

Abstract
Hyperinsulinemia is one of the reported side effects of valproic acid (VPA), a medicine used to treat epilepsy. However, its underlying mechanism remains unknown. The present study was designed to investigate a direct effect of VPA on insulin secretion by using mouse pancreactic islets and β-cells. VPA had no acute effect on insulin secretion from islets, or on cytosolic Ca2+ ([Ca2+]i) in single β-cells. However, following long-term exposure to VPA (48 h), both basal and glucose-stimulated insulin secretion were markedly elevated (5-fold), while the insulin gene expression level was unaltered. Following long-term exposure to VPA, β-cells showed a decrease in whole cell KATP channel current. However, the increase in [Ca2+]i in response to the sulfonylurea drug, tolbutamide was attenuated. The present study shows that VPA has no acute effects, but long-term treatment results in enhancement of both basal and glucose-stimulated insulin secretion. This long-term effect may mediate the KATP channel, while VPA can also attenuate the effect of the KATP channel blocker tolbutamide.
Keywords: Insulin secretion, Pancreatic islet, β-cell, Potassium channel, Valproic acid, Cytosolic Ca2+
Introduction
Valproic acid (VPA) is a simple branched-chain fatty acid and is commonly used for the treatment of epilepsy and bipolar disorder [1]. Up until now, several side effects have been reported. Among these, one of the major side effects is hyperinsulinemia [2]. However, the exact mechanism of this side effect remains unknown. There are two opposing hypotheses about the underlying mechanism. In the past, it was proposed that VPA induces insulin resistance and thereby causes hyperinsulinemia [2]. However, recent reports suggest that VPA has direct effect of enhancing insulin secretion [3, 4].
The ATP-sensitive K+ (KATP) channel has important functions in various organs, but it is well known as being the key factor for regulating insulin secretion in pancreatic β-cells [5, 6]. The KATP channels play a major role in regulating the membrane potential of β-cells, since it is a sensor of intracellular ATP and therefore is able to respond to the metabolic state of the cell. A rise in plasma glucose stimulates glucose uptake and metabolism, causing an increase in intracellular ATP in β-cells. These changes in adenine nucleotide concentrations result in the closure of the KATP channel. The decrease in the membrane’s K+ permeability depolarizes cell membrane and allows the opening of the voltage-dependent Ca2+ channel. The influx of Ca2+ into the cell cytoplasm cause an increase in [Ca2+]i and this increase of [Ca2+]i triggers the release of insulin [5, 7].
Since recent reports suggest a direct action of VPA on insulin secretion, the KATP channel may be a strong candidate that mediates the effect on insulin secretion under the influence of VPA.
In this study, we have investigated the possible involvement of the KATP channel and [Ca2+]i signaling in VPA’s stimulatory effect on insulin secretion.
Materials and methods
Isolation of islets and single β-cells
Islets of Langerhans were isolated from adult (6–8 weeks) male ICR mice using collagenase digestion by the procedures reported previously [8]. The animal protocols for this study were approved by Jichi Medical University Institute of Animal Care and Use Committee. Islets were isolated and washed with Hepes Krebs-Ringer buffer (HKRB) (in mM: 5 CaCl2, 2.8 glucose, 129 NaCl, 4.7 KCl, 1.2 KH2PO4, 5 NaHCO3, 1.2 MgSO4, 10 HEPES, pH 7.4). Single β-cells were prepared from islets by treatment with Ca2+-free HKRB made with 0.1 mM EGTA. Islets or β-cells were cultured in Eagle’s MEM supplemented with 10 % (v/v) fetal bovine serum, 100 mg/ml streptomycin and 100 U/ml penicillin, and cultured at 37 °C in a 95 % air plus 5 % CO2 atmosphere for 48 h with or without VPA 200 μg/ml.
Measurement of insulin release
Insulin release was measured at 37 °C in static incubation experiments using HKRB. Twenty to fifty size-matched islets were incubated in tubes containing 1 ml HKRB with 2.8 mM glucose and VPA 200 μg/ml for 30 or 60 min (preincubation). Addition of VPA did not affect the pH of the solution. Then the incubation medium was removed by aspiration, and 1 ml of fresh HKRB containing various glucose concentrations and VPA 200 μg/ml was introduced. Incubation was then continued for 60 min. At the end of incubation, the medium was aspirated and kept at −80 °C until assayed for insulin. Insulin was measured using a Mouse Insulin ELISA KIT (Shibayagi, Sibukawa, Japan).
[Ca2+]i measurement
Cytoplasmic Ca2+ concentration ([Ca2+]i) of β-cells isolated from islets was measured as previously described [9]. In brief, single β-cells were loaded with 3 mM fura-2/AM (Dojin chemical, Kumamoto, Japan) by incubation with HKRB containing 2.8 mM glucose for 30 min at 37 °C. Following incubation, they were transferred to an open chamber mounted on the stage of fluorescence microscope. The cells were superfused with HKRB at 1 ml/min at 37 °C. Fura-2 was exited alternately at 340 and 380 nm every 5 s, the emissions at 510 nm were detected by a cooled charge-coupled device camera, and ratio images (340/380 nm) were produced by Aquacosmos system (Hamamatsu Photonics, Hamamatsu, Japan). Peak [Ca2+]i values were used for statistical analysis. β-cells were confirmed by the response to 300 μM tolbutamide.
Patch clamp analysis
Whole-cell KATP channel current was measured by voltage clamp recordings. Single β-cells were submerged in a bath with HKRB buffer at room temperature. Glass pipettes were prepared from borosilicate tube glass (Narishige, Tokyo, Japan) and had 4–10 MΩ when filled with pipette solution (in mM: 135 CsCl, 1 MgCl2, 10 HEPES–KOH, 10 EGTA, pH 7.2). After membrane rupture, cell membrane voltage was ramped up from −100 mV to −50 mV for 1,000 ms. Signals were acquired using an Axopatch 200B amplifier (Molecular Devices, Union City, CA) controlled by Clampex 10.2 software via a Digidata 1320A interface (Molecular Devices). Data acquisition and storage were conducted with the use of a pClamp 10.2 (Molecular Devices). Cell capacitances were read directly from the pClamp software.
Real-time RT-PCR experiments
Real-time RT-PCR studies were performed in order to analyze insulin mRNA (INS1, INS2) and KATP channel mRNA (Kir6.2 and SUR1) expression. Total RNA was isolated from cultured islets using TRIzol reagent (Invitrogen, Carlsbad, CA) and treated with RQ1-DNase (Promega, Madison, WI) to eliminate contaminating genomic DNA. The first-strand cDNA synthesis was examined using the ReverTra Ace (Toyobo, Osaka, Japan). PCRs were examined with HotStarTaq DNA polymerase (Qiagen, Valencia, CA), (94 °C for 15 s, 55 °C for 20 s, and 72 °C for 20 s × 35 cycles). As a template, 0.5 μl of cDNA was used. INS1, INS2, Kir6.2, SUR1, GAPDH primers and PCR conditions were purchased from Takara Bio Inc (Takara Bio, Shiga, Japan). Real-time RT-PCR was performed using SYBR premix EX Taq (Takara Bio, Shiga, Japan) with the thermal cycler Dice Real-Time System TP800 (Takara Bio, Shiga, Japan) according to the manufacturerʼs instructions.
Statistical analysis
The data are presented as mean ± SE. Statistical significance was evaluated by unpaired t-test. P values of <0.05 were considered statistically significant.
Results
In static incubation of islets under basal 2.8 mM glucose (2.8G) or elevated 8.3 mM glucose (8.3G), neither the basal nor glucose-stimulated insulin secretion (GSIS) was altered by the presence of VPA (200 μg/ml) compared to the control (Fig. 1a). Challenge with 8.3G increased cytosolic Ca2+ concentration ([Ca2+]i). The elevated level of [Ca2+]i in the presence of 8.3G was not altered by administration of VPA (200 μg/ml) (Fig. 1b). Acute administration of VPA also failed to affect oscillatory [Ca2+]i increase induced by elevated 16.7 mM glucose (16.7G) (Fig. 1c).
Fig. 1.
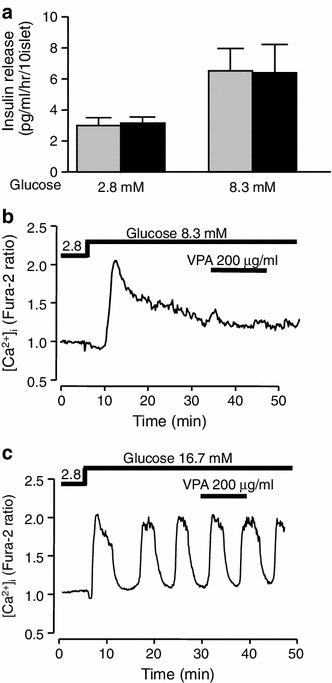
Acute effects of VPA on insulin secretion and [Ca2+]i in islets and β-cells. a Administration of VPA (200 μg/ml) did not alter insulin secretion from islets under static incubation at 2.8 mM (2.8G) and 8.3 mM glucose (8.3G). Islets were isolated from ICR mice. Values are mean ± SE (n = 9–24 for each group). Gray bar control, black bar VPA. b Elevation of glucose concentration to 8.3 mM increased [Ca2+]i and subsequent administration of VPA (200 μg/ml) failed to alter [Ca2+]i in a single β-cell. c Administration of VPA (200 μg/ml) failed to alter oscillatory [Ca2+]i increase induced by 16.7 mM glucose (16.7G) in a single β-cell. The result is representative of 32 β-cells in (b) and 40 β-cells in (c)
In contrast, long-term exposure to VPA (48 h) resulted in stimulation of insulin secretion both at 2.8 G (2.71 vs 6.95 pg/ml/h/10 islets, p < 0.05) and at 8.3G (6.36 vs 33.1 pg/ml/h/10 islets, p < 0.01) (Fig. 2a). We examined the possibility that VPA influenced expression of the insulin genes in mice, INS1 and INS2. Long-term treatment with VPA altered neither INS1 nor INS2 gene expression (Fig. 2b). As presented relative to GAPDH gene expression, the INS1 mRNA level was 527 ± 124 for the control and 559 ± 230 for the VPA-treated group, and the INS2 mRNA level was 1851 ± 581 for the control and 1,724 ± 482 for the VPA-treated group.
Fig. 2.
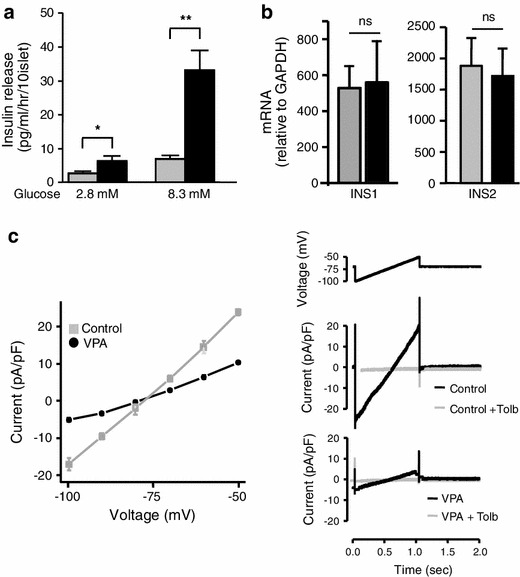
Effects of long-term exposure to VPA for 48 h on insulin secretion, insulin gene expression, and KATP channel currents. a Basal insulin secretion at 2.8G and stimulated insulin secretion at 8.3G were both increased in islets after treatment with VPA (200 μg/ml) compared to the control without VPA. n = 8–15 in each bar. *p < 0.05, **p < 0.01. b Lack of effect of 48 h VPA exposure on mRNA expressions of both INS1 and INS2 in mouse pancreatic islets. Insulin mRNA expression was expressed relative to GAPDH. INS1 insulin 1 gene, which lacks second intron of insulin 2 gene, located in chromosome 19. INS2 insulin 2 gene, which is the murine homologue of the human insulin gene, located in chromosome 7. n = 6 for INS1 and 4 for INS2. Gray bar control, black bar VPA. c Left panel mean current–voltage relationships of long-term VPA treated and control β-cells. Right panel representative recordings of KATP channel currents without and with tolbutamide (Tolb) in long-term VPA treated and control β-cells. n = 5–7 in each group
Next, we measured KATP currents with whole cell recordings in single cells that had been exposed to VPA for 48 h. The membrane voltage was clamped at −75 mV and ramped up from −100 mV to −50 mV for 1,000 ms. The application of tolbutamide (0.3 mM), a blocker of the KATP channel, led to almost complete reduction of the current. Therefore, it is assumed that the currents we detected in this experiment were indeed KATP channel currents. The current from β-cells in the control solution was −17 to +23 pA/pF. Compared with this, the current from β-cells which had been cultured with VPA 200 μg/ml for 48 h appeared to be −5 to +10 pA/pF. The voltage–current curve shown in Fig. 2c is a comparison of mean data obtained from VPA-treated and control single β-cells in response to a voltage ramp. It clearly shows that the currents in response to voltage stimulation have decreased after exposure to VPA (Fig. 2c).
The expression levels of the subunits composing the KATP channel were not altered by the treatment of VPA. Presented relative to GAPDH gene expression, mRNA of Kir6.2 was 0.8 ± 0.2 for the control and 0.6 ± 0.2 for VPA-treated islets (n = 5, relative to GAPDH expression). mRNA of SUR1 was 2.9 ± 0.6 for the control and 3.1 ± 1.2 for VPA-treated islets (n = 5).
[Ca2+]i of β-cells cultured with or without 200 μg/ml VPA for 48 h were measured. Both basal [Ca2+]i and glucose-induced [Ca2+]i increases were not affected by the 48-h treatment with VPA. The basal level of [Ca2+]i was 0.78 ± 0.03 in the control β-cells and 0.74 ± 0.03 in the VPA-treated β-cells. The AUC of Δ[Ca2+]i for 10 min after elevated 8.3 mM glucose application was 0.20 ± 0.01 for control β-cells and 0.22 ± 0.03 for VPA-treated β-cells (Fig. 3a–c). KCl and tolbutamide led to rapid increases in [Ca2+]i (Fig. 3d). Although [Ca2+]i peak value in response to high-KCl was not affected by the presence of VPA (1.39 ± 0.07 vs 1.30 ± 0.07, p = 0.43), peak of tolbutamide-induced [Ca2+]i increase was reduced significantly in the β-cells cultured with VPA for 48 h (1.58 ± 0.05 vs 1.41 ± 0.03, p < 0.05) (Fig. 3e).
Fig. 3.
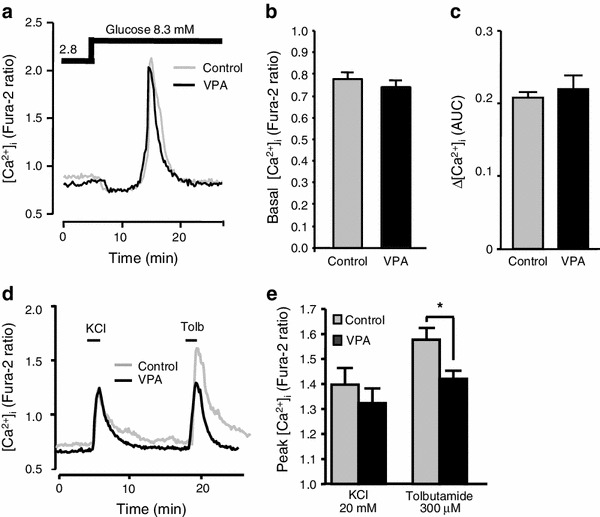
Effects of long-term exposure to VPA on subsequent [Ca2+]i responses in β-cells. a Elevation of glucose concentration from 2.8 mM to 8.3 mM increased [Ca2+]i in control and long-term VPA (200 μg/ml)-treated β-cells in a similar manner. The results are representative of 35 control and 43 VPA-treated cells. b [Ca2+]i levels in the presence of a basal 2.8 mM glucose in β-cells after control culture (gray bar) and after culture with VPA (black bar). c Average area under curve (AUC) of [Ca2+]i increases in response to 8.3 mM glucose for 10 min in β-cells after control culture (gray bar) and after culture with VPA (black bar). d 20 mM KCl and 300 μM tolbutamide (Tolb), a KATP channel blocker, increased [Ca2+]i in β-cells after control culture and after culture with VPA (200 μg/ml). VPA selectively attenuated [Ca2+]i response to Tolb. The result is representative of 35 β-cells. e Average peaks of [Ca2+]i responses to KCl and Tolb in β-cells after control culture (gray bar) and after culture with VPA (black bar). Average peak [Ca2+]i response to Tolb was significantly reduced in β-cells exposed to VPA. n = 4. *p < 0.01
Discussion
In the present study, we investigated the direct effect of VPA on insulin secretion. Short-term exposure to VPA did not significantly alter insulin secretion. In contrast, in islets exposed to VPA for 48 h, insulin secretions were significantly increased at both basal and elevated 8.3 mM glucose conditions. These data were consistent with previous clinical reports showing that a VPA-treated group presented higher postprandial insulin levels compared with the control-treated group [10]. In the past, the mechanisms of VPA-induced hyperinsulinemia have been investigated and several mechanisms have been proposed, including induction of insulin resistance, hyperleptinemia [2, 11], and impaired insulin action in the liver [12]. The present study is the first demonstration of the chronic effect of VPA on islet β-cells that promotes insulin secretion.
In this paper, we have shown that VPA decreases whole-cell KATP channel current after 48-h exposure without affecting the expression of KATP channel mRNA. Since this reduction of whole-cell KATP channel current was only observed after long-term exposure to VPA, it is unlikely that VPA itself has a direct effect on opening and closing of the KATP channel. The chronic exposure to VPA reduced the KATP channel, but unexpectedly failed to significantly alter basal [Ca2+]i and [Ca2+]i responses to elevated glucose (8.3G and 16.7G). The mechanism for the contradicting result that chronic exposure to VPA increased insulin secretion without significantly influencing [Ca2+]i is unclear. However, it may partially be explained by the highly Ca2+-sensitive pool (HCSP) of insulin granules. HCSP insulin granules are localized some distance away from voltage-dependent Ca2+ channels and are reported to be released by small increases of [Ca2+]i. It has been reported that in chronically depolarized β-cells, these HCSP insulin granules cause slow insulin secretion [13]. Chronic VPA treatment may have influenced HCSP that is involved in insulin secretion. However, this model appears not to be sufficient to fully explain this effect and further studies are required to elucidate the mechanism.
It is of interest that VPA showed no change in KCl-induced [Ca2+]i increase but attenuated tolbutamide-induced [Ca2+]i increase. This indicates that long-term treatment with VPA can reduce KATP channel activity but can also attenuate the effect of the sulfonylurea drug, tolbutamide on the KATP channel at the same time.
The KATP channel is a hetero-octameric complex composed of four pore-forming Kir6.2 subunits and four regulatory sulfonylurea receptor 1 (SUR1) subunits [14]. Neither subunit alone can reach the cell membrane. Each subunit must mask the other subunit’s endoplasmic reticulum retention motif which prevents the subunits from expressing in the cell membrane [15]. Since VPA is known to affect expression levels of histone deacetylate 1 [16], it is possible that VPA affects the expression levels of Kir6.2 and SUR1. However, in this study, expressions of both Kir6.2 and SUR1 were not affected by the 48-h exposure to VPA.
Studies of the truncated form of Kir6.2, which produces a functional KATP channel in the absence of SUR1, have shown that the binding site for channel inhibition by ATP is located on Kir6.2 [17]. On the other hand, SUR1 serves as a target for the sulfonylurea drugs such as tolbutamide [18, 19]. Interaction and information transfer from SUR1 to Kir6.2 is considered to be the important process for the proper action of KATP channels [20]. Taking these facts into consideration, several possible mechanisms can be speculated. One possibility is that long-term exposure to VPA may have changed the ATP sensitivity of Kir6.2. Another possibility is that, although VPA has a KATP channel blocking effect when exposed long-term, it may also have changed the property of interaction between Kir6.2 and SUR1, i.e. transfer of the sulfonylurea drug effects from SUR1 to Kir6.2 may have been affected. Further study is required to elucidate the mechanism.
VPA is widely used as an anti-epileptic agent. Its action is considered to involve various mechanisms including sodium channel blocking and activation of GABA. However, its precise mechanism remains complicated and unclear. Recently, it was reported that a gain-of-function mutation in the KATP channel can cause neonatal diabetes. A severe form of neonatal diabetes is known to accompany neurological features such as developmental delay, muscle weakness and epilepsy (DEND syndrome) [14]. These neurological features are caused by the opening of KATP channels in the brain. By using a KATP channel blocker, sulfonylurea, neurological symptoms can be treated in some cases of DEND syndrome. Among neurological symptoms of DEND syndrome, cure of epilepsy is the most prominent feature of sulfonylurea treatment [14]. Therefore, it is obvious that the KATP channel is involved in the development of epilepsy in DEND syndrome patients. In this paper, we showed that long-term exposure to VPA can reduce the KATP channel current. As mentioned above, the mechanism of anti-epileptic effect of VPA is complicated and remains unclear. Our finding indicates that the ability of VPA to reduce the KATP channel current may be one of the complicated mechanisms of VPA’s anti-epileptic effect.
The fact that VPA can inhibit the KATP channel while attenuating the sulfonylurea drug effect is potentially of importance, since many type 2 diabetes patients treated with sulfonylurea drugs are constantly at risk of hypoglycemia [19]. If we could elucidate the mechanism of how VPA inhibits the KATP channel in spite of its ability to attenuate the effect of tolbutamide while still stimulating insulin secretion, it may provide a new potential strategy to treat type 2 diabetic patients. However, in our experiments, VPA stimulated insulin secretion even at basal glucose concentrations. This may indicate the possibility of causing hypoglycemia when VPA was used on patients in a fasting state. Further study to elucidate the detailed mechanism of VPA-induced insulin secretion may provide beneficial information for the treatment of diabetes.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Rosenberg G. The mechanisms of action of valporate in neuropsychiatric disorders: can we see the forest for trees? Cell Mol Life Sci. 2007;64:2090–2103. doi: 10.1007/s00018-007-7079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verrotti A, Basciani F, De Simone M, Trotta D, Morgese G, et al. Insulin resistance in epileptic girls who gain weight after therapy with valproic acid. J Child Neurol. 2002;17:265–268. doi: 10.1177/088307380201700405. [DOI] [PubMed] [Google Scholar]
- 3.Demir E, Aysun S. Weight gain associated with valproate in childhood. Pediatr Neurol. 2000;22:361–364. doi: 10.1016/S0887-8994(00)00133-8. [DOI] [PubMed] [Google Scholar]
- 4.Luef GJ, Lechleitner M, Bauer G, Trinka E, Hengster P. Valproic acid modulates islet cell insulin secretion: a possible mechanism of weight gain in epilepsy patients. Epilepsy Res. 2003;55:53–58. doi: 10.1016/S0920-1211(03)00091-3. [DOI] [PubMed] [Google Scholar]
- 5.Rorsman P. The pancreatic beta-cell as a fuel sensor: an electrophysiologist’s viewpoint. Diabetologia. 1997;40:487–495. doi: 10.1007/s001250050706. [DOI] [PubMed] [Google Scholar]
- 6.Huijie M, Huang X, Li Q, Guan Y, Yuan F, Zhang Y. ATP-sensitive potassium channels and mitochondrial permeability transition pores play roles in the cardioprotection of theaflavin in young rat. J Physiol Sci. 2011;61:337–342. doi: 10.1007/s12576-011-0148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura K, Galvanovskis J, Goldworthy M, Hugill A, Kaizik S, Lee A, Meadows N, Quawalid MM, Teboul L, Rydstrom J, Ashcroft F, Cox RD. Insulin secretion from beta-cells is affected by deletion of nicotinamide nucleotide transhydrogenase. Methods Enzymol. 2009;457:451–480. doi: 10.1016/S0076-6879(09)05025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yada T, Kakei M, Tanaka H. Single pancreatic beta-cells from normal rats exhibit an initial decrease and subsequent increase in cytosolic free Ca2+ in response to glucose. Cell Calcium. 1992;13:69–76. doi: 10.1016/0143-4160(92)90031-M. [DOI] [PubMed] [Google Scholar]
- 10.Luef G, Abraham I, Hoppichler F, Trinka E, Unterberger I, Bauer G, Lechleitner M. Increase in postprandial serum insulin levels in epileptic patients with valproic acid therapy. Metabolism. 2002;51:1274–1278. doi: 10.1053/meta.2002.34708. [DOI] [PubMed] [Google Scholar]
- 11.Verrotti A, Basciani F, Morresi S, de Martino M, Morgese G, Chiarelli F. Serum leptin changes in epileptic patients who gain weight after therapy with valproic acid. Neurology. 1999;53:230–232. doi: 10.1212/WNL.53.1.230. [DOI] [PubMed] [Google Scholar]
- 12.Pylvanen V, Pakarinen A, Knip M, Isojarvi J. Characterization of insulin secretion in Valproate-treated patients with epilepsy. Epilepsia. 2006;47:1460–1464. doi: 10.1111/j.1528-1167.2006.00546.x. [DOI] [PubMed] [Google Scholar]
- 13.Misler S, Silva AM, Barnett D, Dickey AS. Phasic and tonic modes of depolarization-exocytosis coupling in b-cells of porcine islets of Langerhans. Channels. 2009;3:101–109. doi: 10.4161/chan.3.2.7866. [DOI] [PubMed] [Google Scholar]
- 14.Shimomura K. The KATP channel and neonatal diabetes. Endocr J. 2009;20:165–175. doi: 10.1507/endocrj.K08E-160. [DOI] [PubMed] [Google Scholar]
- 15.Zeaangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neurons. 1999;22:233–254. doi: 10.1016/S0896-6273(00)81085-5. [DOI] [PubMed] [Google Scholar]
- 16.Takizawa D, Kakizaki S, Horiuchi N, Tojima H, Yamazaki Y, Ichikawa T, Sato K, Mori M. Histone deacetylate inhibitors induce cytochrome P450 2B by activating nuclear receptor constitutive androstane receptor. Drug Metab Dispos. 2010;38:1493–1498. doi: 10.1124/dmd.110.032854. [DOI] [PubMed] [Google Scholar]
- 17.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP sensitive K+ channels in absence of the sulphonylurea receptor. Nature. 1997;387:179–181. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 18.Gribble FM, Reinmann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- 19.Shimomura K, Ikeda M, Okada S, Kakei M, Matsumoto S, Mori M. Fenofibrate, troglitazone and 15-deoxy-Δ12,14-prostaglandin J2 close KATP channels and induce insulin secretion. J Pharmacol Exp Ther. 2004;310:1273–1280. doi: 10.1124/jpet.104.067249. [DOI] [PubMed] [Google Scholar]
- 20.Craig TJ, Shimomura K, Holl RW, Flanagan SE, Ellard S, Ashcroft FM. An in-frame deletion in Kir6.2 (KCNJ11) causing neonatal diabetes reveals a site of interaction between Kir6.2 and SUR1. J Clin Endocrinol Metab. 2009;94:2251–2257. doi: 10.1210/jc.2009-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]