

Abstract
T-type channels are low-voltage-activated channels that play a role in the cardiovascular system particularly for pacemaker activity. Glycosylation is one of the most prevalent post-translational modifications in protein. Among various glycosylation types, the most common one is asparagine-linked (N-linked) glycosylation. The aim of this study was to elucidate the roles of N-linked glycosylation for the gating properties of the CaV3.1-T-type Ca2+ channel. N-linked glycosylation synthesis inhibitor tunicamycin causes a reduction of CaV3.1-T-type Ca2+ channel current (CaV3.1-ICa.T) when applied for 12 h or longer. Tunicamycin (24 h) significantly shifted the activation curve to the depolarization potentials, whereas the steady-state inactivation curve was unaffected. Use-dependent inactivation of CaV3.1-ICa.T was accelerated, and recovery from inactivation was prolonged by tunicamycin (24 h). CaV3.1-ICa.T was insensitive to a glycosidase PNGase F when the channels were expressed on the plasma membrane. These findings suggest that N-glycosylation contributes not only to the cell surface expression of the CaV3.1-T-type Ca2+ channel but to the regulation of the gating properties of the channel when the channel proteins were processed during the folding and trafficking steps in the cell.
Keywords: Glycosylation, T-type Ca2+ channel, CaV3.1, α1G channel, Tunicamycin
Introduction
T-type calcium channels, known as low-voltage-activated calcium channel or transient-type calcium channel, are predominantly expressed in the atria and nodal cells in the heart. In particular, T-type Ca2+ channels contribute to the generation of action potentials in nodal cells at the phase of slow diastolic depolarization. Genetic analysis demonstrated that two types of T-type Ca2+ channel isoforms, CaV3.1 and CaV3.2, are widely expressed in the heart [1–3]. Studies of genetically modified mice with targeted inactivation of the T-type Ca2+ channel suggest that CaV3.1-T-type Ca2+ channel underlies the functional T-type Ca2+ channel in the heart for the generation of pacemaker rhythm [1, 2, 4] particularly with adult animals [1]. Regarding the automaticity in the pacemaker cells, failure to generate the nodal electrical impulse underlies congenital or acquired bradycardia and bradycardia-associated conditions, such as atrial fibrillation [5]. Sick sinus syndrome is the term used to describe the inability of the sinus node to generate heart rhythms that meet the physiologic needs of an individual. Sinus node dysfunction occurs as a result of disorders in automaticity, conduction, or both. Although several causes of the acquired sick sinus dysfunction including idiopathic degenerative disease have been postulated, molecular mechanisms for the pacemaker dysfunction are mostly unknown.
Ion channels are subject to complex regulation by a wide range of cytosolic modulators [5, 6]. Ion channels are also subject to multiple post-transcription regulatory mechanisms, including glycosylation, formation of disulfide bonds, enzyme digestion, phosphorylation, and ubiquitination [7–9]. Asparagine (Asn, N)-linked glycosylation (N-glycosylation) has been investigated to affect the structural folding, membrane targeting, expression level, stability, and voltage-dependent properties of many ion channels including the T-type Ca2+ channels. For instance, reduction or elimination of N-glycosylation of the CaV3.2-T-type Ca2+ channel was reported to decrease channel activity and expression with altered channel kinetics [10]. In this study, we analyzed an impact of the reduction of N-glycosylation of the CaV3.1-T-type Ca2+ channel, the other isoform of the channel in the heart. We employed patch-clamp techniques to study kinetics of the channel in heterologous expression system with human embryonic kidney cells (HEK293) cells. Tunicamycin has been widely used as a standard method in the study of glycoprotein synthesis in various biological systems [9, 10]. Tunicamycin inhibits N-acetylglucosamine-1-phosphate transferase (GlcNAc phosphotransferase) and inhibits the formation of N-glycosidic linkages in glycoprotein synthesis [9, 10]. Inhibition of N-linked glycosylation synthesis using tunicamycin causes a reduction of CaV3.1-T-type Ca2+ channel current (CaV3.1-ICa.T) density when applied for 12 h or longer, as a long-term effect, although tunicamycin does not affect CaV3.1-ICa.T as an acute effect. Tunicamycin significantly shifted the activation curve toward the depolarization potentials, whereas the steady-state inactivation curve was unaffected, when tunicamycin was applied for 24 h. On the other hand, CaV3.1-ICa.T was insensitive to a glycosidase PNGase F when the glycosylated channels were expressed on the plasma membrane. These findings suggest that N-glycosylation contributes not only to the cell surface expression of the CaV3.1-T-type Ca2+ channel but also to the regulation of the gating properties of the channel when the channel proteins were processed during the folding and trafficking steps in the cell. It is also suggested that deglycosylation of the CaV3.1-T-type Ca2+ channel can be a potential method to reduce heart rate or to treat tachycardia-mediated arrhythmias in the heart.
Materials and methods
Expression of CaV3.1 channel in mammalian cells and cell-culture condition
The CaV3.1-T-type Ca2+ channel isoform (α1G submit) derived from human was stably expressed in human embryonic kidney (HEK)-293 cells with no auxiliary subunits. HEK-293 cells were maintained in DMEM, supplemented with 10% fetal bovine serum, 100 μ/ml penicillin, and 100 mg/l streptomycin, in an atmosphere of 95% O2 and 5% CO2 at 37 °C. This medium was supplemented with 350 mg/l G418 for selection of recombinant HEK-293 cells. For the study on the time-dependent effects of tunicamycin, the HEK-CaV3.1 cells were incubated with or without 10−2 g/l tunicamycin (Wako Chemicals, Japan) for 1 h, 6 h, 12 h, or 24 h prior to the electrophysiological measurements. For the study on chemical cleavage for removal of glycosylation of HEK-CaV3.1 cells, a non-specific glycosidase Peptide-N-Glycosidase F (PNGase F) was applied in the cell-culture medium for 2 h prior to the electrophysiological measurements according to manufacture instruction. For the study on the concentration dependent effects of tunicamycin, HEK-CaV3.1 cells were incubated with 10−6 g/l, 10−5 g/l, 10−4 g/l, 10−3 g/l, or 10−2 g/l tunicamycin for 24 h before electrophysiological measurements.
Electrophysiology
Whole-cell current-clamp and voltage-clamp experiments were performed using EPC-9 amplifier (HEKA Elektronik, Germany) at room temperature (20–24 °C). Acquisition and analysis were performed using Patchmaster software (HEKA). The bath solution contained (in millimolar): TEA-Cl 120, CsCl 6, 4-AP 5, MgCl2 0.5, DIDS 0.5, HEPES 10, CaCl2 1.8, glucose 10 (pH 7.4 with TEA-OH). Patch pipettes were filled with pipette solution containing (in millimolar) CsCl 130, Mg-ATP 2, EGTA 5, HEPES 10 (pH 7.2 with CsOH) and had a resistance of 2–4 MΩ. The T-type calcium channel current (ICa.T) was recorded in response to a series of test steps to depolarized potentials ranging from − 100 to + 50 mV for 250 ms from a holding potential of − 100 mV, with 2-s sweep interval and 10-kHz sampling frequency. The activation curves were fitted with the following modified Boltzmann equation: G/Gmax = [1 + exp (V1/2-act − V)/k]−1, where Gmax is the maximum conductance, V1/2-act is the half-activation potential and k is the slope factor. ICa.T was further characterized by its voltage-dependent inactivation properties. HEK-Cav3.1 cells were held at a range of conditioning potentials (from − 100 to − 20 mV) for 2 s and then stepped to a test potential of − 20 mV. The steady-state inactivation curve was fitted with an equation: I/Imax = [1 + exp (V − V1/2-SSI)/k]−1, where V1/2-SSI is the half-inactivation potential. For a measurement of the use-dependent reduction of ICa,T, repetitive stimulation (a train pulse at − 20 mV for 200-ms duration) of 0.4-Hz, 2.5-Hz, and 4-Hz repetitive voltage pulses were given to HEK-Cav3.1 cells from the holding potentials of − 100 mV. ICa.T recorded from the second test pulses or later (Ix) were normalized to the current recorded from the first pulse (I1 assigned as 1.0), yielding the ratio of Ix/I1. To analyze the recovery phase of the channel inactivation, HEK-Cav3.1 cells were held at − 100 mV and a pair-pulse at − 20 mV with variable interval time was applied. The interval time between the two pulses (interstimulus interval) was adjusted from 2 ms to 2000 ms. Peak ICa.T recorded by the second pulse was normalized by ICa.T at the first pulse. Normalized ICa.T was plotted against the function of interval time and fitted by bi-exponential equation to obtain the recovery time constant, τ-fast and τ-slow.
Statistical analysis
Data were acquired using computer software (Pachmaster, HEKA), and all curve fittings and figures were made on Sigma Plot ver. 10 (Systat Software Inc, USA). All results are presented as mean ± SEM, with the number of observations indicated by n. Values were analyzed by Student’s t test or Holm–Sidak method when multiple comparisons were needed. Differences were considered significant at p < 0.05.
Results
Time-dependent effects of tunicamycin on CaV3.1-ICa.T
Effects of tunicamycin on the HEK-CaV3.1 cells were assessed when applied in the culture medium for 1 to 24 h (Fig. 1). Although tunicamycin was without effects on CaV3.1-ICa.T applied in the culture medium for 1 h or 6 h, CaV3.1-ICa.T was significantly decreased when applied for 12 h or longer. Importantly, reduction ratio of CaV3.1-ICa.T by tunicamycin was time dependent; CaV3.1-ICa.T with the action of tunicamycin for 24 h was significantly smaller than that with tunicamycin for 12 h (Fig. 1). The ratio of the maximal ICa.T density in tunicamycin group to vehicle group (Imax(Tuni)/Imax(Veh)), and the ratio of the maximal conductance in tunicamycin group to vehicle group (Gmax(Tuni)/Gmax(Veh)) also indicate the time-dependent reduction caused by tunicamycin treatment (Fig. 1). These ratios were less than 50% in the 12 h and were even more decreased in the 24 h. Given that tunicamycin inhibits the first step of glycosylation, it may need a certain time to exert an inhibitory effect on CaV3.1-ICa.T, which is consistent with a notion that the reduction of CaV3.1-ICa.T was caused by the inhibition of N-glycosylation synthesis rather than a direct effect on the CaV3.1 channel on the cell surface membrane.
Fig. 1.

Time-dependent effects of glycosylation inhibition with tunicamycin on CaV3.1-ICa.T. Representative current traces with vehicle and tunicamycin treatment, and group data for current (I)-voltage (V) relationship. HEK-CaV3.1 cells were incubated with vehicle or tunicamycin (10−2 g/l) for 1 h (a), 6 h (b), 12 h (c), or 24 h (d). Changes of the maximum ICa.T (Imax) and the maximum conductance (Gmax) by glycosylation inhibition with tunicamycin are shown as group data (e, f). The ordinate indicates the ratio of the maximum inward current (e) and the maximum conductance (f) with tunicamycin normalized to that with vehicle at the same incubation period. The number of cells is indicated in parentheses. Values represent the mean ± SE. *p < 0.01 vs. the ratio at 1 h. #p = 0.06 vs. the ratio at 12 h. †p = 0.12 vs. the ratio at 12 h
Kinetic modulation by inhibition of N-glycosylation
Without glycosylation, some proteins are not folded correctly or not stable. In view of potentially functional effects of N-glycosylation on the CaV3.1-T-type Ca2+ channel, we have analyzed a long-term effect of tunicamycin on the activation and the inactivation properties of the channel. We constructed the activation curve and the steady-state inactivation curve of CaV3.1-ICa.T; the activation curve was shifted toward the depolarization direction when HEK-CaV3.1 cells were incubated with tunicamycin for 12 h, while the inactivation curves was unaffected at the same condition (Fig. 2c). Importantly, the activation curve was even more shifted toward the depolarization direction when HEK-CaV3.1 cells were incubated with tunicamycin for 24 h (Fig. 2d). Analysis and comparison of the potentials for the half-maximum activation (V1/2-activation) and the half-maximum inactivation (V1/2-inactivation) demonstrated a shift of activation curves but not inactivation curves toward the hyperpolarization direction by the treatment of the channel with tunicamycin (Fig. 2). Long-term (24 h) application of tunicamycin decreased CaV3.1-ICa.T in a dose-dependent manner with IC50 of 2.01 × 10−3 g/l or 10−2.697 g/l (Fig. 3). Tunicamycin at 10−3 g/l concentration significantly decreased the maximum inward current by 37% (Fig. 3b). Consistently, V1/2-activation was appreciably shifted toward the depolarization direction by tunicamycin with the concentration of 10−3 g/l, and the shift became significant with the concentration of 10−2 g/l (Fig. 4). To evaluate the tonic and the use-dependent effect of tunicamycin on CaV3.1-ICa.T, a train of pulses (0.4 Hz, 2.5 Hz and 4.0 Hz) was applied (Fig. 5a). Repetitive voltage-clamp pulses with or without a treatment of tunicamycin resulted in a negligible changes of CaV3.1-ICa.T at the stimulation frequency of 0.4 Hz. At the stimulation ratio of 2.5 Hz, however, ICa.T was reduced by 9.2% without tunicamycin treatment and by 16.9% with tunicamycin treatment at the second pulse (I2/I1). At the end of the train pulse (28th pulse), ICa.T ratio (I28/I1) was reduced to 87.4 ± 1.1% without tunicamycin treatment and to 80.0 ± 1.3% with tunicamycin treatment. At the stimulation ratio of 4.0 Hz, ICa.T was reduced by 16.8% without tunicamycin treatment and by 39.9% with tunicamycin treatment at the second pulse (I2/I1). At the end of the train pulse (28th pulse), ICa.T ratio (I28/I1) was reduced to 70.0 ± 1.2% without tunicamycin treatment and to 57.3 ± 1.5% with tunicamycin treatment. These results indicate that tunicamycin treatment accelerate the use-dependent reduction of the current particularly at the high stimulation frequency.
Fig. 2.

Changes of the activation (G/Gmax) and the steady-state inactivation (availability) curves by glycosylation inhibition with tunicamycin, and their parameters of half-activation potentials (V1/2-activation) and half-inactivation potentials (V1/2-inactivation). Both curves were obtained by Boltzmann fittings with data when cells were incubated with vehicle or tunicamycin (10−2g/l) for 1 h (a), 6 h (b), 12 h (c), or 24 h (d). The number of cells is indicated in parentheses. Values represent the mean ± SE. *p < 0.01 vs. vehicle
Fig. 3.
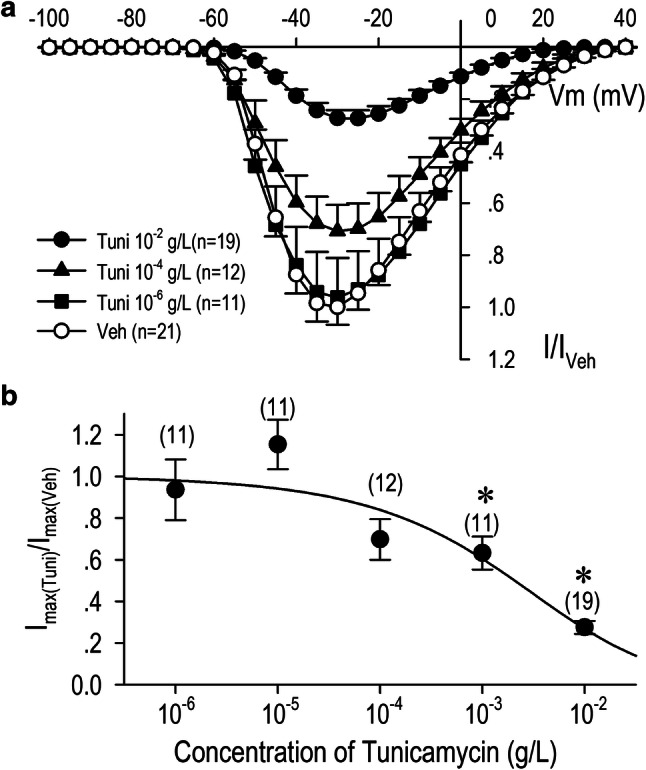
Concentration-dependent effects of tunicamycin on CaV3.1-ICa.T. HEK-CaV3.1 cells were incubated for 24 h with vehicle or tunicamycin at the concentration of 10−6 g/l, 10−5 g/l, 10−4 g/l, 10−3 g/l, or 10−2 g/l. a Current (I)-voltage (V) relation were constructed by normalizing each ICa.T to that of the maximum inward current with vehicle (I/Iveh). b Dose–response relationship of tunicamycin on ICa.T was evaluated by a pulse protocol composed of a holding potential of − 100 mV to a test potential for the maximum current (− 30 or − 25 mV) at the stimulation frequency of 0.2 Hz. Data were fitted by the Hill equation yielding IC50 value of 2.01 × 10−3 g/l. Values represent the mean ± SE. *p < 0.01 vs. vehicle
Fig. 4.

Concentration-dependent effects of tunicamycin on half-activation potentials (V1/2-activation) and half-inactivation potentials (V1/2-inactivation). HEK-CaV3.1 cells were incubated for 24 h with vehicle or tunicamycin at the concentration of 10−6 g/l (a), 10−5 g/l (b), 10−4 g/l (c), 10−3 g/l (d), or 10−2 g/l (e). Values for V1/2-activation and V1/2-inactivation were obtained by Boltzmann fittings with the same pulse protocol in Fig. 2. The number of cells is indicated in parentheses. Values represent the mean ± SE. *p < 0.01 vs. vehicle
Fig. 5.

Use-dependent reduction and recovery from inactivation of ICa.T modified by glycosylation inhibition with tunicamycin. a Time course for development of use-dependent reduction of ICa.T with or without tunicamycin (10−6 g/l) treatment for 24 h. ICa.T was recorded by a train of 28 pulses consisting of 100-ms duration at a test potential − 10 mV from the holding potential of − 100 mV with the stimulation frequency of 0.4, 2.5, or 4.0 Hz. The current amplitude of the first pulse of train stimulation was assigned as 1.0. b Time course of recovery from inactivation of ICa.T. Pulse protocol is shown in the inset. The current amplitude at the conditioning pulse (Ic) was assigned as 1.0, and the current amplitudes at the test pulse (It) were normalized to the ratio of Ic (It/Ic). Curves were fitted to the data using a double-exponential equation shown in the inset. c Time constants for the fast and slow components (Tfast, Tslow) and d their amplitudes in equation (Ampfast, Ampslow). The number of cells is indicated in parentheses. Values represent the mean ± SE. *p < 0.01 vs. vehicle
To further study the effect of tunicamycin treatment on the gating property of the CaV3.1 channel, we examined whether the time of ICa.T recovery from inactivation was modulated by tunicamycin treatment. Thus, we examined the recovery time course from inactivation by using the pulse protocol illustrated in Fig. 5b. Recovery from inactivation was significantly delayed by tunicamycin treatment [fast time constant (Tfast): vehicle, 55.9 ± 2.0 ms; tunicamycin, 92.7 ± 5.1 ms] [slow time constant (Tslow): vehicle, 278.1 ± 26.0 ms; tunicamycin, 432.6 ± 48.8 ms], which is consistent with the findings shown in Fig. 5a, although amplitudes of fast and slow components were substantially unchanged. These results indicate the retarded inactivation process of the channel gating produced by tunicamycin treatment.
Effects of chemical cleavage for removal of glycosylation
To investigate whether the removal of glycosylation has effects on the channel function when the channel proteins are expressed with the full glycosylation steps on the plasma membrane, we applied a non-specific glycosidase PNGase F in the cell-culture medium prior to the electrophysiological experiments. CaV3.1-ICa.T was insensitive to PNGase F (Fig. 6), suggesting that N-glycosylation contributes to the regulation of the gating properties of the channel only when the channel proteins were processed during the folding and trafficking steps in the cell.
Fig. 6.

Effects of chemical cleavage for removal of glycosylation on CaV3.1-ICa.T. Changes of the activation (G/Gmax) and the steady-state inactivation (availability) curves were evaluated by a non-specific glycosidase PNGase F in the cell-culture medium prior to the electrophysiological experiments for 2 h. Representative current traces with vehicle or PNGase F (a), group data for current (I)-voltage (V) relationship (b), and changes of the activation (G/Gmax) and the steady-state inactivation (Availability) curves as group data (c). The number of cells is indicated in parentheses. Values represent the mean ± SE
Discussion
The biosynthesis of N-linked glycans needs three steps: (1) synthesis of dolichol-linked precursor oligosaccharide, (2) en bloc transfer of precursor oligosaccharide to protein, and (3) processing of the oligosaccharide [9]. Tunicamycin is widely used as an inhibitor of N-linked glycosylation, because tunicamycin inhibits N-acetylglucosamine (GlcNAc) phosphotransferase, which catalyzes the transfer of N-acrtylglucosamine-1-phosphase from uridine diphosphate (UDP)-GlcNAc to dolichol phosphate in the first step of glycosylation synthesis [10]. In this regard, it is rightly assumed that tunicamycin triggers a halt of the channel maturation by inhibiting the N-linked glycosylation not as a short-term effect but as a long-term effect prior to the trafficking to the plasma membrane. This process probably needs several hours concerning all the synthesis steps of the channel from the gene transcription to the trafficking of proteins to cell surface membrane [8, 9]. In this context, a negligible acute effect of tunicamycin on CaV3.1-ICa.T would be consistent to the postulated function of N-glycosylation to the ion channel. In this study, we have successfully demonstrated that a lack of proper glycosylation not only decreased the channel current density but also modified the gating properties of the CaV3.1-T-type Ca2+ channel.
Although excitation of cardiomyocyte is highly integratively regulated [6, 11–15], the trigger of the action potentials mainly depends on the function of voltage-gated ion channels; Na+ channels and Ca2+ channels. In many cells, including cardiomyocytes, though, the most important role of Ca2+ channels is to transduce the electrical signals derived from action potentials into the intracellular chemical signals by an increase in [Ca2+]i. Two different Ca2+ channels exist in cardiomyocytes; the L-type Ca2+ channels and T-type Ca2+ channels [5]. The current density of the T-type Ca2+ channel (ICa.T) is relatively low in comparison to those of L-type Ca2+ channel current (ICa.L) in cardiomyocytes, however ICa.T plays important roles in the heart particularly for the pacemaking activity in nodal cells. At generating action potentials, ICa.T regulates phase 4 slow diastolic potentials together with currents by ICa.L, hyperpolarization-activated inward current (If), transient inward current caused by Na+–Ca2+ exchanger, and others [16]. Regarding the isoform-specific roles of the T-type Ca2+ channel, CaV3.1-ICa.T plays a definite role in the adult heart according to a study with CaV3.1(−/−) mice; a significantly slow heart rate and a prolonged the sinus node recovery time [17]. On the other hand, CaV3.2-ICa.T plays a different role in the heart. Coronary arteries isolated from CaV3.2 (−/−) mice demonstrate reduced relaxation in response to acetylcholine and nitroprusside, which indicates a function of the CaV3.2-T-type Ca2+ channel as essential element for normal relaxation of coronary arteries [18].
Although impacts of glycosylation of the voltage-gated Ca2+ channels have been investigated, it is only lately that the functional significance of N-glycosylation on the function of the T-type Ca2+ channel was investigated. Recent investigations reported potential glycosylation motif within the T-type Ca2+ channels [9]. According to their study, some potential glycosylation sites in the CaV3.1-T-type Ca2+ channel are conserved, and others are not conserved in the CaV3.2-T-type Ca2+ channel, which suggests that glycosylation/deglycosylation effects of the CaV3.1 channel and the CaV3.2 channel may not be identical. The role of glycosylation/deglycosylation of the CaV3.2 channel is somewhat controversial among the studies [7, 19–21]. Acute enzymatic deglycosylation of the CaV3.2 channel displayed a reduction of the channel conductance without affecting the voltage dependency of the activation in heterologous expression system with HEK-293 cells [7, 18]. Also ablation of the potential N-link glycosylation of the CaV3.2 channel displayed a reduction of the channel conductance and a positive shift in the activation kinetics without affecting the fast inactivation of the channel decay [20]. Unlike the case of the CaV3.2-channel, potential effect of glycosylation/deglycosylation on the CaV3.1 channel is largely unknown. In our study, in contrast, elimination of N-link glycosylation in the CaV3.1-T-type Ca2+ channel resulted in a reduction of the maximum conductance, a positive shift in the activation kinetics, and a delayed recovery from the inactivation in a manner consistent with an acceleration of the use-dependent reduction of the current. Given the diversity of the functional results caused by ablations of N-linked glycosylation with tunicamycin, it is assumed that multiple N-linked glycosylation sites in the CaV3.1 channel distinctly act for the channel maturation prior to the sorting into cell surface plasma membrane. It is also important to note that cleavage of N-linked glycosylation by PNGase F, a non-specific enzyme which cleaves N-linked glycosylation [22], was without effect on CaV3.1-ICa.T density nor kinetics. These observations support out conclusion that N-glycosylation contributes not only to the cell surface expression of the CaV3.1-T-type Ca2+ channel but also to the regulation of the gating properties of the channel only when the channel proteins were processed during the folding and trafficking steps possibly in the endoplasmic reticulum and in the Golgi by the final settling-down process to the plasma membrane.
Functional abnormalities in N-glycosylation were also observed in other voltage-gated ion channels [23, 24]; modifications differ from channels to channels. For instance, the activation curve and the steady-state inactivation curve of the NaV1.4 channel shift toward the depolarization direction when N-glycosylation was blocked by shortage of N-linked sialic acid, although the NaV1.2, NaV1.5, and NaV1.7 were unaffected [23]. When β-subunits were simultaneously expressed, activation curves and steady-state inactivation curves of the NaV1.2, NaV1.5, and NaV1.7 shifted toward the hyperpolarization direction, while both curves shifted toward the depolarization direction in the NaV1.4 channel [23]. Meanwhile, ablation of four potential N-linked glycosylation sites in the CaV1.2 channel resulted in a decrease in current density and a depolarization shift of the activation curve [24], which suggests a functional similarity with the CaV3.1 channel in this study.
The role of glycosylation in the pathophysiological aspect of ion channel has remained largely overleaped. Defect of glycosylation in the CaV3.1 channel would lead to the pathology of the heart. A reduction of CaV3.1-ICa.T would indicate many physiological aberrations in the heart. A reduction of ICa.T leads a decrement of window current of T-type calcium channel (ICa.T-window). ICa.T-window is the constant inward current providing by the overlap of the activation and the steady-state inactivation near the activation threshold, and is a main way of working for the T-type Ca2+ channel. ICa.T-window stands in phase 4 at action potentials in sinus node cells, and could be speculated to facilitate the slow diastolic depolarization of sinus node cells, which contributes to their electrical automaticity [16, 25]. About 20% of patients with congenital disorders of glycosylation (CDG) have cardiac complications, such as arrhythmias, cardiomyopathies, and structural defects [26]. Patients with CDG-1a who have N-glycosylation defect always have neurological alterations [10]. So, reduction of peak ICav3.1-T and IT-window may participate in such CDG symptoms.
In conclusion, we have analyzed the long-term effect of N-glycosylation synthesis inhibition on CaV3.1-ICa.T. The inhibition of N-glycosylation synthesis not only decreased the current density of the CaV3.1 channel but also modified the gating kinetics of the channel including a shift of the activation curve to depolarization direction, acceleration of the use-dependent reduction, and slow recovery from inactivation. Although the implication of N-linked glycosylation in the CaV3.1 channel is a relatively new notion, emerging evidence indicate that altered glycosylation of the channel proteins may contribute to a number of pathological condition of the heart particularly for arrhythmias and pacemaking abnormalities.
Acknowledgements
We are grateful to Ms. Watanabe and Matsuno for their secretary assistance.
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Research involving human participants and/or animals
This study does not contain any studies with human participants performed by any of the authors. All procedures performed in studies involving animals were in accordance with the ethical standards of Oita University School of Medicine.
Informed consent
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mesirca P, Torrente AG, Mangoni ME. Functional role of voltage gated Ca2+ channels in heart automaticity. Front Physiol. 2015;6:19. doi: 10.3389/fphys.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mesirca P, Torrente AG, Mangoni ME. T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Archy. 2014;466:791–799. doi: 10.1007/s00424-014-1482-6. [DOI] [PubMed] [Google Scholar]
- 3.Curran J, Musa H, Kline CF, Makara MA, Little SC, et al. Eps15 homology domain-containing protein 3 regulates cardiac T-type Ca2+ channel targeting and function in the atria. J Biol Chem. 2015;290:12210–12221. doi: 10.1074/jbc.M115.646893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Quang K, Benito B, Naud P, Qi X-Y, Shi YF, et al. T-type calcium current contributes to escape automaticity and governs the occurrence of lethal arrhythmias following atrioventricular block in mice. Circ Arrhythm Electrophysiol. 2013;6:799–808. doi: 10.1161/CIRCEP.113.000407. [DOI] [PubMed] [Google Scholar]
- 5.Gillis AM, Russo AM, Ellenbogen KA, Swerdlow CD, Olshansky B, et al. HRS/ACCF expert consensus statement on pacemaker device and mode selection. J Am Coll Cardiol. 2012;9:1344–1365. doi: 10.1016/j.jacc.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Jin H, Fujita T, Jin M, Kurotani R, Hidaka Y, et al. Epac activation inhibits IL-6-induced cardiac myocyte dysfunction. J Physiol Sci. 2018;68:77–87. doi: 10.1007/s12576-016-0509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orestes P, Osuru HP, McIntire WE, Jacus MO, Salajegheh R, et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of CaV3.2 T-type calcium channels. Diabetes. 2013;62:3828–3838. doi: 10.2337/db13-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baycin-Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, et al. Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem Biophys Res Commun. 2014;453:243–253. doi: 10.1016/j.bbrc.2014.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazniewska J, Weiss N. Glycosylation of voltage-gated calcium channels in health and disease. Biochim Biophys Acta. 2017;1859:662–668. doi: 10.1016/j.bbamem.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 10.Aebi M, Hennet T. Congenital disorders of glycosylation: genetic model systems lead the way. Trends Cell Biol. 2001;11:136–141. doi: 10.1016/S0962-8924(01)01925-0. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama U, Ichikawa Y, Minamisawa S, Ishikawa Y. Pathology and molecular mechanisms of coarctation of the aorta and its association with the ductus arteriosus. J Physiol Sci. 2017;67:259–270. doi: 10.1007/s12576-016-0512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee Y, Kwon I, Jang Y, Song W, Cosio-Lima LM, et al. Potential signaling pathways of acute endurance exercise-induced cardiac autophagy and mitophagy and its possible role in cardioprotection. J Physiol Sci. 2017;67:639–654. doi: 10.1007/s12576-017-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khokhlova A, Balakina-Vikulova N, Katsnelson L, Iribe G, Solovyova O. Transmural cellular heterogeneity in myocardial electromechanics. J Physiol Sci. 2017;68:387–413. doi: 10.1007/s12576-017-0541-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Argenziano M, Tiscornia G, Moretta R, Casal L, Potilinski C, et al. Arrhythmogenic effect of androgens on the rat heart. J Physiol Sci. 2017;67:217–225. doi: 10.1007/s12576-016-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masuda K, Takanari H, Morishima M, Ma F, Wang Y, et al. Testosterone-mediated upregulation of delayed rectifier potassium channel in cardiomyocytes causes abbreviation of QT intervals in rats. J Physiol Sci. 2018;68:1–9. doi: 10.1007/s12576-017-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ono K, Iijima T. Pathophysiological significance of T-type Ca2+ channels: properties and functional roles of T-type Ca2+ channels in cardiac pacemaking. J Pharmacol Sci. 2005;99:197–204. doi: 10.1254/jphs.FMJ05002X2. [DOI] [PubMed] [Google Scholar]
- 17.Mangoni ME, Traboulsie A, Leoni A-L, Couette B, Marger L, et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-type calcium channels. Circ Res. 2006;98:1422–1430. doi: 10.1161/01.RES.0000225862.14314.49. [DOI] [PubMed] [Google Scholar]
- 18.Chen C-C, Lamping KG, Nuno DW, Barresi R, Prouty SJ, et al. Abnormal coronary function in mice deficient in α1H T-type Ca2+ channels. Science. 2003;302:1416–1418. doi: 10.1126/science.1089268. [DOI] [PubMed] [Google Scholar]
- 19.Weiss N, Black SA, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465:1159–1170. doi: 10.1007/s00424-013-1259-3. [DOI] [PubMed] [Google Scholar]
- 20.Lazniewska J, Rzhepetskyy Y, Zhang F-X, Zamponi GW, Weiss N. Cooperative roles of glucose and asparagine-linked glycosylation in T-type calcium channel expression. Pflugers Arch. 2016;468:1837–1851. doi: 10.1007/s00424-016-1881-y. [DOI] [PubMed] [Google Scholar]
- 21.Fukami K, Asano E, Ueda M, Sekiguchi F, Yoshida S, et al. High glucose induces N-linked glycosylation-mediated functional upregulation and overexpression of CaV3.2 T-type calcium channels in neuroendocrine-like differentiated human prostate cancer cells. J Pharmacol Sci. 2017;133:57–60. doi: 10.1016/j.jphs.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 23.Johnson D, Montpetit ML, Stocker PJ, Bennett ES. The sialic acid component of the β1 subunit modulates voltage-gated sodium channel function. J Biol Chem. 2004;279:44303–44310. doi: 10.1074/jbc.M408900200. [DOI] [PubMed] [Google Scholar]
- 24.Park H-J, Min S-H, Won Y-J, Lee J-H. Asn-linked glycosylation contributes to surface expression and voltage-dependent gating of CaV1.2 Ca2+ channel. J Microbiol Biotechnol. 2015;25:1371–1379. doi: 10.4014/jmb.1501.01066. [DOI] [PubMed] [Google Scholar]
- 25.Vassort G, Talavera K, Alvarez JL. Role of T-type Ca2+ channels in the heart. Cell Calcium. 2006;40:205–220. doi: 10.1016/j.ceca.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 26.D’Amico A, Petrini S, Parisi F, Tessa A, Francalanci P, et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord. 2008;18:153–155. doi: 10.1016/j.nmd.2007.09.013. [DOI] [PubMed] [Google Scholar]