

Abstract
Thyroid hormone receptors (TRs) play a critical role in the expression of genes that are major determinants of myocardial contractility, including α-myosin heavy chain (α-MHC) and β-MHC. After myocardial infarction (MI), changes in myocardial TRs consistently correlate with changes in thyroid hormone (TH) target gene transcription, and this is thought to play a key role in the progression to end-stage heart failure. Interestingly, post-MI exercise training has been shown to beneficially alter TH-target gene transcription and preserve cardiac function without changing serum TH. Therefore, in this study, we investigated whether mild exercise training alters expression of α1 and β1 TR isoforms in post-MI rats. Seven-week-old male Sprague–Dawley rats underwent coronary ligation or sham operation, and were assigned to 3 groups (n = 10): sham, sedentary MI (MI-Sed), and exercise MI (MI-Ex). Treadmill training was initiated 1 week post-MI, and gradually increased up to 16 m/min, 5° incline, 50 min/day, 5 days/week, and lasted for a total of 8 weeks. Real-time polymerase chain reaction and gel electrophoresis were performed to quantify changes in TR isoforms. Our results illustrated that mRNA expression of TR-α1 and TR-β1 was higher in both MIs; however, protein electrophoresis data showed that TR-α1 was 1.91-fold higher (P < 0.05) and TR-β1 was 1.62-fold higher (P < 0.05) in the MI-Ex group than in the MI-Sed group. After MI, TR-α1 and TR-β1 protein levels are significantly decreased in the surviving non-infarcted myocardium. Moderate-intensity exercise training significantly increases TR-α1 and TR-β1 protein expression, which in turn may upregulate α-MHC and improve myocardial contractile function and prognosis.
Keywords: Myocardial infarction, Exercise training, Thyroid hormone receptors
Introduction
Thyroid hormone (TH) critically regulates a wide range of genes, and has profound effects on the cardiovascular system [7, 16]. Triiodo-l-thyronine (T3), the cellular-active metabolite of TH, mediates its genomic effects upon binding to thyroid hormone nuclear receptors (TRs) [7, 22]; however, recent evidence suggests that unliganded TRs are also transcriptionally active [3, 22]. T3-responsive genes in cardiac muscle include α-myosin heavy chain (α-MHC), β-MHC, and sarcoplasmic reticulum calcium-activated ATPase (SERCA) [15]. The relative cardiac expression of α- and β-MHC heavily influences myocardial contractility, and is determined by factors including, but not limited to, serum T3, TR isoform specificity, and the liganded or unliganded state of TR [11, 12].
Acute myocardial infarction (MI) is the major cause of heart failure in the adult American population [10]. Compelling evidence based on human studies and extensive research on rodent myocardium suggests that pathological remodeling following acute MI results in a hypothyroid-like molecular phenotype, also called the fetal program [22, 24]; this re-expression of the protein isoforms normally expressed in fetal life is characterized by the downregulation of α-MHC and SERCA, and the reciprocal upregulation of β-MHC [16], and is thought to play a major role in the progressive decompensation to chronic heart failure (CHD) and mortality [26, 34]. Although TH treatment has been shown to reverse most of these changes and significantly improve cardiac function [21, 24], accumulating evidence suggests that reduced TH-signaling responsible for fetal gene induction in the failing myocardium may be attributed to altered TR isoform expression [14, 15, 25, 26].
Previous studies by our group and others have demonstrated that exercise training positively influences cardiac function and attenuates myocardial remodeling in rats with MI or congestive heart failure [1, 4, 13, 32, 33]. It has also been reported that exercise training significantly increases α-MHC expression in the anterior wall (AW) and posterior wall (PW) in both sham and MI rats [24]. Interestingly, one of our previous studies demonstrated that post-MI exercise training upregulated α-MHC and downregulated β-MHC without restoring resting TH [30]. Therefore, it is conceivable that upregulation of α-MHC in response to exercise is mediated by changes in TR isoform expression. In fact, studies in the aging rat and in human end-stage heart failure reveal changes in myocardial TR levels that correlate with changes in TH target gene transcription [14, 19]. To date, however, the effects of post-MI exercise on cardiac TR isoform expression have not been addressed. Therefore, the purpose of this study was to investigate the exercise-induced gene and protein alterations of α1 and β1 TR isoforms in post-myocardial infarction rats.
Materials and methods
Animal preparation
Seven-week-old (185–200 g) male Sprague–Dawley rats (Harlan, Indianapolis, IN, USA) were treated in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals, and study protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas at San Antonio. To ensure the rats were accustomed to running, they were habitually trained on a rodent treadmill for 5 min at 5 m/min for 5 days prior to surgery. MI was surgically induced by ligation of the left anterior descending coronary artery as described previously [32].
One week after surgery, the surviving rats were assigned to three experimental groups (n = 10/group): a sham-operated control (Sham), a sedentary group with MI (MI-Sed) and an exercise group with MI (MI-Ex). The MI-Ex group started exercising 1 week post-MI using a motorized rodent treadmill, while the Sham and MI-Sed groups remained sedentary throughout the entire experiment. To allow gradual adaptation to exercise stress, training was initiated at 10 m/min, 5° incline for 10 min per session. The speed and duration were gradually increased to 16 m/min and 50 min per session (including a 5-min warm-up at 10 m/min) and maintained constant throughout the experiment. Based on the regression formula described by Lawler et al. [18], the exercise intensity used in this study was about 55% of the maximal oxygen consumption. This is equivalent to moderate exercise intensity in human exercise. The exercise training was performed 5 days per week for 8 weeks. The exercise intensity and duration used in the present study was based on our previous studies [31–33]. The exercise duration was gradually increased to allow for exercise adaptation. This exercise regimen suited the rats with MI very well and there were no exercise training-related mortalities.
Tissue collection
The rats were anesthetized 48 h after the last exercise session, and their hearts were quickly harvested and rinsed in cold saline. The myocardial tissue of the non-infarcted left ventricle (LV) was collected and immediately frozen in isopentane with dry ice. Tissues were stored at – 80 °C until use.
Infarct size determination
The LV was cut from apex to base into 3 transverse sections. Sections 6-μm thick were cut and stained with Masson’s trichrome. Infarct size was calculated by dividing the sum of the planimetered endocardial and epicardial circumferences of the infarcted area by the sum of the total epicardial and endocardial circumferences of the LV [31]. Total epicardial and endocardial lengths occupied by the infarct as identified by Masson’s trichrome staining was measured using Image Pro Plus program (Media Cybernetics, Silver Spring, MD, USA).
Total RNA isolation
Total RNA was isolated using the TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Briefly, a noninfarcted LV tissue sample was ground, and the resulting powder was re-suspended in 1 ml of TRIZOL. The suspension was then homogenized and incubated for 5 min at room temperature. The homogenate was extracted with 0.2 ml of chloroform, and upon centrifugation (12,000g, 15 min, 4 °C) the aqueous phase was mixed with 0.5 ml of isopropyl alcohol. The resulting pellet was washed with 1 ml of 75% ethanol and re-suspended in 50 µl of RNase-free water. Total RNA samples were stored at − 80 °C until use.
Real-time PCR
All RNA samples were DNase treated and 1 µg of total RNA samples were reverse-transcribed with oligo (dT) primers and MMLV reverse transcriptase (Promega, Madison, WI, USA). Quantification of cardiac gene expression was determined by real-time polymerase chain reaction (PCR). The relative expression of TR-α and TR-β mRNA was normalized to the amount of β-actin in the same cDNA sample by using the standard curve method. Analysis of each specific mRNA was conducted in triplicate. The primers and probes used in this study were Assay-on-Demand gene expression products (Applied Biosystems, Foster City, CA, USA). Because of proprietary issues and the policy of Applied Biosystems, the exact primer sequences used for the real-time PCR experiments are not provided but can be requested from the company based on the information in Table 1.
Table 1.
Primers and probes used for real-time PCR
| Gene | Assay IDa | Reference sequence (GenBank) |
|---|---|---|
| β-actin | Rn00667869_m1 | NM_031144.2 |
| TR-α | Rn01464139_m1 | NM_031134.2 |
| TR-β | Rn01640361_m1 | NM_012672.1 |
aGene expression assays product ID of Applied Biosystems (Foster City, CA, USA)
Western blot
Twenty micrograms of each protein tissue sample was separated by sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE), and transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). The primary antibodies used were anti-TR-α1 and anti-TR-β1 (Santa Cruz). The membranes were detected with enhanced chemiluminescence (Amersham, Little Chalfont, Buckinghamshire, UK) followed by exposure to an X-ray film. The protein bands on the X-ray film were scanned and band densities were calculated using Quantity One software (Bio-Rad). GAPDH was used as an internal control.
Statistics
One-way analyses of variance (ANOVA) were carried out to determine whether there were significant mean differences among the experiment groups. An ANOVA with significant F ratios (P < 0.05) was followed by Student–Newman–Keuls post hoc comparisons. A P value of less than 0.05 was considered statistically significant with the values being expressed as mean ± SEM.
Results
General characteristics of experimental groups
Table 2 describes the general characteristics of the experimental groups. MI was associated with ~ 45% mortality during the first 48 h following ligation. No deaths occurred during the 8-week experimental period. Infarct sizes were comparable between MI-Sed (41.2 ± 2.94%, n = 10) and MI-Ex (40.1 ± 1.37, n = 10). There was no significant difference for the body weight among the experimental groups (P > 0.05). The MI-Sed and MI-Ex groups had similar heart weight and the ratio of heart weight to body weight (P > 0.05), which were higher than their Sham counterparts (P < 0.05).
Table 2.
General characteristics of the experimental groups
| Group | Sham (n = 10) | MI-Sed (n = 10) | MI-Ex (n = 10) |
|---|---|---|---|
| Infarct size (%) | – | 41.2 ± 2.94 | 40.1 ± 1.37 |
| BW (g) | 392.63 ± 12.04 | 392.38 ± 8.75 | 401.5 ± 5.88 |
| Ht Wt (g) | 1.28 ± 0.09* | 1.54 ± 0.24 | 1.52 ± 0.18 |
| Ht Wt (g)/BW (kg) | 3.35 ± 0.06* | 3.94 ± 0.15 | 3.85 ± 0.13 |
Values are expressed as mean ± SEM
BW body weight when sacrificed, Ht Wt heart weight
*P < 0.05 compared with MI-Sed and MI-Ex groups
TR-α1 and TR-β1 gene and protein expressions
To determine whether exercise training altered TR-mRNA levels, quantitative RT-PCR was performed to quantify cardiac gene expression among groups. Our results illustrated that mRNA expression (Fig. 1) of TR-α1 was elevated in both MI groups compared to Sham baseline, yet was virtually the same between MI-Sed and MI-Ex (1.65 vs 1.64). Similarly, TR-β1 expression (Fig. 1) was also higher in rats with MI, but once again, was not statistically different (MI-Sed, 1.4 vs MI-Ex, 1.65). Figure 2 illustrates the representative separations of TR-α1 and TR-β1 relative to GAPDH. Compared to the Sham group, both MI groups expressed lower levels of TR-α1 and TR-β1; however, TR-α1 was 1.91-fold higher and TR-β1 was 1.62-fold higher (P < 0.05) in the MI-Ex group than in the MI-Sed group, suggesting that exercise training significantly upregulates protein expression of TR-α1 and TR-β1.
Fig. 1.

Exercise-induced TR isoform gene expressions in post-infarcted rat heart. There were no significant differences in both TR-α1 and TR-β1 between MI-Ex and MI-Sed groups. Data are expressed as ratios of target genes to β-actin relative to Sham. Values are mean ± SE
Fig. 2.
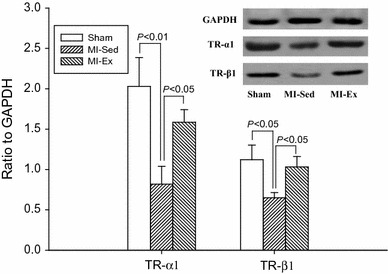
Electrophoresis results of TR-α1 and TR-β1 protein expression. Both MI-Ex and Sham animals had significantly higher TR-α1 and TR-β1 protein expression than MI-Sed group. There were no significant differences in TR-α1 and TR-β1 between Sham and MI-Ex rats
Discussion
In the present study, we demonstrated that moderate-intensity exercise training markedly increased cardiac protein expression of TR-α1 and TR-β1 9 weeks after MI. These data provide novel insights into the mechanisms underlying the improvement in morbidity and mortality produced by exercise training in patients with MI, and further elucidate the complexities of TH-signaling pathways in pathological models of hypertrophy.
Our previous studies (utilizing the same post-MI exercise model as the present study) demonstrated that post-MI exercise training not only attenuates the plasma renin–angiotensin–aldosterone-system (RAAS) [29], but also downregulates cardiac tissue angiotensin converting enzyme [32]. These beneficial changes lead to the decrease in myocardial fibrosis of the infarcted heart [31] and the improvement of cardiac function [29, 31, 32]. In a recent study, we found that post-MI exercise training significantly upregulated α-MHC, downregulated β-MHC, and improved the ratio of cardiac α-MHC to β-MHC. However, exercise training did not normalize T3 and T4 from MI-induced low plasma concentrations. Nevertheless, these beneficial changes in MHC subunits of the exercise trained rats were accompanied with enhanced cardiac function compared to the sedentary animals [30]. This finding leads to the hypothesis that the post-MI exercise-induced MHC modification may be due to the improved TRs.
T3 is an important mediator for upregulating the α-MHC isoform in the myocardium and downregulating the β-isoform [2, 23, 24, 27]. Without T3, the α-MHC gene cannot be transcribed [23]. After MI, both circulating and cardiac levels of T3 are significantly decreased despite the presence of normal serum T4 [5, 6, 9]. Our previous study demonstrated that post-MI exercise training did not normalize both T3 and T4, but the exercise training upregulated α-MHC and downregulated β-MHC with improved cardiac function [30]. This finding leads to the hypothesis that the post-MI exercise-induced MHC modification may be due to the improved TRs.
There are two genes that encode for TR, TR-α and TR-β, each with distinct patterns of expression in development and in adult tissue. TR isoforms α1, β1, β2, and β3 have similar affinities for endogenous TH and characteristics for DNA binding, while TR-α2 and -α3 do not bind TH, and therefore function as dominant-negative receptors. In the present study, we focused our efforts on T3-binding splice products predominantly expressed in cardiac muscle, namely TR-α1 and TR-β1 [20, 28].
Cardiac remodeling after MI involves several changes in TH signaling. Among the most widely observed are the downregulation of MHC-α and SERCA, and upregulation of MHC-β and phospholamban (PLB). Because α-MHC elicits 2 to 3 times faster actin-activated ATPase activity and actin filament sliding velocity than MHC-β [12, 17], it appears that MI-induced shifts in MHC isoforms is a regulatory response necessary to preserve myocardial work efficiency under pathological conditions, but does so at the expense of cardiac contractility. Furthermore, the downregulation of SERCA mediated by decreased TH signaling is also known to negate contractile function by reducing the rate of calcium reuptake into the lumen of the sarcoplasmic reticulum during diastole [7].
In the present study, MI significantly decreased expression of TR-α1 (0.85) and TR-β1 (0.65) in MI-Sed compared to Sham (2.05 and 1.15, respectively) as shown in Fig. 2. Likewise, previous studies have also demonstrated post-MI decreases in TR expression. Pantos et al. reported that after myocardial infarction, TR-α1 and TR-β1 expression were 1.3- and 1.8-fold lower than in the sham hearts (P < 0.05) [25]. Similarly, a study on the human heart revealed that compared to non-failing hearts, TR-α1 was downregulated in failing left ventricles, whereas TR-α2, a splice variant that inhibits responses to liganded TRs, was increased. Although TR-β1 did not differ significantly between groups, linear regression analysis demonstrated that TR-α1 was positively and TR-α2 was negatively correlated with MHC-α gene expression [14]. Thus, although the specific mechanisms for altered TR levels may differ among species, human and rat studies both suggest that TR-α1 downregulation is a mechanism for hypothyroid-like signaling in pathological hypertrophy.
Although not fully elucidated, much is recognized about the transcriptional functions of TR isoforms in rat and human models; for example, increased MHC-β transcription is induced by decreased expression of TR-β1, while reduced MHC-α is mediated by decreased TR-α1 expression. These TR isoform specific transcriptional functions are consistent with previous reports that exercise training significantly increases MHC-α expression while repressing the β-isoform at gene and protein levels in both sham and MI rats [8, 11, 30]. In the present study, post-MI exercise training significantly altered cardiac expression of TR isoforms. Compared to MI-Sed, TR-α1 was 1.91-fold higher (P < 0.05) and TR-β1 was 1.62-fold higher (P < 0.05) in the MI-Ex group. Increased expression of TR isoforms may prove beneficial in the surviving myocardium by increasing MHC-α and decreasing MHC-β. In fact, physiological hypertrophy in the healthy rat has been shown to alter cardiac TR expression and induce a hyperthyroid-like molecular phenotype, thus improving myocardial contractility and overall cardiac functioning. For example, Kinugawa et al. reported that physiological hypertrophy stimulated by volunteer treadmill running in rats significantly increased TR-β1 but elicited no change in TR-α1; furthermore, MHC-α was increased and MHC-β was decreased, resulting in enhanced contractile function [15].
Since there were no significant differences in the body weight and heart weight between the MI-Sed and MI-Ex groups, we didn’t detect any differences in the ratio of heart weight to body weight. However, the heart weight in both the MI-Sed and MI-Ex were significantly heavier than that of the Sham group, indicating MI-induced cardiac hypertrophy. The lack of differences in heart weight between the MI-Sed and MI-Ex groups may indicate that post-MI exercise may not significantly attenuate the MI-induced cardiac hypertrophy, although the heart weight in the MI-Ex group tended to be lighter (P > 0.05). Additionally, the lack of significant difference in heart weight may be attributed to the small sample size (n = 10 for each group).
In summary, TR downregulation is widely observed across various models of pathological hypertrophy, and is associated with reduced transcription of TH target genes and the return of the heart to fetal gene programming; however, we found that post-MI exercise training significantly alters cardiac expression of TR isoforms, and in turn may beneficially influence the course of myocardial remodeling.
Compliance with ethical standards
Funding
This study was funded by the National Heart, Lung, and Blood Institute (RO1-HL074273).
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflict of interest
Xiaohua Xu declares that he has no conflict of interest. Wenhan Wan declares that she has no conflict of interest. Michael A. Garza declares that he has no conflict of interest. John Q. Zhang declares that he has no conflict of interest.
References
- 1.Alhaddad IA, Hakim I, Siddiqi F, Lagenback E, Mallavarapu C, Nethala V, Mounce D, Ross PL, Brown EJ., Jr Early exercise after experimental myocardial infarction: effect on left ventricular remodeling. Coron Artery Dis. 1998;9:319–327. doi: 10.1097/00019501-199809060-00001. [DOI] [PubMed] [Google Scholar]
- 2.Beck-Peccoz P, Chatterjee VK. The variable clinical phenotype in thyroid hormone resistance syndrome. Thyroid Off J Am Thyroid Assoc. 1994;4:225–232. doi: 10.1089/thy.1994.4.225. [DOI] [PubMed] [Google Scholar]
- 3.Brent GA. Mechanisms of thyroid hormone action. J Clin Investig. 2012;122:3035–3043. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia-reperfusion in rat heart. J Appl Physiol. 2003;95:2510–2518. doi: 10.1152/japplphysiol.00487.2003. [DOI] [PubMed] [Google Scholar]
- 5.Chopra IJ. Clinical review 86: Euthyroid sick syndrome: is it a misnomer? J Clin Endocrinol Metab. 1997;82:329–334. doi: 10.1210/jcem.82.2.3745. [DOI] [PubMed] [Google Scholar]
- 6.De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999;84:151–164. doi: 10.1210/jcem.84.1.5364. [DOI] [PubMed] [Google Scholar]
- 7.Fazio S, Palmieri EA, Lombardi G, Biondi B. Effects of thyroid hormone on the cardiovascular system. Recent Prog Horm Res. 2004;59:31–50. doi: 10.1210/rp.59.1.31. [DOI] [PubMed] [Google Scholar]
- 8.Fernandes T, Soci UP, Oliveira EM. Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants. Braz J Med Biol Res (Revista brasileira de pesquisas medicas e biologicas/Sociedade Brasileira de Biofisica [et al]) 2011;44:836–847. doi: 10.1590/s0100-879x2011007500112. [DOI] [PubMed] [Google Scholar]
- 9.Franklyn JA, Gammage MD, Ramsden DB, Sheppard MC. Thyroid status in patients after acute myocardial infarction. Clin Sci (Lond) 1984;67:585–590. doi: 10.1042/cs0670585. [DOI] [PubMed] [Google Scholar]
- 10.Gheorghiade M, Bonow RO. Chronic heart failure in the United States: a manifestation of coronary artery disease. Circulation. 1998;97:282–289. doi: 10.1161/01.CIR.97.3.282. [DOI] [PubMed] [Google Scholar]
- 11.Hashimoto T, Kambara N, Nohara R, Yazawa M, Taguchi S. Expression of MHC-beta and MCT1 in cardiac muscle after exercise training in myocardial-infarcted rats. J Appl Physiol (Bethesda, Md: 1985) 2004;97:843–851. doi: 10.1152/japplphysiol.01193.2003. [DOI] [PubMed] [Google Scholar]
- 12.Herron TJ, McDonald KS. Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res. 2002;90:1150–1152. doi: 10.1161/01.RES.0000022879.57270.11. [DOI] [PubMed] [Google Scholar]
- 13.Hochman JS, Healy B. Effect of exercise on acute myocardial infarction in rats. J Am Coll Cardiol. 1986;7:126–132. doi: 10.1016/S0735-1097(86)80269-8. [DOI] [PubMed] [Google Scholar]
- 14.Kinugawa K, Minobe WA, Wood WM, Ridgway EC, Baxter JD, Ribeiro RC, Tawadrous MF, Lowes BA, Long CS, Bristow MR. Signaling pathways responsible for fetal gene induction in the failing human heart: evidence for altered thyroid hormone receptor gene expression. Circulation. 2001;103:1089–1094. doi: 10.1161/01.CIR.103.8.1089. [DOI] [PubMed] [Google Scholar]
- 15.Kinugawa K, Yonekura K, Ribeiro RC, Eto Y, Aoyagi T, Baxter JD, Camacho SA, Bristow MR, Long CS, Simpson PC. Regulation of thyroid hormone receptor isoforms in physiological and pathological cardiac hypertrophy. Circ Res. 2001;89:591–598. doi: 10.1161/hh1901.096706. [DOI] [PubMed] [Google Scholar]
- 16.Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- 17.Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 18.Lawler JM, Powers SK, Hammeren J, Martin AD. Oxygen cost of treadmill running in 24-month-old Fischer-344 rats. Med Sci Sports Exerc. 1993;25:1259–1264. doi: 10.1249/00005768-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Long X, Boluyt MO, O’Neill L, Zheng JS, Wu G, Nitta YK, Crow MT, Lakatta EG. Myocardial retinoid X receptor, thyroid hormone receptor, and myosin heavy chain gene expression in the rat during adult aging. J Gerontol Ser A Biol Sci Med Sci. 1999;54:B23–B27. doi: 10.1093/gerona/54.1.B23. [DOI] [PubMed] [Google Scholar]
- 20.Luongo C, Trivisano L, Alfano F, Salvatore D. Type 3 deiodinase and consumptive hypothyroidism: a common mechanism for a rare disease. Front Endocrinol. 2013;4:115. doi: 10.3389/fendo.2013.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morkin E, Pennock G, Spooner PH, Bahl JJ, Underhill Fox K, Goldman S. Pilot studies on the use of 3,5-diiodothyropropionic acid, a thyroid hormone analog, in the treatment of congestive heart failure. Cardiology. 2002;97:218–225. doi: 10.1159/000063110. [DOI] [PubMed] [Google Scholar]
- 22.Mourouzis I, Forini F, Pantos C, Iervasi G. Thyroid hormone and cardiac disease: from basic concepts to clinical application. J Thyroid Res. 2011;2011:958626. doi: 10.4061/2011/958626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadal-Ginard B, Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Investig. 1989;84:1693–1700. doi: 10.1172/JCI114351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojamaa K, Kenessey A, Shenoy R, Klein I. Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am J Physiol Endocrinol Metab. 2000;279:E1319–E1324. doi: 10.1152/ajpendo.2000.279.6.E1319. [DOI] [PubMed] [Google Scholar]
- 25.Pantos C, Mourouzis I, Saranteas T, Paizis I, Xinaris C, Malliopoulou V, Cokkinos DV. Thyroid hormone receptors alpha1 and beta1 are downregulated in the post-infarcted rat heart: consequences on the response to ischaemia-reperfusion. Basic Res Cardiol. 2005;100:422–432. doi: 10.1007/s00395-005-0545-4. [DOI] [PubMed] [Google Scholar]
- 26.Pantos C, Mourouzis I, Xinaris C, Kokkinos AD, Markakis K, Dimopoulos A, Panagiotou M, Saranteas T, Kostopanagiotou G, Cokkinos DV. Time-dependent changes in the expression of thyroid hormone receptor alpha 1 in the myocardium after acute myocardial infarction: possible implications in cardiac remodelling. Eur J Endocrinol. 2007;156:415–424. doi: 10.1530/EJE-06-0707. [DOI] [PubMed] [Google Scholar]
- 27.Rafalski K, Abdourahman A, Edwards JG. Early adaptations to training: upregulation of alpha-myosin heavy chain gene expression. Med Sci Sports Exerc. 2007;39:75–82. doi: 10.1249/01.mss.0000240324.08406.3d. [DOI] [PubMed] [Google Scholar]
- 28.Sussman MA. When the thyroid speaks, the heart listens. Circ Res. 2001;89:557–559. doi: 10.1161/res.89.7.557. [DOI] [PubMed] [Google Scholar]
- 29.Wan W, Powers AS, Li J, Ji L, Erikson JM, Zhang JQ. Effect of post-myocardial infarction exercise training on the renin–angiotensin–aldosterone system and cardiac function. Am J Med Sci. 2007;334:265–273. doi: 10.1097/MAJ.0b013e318068b5ed. [DOI] [PubMed] [Google Scholar]
- 30.Wan W, Xu X, Zhao W, Garza MA, Zhang JQ. Exercise training induced myosin heavy chain isoform alteration in the infarcted heart. Appl Physiol Nutr Metab (Physiologie appliquee, nutrition et metabolisme) 2014;39:226–232. doi: 10.1139/apnm-2013-0268. [DOI] [PubMed] [Google Scholar]
- 31.Xu X, Wan W, Ji L, Lao S, Powers AS, Zhao W, Erikson JM, Zhang JQ. Exercise training combined with angiotensin II receptor blockade limits post-infarct ventricular remodelling in rats. Cardiovasc Res. 2008;78:523–532. doi: 10.1093/cvr/cvn028. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Wan W, Powers AS, Li J, Ji LL, Lao S, Wilson B, Erikson JM, Zhang JQ. Effects of exercise training on cardiac function and myocardial remodeling in post myocardial infarction rats. J Mol Cell Cardiol. 2008;44:114–122. doi: 10.1016/j.yjmcc.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu X, Zhao W, Wan W, Ji LL, Powers AS, Erikson JM, Zhang JQ. Exercise training combined with angiotensin II receptor blockade reduces oxidative stress after myocardial infarction in rats. Exp Physiol. 2010;95:1008–1015. doi: 10.1113/expphysiol.2010.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yue P, Long CS, Austin R, Chang KC, Simpson PC, Massie BM. Post-infarction heart failure in the rat is associated with distinct alterations in cardiac myocyte molecular phenotype. J Mol Cell Cardiol. 1998;30:1615–1630. doi: 10.1006/jmcc.1998.0727. [DOI] [PubMed] [Google Scholar]