

Abstract
Extended periods of skeletal muscle disuse results in muscle atrophy and weakness. Currently, no therapeutic treatment is available for the prevention of this problem. Nonetheless, growing evidence suggests that prevention of disuse-induced oxidative stress in inactive muscle fibers can delay inactivity-induced muscle wasting. Therefore, this study tested the hypothesis that dietary supplementation with the antioxidant astaxanthin would protect against disuse muscle atrophy, in part, by prevention of myonuclear apoptosis. Wistar rats (8 weeks old) were divided into control (CT, n = 9), hindlimb unloading (HU, n = 9), and hindlimb unloading with astaxanthin (HU + AX, n = 9) groups. Following 2 weeks of dietary supplementation, rats in the HU and HU + AX groups were exposed to unloading for 7 days. Seven-day unloading resulted in reduced soleus muscle weight and myofiber cross-sectional area (CSA) by ~30 and ~47 %, respectively. Nonetheless, relative muscle weights and CSA of the soleus muscle in the HU + AX group were significantly greater than those of the HU group. Moreover, astaxanthin prevented disuse-induced increase in the number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei. We conclude that astaxanthin supplementation prior to and during hindlimb unloading attenuates soleus muscle atrophy, in part, by suppressing myonuclear apoptosis.
Keywords: Antioxidant, Apoptosis, Disuse muscle atrophy, Protein degradation
Introduction
It is well established that prolonged skeletal muscle inactivity results in muscle atrophy and a decrease in muscle force production (i.e., weakness). Disuse-induced muscle atrophy is the consequence of a loss in muscle protein resulting from increased protein degradation and decreased protein synthesis [1, 2]. Protein synthesis and breakdown are regulated through multiple signaling pathways [3–5], but the molecular mechanisms are not completely understood. Previous studies have reported that increased production of reactive oxygen species (ROS) in muscle fibers could be important as signaling molecules contributing to disuse muscle atrophy, by promoting protease activation and depressing protein synthesis [4, 6]. Therefore, it has been hypothesized that antioxidant supplementation is a potential therapeutic countermeasure to protect against inactivity-induced skeletal muscle atrophy [7].
During the past two decades, the effects of antioxidants in disuse muscular atrophy have been investigated [7]. For example, evidence reveals that disuse muscle atrophy can be partially prevented by treatment with vitamin E supplementation. Vitamin E is a potent antioxidative nutrient, which has been reported to decrease the rate of muscle atrophy induced by immobilization and hindlimb unloading [8, 9]. Azzi et al. demonstrated that vitamin E mediates cell signaling and regulates the expression of several genes [10]. However, Koesterer et al. [11] and Ikemoto et al. [12] reported that vitamin E supplementation failed to attenuate skeletal muscle atrophy in similar contexts. Based on these findings, although supplementation of antioxidative nutrients may be beneficial in unloading-induced muscle atrophy, effectiveness in disuse muscle atrophy and underlying mechanisms remain unclear. Therefore, it may be necessary to apply a more potent antioxidant formulation to prevent disuse muscle atrophy.
Recently, astaxanthin (AX) has attracted attention as an effective treatment against disuse-induced muscle atrophy. AX, a red carotenoid pigment, is a biological antioxidant that is found in some marine flora and fauna, such as fish, shrimp, and algae [13–15]. AX has potent antioxidant and anti-inflammatory properties, with an antioxidant capacity that is 100 times greater than that of alpha-tocopherol (i.e., vitamin E) [16, 17]. A previous study reported that high dose (100 mg/kg per day) AX administered orally during hindlimb unloading reduced capillary regression [18]. However, the preventive effects of AX in disuse muscle atrophy remain unclear. Recent data demonstrated that a combination of daily AX administration (100 mg/kg per day) and intermittent muscle reloading attenuated unloading-induced muscle atrophy; however, AX had no effect on the decrease in muscle mass and fiber cross-sectional area [19]. The lack of efficacy in preventing muscle atrophy noted in this previous study may have been a product of the timing of AX administration; it is possible that oxidative stress from disuse could have been prevented with a different administration schedule. In this regard, Appell et al. argued that oxidative stress plays a role in initiating muscle atrophy, and antioxidant supplementation prior to and during the early phase of immobilization should therefore be recommended [8]. Importantly, numerous studies have indicated that apoptosis-related proteins and gene expression increase during the early stages of disuse-induced skeletal muscle atrophy [20–22]. Indeed, Leeuwenburgh et al. [22] showed that a significant increase in apoptosis, as evidenced by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei, could be observed within 2 days of muscle unloading. Maintaining the number of myonuclei in the myofiber is important, because the myonuclei play an important role in skeletal muscle size adaptation [23]. This finding suggests that preventing myonuclei loss may be important in effectively attenuating disuse-induced muscle atrophy.
In reference to antioxidant protection against ROS-induced apoptosis, Ikeda et al. [24] demonstrated that AX treatment abolished 6-hydroxydopamine-induced ROS generation and apoptosis in SH-SY5Y cells. They suggest that the protective effect observed in association with AX may be the product of modulation of apoptosis-related factors, which is necessary to suppress the early stages of apoptosis and consequently attenuate disuse muscle atrophy. Therefore, the purpose of this study was to examine the effect of AX supplementation, prior to and during the phase of unloading, on unloading-induced soleus muscle atrophy. We hypothesized that dietary AX supplementation would attenuate the apoptosis induced by skeletal muscle disuse, and thereby suppress atrophy of the soleus muscle.
Materials and methods
Experimental animals
Twenty-seven male Wistar rats (age, 8 weeks; body weight, 317.7 ± 2.6 g) were used. The rats were housed in a climate-controlled room (24.0 ± 1 °C, 50–60 % relative humidity, 12:12 h light–dark photoperiod) and were given standard rat chow and water ad libitum. Rats were divided randomly into three groups: control (CT; 316.6 ± 1.9 g, n = 9), hindlimb unloading (HU; 319.7 ± 4.8 g, n = 9), and hindlimb unloading with astaxanthin administration (HU + AX; 316.3 ± 6.1 g, n = 9). This study was approved by the Yamaguchi University Animal Care Committee and was performed in accordance with the guiding principles for the care and use of animals, as set forth by the Physiological Society of Japan.
Astaxanthin supplementation
The base diet was a closed-formula commercial powder for rodents (CE-2, CLEA Japan, Inc., Japan). The diet for the AX administration group was prepared by mixing CE-2 powder with AX (BioAstin, Toyo Koso Kagaku, Co. Ltd, Japan) at 0.04 % (wt/wt). Rats in the AX group were fed the AX diet every day for 3 weeks, beginning 2 weeks prior to unloading. Rats in the CT and HU groups received a control diet without AX. The composition of both the AX and control diets is shown in Table 1. Daily measurements of food intake and body weight were performed throughout the study duration.
Table 1.
Composition (%) of AX and control diets
| AX | Control | |
|---|---|---|
| CLEA CE-2 powder | 98.38 | 98.42 |
| BioAstin SCE7 | 0.04 | 0 |
| Oil | 0.11 | 0.11 |
| Ester F160 | 0.15 | 0.15 |
| Quillayanin C-100 | 0.04 | 0.04 |
| Amycol C | 0.23 | 0.23 |
| Trehalose | 0.97 | 0.97 |
| Zein DP | 0.08 | 0.08 |
The diet for the astaxanthin administration group (AX) was prepared by mixing CE-2 powder with astaxanthin at 0.04 % (wt/wt). Rats in the CT and HU groups were fed a control diet that did not contain astaxanthin (control)
Hindlimb unloading
The HU and HU + AX groups were unloaded for 7 days, as described previously [25]. Briefly, a tail cast was applied to each rat, leaving the distal one-third of the tail free to allow for proper thermoregulation. The tail cast was attached to a hook on the ceiling of the cage, and the height of the hook was adjusted at an inclination of approximately 35° in a head-down orientation. The rat was free to move around the cage on its front feet. Rats were checked daily for signs of tail lesions or discoloration. After the experimental period, all rats were anesthetized with ether, the soleus muscle was removed from both legs, frozen rapidly in liquid nitrogen, and stored at −80 °C until analysis.
Histology and immunohistochemistry
Cross-sectional area (CSA) determination
Cross sections of the soleus muscle (n = 5, per group) were cut on a cryostat (8 μm), before being rehydrated in phosphate buffered saline with 0.1 % Tween-20 (T-PBS), and stained with a standard hematoxylin and eosin protocol to measure the total myofiber CSA, as previously described [26]. Muscle sections were visualized and captured as digital images using a fluorescence microscope (Y-FL 077907, Nikon, Tokyo, Japan), equipped with a CCD camera (C120-PC, Leica, Wetzlar, Germany). CSA was determined on 400–500 fibers from five different sections of the mid-belly region of the soleus muscle; subsequently, the mean myofiber CSA was calculated using the Scion Image software package (NIH, Bethesda, MD, USA).
Determination of myonuclear apoptosis
Nuclei exhibiting apoptotic changes were identified with the TUNEL assay, using the in situ cell death detection kit, TMRred (Roche Applied Science, Indianapolis, IN, USA), as described previously [26]. Briefly, sections were rehydrated in PBS with 0.1 % Tween 20, fixed in 4 % paraformaldehyde at room temperature for 20 min, and then fixed in 0.1 % (w/v) sodium citrate (0.1 % C5H3O7Na3∙2H2O, 0.1 % Triton X-100) at room temperature for 2 min. After several washes in T-PBS, sections were immunoreacted with anti-laminin antibody (1:500, Dako Japan, Tokyo, Japan) diluted in a solution of 1 % (w/v) blocking reagent (Roche) in 1 % (w/v) maleic acid buffer (0.1 M maleic acid, 0.15 M NaCl, pH 7.5) at room temperature for 1 h. After washing, a fluorescein isothiocyanate (FITC) conjugated goat anti-rabbit IgG secondary antibody (1:250, Sigma, St. Louis, MO, USA) in blocking buffer (Roche) was applied for 1 h at room temperature. After several washes in T-PBS, a TUNEL reaction mix diluted in TUNEL dilution buffer (Roche) was applied and incubated at 37 °C for 60 min in the dark. After several washes in T-PBS, sections were coverslipped using the Vectashield mounting medium with 4′, 6-diamidino-2-phenylindole (DAPI; Vector Labs, Burlingame, CA). TUNEL-positive nuclei were located on the laminin basement membrane, overlapping the nucleus, and were counted as apoptotic muscle nuclei. The number of TUNEL positive nuclei was expressed per muscle section and CSA.
Protein isolation and western blot analysis
Muscle preparation
Frozen soleus muscle samples (~90 mg) were minced and homogenized in 10 volumes of ice-cold homogenization buffer (20 mM Tris–HCl, pH 7.4, 25 mM KCl, 5 mM EDTA, 5 mM EGTA, and 1 mM dithiothreitol) containing a protease inhibitor cocktail (Nacalai Tesque, Inc., Tokyo, Japan). Homogenates were centrifuged at 3000 rpm for 10 min at 4 °C, and the supernatant was collected as the soluble fraction. The protein concentration in the supernatant was determined using a protein assay kit (Bio-Rad, Hercules, CA, USA). The insoluble pellets, corresponding to the particulate fraction, were suspended in lysis buffer using the method of Solaro et al. [14]. Subsequently, the protein concentration of the solubilized particulate fraction was determined using the Biuret method.
Immunoblotting
Proteins were loaded onto 10 % SDS-PAGE gels and transferred onto a polyvinylidene difluoride membrane (Amersham, Buckinghamshire, UK). After transferring the proteins, membranes were blocked with 5 % nonfat dry milk and incubated with primary antibodies overnight at 4 °C. The primary antibodies used for the assay included: superoxide dismutase (SOD) 1 (1:10,000; Stressgen, Victoria, BC, CAN), caspase-3 (1:500; Stressgen), Bcl-2 (1:200; Santa Cruz, Dallas, TX, USA), multi-ubiquitin (1:500; Stressgen), and actin (1:5000; Sigma). Subsequently, membranes were incubated with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies (1:20,000; Sigma) for 2 h at room temperature. Each band was revealed using ECL Plus reagents (GE Healthcare, Piscataway, NJ, USA), and the signal was recorded by an ATTO Light Capture system (ATTO, Tokyo, Japan). Band intensities were quantified with the CS ANALYZER 3.0 software (ATTO). Protein expression is given as a percentage of that observed in the CT group.
To analyze heat shock protein (HSP) 72 expression levels, membranes were incubated with anti-Hsp70 alkaline phosphatase conjugate (1:2000; Stressgen) in T-TBS, with 5 % nonfat dry milk, for 1 h at room temperature. Subsequently, the membranes were reacted with alkaline phosphatase substrate (1:6000; Bio-Rad) at room temperature. Band quantification was performed using computerized densitometry (Scion Image).
Statistical analysis
Values are presented as mean ± standard error (SE). Group differences were detected using one-way analysis of variance (ANOVA). When ANOVA returned a significant result, group differences were identified using Sidak’s post hoc test. Food consumption data were analyzed by two-way (time, group) ANOVA. P values < 0.05 were considered significant. All analyses were made using Prism ver. 6.0 (GraphPad Software Inc., CA, USA).
Results
Body weight and food consumption
Mean body weights for the HU and HU + AX groups were significantly lower than that of the CT group, but no differences were observed between the two hindlimb unloading groups (Fig. 1a). Although no significant changes were observed between the two unloaded groups (days 2–14: CT; 23.1 ± 0.3, HU; 22.7 ± 0.7, HU + AX; 24.1 ± 0.8 g/day, during hindlimb unloading days 2–6: CT; 21.7 ± 0.8, HU; 19.8 ± 0.8, HU + AX; 19.7 ± 1.0 g/day), the observed food intake in the HU and HU + AX groups was significantly lower than that of the CT group, at approximately 90 % of the total intake in the CT group (Fig. 1b). The decline in food consumption in the HU and HU + AX groups (10.3 and 10.8 %, respectively) was relatively small compared to a previous study, which indicated that a 20 % food restriction (for 10–14 weeks) did not result in changes to the weight of the soleus muscle, when compared with non-food restricted sedentary control rats. Therefore, it is thought that soleus muscle atrophy in the present study was not a product of starvation-induced atrophy, but rather unloading-induced skeletal muscle atrophy.
Fig. 1.
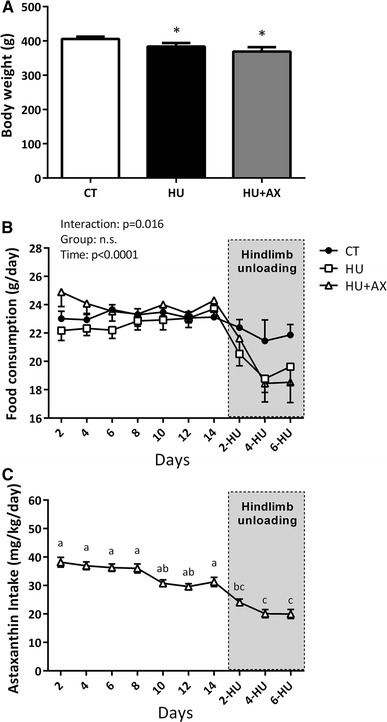
Body weight (a), food consumption (b) and astaxanthin intake (c) during experimental periods. CT control, HU hindlimb unloading, HU + AX hindlimb unloading with astaxanthin administration. Values are mean ± standard error (SE); n = 9 per group. *p < 0.05 versus CT. The results of two-way ANOVA are displayed in Fig. 1b. Notations a–c in (c) indicate significant differences at p < 0.05
The mean dietary AX intake for 3 weeks in the HU + AX group was 30.3 ± 0.1 mg/kg/day (prior to hindlimb unloading days 2–14; 34.1 ± 1.2 mg/kg/day, during hindlimb unloading days 2–6; 21.4 ± 0.9 mg/kg/day) (Fig. 1c). The mean dietary AX intake was significantly decreased during unloading (days 2-, 4-, and 6-HU), when compared with days 2, 4, 6, 8, and 14. Moreover, mean dietary AX intake on hindlimb unloading days 4 and 6 (days 4- and 6-HU) was significantly lower than days 10 and 12.
Soleus muscle weight and myofiber cross-sectional area (CSA)
Although 7 days of hindlimb unloading resulted in a significant reduction in soleus muscle weights in both hindlimb unloaded groups (HU; −30.7 %, HU + AX; −23.6; +10.2 % versus HU, Fig. 2a), the relative muscle weight in the HU + AX group was significantly greater than that in the HU group (HU; −26.7 %, HU + AX; −14.4; +16.7 % versus HU, Fig. 2b). Moreover, hindlimb unloading resulted in a significant reduction in soleus myofiber CSA (HU; −47.3 %, HU + AX; −32.0; +29.1 % versus HU); however, the decrease observed in the HU + AX group was somewhat attenuated by AX supplementation (Fig. 2c).
Fig. 2.
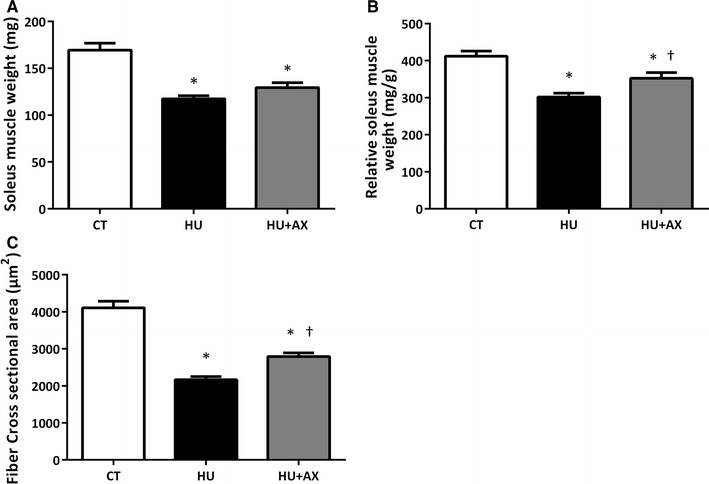
Soleus muscle weight (a), relative soleus muscle weight (b), and fiber CSA (c) after 7 days of hindlimb unloading. CSA cross-sectional area. Values are mean ± standard error (SE); n = 9 (n = 5 for fiber CSA) per group. *p < 0.05 versus CT; †p < 0.05 versus HU
TUNEL-positive nuclei
To identify apoptotic nuclei, a TUNEL assay was implemented to identify TUNEL-positive nuclei located on the laminin basement membrane that were overlapping the nucleus; these nuclei were taken to indicate apoptotic muscle nuclei (Fig. 3a). Compared to the CT group, the number of TUNEL-positive nuclei per section of soleus muscle fibers increased significantly in the HU group after 7 days of hindlimb unloading. However, this increase in TUNEL-positive nuclei was significantly suppressed in the HU + AX group (Fig. 3b). Similarly, the number of TUNEL-positive nuclei per CSA in the HU group was significantly higher than the CT group, but this increase was attenuated by AX supplementation in the HU + AX group (Fig. 3c).
Fig. 3.
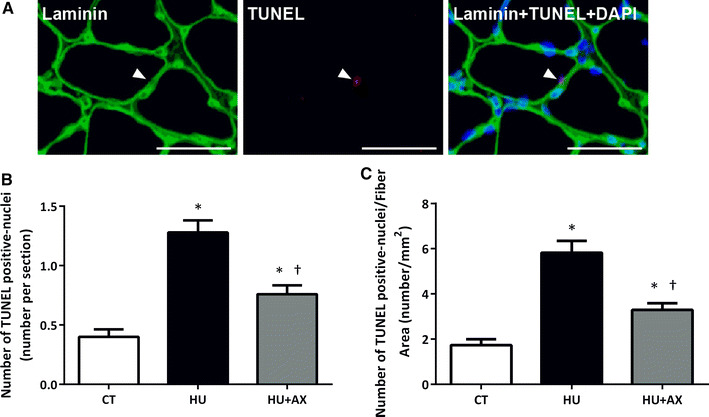
Localization of TUNEL positive nuclei. Soleus muscle cross sections were immunoreacted with laminin (green), TUNEL-positive nuclei (red), and DAPI (blue) (a). TUNEL positive interstitial cells were observed in the soleus muscles (arrow). The scale bar in the picture is 50 μm. The number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei (per section and fiber CSA) in the soleus muscle after 7 days of hindlimb unloading (b, c). Values are mean ± SE; n = 5 per group. *p < 0.05 versus CT; †p < 0.05 versus HU
HSP72 and SOD1 expression
Expression levels of HSP72 in the soleus muscle were significantly increased in the HU + AX group, when compared with the CT group (Fig. 4a). SOD1 expression exhibited a significant increase in only the HU group (Fig. 4b).
Fig. 4.
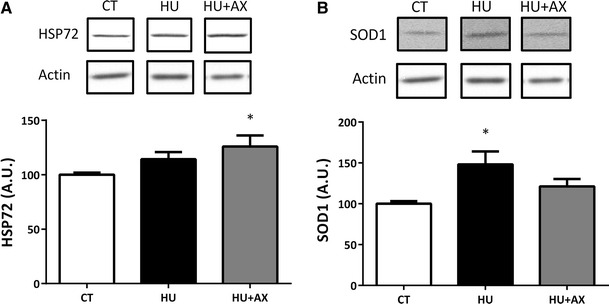
Representative western blots, and HSP72 (a) and SOD1 (b) protein expression in the soleus muscle after 7 days of hindlimb unloading. Experimental group data are presented as a percentage of the mean CT value, which was set to 100 %. Values are mean ± SE; n = 9 per group. *p < 0.05 versus CT. Actin served as a loading control
Caspase-3 and Bcl-2 expression
Caspase-3 expression was significantly increased in the HU group, when compared with the CT group; however, no such increase was observed in the HU + AX group (Fig. 5a). There were no significant differences observed in Bcl-2 protein expression in soleus muscles from any group (Fig. 5b).
Fig. 5.
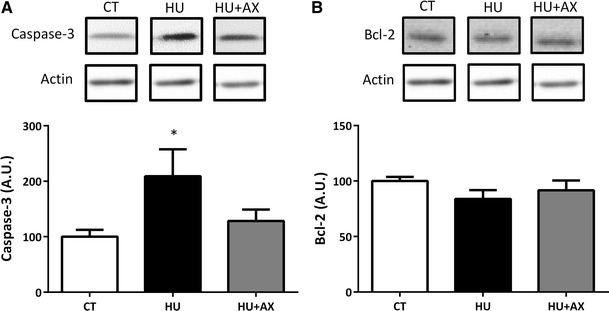
Representative western blots, and caspase-3 (a) and Bcl-2 (b) protein expression in the soleus muscle after 7 days of hindlimb unloading. Experimental group data are presented as a percentage of the mean CT value, which was set to 100 %. Values are mean ± SE; n = 9 per group. *p < 0.05 versus CT. Actin served as a loading control
Ubiquitinated protein expression
No significant differences were observed between the groups with respect to ubiquitinated protein expression in the soluble fraction. In contrast, ubiquitinated protein expression in the particulate fraction from the HU group was significantly increased, compared with the CT group, after 7 days of hindlimb unloading (Fig. 6a, b). However, this increase was significantly attenuated in the HU + AX group.
Fig. 6.
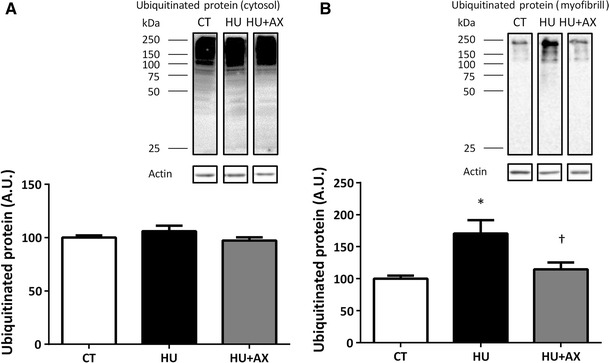
Representative western blots, and ubiquitinated protein expression in the soleus muscle soluble (a) and particulate (b) fractions after 7 days of hindlimb unloading. Experimental group data are presented as a percentage of the mean CT value, which was set to 100 %. Values are mean ± SE; n = 9 per group. *p < 0.05 versus CT; †p < 0.05 versus HU
Discussion
Overview of principal findings
This is the first study to demonstrate the protective effects of dietary AX supplementation in the context of apoptosis in an atrophied rat soleus muscle after 7 days of unloading. Our data reveal that: (1) dietary AX supplementation prior to and during hindlimb unloading attenuates soleus muscle atrophy, and (2) unloading-induced apoptotic myonuclei and protein ubiquitination are suppressed by AX. A brief discussion of these issues follows.
Preventive effect of astaxanthin on unloading-induced muscle atrophy via prevention of apoptosis
The main finding of this investigation is that dietary AX supplementation 2 weeks prior to and during hindlimb unloading attenuates soleus muscle atrophy, in part, via suppression of apoptotic activation and the loss of myonuclei. It is well established that skeletal muscle atrophy is associated with increased apoptosis [5, 27, 28]. Consistent with previous studies using the hindlimb unloading model [20, 22], the current results reveal that the number of TUNEL-positive nuclei in the soleus muscle increases significantly with disuse. Our findings suggest that the number of TUNEL-positive nuclei can be suppressed with dietary AX supplementation (Fig. 3); most importantly, AX supplementation attenuates inactivity-induced decreases in relative muscle weight and fiber CSA (Fig. 2). Unfortunately, the underlying mechanisms responsible for AX-induced protection against disuse muscle atrophy are currently unknown. Nonetheless, it appears likely that many of these effects can be attributed to the action of AX as a scavenger for free radicals and oxidants. Previous studies have demonstrated that AX treatment abolishes 6-hydroxydopamine-induced ROS generation and p38 MAPK activation, including the release of cytochrome c, and the cleavage of caspase-9 and caspase-3 in SH-SY5Y cells [24]. Moreover, AX supplementation has been associated with attenuation of strenuous exercise-induced oxidative damage in the gastrocnemius and cardiac muscle [29], and inhibition of hexanoyl-lysine modification of carnitine palmitoyltransferase I (CPT I) in skeletal muscle [30]. As shown in Fig. 4b, we found that AX protects the soleus muscle against unloading-induced oxidative stress. In general, SOD1 (Cu, Zn-SOD) is induced by superoxide anions; therefore, SOD1 levels have been thought to reflect the generation of superoxide anions in the cytoplasm during disuse-induced muscle atrophy [31, 32]. Moreover, a disuse-induced increase in caspase-3 was not observed in the AX supplementation group (Fig. 5a). Therefore, AX may reduce the disuse-induced production of superoxide anions, production of hydrogen peroxide (as catalyzed by SOD1), and subsequent activation of caspases in the atrophied soleus muscle. It follows that ROS-mediated apoptosis may be prevented in the rat soleus muscle during hindlimb unloading due to the ROS scavenging effects of AX supplementation. Furthermore, no changes in Bcl-2 levels in either unloaded groups were observed (Fig. 5b). The Bcl-2 family includes anti-apoptotic members that prevent apoptosis by either sequestering proforms of death-driving caspases (a complex called the apoptosome), or by preventing the release of mitochondrial apoptogenic factors such as cytochrome c into the cytoplasm [21]. However, previous research has demonstrated that Bcl-2 protein expression is decreased in the rat soleus muscle following 6–12 h of hindlimb unloading, and returns to basal levels 48 h after unloading [21]. Moreover, Dupont-Versteegden et al. [20] have shown that significant changes in apoptosis-related signaling occur within 2 days of unloading, suggesting that the marked changes in apoptosis markers, such as Bcl-2, may have been observed during the early phase of disuse muscle atrophy in our study.
It was also noted that an increase in ubiquitinated protein content in the atrophied soleus muscle was prevented by AX supplementation (Fig. 6b). Ubiquitination of myofibrillar proteins reflects enhancement of muscle protein degradation (protein catabolism) via the ubiquitin–proteasome pathway; with elevated ubiquitination, consequent decreases in muscle weight and CSA are observed [33]. Ikemoto et al. [34] have demonstrated that endogenous cysteine antioxidant supplementation (140 mg/rat) prevents skeletal muscle atrophy induced by hindlimb unloading, via suppression of oxidative stress and subsequent protein ubiquitination. Given that skeletal muscle myofibrillar protein degradation during disuse is primarily due to the activity of the ubiquitin–proteasome system [35, 36], this may indicate that AX supplementation is effective in reducing myofibrillar protein ubiquitination induced by oxidative stress and, therefore, protects against increased muscle protein degradation via the ubiquitin–proteasome system [35, 36]. Nonetheless, because we did not assess muscle ROS production and oxidative stress makers directly in the present study, further study will be required to reveal the specific effects of dietary AX supplementation on oxidative stress during disuse muscle atrophy.
It has been demonstrated that HSPs also play a role in suppression of cellular apoptosis. Li et al. [37] reported that HSP70/HSP72 inhibited apoptosis downstream of cytochrome c release and upstream of caspase-3 activation in vitro (U937/HSP70 cells). HSP72, the most inducible cytoprotective HSP, has molecular chaperone and protein repair functions to protect against muscle atrophy [38, 39]. Servais et al. reported that antioxidant vitamin E supplementation induces an upregulation of the inducible form of HSP70 (HSP72) expression in unloaded rats [9]. Furthermore, Lee et al. reported that AX treatment increased HSP72 expression and was associated with protective effects against cerebral ischemia in SH-SY5Y cells [40]. As expected, we found that AX supplementation prior to (2 weeks) and during unloading increased HSP72 expression in the atrophied soleus muscle (Fig. 4a). Thus, these results suggest that AX supplementation is useful in attenuating the loss of muscle mass and size induced by disuse in vivo. Although the cellular mechanism responsible for AX-induced suppression of muscle atrophy remains unclear, HSPs may play an important role in facilitating protein translation as molecular chaperones to counter disuse muscle atrophy. Recent evidence has reported that high dose AX supplementation (50 mg/kg, with a 6-h interval between two doses; total of 100 mg/kg/day) attenuates capillary regression after 1 or 2 weeks of unloading, but AX supplementation alone did not suppress soleus muscle atrophy [18, 19]. Interestingly, we demonstrate herein that relatively low dose dietary AX supplementation (30.3 ± 0.1 mg/kg/day, prior to hindlimb unloading; 34.1 ± 1.2 mg/kg/day, during hindlimb unloading; 21.4 ± 0.9 mg/kg/day) can attenuate unloading-induced soleus muscle atrophy. When comparing AX intake in the present study with that of a previous study, the total intake observed herein was 606.5 ± 20.3 mg/kg (prior to hindlimb unloading; 478.1 ± 16.9 mg/kg, during hindlimb unloading; 128.4 ± 5.5 mg/kg) for 3 weeks (prior to and during hindlimb unloading). In contrast, the aforementioned previous study reported that 700 and 1400 mg/kg AX administration alone failed to suppress soleus muscle atrophy. This may be due to the fact that apoptosis is a very early event in disuse muscle atrophy [21, 22]. Previous studies have indicated that apoptosis and proteolysis related proteins and genes increase during the early stages of disuse-induced skeletal muscle atrophy, with an increase in apoptosis occurring within 2 days of unloading [20, 21]. Notably, whilst food consumption (AX intake) was decreased by hindlimb unloading, 2 weeks of AX supplementation prior to unloading may contribute to the suppression of muscle atrophy-related protein expression. These facts indicate that suppressing increases in the early phase of apoptotic and proteolytic activation may be important in effectively attenuating disuse-induced muscle atrophy. Consequently, it is necessary to enhance the pre-antioxidant capacity for prevention of muscle atrophy, even with low dose treatment, rather than ingesting a high dose of AX during hindlimb unloading.
Conclusions
In conclusion, our data demonstrate that dietary AX supplementation prior to and during hindlimb unloading attenuates soleus muscle atrophy. Similarly, increases in unloading-induced apoptotic myonuclei and protein ubiquitination appear to be suppressed by AX supplementation. Future research is required to improve our understanding of the mechanism(s) responsible for the protective effect observed with AX administration for disuse-induced muscle atrophy.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. 2050057 (T. Sugiura). Grants from the MEXT-Supported Program for the Strategic Research Foundation at Private Universities also supported this research. The authors thank Tatsuya Furukawa for experimental assistance.
Compliance with ethical standards
Conflict of interest
No financial or other conflicts of interest are declared by the authors.
References
- 1.Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol. 1990;68(1):1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Booth FW, Criswell DS. Molecular events underlying skeletal muscle atrophy and the development of effective countermeasures. Int J Sports Med. 1997;18(Suppl 4):S265–S269. doi: 10.1055/s-2007-972723. [DOI] [PubMed] [Google Scholar]
- 3.Taillandier D, Aurousseau E, Meynial-Denis D, Bechet D, Ferrara M, Cottin P, Ducastaing A, Bigard X, Guezennec CY, Schmid HP, et al. Coordinate activation of lysosomal, Ca 2+-activated and ATP-ubiquitin-dependent proteinases in the unweighted rat soleus muscle. Biochem J. 1996;316(Pt 1):65–72. doi: 10.1042/bj3160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R337–R344. doi: 10.1152/ajpregu.00469.2004. [DOI] [PubMed] [Google Scholar]
- 5.Powers SK, Kavazis AN, McClung JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. 2007;102(6):2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- 6.Powers SK, Smuder AJ, Judge AR. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr Opin Clin Nutr Metab Care. 2012;15(3):240–245. doi: 10.1097/MCO.0b013e328352b4c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers SK. Can antioxidants protect against disuse muscle atrophy? Sports Med. 2014;44(Suppl 2):S155–S165. doi: 10.1007/s40279-014-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appell HJ, Duarte JA, Soares JM. Supplementation of vitamin E may attenuate skeletal muscle immobilization atrophy. Int J Sports Med. 1997;18(3):157–160. doi: 10.1055/s-2007-972612. [DOI] [PubMed] [Google Scholar]
- 9.Servais S, Letexier D, Favier R, Duchamp C, Desplanches D. Prevention of unloading-induced atrophy by vitamin E supplementation: links between oxidative stress and soleus muscle proteolysis? Free Radic Biol Med. 2007;42(5):627–635. doi: 10.1016/j.freeradbiomed.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azzi A, Gysin R, Kempna P, Munteanu A, Negis Y, Villacorta L, Visarius T, Zingg JM. Vitamin E mediates cell signaling and regulation of gene expression. Ann N Y Acad Sci. 2004;1031:86–95. doi: 10.1196/annals.1331.009. [DOI] [PubMed] [Google Scholar]
- 11.Koesterer TJ, Dodd SL, Powers S. Increased antioxidant capacity does not attenuate muscle atrophy caused by unweighting. J Appl Physiol. 2002;93(6):1959–1965. doi: 10.1152/japplphysiol.00511.2002. [DOI] [PubMed] [Google Scholar]
- 12.Ikemoto M, Okamura Y, Kano M, Hirasaka K, Tanaka R, Yamamoto T, Sasa T, Ogawa T, Sairyo K, Kishi K, Nikawa T. A relative high dose of vitamin E does not attenuate unweighting-induced oxidative stress and ubiquitination in rat skeletal muscle. J Physiol Anthropol Appl Hum Sci. 2002;21(5):257–263. doi: 10.2114/jpa.21.257. [DOI] [PubMed] [Google Scholar]
- 13.Petri D, Lundebye AK. Tissue distribution of astaxanthin in rats following exposure to graded levels in the feed. Comp Biochem Physiol C: Toxicol Pharmacol. 2007;145(2):202–209. doi: 10.1016/j.cbpc.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Page GI, Davies SJ. Tissue astaxanthin and canthaxanthin distribution in rainbow trout (Oncorhynchus mykiss) and Atlantic salmon (Salmo salar) Comp Biochem Physiol A: Mol Integr Physiol. 2006;143(1):125–132. doi: 10.1016/j.cbpa.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Matthews SJ, Ross NW, Lall SP, Gill TA. Astaxanthin binding protein in Atlantic salmon. Comp Biochem Physiol B: Biochem Mol Biol. 2006;144(2):206–214. doi: 10.1016/j.cbpb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Kurashige M, Okimasu E, Inoue M, Utsumi K. Inhibition of oxidative injury of biological membranes by astaxanthin. Physiol Chem Phys Med NMR. 1990;22(1):27–38. [PubMed] [Google Scholar]
- 17.Miki W. Biological functions and activities of animal carotenoids. Pure Appl Chem. 1991;63(1):141–146. doi: 10.1351/pac199163010141. [DOI] [Google Scholar]
- 18.Kanazashi M, Tanaka M, Murakami S, Kondo H, Nagatomo F, Ishihara A, Roy RR, Fujino H. Amelioration of capillary regression and atrophy of the soleus muscle in hindlimb-unloaded rats by astaxanthin supplementation and intermittent loading. Exp Physiol. 2014;99(8):1065–1077. doi: 10.1113/expphysiol.2014.079988. [DOI] [PubMed] [Google Scholar]
- 19.Kanazashi M, Okumura Y, Al-Nassan S, Murakami S, Kondo H, Nagatomo F, Fujita N, Ishihara A, Roy RR, Fujino H. Protective effects of astaxanthin on capillary regression in atrophied soleus muscle of rats. Acta Physiol (Oxf) 2013;207(2):405–415. doi: 10.1111/apha.12018. [DOI] [PubMed] [Google Scholar]
- 20.Dupont-Versteegden EE, Strotman BA, Gurley CM, Gaddy D, Knox M, Fluckey JD, Peterson CA. Nuclear translocation of EndoG at the initiation of disuse muscle atrophy and apoptosis is specific to myonuclei. Am J Physiol Regul Integr Comp Physiol. 2006;291(6):R1730–R1740. doi: 10.1152/ajpregu.00176.2006. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira R, Neuparth MJ, Vitorino R, Appell HJ, Amado F, Duarte JA. Evidences of apoptosis during the early phases of soleus muscle atrophy in hindlimb suspended mice. Physiol Res. 2008;57(4):601–611. doi: 10.33549/physiolres.931272. [DOI] [PubMed] [Google Scholar]
- 22.Leeuwenburgh C, Gurley CM, Strotman BA, Dupont-Versteegden EE. Age-related differences in apoptosis with disuse atrophy in soleus muscle. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1288–R1296. doi: 10.1152/ajpregu.00576.2004. [DOI] [PubMed] [Google Scholar]
- 23.Allen DL, Roy RR, Edgerton VR. Myonuclear domains in muscle adaptation and disease. Muscle Nerve. 1999;22(10):1350–1360. doi: 10.1002/(SICI)1097-4598(199910)22:10<1350::AID-MUS3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda Y, Tsuji S, Satoh A, Ishikura M, Shirasawa T, Shimizu T. Protective effects of astaxanthin on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Neurochem. 2008;107(6):1730–1740. doi: 10.1111/j.1471-4159.2008.05743.x. [DOI] [PubMed] [Google Scholar]
- 25.Sugiura T, Abe N, Nagano M, Goto K, Sakuma K, Naito H, Yoshioka T, Powers SK. Changes in PKB/Akt and calcineurin signaling during recovery in atrophied soleus muscle induced by unloading. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1273–R1278. doi: 10.1152/ajpregu.00688.2004. [DOI] [PubMed] [Google Scholar]
- 26.Yoshihara T, Sugiura T, Yamamoto Y, Shibaguchi T, Kakigi R, Naito H. The response of apoptotic and proteolytic systems to repeated heat stress in atrophied rat skeletal muscle. Physiol Rep. 2015;3(10):e12597. doi: 10.14814/phy2.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen DL, Yasui W, Tanaka T, Ohira Y, Nagaoka S, Sekiguchi C, Hinds WE, Roy RR, Edgerton VR. Myonuclear number and myosin heavy chain expression in rat soleus single muscle fibers after spaceflight. J Appl Physiol. 1996;81(1):145–151. doi: 10.1152/jappl.1996.81.1.145. [DOI] [PubMed] [Google Scholar]
- 28.Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113(1):115–123. doi: 10.1172/JCI18330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoi W, Naito Y, Sakuma K, Kuchide M, Tokuda H, Maoka T, Toyokuni S, Oka S, Yasuhara M, Yoshikawa T. Astaxanthin limits exercise-induced skeletal and cardiac muscle damage in mice. Antioxid Redox Signal. 2003;5(1):139–144. doi: 10.1089/152308603321223630. [DOI] [PubMed] [Google Scholar]
- 30.Aoi W, Naito Y, Takanami Y, Ishii T, Kawai Y, Akagiri S, Kato Y, Osawa T, Yoshikawa T. Astaxanthin improves muscle lipid metabolism in exercise via inhibitory effect of oxidative CPT I modification. Biochem Biophys Res Commun. 2008;366(4):892–897. doi: 10.1016/j.bbrc.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 31.Kondo H, Miura M, Itokawa Y. Antioxidant enzyme systems in skeletal muscle atrophied by immobilization. Pflugers Arch. 1993;422(4):404–406. doi: 10.1007/BF00374299. [DOI] [PubMed] [Google Scholar]
- 32.Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radical Biol Med. 2003;35(1):9–16. doi: 10.1016/S0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 33.Vermaelen M, Marini JF, Chopard A, Benyamin Y, Mercier J, Astier C. Ubiquitin targeting of rat muscle proteins during short periods of unloading. Acta Physiol Scand. 2005;185(1):33–40. doi: 10.1111/j.1365-201X.2005.01446.x. [DOI] [PubMed] [Google Scholar]
- 34.Ikemoto M, Nikawa T, Kano M, Hirasaka K, Kitano T, Watanabe C, Tanaka R, Yamamoto T, Kamada M, Kishi K. Cysteine supplementation prevents unweighting-induced ubiquitination in association with redox regulation in rat skeletal muscle. Biol Chem. 2002;383(3–4):715–721. doi: 10.1515/BC.2002.074. [DOI] [PubMed] [Google Scholar]
- 35.Furuno K, Goodman MN, Goldberg AL. Role of different proteolytic systems in the degradation of muscle proteins during denervation atrophy. J Biol Chem. 1990;265(15):8550–8557. [PubMed] [Google Scholar]
- 36.Tiao G, Fagan JM, Samuels N, James JH, Hudson K, Lieberman M, Fischer JE, Hasselgren PO. Sepsis stimulates nonlysosomal, energy-dependent proteolysis and increases ubiquitin mRNA levels in rat skeletal muscle. J Clin Invest. 1994;94(6):2255–2264. doi: 10.1172/JCI117588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem. 2000;275(33):25665–25671. doi: 10.1074/jbc.M906383199. [DOI] [PubMed] [Google Scholar]
- 38.Locke M. The cellular stress response to exercise: role of stress proteins. Exerc Sport Sci Rev. 1997;25:105–136. doi: 10.1249/00003677-199700250-00007. [DOI] [PubMed] [Google Scholar]
- 39.Naito H, Powers SK, Demirel HA, Sugiura T, Dodd SL, Aoki J. Heat stress attenuates skeletal muscle atrophy in hindlimb-unweighted rats. J Appl Physiol. 2000;88(1):359–363. doi: 10.1152/jappl.2000.88.1.359. [DOI] [PubMed] [Google Scholar]
- 40.Lee DH, Lee YJ, Kwon KH. Neuroprotective effects of astaxanthin in oxygen-glucose deprivation in SH-SY5Y cells and global cerebral ischemia in rat. J Clin Biochem Nutr. 2010;47(2):121–129. doi: 10.3164/jcbn.10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]