

Abstract
Oxidative stress contributes to acceleration of muscle atrophy. However, it is still not completely understood what triggers the production of reactive oxygen species (ROS) during muscle atrophy. The objective of this study was to investigate redox balance during muscle atrophy. ROS generators and antioxidants were analyzed in atrophied soleus muscles after 2 weeks of hindlimb suspension (HLS) in mice. The HLS group showed an increase in lipid peroxidation, upregulated NOX1 and NOXO1, and downregulated mitochondrial complex I subunits NDUFS5 and NDUFV2. Additionally, HLS mice demonstrated a decrease in Prdx5 and MnSOD, but an increase in GPX2 and GPX3 in both mRNA and protein levels. As expected, MnSOD activity declined in the HLS group, while GPX activity was enhanced. These results suggest that redox imbalance occurs during muscle atrophy through NOX1 activation, mitochondrial complex I deficiency, and disturbance of antioxidants. Antioxidants altered by HLS may represent potential therapeutic targets for the protection against muscle atrophy.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-016-0444-5) contains supplementary material, which is available to authorized users.
Keywords: Oxidative stress, Muscle atrophy, Antioxidant, Hindlimb suspension
Introduction
Skeletal muscle is one of the most abundant tissue types in the vertebrate body, and it plays a crucial role in the movements of humans and other animals. Following long-term inactivity, skeletal muscle can easily lose weight, fiber size, and contractile function—a process which is defined as muscle atrophy. This is especially noticeable in astronauts who have experienced long periods of space flight or in patients who were confined to bed after a severe surgery [1]. Hindlimb suspension (HLS) animal models which mimic the muscle atrophy that occurs during muscle disuse in human have been developed to further our understanding of this process. Tail-HLS has been the main method to induce muscle atrophy in rodent models, but more recently pelvic-HLS has been suggested as an appropriate alternative to the tail-HLS method in rat [2]. However, pelvic-HLS-induced muscle atrophy in the mouse remains unclarified. It would be beneficial to develop a mouse HLS system for studying the mechanism of muscle atrophy using various kinds of transgenic mice.
It has been demonstrated that disuse-induced muscle atrophy occurs as a combined result of a decrease in muscle protein synthesis and an increase in protein degradation [3, 4]. Long-term inactivation of skeletal muscle has also been shown to result in the increased production of reactive oxygen species (ROS), as well as increased redox signaling and oxidative damage [4–6]. Oxidative stress may be a crucial contributor to inactivity-induced muscle atrophy, as it activates key proteases (e.g., calpains and caspase-3) or triggers the ubiquitin-related proteasome system in skeletal muscle [6, 7]. There are various sources of ROS leading to oxidative stress. NADPH oxidase (NOX) and xanthine oxidase (XO) are the main generators of ROS in inactive muscles [8, 9], and a number of recent studies have highlighted the importance of mitochondrial ROS production in prolonged disuse-induced muscle atrophy [5, 10, 11]. However, the specific targets that are responsible for ROS production during HLS remain to be determined.
Antioxidants, including enzymes controlling cellular redox status, such as glutathione (GSH), play a vital role in the maintenance of cellular redox homeostasis. Antioxidants can respond quickly to specific ROS. Therefore, oxidative stress, when the redox status becomes imbalanced, can lead to the upregulation or depletion of various antioxidant systems. Girten et al. showed that after hindlimb unloading, antioxidant enzyme catalase and total superoxide dismutase (SOD) activities decrease in the rat [12]. More specifically, Lawler et al. reported a slight decrease in MnSOD activity, a significant increase in Cu/ZnSOD activity, and a remarkable reduction in catalase and glutathione peroxidase (GPX) activity, two enzymes which remove hydroperoxides in hindlimb-unloaded rat muscle [13]. However, the antioxidant defense mechanisms present in atrophied muscle have still not been entirely elucidated and are consequently not completely understood in the mouse.
The aims of this study were to screen and identify specific antioxidants whose expression is affected during 2 weeks of muscle atrophy as simulated in the mouse pelvic-HLS model and to investigate the oxidative stress and ROS-generating system involved. The results of this study will provide helpful information on potential targets for the treatment of disuse-induced muscle atrophy by identifying the specific antioxidants and ROS generators which respond to HLS-induced muscle atrophy.
Materials and methods
Animals
Animal experiments were conducted in accordance with protocol approved by the Chonnam National University Animal Care and Use Committee, South Korea. The mice were housed in a specific pathogen-free facility following the guidelines in the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, Washington DC). Adult male C57BL/6J mice (10 weeks old) were used for this study.
Pelvis-HLS system
The pelvis-HLS system used for this study is shown in Electronic Supplementary Material (ESM) Fig. 1. This is a modified system that has been used previously for rat hindlimb unloading [2]. Mice were fitted into the HLS system and unloaded for 14 days, at approximately a 30° head-down tilt. The angle of suspension was adjusted to ensure that the hind limbs were unable to touch the ground when the animals completely stretched. Mice that were freely placed in a cage without a restriction of behavior were assigned to the control (CTL) group. Body weight and food and water consumption were measured at baseline and at 3-day intervals for 2 weeks. At 2 weeks of HLS, blood was collected by a ventricular puncture under anesthesia, and soleus muscle and femur tissue were obtained from both the HLS and control mice for further study.
Muscle weight measurements
Left and right leg soleus muscles were dissected from mice and their weight measured and expressed in relative units (mg/g of body weight).
Insulin and glucose analysis
Blood glucose level was measured using a blood glucose meter (Omnitest Plus; B. Braun Melsungen AG, Melsungen, Germany). The blood samples were centrifuged at 3000 g for 15 min to separate the plasma, following which plasma insulin levels were measured using the Rat/Mouse Insulin ELISA kit (EMD Millipore Co., Billerica, MA) according to manufacturer’s instructions.
C-terminal cross-linking telopeptide of type I collagen measurements
The plasma C-terminal cross-linking telopeptide of type I collagen (CTX-1) concentration was determined using the RatLaps™ EIA kit (Immunodiagnostic Systems Ltd., Boldon, UK) according to the manufacturer’s instructions.
Micro-computed tomography analysis
For the micro-computed tomography analysis, the femur was first fixed in 10 % neutral formalin solution (Sigma-Aldrich, St. Louis, MO), and then the distal femurs were analyzed using a high-resolution Skyscan 1172 system (Bruker, Kontich, Belgium) with a X-ray source at 50 kV, at 201 mA with a 0.5-mm aluminum filter. Images were captured every 0.7° with a pixel size of 11 mm, over an angular range of 180°. Raw images and three-dimensional images were reconstructed into serial cross-section images using image reconstruction software (CTAn; Bruker, Mimics 14.0; Leuven, Materialise, Belgium). Bone volume fraction (BV/TV) was then determined.
Cross-sectional area muscle fiber analysis
Soleus muscle was fixed in 10 % neutral formalin solution, dehydrated with ethanol, and embedded in paraffin. Serial transverse sections (5-µm thick) of muscle fibers were prepared and stained with hematoxylin–eosin. The images of the stained sections were captured on a digital microscope camera (Leica DFC450C; Leica Inc., Mannheim, Germany) using a 20× objective lens. Cross-sectional areas (CSA) of soleus muscles were analyzed using ImageJ analytical software (NIH, Bethesda, MD).
Western blot analysis
Soleus muscles were lysed in chilled RIPA buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 % NP-40, 0.5 % sodium deoxycholate, 0.1 % sodium dodecyl sulfate (SDS), 2 mM EDTA, and protease inhibitors], and supernatants were collected by centrifugation (13,000 rpm, 4 °C, 30 min).
Protein samples (30 µg) were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto PVDF membranes (Bio-Rad Laboratories Inc., Hercules, CA). The membranes were immunoblotted with following primary antibodies: peroxiredoxin 1 (Prdx1; lab-made), Prdx2 (Abfrontier, Seoul, Korea), Prdx3 (Abcam, Cambridge, UK), Prdx4 (lab-made), Prdx5 (lab-made), Prdx6 (Abfrontier), SOD1 (Cu/ZnSOD; Abfrontier), SOD2 (MnSOD; Abfrontier), SOD3 (EcSOD; Abcam), GPX1 (Abfrontier), GPX2 (R&D systems, Minneapolis, MN), GPX3 (R&D systems), catalase (Abfrontier), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Abfrontier). This was followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody and detected using an ECL system (iNtRON, Seoul, Korea). The density of detected bands was measured using CS Analyzer software (version 4.0; ATTO Software Inc, Tokyo, Japan) and normalized to GAPDH.
RNA extraction and quantitative real-time PCR
Soleus muscles were homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA) and extracted following the manufacturer’s instructions. RNA quantity and quality were assessed on an Epoch Microplate Spectrophotometer (Biotek®, Winooski, VT).
Total RNA was reverse-transcribed using the PrimeScript™ RT Reagent kit (Takara, Shiga, Japan), and quantitative real-time PCR was performed using an ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster City, CA) with SYBR premix Ex Taq (Takara). Reactions were performed in triplicate, and all data were normalized to GAPDH. Relative mRNA expression was determined using the 2−ΔΔCT method. The primer sets for this study are listed in ESM Table 1.
Malondialdehyde measurement
The concentration of the lipid peroxidation product malondialdehyde (MDA) was determined using the Parameter TBARS Assay kit (R&D Systems) according to the manufacturer’s instructions. The supernatant of the soleus muscle homogenate was prepared with ice-cold Cell Lysis Buffer 3 (R&D System), and the measured MDA levels were normalized to the total protein amount of supernatant.
Protein carbonylation assay
Protein carbonylation is a type of protein oxidation that can be promoted by ROS [14]. The protein carbonyl group was evaluated using the OxyBlot Protein Oxidation Detection kit (EMD Millipore Co.). Briefly, the carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNP) and then were separated in 12 % SDS-PAGE gels. After the gels were transferred to PVDF membrane, the membranes were incubated with primary antibody (rabbit anti-DNP antibody), followed by incubation with HRP-conjugated secondary antibody, and detected using an ECL system (iNtRON).
Determination of SOD activity
Total SOD and MnSOD activities were determined using the Superoxide Dismutase Assay kit (Cayman Chemical Co., Inc., Ann. Arbor, MI) according to the manufacturer’s instructions. Briefly, soleus muscles were homogenized in cold buffer [20 mM HEPES buffer (pH 7.2), 1 mM EGTA, 210 mM mannitol, 70 mM sucrose]. After centrifugation at 1500×g for 5 min at 4 °C, the supernatants were collected and measured for total SOD activity. MnSOD activity was determined in the presence of potassium cyanide.
Determination of GPX activity
Glutathione peroxidase activity was measured using the Glutathione Peroxidase Assay kit (Cayman Chemical Company, Inc.) according to the manufacturer’s instructions. Briefly, soleus muscles were homogenized in cold buffer [50 mM Tris–HCl (pH 7.5), 5 mM EDTA, 1 mM DTT], followed by a centrifugation at 10,000×g for 15 min at 4 °C. GPX activity was then measured in the supernatants.
Measurement of GSH content
Total and oxidized GSH levels were measured using the Glutathione Assay kit (Cayman Chemical Company, Inc.). Briefly, soleus muscles were homogenized in cold buffer [50 mM MES (pH 6–7), 1 mM EDTA], and centrifuged at 10,000×g for 15 min at 4 °C. The supernatants were deproteinized by adding MPA Reagent, centrifuged at 3000 g for 2 min, and treated with TEAM Reagent before being measured for total GSH content. Oxidized GSH (GSSG) was measured in the presence of 2-vinylpyridine and reduced GSH was then determined by subtracting GSSG from total GSH.
Statistical analysis
All statistical analyses were conducted using SPSS ver. 20.0 software (SPSS Inc, Chicago, IL). The Shapiro–Wilk test was utilized for normality analysis of the parameters. The paired t test or Wilcoxon signed-ranks test was used according to a normal distribution. The CSA of muscle fibers was analyzed using the Mann–Whitney test. A p value of <0.05 was considered to be statistically significant. Data are expressed as the mean ± standard error.
Results
Physiological characteristics in HLS mice during 2 weeks of suspension
The HLS mice underwent a significant decline in body weight during the suspension period, with the highest decrease observed in the first week (from approx. 25 to approx. 23 g) (Fig. 1a). In the second week, the body weight of the HLS group was maintained, with no change in the rate of body weight loss, indicating that the HLS mice may have recovered to some extent from the stress induced by the early phase of suspension. However, HLS eventually resulted in a significant growth retardation during the 2-week study period, despite an increase in food consumption (Fig. 1b). There was no difference between the HLS and CTL groups in terms of the total amount of water consumed during the 2 weeks, although at specific time-points, namely, day 3 and day 6, water consumption was higher in the HLS group than in the CTL group (Fig. 1c).
Fig. 1.
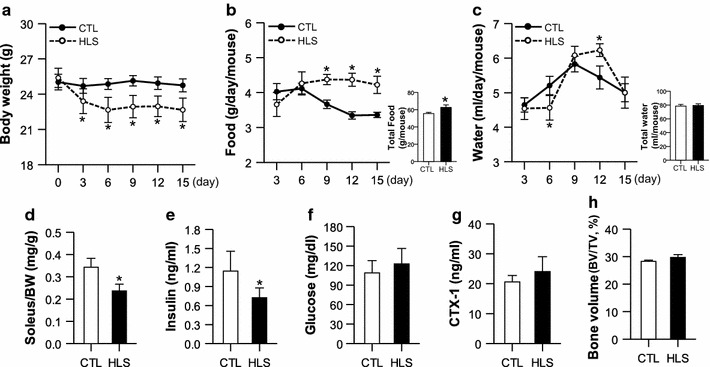
Changes in the physiological characteristics of mice subjected hindlimb suspension (HLS) and control mice (CTL) during the 2-week study period. a Mouse body weight (n = 8). b, c Daily and total food intake (b; n = 8) and water consumption (c; n = 8). d–h Changes in soleus muscle weight (d; n = 8), plasma insulin (e; n = 8), blood glucose (f; n = 8), plasma C-terminal cross-linking telopeptide of type I collagen (CTX-1) (g; n = 8), and bone volume (BV) of distal femur (h; n = 4) after the suspension period. BW Body weight, TV total volume. Values are presented as the mean ± standard error (SE). Asterisk indicates a significant difference between HLS and CTL mice at p < 0.05
The muscle-to-body weight ratio of the soleus muscles decreased significantly in the HLS mice after the suspension period in comparison with the CTL mice (Fig. 1d). Our analysis of the insulin and glucose levels in HLS mice to determine the muscle disuse-associated insulinopenia that has been observed in the HLS rat [2] revealed that the plasma insulin level was lower in the HLS mice after 2 weeks of suspension than in the CTL mice, whereas the glucose level did not differ between the two groups (Fig. 1e, f). The CTX-1 level and BV were measured in HLS and CTL mice to determine the association of disuse-related muscle atrophy and bone loss. There were no significant differences in CTX-1 level and BV between the two groups (Fig. 1g, h). These results indicate that the HLS system induced muscle atrophy and mild hypoinsulinemia in mice but did not affect the glucose level and bone volume.
Muscle fiber atrophy was also determined in HLS mice by staining and measurement of the CSA of the soleus muscle fibers (Fig. 2). We found that the size of the soleus muscle fibers decreased in the HLS mice (1931.52 ± 80.69 µm2) after 2 weeks of treatment in comparison to the CTL mice (2635.61 ± 77.54 µm2), with a reduction of approximately 20 % (Fig. 2b).
Fig. 2.
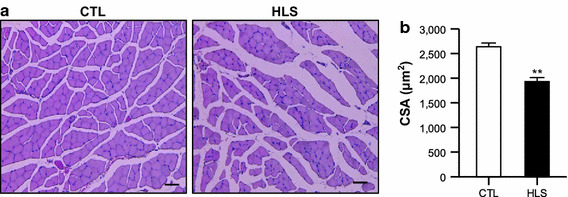
Cross-sectional area (CSA) of soleus muscle of HLS mice at 2 weeks of treatment and CTL mice. a Representative hematoxylin–eosin staining of soleus muscle from a CTL (left) and HLS mouse (right). b Average fiber CSA were measured in the stained soleus muscles (n = 4). Values are presented as the mean ± SE. Double asterisk indicates a significant difference between HLS and CTL mice at p < 0.01. Scale bar 100 µm
ROS-generating system and oxidative stress in HLS-induced muscle atrophy
Protein carbonylation and lipid peroxidation were evaluated to determine whether the soleus muscles of HLS mice suffered from oxidative damage. The levels of carbonylated proteins in the soleus muscles of HLS mice did not differ significantly from those of the CTL mice (Fig. 3a). In contrast, there was a significant increase in lipid peroxidation levels in the HLS mice (Fig. 3b), possibly implying that HLS soleus muscles have been oxidatively damaged.
Fig. 3.
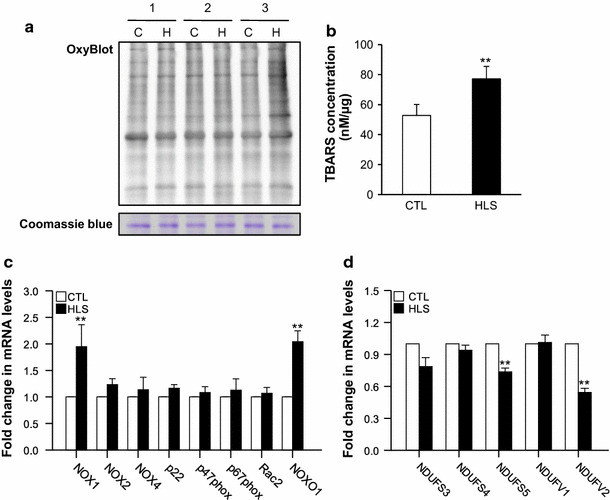
Oxidative stress in soleus muscles of HLS mice after 2 weeks of treatment and CTL mice. a Evaluation of protein oxidation based on the detection of carbonylated protein using the OxyBlot Protein Oxidation Detection kit (upper part), with Coomassie Blue staining (bottom part) used for the loading control (The numbers [1, 2, and 3] indicate biological replicates, n = 3). H HLS mice, C CTL mice. b Lipid peroxidation levels, determined using the TBARS assay (n = 4). c mRNA expression of the NADPH oxidase (NOX) family and subunits [n = 5 for NOX1 and NADPH oxidase organizer 1 (NOXO1); n = 3 for the other targets). d mRNA expression of mitochondrial complex I subunits [n = 5 for NADH dehydrogenase (ubiquinone) iron-sulfur protein subunits 3, 4, 5 (NDUFS3, 4, 5) and NADH dehydrogenase (ubiquinone) flavoprotein 1 and 2 (NDUFV1, 2); n = 3 for the other targets). mRNA levels are expressed as relative fold change compared with the CTL. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a reference gene. Values are presented as the mean ± SE. Double asterisk indicates a significant difference between HLS and CTL mice at p < 0.01
NOX and mitochondrial complex I subunits are major ROS generators within the membrane and mitochondria, respectively, and, therefore, knowledge of their contribution to oxidative stress in HLS mice is essential. We found that NOX1 and NADPH oxidase organizer 1 (NOXO1) mRNA levels in the soleus muscles of HLS mice were significantly upregulated compared with those of the CTL mice, while there were no significant changes in the mRNA expression of other NOX family members and subunits (Fig. 3c). Regarding mitochondrial complex 1 subunits, NDUFS5 and NDUFV2 were downregulated in HLS mice relative to CTL mice (Fig. 3d), which may lead to the loss of mitochondrial complex 1 function or disruption of the assembly with other mitochondrial complex I subunits. These data suggest that activated NOX1 and NOXO1 combined with mitochondrial dysfunction may play a crucial role in the production of ROS, eventually inducing oxidative stress, such as lipid peroxidation in HLS.
Alterations in the antioxidant system in HLS-induced muscle atrophy
The mRNA and protein levels of SODs, Prdxs, GPXs, and catalase were analyzed to assess the antioxidant defense system in HLS mice. The mRNA and proteins levels of SOD2 and Prdx5, both localized in mitochondria, fell in the HLS group as compared to the CTL group (Fig. 4a–c), while those of GPX2 and GPX3 were substantially enhanced (Fig. 4a, d). These results suggest that HLS can cause ROS (i.e., superoxide) accumulation in mitochondria accompanied by SOD2 and Prdx5 deficiency. The excess mitochondrial ROS may result in muscle cell damage by affecting mitochondrial function.
Fig. 4.
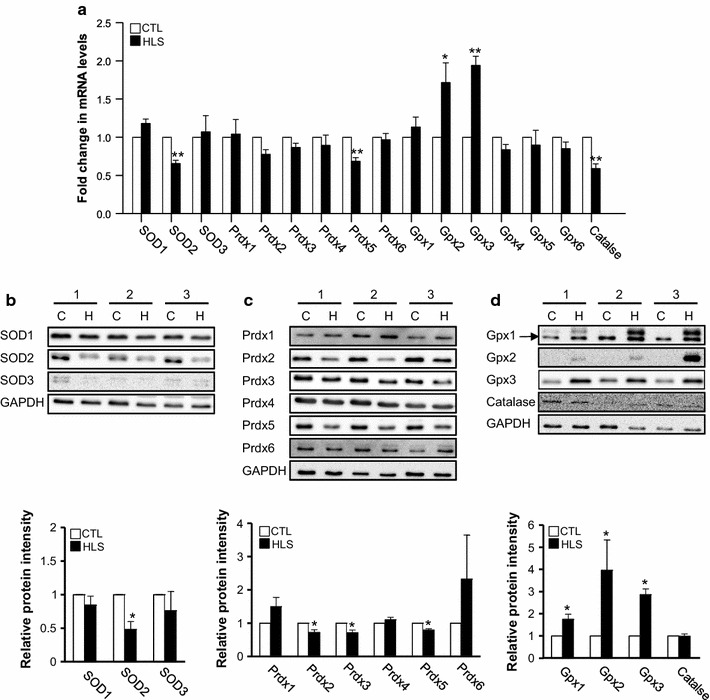
Changes in the antioxidant defense system in HLS mice after 2 weeks of treatment and CTL mice. a mRNA expression level of antioxidants determined using quantitative real-time PCR [n = 5 for the superoxide dismutases (SOD) SOD2 and SOD3, peroxiredoxins (Prdx) Prdx4 and Prdx5, glutathione peroxidases (GPX) GPX2 and GPX3, and catalase; n = 3 for the other targets]. b–d Representative immunoblots (upper, the numbers [1, 2, and 3] indicate biological replicates) and densitometric analysis (bottom) of antioxidant protein levels (n = 3 for Prdx1, Prdx4, Prdx6, and SOD3; n = 4 for SOD1, SOD2, and GPX3; n = 7 for Prdx2, Prdx3, Prdx5, GPX1, GPX2 and catalase. Values are presented as the mean ± SE. Single asterisk and double asterisk indicate a significant difference between HLS and CTL mice at p < 0.05 and p < 0.01, respectively
The ROS scavenging capability of antioxidants was also confirmed by measuring SOD and GPX activity and glutathione (GSH) content. Total SOD activity was not significantly different between the HLS and CTL mice, whereas mitochondrial MnSOD activity was lower in the HLS mice than in the CTL mice (Fig. 5a). Interestingly, GPX activity was higher in the HLS mice despite a decrease in total GSH of the HLS group (Fig. 5b, c). However, there was no difference between the HLS and CTL mice in terms of reduced GSH (rGSH), oxidized GSH content (GSSG), and the ratio of GSH/GSSG (Fig. 5c). Similar to the expression levels of antioxidant proteins observed in the soleus muscle of HLS mice (Fig. 4), these data support a possibility of ROS formation within mitochondria and the importance of mitochondrial antioxidants in HLS-induced muscle atrophy.
Fig. 5.
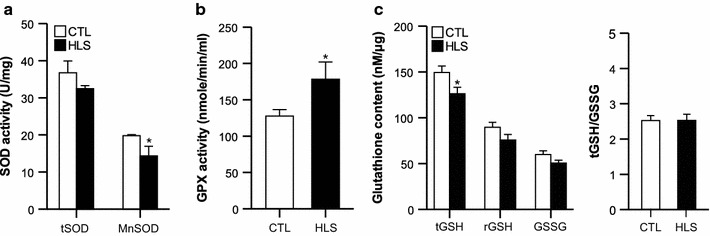
The activities of antioxidant enzymes in HLS mice after 2 weeks of treatment and CTL mice. a SOD activity, b GPX activity, c glutathione content (n = 5 for CTL, n = 6 for HLS group). tSOD total SOD, tGSH total GSH, rGSH reduced GSH. Values are presented as the mean ± standard error (SE). Asterisk indicates a significant difference between HLS and CTL mice at p < 0.05
Discussion
The aim of our study was to use the pelvic-HLS mouse model to determine muscle atrophy-associated disorders, including induction of muscle atrophy, by elucidating the mouse physiological properties (e.g., food and water consumption, body and muscle weight, and bone loss) previously reported in the tail-HLS rat and mouse model [2, 15]. We observed a notable decrease in the body weight of the HLS mice during the first week of suspension, but the body weight of the HLS mice did not change even with an increase in food intake during the second week of suspension (Fig. 1b). One possible explanation is that the HLS mice adapted to some degree to the environmental conditions. We observed a decrease in insulin level in HLS mice. Similarly, the concentration of insulin has been reported to decrease in the circulation of healthy humans during space flights or prolonged bed rest and physical inactivity [16–18], as well as in skeletal muscles of tail-HLS rat models [15]. It has been suggested that insulin resistance in the disused skeletal muscles may accelerate protein degradation through activation of the ubiquitin–proteasome signaling pathway [19, 20].
Major classes of ROS in inactive muscles could be produced through the activation of NOX [8] and mitochondrial enzymes [5, 21]. Our results indicate that mRNA levels of NOX1 and NOXO1 in HLS mice were significantly higher than those in the CTL group (Fig. 3c). Phosphorylation of NOXO1 enhances the full assembly and activation of NOX1, leading to increased ROS production [22, 23]. Although we did not detect of NOX1 and NOXO1 protein levels in HLS mice, we consider that the high levels of ROS generated by the activated NOX1 may cause increased lipid peroxidation these mice (Fig. 3b).
The mitochondrial respiratory chain (MRC) consists of four membrane-bound electron-transporting protein complexes (I–IV) and the ATP synthase (complex V), generating ATP for cellular processes. A deficiency in complex I, NADH ubiquinone oxidoreductase is the most common form of MRC dysfunction and has been linked to various diseases [24, 25]. In our study, the considerable decline in NDUFS5 and NDUFV2 mRNA levels in HLS mice (Fig. 3d) indicates that they could be deficient in MRC complex I assembly. Complex I deficiency leads to various physiological disorders, such as ATP depletion, imbalanced calcium homeostasis, accumulation of ROS [26], and induction of apoptosis [27].
The antioxidant defense system plays a role against cellular damage from exposure to free radicals. We observed two different alterations in the expression of specific antioxidants during HLS. HLS led to a significant decrease in the mRNA and protein levels of Prdx5 and SOD2, both of which are localized in mitochondria (Fig. 4a–c). The accumulation of ROS in mitochondria due to complex I deficiency may cause the depletion of antioxidants, with scavenging of mitochondrial ROS in pelvic-HLS mouse system. The decline in MnSOD enzyme activity is supported by the observed decrease in MnSOD mRNA and protein levels in HLS mice, while no difference in total SOD activity was observed between HLS and CTL mice (Fig. 5a). In contrast to our results, Brocca et al. observed an increase in SOD1 and a decrease in Prdx6 in tail-HLS in mice [28]. The tail-HLS system appears to result in the cytosolic accumulation of hydrogen peroxide and promotes oxidative stress via SOD1 upregulation and downregulation of Prdx6. It is thought that the differences in HLS system may produce different kinds of ROS with different mechanisms in the process of muscle atrophy.
Interestingly, GPX2 and GPX3 mRNA and protein expression increased remarkably with enhanced GPX activity (Fig. 4a, c). GPX2 is known as a gut epithelium-specific peroxidase and GPX3 [an extracellular (plasma) peroxidase] is expressed mainly in the kidney and released into the blood [29]. Therefore, we evaluated the protein expression of GPX2 and GPX3 in the intestine, kidney, and plasma to identify whether the increase in GPX2 and GPX3 observed in atrophied soleus muscle was due to the production of GPX2 and GPX3 from other tissues/organs during HLS. However, GPX2 and GPX3 expression in the kidney, intestine, and plasma did not differ between CTL and HLS mice (data not shown). Further study will be required to determine if GPX2 and GPX3 provide protection against oxidative stress in the soleus muscle during HLS and whether they are potential targets in muscle atrophy.
In conclusion, the results from this study support the hypothesis that oxidative stress may cause HLS-induced muscle atrophy, where NOX1 activation and mitochondrial complex I deficiency can lead to increased ROS generation. This process may also cause significant alterations in the mitochondrial antioxidant defense system, such as in Prdx5 and SOD2 activity. Our results indicate that antioxidants altered by HLS (Prdx5, SOD2, Gpx2, and Gpx3) are potential therapeutic targets for the prevention and treatment of disuse-induced muscle atrophy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by Korea Mouse Phenotyping Project (NRF-2014M3A9D5073721) of the Ministry of Science, ICT and Future Planning through the National Research Foundation and by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (2011-0030121).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflicts of interest.
Footnotes
T. N. Nuoc and S. Kim have contributed equally to this study.
References
- 1.Ohira T, Kawano F, Ohira T, Goto K, Ohira Y. Responses of skeletal muscles to gravitational unloading and/or reloading. J Physiol Sci. 2015;65:293–310. doi: 10.1007/s12576-015-0375-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chowdhury P, Long A, Harris G, Soulsby ME, Dobretsov M. Animal model of simulated microgravity: a comparative study of hindlimb unloading via tail versus pelvic suspension. Physiol Rep. 2013;1:e00012. doi: 10.1002/phy2.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondo H, Miura M, Nakagaki I, Sasaki S, Itokawa Y. Trace element movement and oxidative stress in skeletal muscle atrophied by immobilization. Am J Physiol. 1992;262:E583–E590. doi: 10.1152/ajpendo.1992.262.5.E583. [DOI] [PubMed] [Google Scholar]
- 4.Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- 5.Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whidden MA, Smuder AJ, Wu M, Hudson MB, Nelson WB, Powers SK. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol. 2010;108:1376–1382. doi: 10.1152/japplphysiol.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J Appl Physiol. 2011;111:1190–1198. doi: 10.1152/japplphysiol.00429.2011. [DOI] [PubMed] [Google Scholar]
- 8.McClung JM, Van Gammeren D, Whidden MA, Falk DJ, Kavazis AN, Hudson MB, Gayan-Ramirez G, Decramer M, DeRuisseau KC, Powers SK. Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med. 2009;37:1373–1379. doi: 10.1097/CCM.0b013e31819cef63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, Powers SK. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol. 2009;106:385–394. doi: 10.1152/japplphysiol.91106.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kikusato M, Toyomizu M. Moderate dependence of reactive oxygen species production on membrane potential in avian muscle mitochondria oxidizing glycerol 3-phosphate. J Physiol Sci. 2015;65:555–559. doi: 10.1007/s12576-015-0395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girten B, Oloff C, Plato P, Eveland E, Merola AJ, Kazarian L. Skeletal muscle antioxidant enzyme levels in rats after simulated weightlessness, exercise and dobutamine. Physiologist. 1989;32:S59–S60. [PubMed] [Google Scholar]
- 13.Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic Biol Med. 2003;35:9–16. doi: 10.1016/S0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki YJ, Carini M, Butterfield DA. Protein carbonylation. Antioxid Redox Signal. 2010;12:323–325. doi: 10.1089/ars.2009.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowdhury P, Soulsby ME, Jayroe J, Akel NS, Gaddy D, Dobretsov M. Pressure hyperalgesia in hind limb suspended rats. Aviat Space Environ Med. 2011;82:988–991. doi: 10.3357/ASEM.3063.2011. [DOI] [PubMed] [Google Scholar]
- 16.Cree MG, Paddon-Jones D, Newcomer BR, Ronsen O, Aarsland A, Wolfe RR, Ferrando A. Twenty-eight-day bed rest with hypercortisolemia induces peripheral insulin resistance and increases intramuscular triglycerides. Metabolism. 2010;59:703–710. doi: 10.1016/j.metabol.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamburg NM, McMackin CJ, Huang AL, Shenouda SM, Widlansky ME, Schulz E, Gokce N, Ruderman NB, Keaney JF, Jr, Vita JA. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler Thromb Vasc Biol. 2007;27:2650–2656. doi: 10.1161/ATVBAHA.107.153288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobin BW, Uchakin PN, Leeper-Woodford SK. Insulin secretion and sensitivity in space flight: diabetogenic effects. Nutrition. 2002;18:842–848. doi: 10.1016/S0899-9007(02)00940-1. [DOI] [PubMed] [Google Scholar]
- 19.Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K, Kishi K. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 2001;15:1279–1281. doi: 10.1096/fj.00-0629fje. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology. 2006;147:4160–4168. doi: 10.1210/en.2006-0251. [DOI] [PubMed] [Google Scholar]
- 21.Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debbabi M, Kroviarski Y, Bournier O, Gougerot-Pocidalo MA, El-Benna J, Dang PM. NOXO1 phosphorylation on serine 154 is critical for optimal NADPH oxidase 1 assembly and activation. FASEB J. 2013;27:1733–1748. doi: 10.1096/fj.12-216432. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto A, Takeya R, Matsumoto M, Nakayama KI, Sumimoto H. Phosphorylation of Noxo1 at threonine 341 regulates its interaction with Noxa1 and the superoxide-producing activity of Nox1. FEBS J. 2013;280:5145–5159. doi: 10.1111/febs.12489. [DOI] [PubMed] [Google Scholar]
- 24.Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, van den Heuvel LP. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 25.Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. Am J Hum Genet. 1999;64:1505–1510. doi: 10.1086/302432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH, Smeitink JA. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. doi: 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- 27.Perier C, Tieu K, Guegan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brocca L, Pellegrino MA, Desaphy JF, Pierno S, Camerino DC, Bottinelli R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp Physiol. 2010;95:331–350. doi: 10.1113/expphysiol.2009.050245. [DOI] [PubMed] [Google Scholar]
- 29.Schnabel D, Salas-Vidal E, Narvaez V, Sanchez-Carbente Mdel R, Hernandez-Garcia D, Cuervo R, Covarrubias L. Expression and regulation of antioxidant enzymes in the developing limb support a function of ROS in interdigital cell death. Dev Biol. 2006;291:291–299. doi: 10.1016/j.ydbio.2005.12.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.