
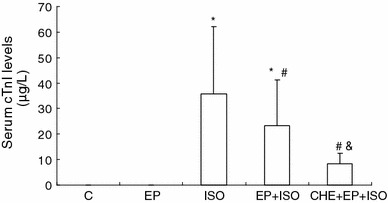
Serum cTnI levels among different groups. Significant differences (P < 0.05) are indicated as follows: from group C (*), from group ISO (#), from group EP + ISO (&)
HBFP staining of cardiomyocytes
HBFP staining of cardiomyocytes of the left ventricular free wall at the level of the near apex and image analysis are shown in Fig. 2. The ischemic cardiomyocytes stain a vivid crimson red color in contrast to the light brown color of nonischemic tissue. Cardiomyocytes in group C (Fig. 2a) and group EP (Fig. 2b) displayed a light brown color; no positive case was found. In group ISO (Fig. 2c), crimson red stain was seen in the majority of cases. In group EP + ISO (Fig. 2d), the red patchy stain was scattered across the cardiomyocytes, some of which were partly or not stained. In group CHE + EP + ISO (Fig. 2e), the red spot stain was only seen in a small fraction of cardiomyocytes.
Fig. 2.
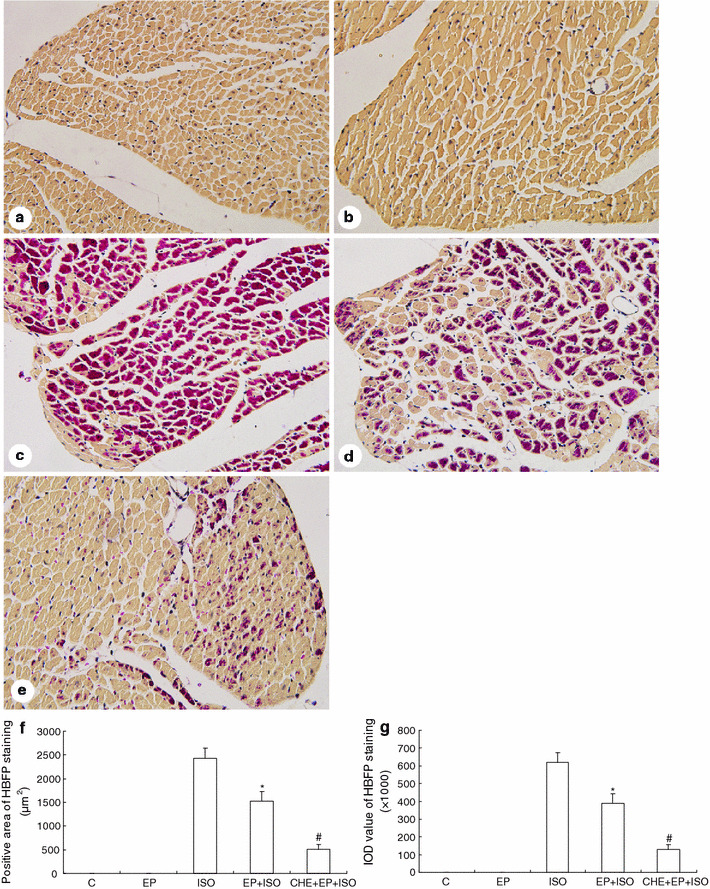
HBFP staining of cardiomyocytes of the left ventricular free wall at the level of the near apex and image analysis. a Group C showed all cardiomyocytes with a light brown color. b Group EP showed results similar to group C. c Group ISO showed crimson red stain in the majority of the cases. d Group EP + ISO showed the crimson red patchy stain scattered across the cardiomyocytes, some of which were partly or not stained. e For group CHE + EP + ISO, the red spot stain was only seen in a small fraction of cardiomyocytes. Original magnification, ×400. f Cardiomyocytes in group C and group EP had no positive area. The positive area in group EP + ISO was significantly lower than that in group ISO, and the positive area in group CHE + EP + ISO was significantly lower than that in group EP + ISO. g Cardiomyocytes in group C and group EP had no IOD value. IOD value in group EP + ISO was significantly lower than that in group ISO, and IOD value in group CHE + EP + ISO was significantly lower than that in group EP + ISO. Significant differences (P < 0.05) are indicated as follows: from group ISO (*), from group EP + ISO (#)
Figure 2f shows that the positive area of HBFP staining in group EP + ISO was significantly lower than that in group ISO (1,528 ± 203 vs. 2,430 ± 215 μm2, P < 0.05; 25 visual fields per group), and the positive area in group CHE + EP + ISO was significantly lower than that in group EP + ISO (508 ± 104 vs. 1,528 ± 203 μm2, P < 0.05; 25 visual fields per group). Figure 2g shows that the value of IOD, representing the intensity of the positive staining, was in good agreement with the positive area. IOD in group EP + ISO was significantly lower than that in group ISO (389.7 ± 51.8 × 103 vs. 619.7 ± 54.7 × 103, P < 0.05; 25 visual fields per group), and IOD in group CHE + EP + ISO was significantly lower than that in group EP + ISO (129.6 ± 26.4 × 103 vs. 389.7 ± 51.8 × 103, P < 0.05; 25 visual fields per group).
The ultrastructure of cardiomyocytes
The ultrastructure of cardiomyocytes from the left ventricular anterior free wall at the level of the near apex is shown in Fig. 3. Normal cardiomyocytes in group C (Fig. 3a) were mainly characterized by finely dispersed nuclear chromatin in the nucleus and slightly contracted myofibrils. Cristae in mitochondria were tightly packed. The sarcolemma was attached to the underlying myofibrils at each Z disk. All myofibrils and mitochondria were well arranged. The ultrastructures of cardiomyocytes in group EP (Fig. 3b) were similar to those described in group C.
Fig. 3.

The ultrastructure of cardiomyocytes from the left ventricular anterior free wall at the level of the near apex. a Group C showed finely dispersed nuclear chromatin, slightly contracted myofibrils (F), well arranged Z disk (Z), mitochondria (M) with tightly packed cristae, and an intact sarcolemma. b Group EP showed results similar to those described in group C. c Group ISO showed aggregated chromatin of the nucleus (N), disarrayed myofibrils, swollen mitochondria with obvious amorphous matrix densities (arrows) and disorganized cristae, ruptured sarcolemma, and edema of sarcoplasm (asterisk). d Group EP + ISO showed some aggregated chromatin, stretched myofibrils with prominent I-bands (I), slightly swollen mitochondria with cristae visible but not packed, and well preserved sarcolemma with only occasional subsarcolemmal blebs (arrows). e Group CHE + EP + ISO showed finely dispersed nuclear chromatin, well organized myofibrils, slightly swollen mitochodria with disarrayed cristae, and increased dilation of sarcoplasmic reticulum (D). Original magnification, ×10,000
The ultrastructural changes that occurred in group ISO (Fig. 3c) were the most severe observed in this experiment. The chromatin of the nucleus was markedly aggregated peripherally. Mitochondria were severely swollen with obvious amorphous matrix densities and disorganized cristae. Myofibrils were in disarray and accompanied by a ruptured sarcolemma. The edema of cardiomyocytes resulted in clearing of the sarcoplasm.
The cardiomyocytes in group EP + ISO demonstrated injury of a lesser extent (Fig. 3d). Myofibrils were stretched with prominent I-bands on either side of the Z-line. The chromatin in the nucleus was dispersed evenly, sometimes aggregated peripherally. Mitochondria were slightly swollen with visible cristae but not packed, and amorphous matrix densities were not common. The membrane damage was occasionally displayed, characterized by the appearance of subsarcolemmal blebs, and the edema was clearly alleviated.
In contradiction with our initial hypothesis, PKC inhibitor CHE did not abolish the cardioprotective effect of EP, with group CHE + EP + ISO presenting a slight myocardial injury (Fig. 3e). The cardiomyocytes were mainly characterized by finely dispersed nuclear chromatin in the nucleus, well organized myofibrils, and intact sarcolemma. The signs of injury were just the slightly swollen mitochondria with disarrayed cristae and the increased dilation of sarcoplasmic reticulum. Generally, the cardiomyocytes in group CHE + EP + ISO demonstrated a well-preserved ultrastructure when compared to group EP + ISO.
Discussion
Cardioprotective effect of exercise preconditioning
In previous EP research, the animal models of myocardial injury mostly consisted of mechanical occlusion of the coronary artery under anesthetic in vivo or of isolated buffer-perfused hearts in vitro, using infarct size as an injury marker. In the present work, we prepared an ISO-induced myocardial injury model in vivo. ISO induced a severe myocardial injury including significantly increased cTnI levels in serum, large myocardial ischemia/hypoxia area, and high IOD value under HBFP staining, indicating a high concentration of damaged protein. These indications of substantial injury were confirmed by the disturbed myocardial ultrastructure. Bertinchant et al. [14] and York et al. [17] also found serum cTnI levels increased significantly 2 h after ISO injection. Consistent with the high serum cTnI levels, mitochondrial amorphous matrix densities, and broken sarcolemma, signs of the irreversible ischemic injury were found in cardiomyocytes treated with ISO. Similarly, using H&E staining, Zhang et al. [18] observed hypercontraction bands, edema, degeneration, and necrosis in the ISO-injured hearts of rats. A comparison of H&E- and HBFP-stained myocardial sections showed a rather constant correlation between eosinophilia and uptake of basic fuchsin when myocardial ischemia occurred [19]. With EP, we found that the injury was markedly attenuated, resulting in lower serum cTnI levels, decreased myocardial ischemia/hypoxia area and IOD values, and lesser extent of ultrastructural damage. The cTnI levels were consistent with the alteration of myocardial ultrastructure during injury [14, 17]. The occurrence of the protective effect at 24 h post-exercise also agreed with the observations of other researchers [2, 3].
According to the results gleaned from the myocardial ultrastructure, in group EP + ISO, the edema was clearly alleviated and mitochondria—believed to be involved in the altered ion channel permeability—were slightly swollen. Brown et al. [20] demonstrated that increased sarcolemmal ATP-sensitive K+ (KATP) channel subunit protein expression (SUR2A and/or Kir6.2) in rat heart correlated closely with exercise-acquired protection against myocardial infarction. Chicco et al. [21] found that sarcolemmal KATP channel antagonist HMR-1098 abolished the late cardioprotection of EP in rats. Most recently, Parra et al. [22] also noted that the mitochondrial KATP channel antagonist 5-hydroxydecanoate blocked the late cardioprotection of EP in dogs. Sarcolemmal and mitochondrial KATP channel opening are believed to limit intracellular Ca2+ accumulation, enabling ionic homeostasis to be preserved [23], which may prevent cardiomyocytes and mitochondria from swelling. Moreover, less amorphous matrix densities in mitochondria and lower IOD values following HBFP staining in group EP + ISO suggest less damaged protein in cardiomyocytes, which may be related to enhanced antioxidant ability or increased heat shock protein (HSP) generation. Demirel et al. [24] confirmed that EP improved myocardial contractile performance of rats during in vivo I/R in the late phase and that this exercise-induced myocardial protection was associated with an increase in myocardial HSP72, glutathione levels, and manganese superoxide dismutase (MnSOD) activity. Further investigation by French et al. [25] found that MnSOD provided exercise-induced cardioprotection by attenuating I/R-induced oxidation and calpain-mediated degradation of myocardial Ca2+-handling proteins (L-type Ca2+ channels, phospholamban, and sarcoplasmic/endoplasmic reticulum calcium ATPase), thereby preventing myocardial apoptosis and necrosis. Importantly, activation of these end-protective effectors including ion channels, antioxidant enzymes, and HSP in EP are supposed to be carried by a series of signal transduction pathways. Several metabolites are known to be released during exercise, such as the adrenergic agonists, bradykinin, opioids, nitric oxide, and reactive oxygen species (ROS). Among them, opioids [26] and nitric oxide [27] have been shown to serve as triggers inducing the cardioprotection of EP. These triggers are proposed to exert their effects on membrane ion channels and receptors, which activate mediators such as protein kinase A (PKA), PKC [4, 6, 28], and mitogen-activated protein kinases (MAPK) [29] in the cytoplasm and terminate the signal at the above-mentioned end effectors, finally rendering cardiomyocytes very resistant to injury.
Currently, some researchers are of the opinion that both long term training and a single bout of exercise are EP [30]. Our EP model involved a single bout of interval exercise, mainly based on the concept of IPC (a brief episode of repetitive cardiac I/R). We speculate that the high-intensity period of the interval exercise may induce a relative ischemic status, and the rest period enables the heart to be fully perfused with the blood, which can mimic “reperfusion.” It is worth noting that the exercise protocol in our study induced no alteration in serum cTnI levels, myocardial ischemia/hypoxia area, or ultrastructure, which may give evidence of the noninjurious nature of the interval exercise, whereas prolonged strenuous exercise has been demonstrated to induce subclinical myocardial damage characterized by biomarker release and impaired myocardial function [31, 32], which can even be sustained for several days [33]. Although both a single bout of continuous exercise [4, 6, 27] and interval exercise [2, 3, 5, 22] have been associated with the cardioprotective effects similar to IPC, more and more evidence shows that aerobic interval exercise at a relatively high intensity can be used in experimental and clinical settings and that this type of training induces more beneficial effects in the heart compared with continuous exercise at moderate or low intensity [34–36]. The molecular mechanisms involved in this difference are currently unclear. The optimal intensity, duration, and number of interval bouts and duration of the recovery between exercise bouts in interval exercise deserve further investigations to elicit the greatest cardioprotective effect induced by EP.
Role of PKC inhibitor CHE in cardioprotection of exercise preconditioning
In cardiomyocytes, PKC is a family of isozymes consisting of three major subgroups [37]: the conventional calcium-dependent (α, βI, βII, and γ), the novel calcium-independent (δ, ε, η, θ, and possibly μ), and the atypical PKCs (ζ and τ/λ). Most studies have focused on isozymes εPKC, δPKC, and αPKC. Carson and Korzick [28] demonstrated that εPKC, δPKC, and αPKC activation and translocation occur in rat heart following a single bout of exercise. Yamashita et al. [6] and Melling et al. [4] reported that myocardial infarct size limitation induced by EP at 24 h post-exercise was abolished by the PKC inhibitor CHE in the isolated rat heart subjected to I/R injury on the Langendorff apparatus, and CHE (5 mg/kg) suppressed the exercise-induced translocations of εPKC, δPKC, and αPKC. In contrast, we failed to find that CHE attenuated the cardioprotective effect of EP against the ISO-induced myocardial injury at 24 h post-exercise. Unexpectedly, CHE treatment before EP induced a stronger resistance to the ISO-induced myocardial injury than EP alone, presenting lower serum cTnI levels, mild myocardial ischemia/hypoxia, and well-preserved ultrastructure. This finding is different from the previous result showing PKC as a pivotal mediator for the cardioprotective effect of EP.
The discrepancy is likely due to the different models of myocardial injury. The Langendorff-perfused heart preparation in vitro used by other researchers has the advantage of allowing the intrinsic factors of the heart in cardioprotection against I/R injury to be studied in the absence of cardiac innervation, blood-borne inflammatory cell types, or other extrinsic factors. Unlike in the isolated Langendorff-perfused heart, ISO mainly stimulates β-adrenergic receptors on cardiomyocytes in vivo, resulting in cardiac tissue anoxia/hypoxia due to elevated oxygen demand and bringing about a number of complex physiological and pathological changes including tachycardia, neurohormone production, coronary hypotension, inflammation, and severe oxidative stress [10, 14, 17, 18]. Therefore, under this integrated condition, PKC inhibition may have a different effect. Moreover, stimulation of β-adrenergic receptors primarily activates PKA signaling cascades, which influence cardiomyocyte contraction, hypertrophy, and apoptosis [38]. Evidence showed that a cross-talk exists between PKA and PKC [39]. Thus, it is reasonable to believe that PKC inhibitor may block some of the PKA signaling, and the effect of ISO will also be partially attenuated.
In cultured neonatal rat cardiomyocytes, Johnson et al. [40] demonstrated that pretreatment with εPKC inhibitor completely prevented ISO-induced cardiomyocyte apoptosis, and inhibition of εPKC translocation specifically blocked phorbol ester-mediated or norepinephrine-mediated regulation of contraction. Similarly, Kang et al. [41] found that εPKC produced a strong positive inotropic response upon accumulation at the Golgi or other intracellular sites in adult rat ventricular myocytes. Evidence also supported that αPKC functions as a proximal regulator of Ca2+ handling and myocardial contractility in cardiomyocytes [42–44]. Most recently, a study by Zheng et al. [45] found that PMA (a PKC activator) increased the Ca2+ transient and accelerated the beating rates of neonatal rat cardiomyocytes. These studies suggest that PKC inhibition may attenuate the ISO-induced positive inotropic effect in rat heart. In fact, CHE abolished ECG-epicatechin-3-gallate (the most effective tea catechin)-induced and intermedin (adrenomedullin-2)-induced inotropic effects on the isolated murine cardiomyocytes [46, 47]; the effect may alleviate the myocardial ischemia/hypoxia and prevent the heart from experiencing ultrastructural damage induced by ISO. Furthermore, δPKC is proapoptotic in myocardial I/R injury and involved in several aspects of mitochondrial function including cytochrome c release, ATP synthesis, and ROS generation [7]. Simonis et al. [48] reported CHE influenced the balance of pro- and anti-apoptotic pathways in the remote myocardium of rats after infarction, with an inhibition of proapoptotic and an activation of anti-apoptotic signals. Inhibition of δPKC protects ischemic myocardium by preventing both oncotic and apoptotic cell death [7], which is also responsible for our current result.
Taken together, as a nonselective inhibitor, CHE may block the activity of PKC isozymes during EP and attenuate the ISO-induced myocardial injury by modulating Ca2+ handling and myocardial contractility through εPKC and αPKC, and reducing the δPKC-mediated apoptosis or necrosis. Studies on I/R injury also showed that, although CHE blocked the cardioprotection of IPC, CHE by itself could directly reduce the infarct size during I/R injury in situ and in isolated perfused rabbit hearts [49]. A new specific PKC inhibitor, Gö 6983, also showed a cardioprotective effect against I/R injury [50]. These results indicate PKC inhibition may also have a beneficial effect even in I/R injury; thus the role of PKC in preconditioning is complicated and needs further investigation.
In conclusion, the present work demonstrated that EP attenuated the ISO-induced myocardial injury at 24 h post-exercise in rats. Injection of the nonselective PKC inhibitor CHE before EP failed to attenuate the cardioprotective effect of EP and in fact resulted in an unexpectedly mild myocardial injury. These findings suggest that EP can exert a late cardioprotective effect against ISO-induced myocardial injury, and that injection of the nonselective PKC inhibitor CHE before EP may have a different effect on ISO-induced myocardial injury. Further investigation needs to be conducted to determine the role of different PKC isozymes in EP by using isozyme-selective inhibitors.
Acknowledgments
This work was supported by the Science and Technology Commission of Shanghai Municipality (No. 09490503300) and Shanghai Leading Academic Discipline Project (No. S30802).
Conflict of interest
The authors declare no conflicts of interest.
Footnotes
Y.-J. Shen, S.-S. Pan, and T. Zhuang have equally contributed to this article.
References
- 1.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Domenech R, Macho P, Schwarze H, Sánchez G. Exercise induces early and late myocardial preconditioning in dogs. Cardiovasc Res. 2002;55:561–566. doi: 10.1016/S0008-6363(02)00334-6. [DOI] [PubMed] [Google Scholar]
- 3.Lennon SL, Quindry JC, French JP, Kim S, Mehta JL, Powers SK. Exercise and myocardial tolerance to ischaemia–reperfusion. Acta Physiol Scand. 2004;182:161–169. doi: 10.1111/j.1365-201X.2004.01346.x. [DOI] [PubMed] [Google Scholar]
- 4.Melling CW, Thorp DB, Milne KJ, Noble EG. Myocardial Hsp70 phosphorylation and PKC-mediated cardioprotection following exercise. Cell Stress Chaperones. 2009;14:141–150. doi: 10.1007/s12192-008-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sánchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Härtel S, Hidalgo C, Donoso P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–386. doi: 10.1093/cvr/cvm011. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita N, Baxter GF, Yellon DM. Exercise directly enhances myocardial tolerance to ischaemia–reperfusion injury in the rat through a protein kinase C mediated mechanism. Heart. 2001;85:331–336. doi: 10.1136/heart.85.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia–reperfusion injury. Pharmacol Res. 2007;55:523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Simkhovich BZ, Przyklenk K, Kloner RA. Role of protein kinase C as a cellular mediator of ischemic preconditioning: a critical review. Cardiovasc Res. 1998;40:9–22. doi: 10.1016/S0008-6363(98)00142-4. [DOI] [PubMed] [Google Scholar]
- 9.Devika PT, Stanely Mainzen Prince P. (−) Epigallocatechin-gallate (EGCG) prevents mitochondrial damage in isoproterenol-induced cardiac toxicity in albino Wistar rats: a transmission electron microscopic and in vitro study. Pharmacol Res. 2008;57:351–357. doi: 10.1016/j.phrs.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Rona G, Kahn DS, Chappel CI. Studies on infarct-like myocardial necrosis produced by isoproterenol: a review. Rev Can Biol. 1963;22:241–255. [PubMed] [Google Scholar]
- 11.Lie JT, Holley KE, Kampa WR, Titus JL. New histochemical method for morphologic diagnosis of early stages of myocardial ischemia. Mayo Clin Proc. 1971;46:319–327. [PubMed] [Google Scholar]
- 12.Li S, Zhong S, Zeng K, Luo Y, Zhang F, Sun X, Chen L. Blockade of NF-kappaB by pyrrolidine dithiocarbamate attenuates myocardial inflammatory response and ventricular dysfunction following coronary microembolization induced by homologous microthrombi in rats. Basic Res Cardiol. 2010;105:139–150. doi: 10.1007/s00395-009-0067-6. [DOI] [PubMed] [Google Scholar]
- 13.Bedford TG, Tipton CM, Wilson NC, Oppliger RA, Gisolfi CV. Maximum oxygen consumption of rats and its changes with various experimental procedure. J Appl Physiol. 1979;47:1278–1283. doi: 10.1152/jappl.1979.47.6.1278. [DOI] [PubMed] [Google Scholar]
- 14.Bertinchant JP, Robert E, Polge A, Marty-Double C, Fabbro-Peray P, Poirey S, Aya G, Juan JM, Ledermann B, de la Coussaye JE, Dauzat M. Comparison of the diagnostic value of cardiac troponin I and T determinations for detecting early myocardial damage and the relationship with histological findings after isoprenaline-induced cardiac injury in rats. Clin Chim Acta. 2000;298:13–28. doi: 10.1016/S0009-8981(00)00223-0. [DOI] [PubMed] [Google Scholar]
- 15.Pan SS. Alterations of atrial natriuretic peptide in cardiomyocytes and plasma of rats after different intensity exercise. Scand J Med Sci Sports. 2008;18:346–353. doi: 10.1111/j.1600-0838.2007.00684.x. [DOI] [PubMed] [Google Scholar]
- 16.Scherer AT, Masi AT. Technical aspects of the haematoxylin–basic fuchsin–picric acid (HBFP) stain applied to skeletal muscle. Histochem J. 1975;7:335–341. doi: 10.1007/BF01007018. [DOI] [PubMed] [Google Scholar]
- 17.York M, Scudamore C, Brady S, Chen C, Wilson S, Curtis M, Evans G, Griffiths W, Whayman M, Williams T, Turton J. Characterization of troponin responses in isoproterenol-induced cardiac injury in the Hanover Wistar rat. Toxicol Pathol. 2007;35:606–617. doi: 10.1080/01926230701389316. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Knapton A, Lipshultz SE, Weaver JL, Herman EH. Isoproterenol-induced cardiotoxicity in Sprague-Dawley rats: correlation of reversible and irreversible myocardial injury with release of cardiac troponin T and roles of iNOS in myocardial injury. Toxicol Pathol. 2008;36:277–288. doi: 10.1177/0192623307313010. [DOI] [PubMed] [Google Scholar]
- 19.Rajs J, Jakobsson S. Experiences with the hematoxylin basic fuchsin picric acid staining method for morphologic diagnosis of myocardial ischemia—an experimental study in forensic pathology. Forensic Sci. 1976;8:37–48. doi: 10.1016/0300-9432(76)90045-5. [DOI] [PubMed] [Google Scholar]
- 20.Brown DA, Lynch JM, Armstrong CJ, Caruso NM, Ehlers LB, Johnson MS, Moore RL. Susceptibility of the heart to ischaemia–reperfusion injury and exercise-induced cardioprotection are sex-dependent in the rat. J Physiol. 2005;564:619–630. doi: 10.1113/jphysiol.2004.081323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chicco AJ, Johnson MS, Armstrong CJ, Lynch JM, Gardner RT, Fasen GS, Gillenwater CP, Moore RL. Sex-specific and exercise-acquired cardioprotection are abolished by sarcolemmal KATP channel blockade in the rat heart. Am J Physiol Heart Circ Physiol. 2007;292:H2432–H2437. doi: 10.1152/ajpheart.01301.2006. [DOI] [PubMed] [Google Scholar]
- 22.Parra VM, Macho P, Domenech RJ (2010) Late cardiac preconditioning by exercise in dogs is mediated by mitochondrial potassium channels. J Cardiovasc Pharmacol. doi: 10.1097/FJC.0b013e3181eb3049 [DOI] [PubMed]
- 23.Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- 24.Demirel HA, Powers SK, Zergeroglu MA, Shanely RA, Hamilton K, Coombes J, Naito H. Short-term exercise improves myocardial tolerance to in vivo ischemia–reperfusion in the rat. J Appl Physiol. 2001;91:2205–2212. doi: 10.1152/jappl.2001.91.5.2205. [DOI] [PubMed] [Google Scholar]
- 25.French JP, Hamilton KL, Quindry JC, Lee Y, Upchurch PA, Powers SK. Exercise-induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-handling proteins, and calpain. FASEB J. 2008;22:2862–2871. doi: 10.1096/fj.07-102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson EW, Hogrefe CP, Ludwig PS, Ackermann LW, Stoll LL, Denning GM. Exercise enhances myocardial ischemic tolerance via an opioid receptor-dependent mechanism. Am J Physiol Heart Circ Physiol. 2008;294:H402–H408. doi: 10.1152/ajpheart.00280.2007. [DOI] [PubMed] [Google Scholar]
- 27.Babai L, Szigeti Z, Parratt JR, Végh A. Delayed cardioprotective effects of exercise in dogs are aminoguanidine sensitive: possible involvement of nitric oxide. Clin Sci (Lond) 2002;102:435–445. doi: 10.1042/CS20010296. [DOI] [PubMed] [Google Scholar]
- 28.Carson LD, Korzick DH. Dose-dependent effects of acute exercise on PKC levels in rat heart: is PKC the heart’s prophylactic? Acta Physiol Scand. 2003;178:97–106. doi: 10.1046/j.1365-201X.2003.01131.x. [DOI] [PubMed] [Google Scholar]
- 29.Iemitsu M, Maeda S, Jesmin S, Otsuki T, Kasuya Y, Miyauchi T. Activation pattern of MAPK signaling in the hearts of trained and untrained rats following a single bout of exercise. J Appl Physiol. 2006;101:151–163. doi: 10.1152/japplphysiol.00392.2005. [DOI] [PubMed] [Google Scholar]
- 30.Kavazis AN. Exercise preconditioning of the myocardium. Sports Med. 2009;39(11):923–935. doi: 10.2165/11317870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 31.Neilan TG, Januzzi JL, Lee-Lewandrowski E, Ton-Nu TT, Yoerger DM, Jassal DS, Lewandrowski KB, Siegel AJ, Marshall JE, Douglas PS, Lawlor D, Picard MH, Wood MJ. Myocardial injury and ventricular dysfunction related to training levels among nonelite participants in the Boston marathon. Circulation. 2006;114:2325–2333. doi: 10.1161/CIRCULATIONAHA.106.647461. [DOI] [PubMed] [Google Scholar]
- 32.Scharhag J, Herrmann M, Urhausen A, Haschke M, Herrmann W, Kindermann W. Independent elevations of N-terminal pro-brain natriuretic peptide and cardiac troponins in endurance athletes after prolonged strenuous exercise. Am Heart J. 2005;150:1128–1134. doi: 10.1016/j.ahj.2005.01.051. [DOI] [PubMed] [Google Scholar]
- 33.Sahlén A, Rubulis A, Winter R, Jacobsen PH, Ståhlberg M, Tornvall P, Bergfeldt L, Braunschweig F. Cardiac fatigue in long-distance runners is associated with ventricular repolarization abnormalities. Heart Rhythm. 2009;6:512–519. doi: 10.1016/j.hrthm.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 34.Haram PM, Kemi OJ, Lee SJ, Bendheim MO, Al-Share QY, Waldum HL, Gilligan LJ, Koch LG, Britton SL, Najjar SM, Wisløff U. Aerobic interval training vs. continuous moderate exercise in the metabolic syndrome of rats artificially selected for low aerobic capacity. Cardiovasc Res. 2009;81:723–732. doi: 10.1093/cvr/cvn332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tjønna AE, Lee SJ, Rognmo Ø, Stølen TO, Bye A, Haram PM, Loennechen JP, Al-Share QY, Skogvoll E, Slørdahl SA, Kemi OJ, Najjar SM, Wisløff U. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: a pilot study. Circulation. 2008;118:346–354. doi: 10.1161/CIRCULATIONAHA.108.772822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wisløff U, Støylen A, Loennechen JP, Bruvold M, Rognmo Ø, Haram PM, Tjønna AE, Helgerud J, Slørdahl SA, Lee SJ, Videm V, Bye A, Smith GL, Najjar SM, Ellingsen Ø, Skjaerpe T. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115:3086–3094. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- 37.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.43.25526. [DOI] [PubMed] [Google Scholar]
- 38.Tilley DG, Rockman HA. Role of beta-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther. 2006;4:417–432. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- 39.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 40.Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 41.Kang M, Walker JW. Protein kinase C delta and epsilon mediate positive inotropy in adult ventricular myocytes. J Mol Cell Cardiol. 2005;38:753–764. doi: 10.1016/j.yjmcc.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 42.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity towards heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 43.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW., 2nd Protein kinase Cα negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res. 2003;93:1111–1119. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- 44.Johnsen DD, Kacimi R, Anderson BE, Thomas TA, Said S, Gerdes AM. Protein kinase C isozymes in hypertension and hypertrophy: insight from SHHF rat hearts. Mol Cell Biochem. 2005;270:63–69. doi: 10.1007/s11010-005-3781-x. [DOI] [PubMed] [Google Scholar]
- 45.Zheng M, Wang Y, Kang L, Shimaoka T, Marni F, Ono K. Intracellular Ca2+- and PKC-dependent upregulation of T-type Ca2+ channels in LPC-stimulated cardiomyocytes. J Mol Cell Cardiol. 2010;48:131–139. doi: 10.1016/j.yjmcc.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 46.Dong F, Taylor MM, Samson WK, Ren J. Intermedin (adrenomedullin-2) enhances cardiac contractile function via a protein kinase C- and protein kinase A-dependent pathway in murine ventricular myocytes. J Appl Physiol. 2006;101:778–784. doi: 10.1152/japplphysiol.01631.2005. [DOI] [PubMed] [Google Scholar]
- 47.Li D, Yang C, Chen Y, Tian J, Liu L, Dai Q, Wan X, Xie Z. Identification of a PKCepsilon-dependent regulation of myocardial contraction by epicatechin-3-gallate. Am J Physiol Heart Circ Physiol. 2008;294:H345–H353. doi: 10.1152/ajpheart.00785.2007. [DOI] [PubMed] [Google Scholar]
- 48.Simonis G, Wiedemann S, Schwarz K, Christ T, Sedding DG, Yu X, Marquetant R, Braun-Dullaeus RC, Ravens U, Strasser RH. Chelerythrine treatment influences the balance of pro- and anti-apoptotic signaling pathways in the remote myocardium after infarction. Mol Cell Biochem. 2008;310:119–128. doi: 10.1007/s11010-007-9672-6. [DOI] [PubMed] [Google Scholar]
- 49.Lasley RD, Noble MA, Mentzer RM., Jr Effects of protein kinase C inhibitors in in situ and isolated ischemic rabbit myocardium. J Mol Cell Cardiol. 1997;29:3345–3356. doi: 10.1006/jmcc.1997.0559. [DOI] [PubMed] [Google Scholar]
- 50.Young LH, Balin BJ, Weis MT. Gö 6983: a fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc Drug Rev. 2005;23:255–272. doi: 10.1111/j.1527-3466.2005.tb00170.x. [DOI] [PubMed] [Google Scholar]