

Abstract
The currents through the volume-regulated outwardly rectifying anion channel (VRAC) were measured in single ventricular myocytes obtained from streptozotocin (STZ)-induced diabetic mice, using whole-cell voltage-clamp method. In myocytes from STZ-diabetic mice, the density of VRAC current induced by hypotonic perfusion was markedly reduced, compared with that in the cells form normal control mice. Video-image analysis showed that the regulatory volume decrease (RVD), which was seen in normal cells after osmotic swelling, was almost lost in myocytes from STZ-diabetic mice. Some mice were pretreated with 3-O-methylglucose before STZ injection, to prevent the STZ’s β cell toxicity. In the myocytes obtained from such mice, the magnitude of VRAC current and the degree of RVD seen during hypotonic challenge were almost normal. Incubation of the myocytes from STZ-diabetic mice with insulin reversed the attenuation of VRAC current. These findings suggested that the STZ-induced chronic insulin-deficiency was an important causal factor for the attenuation of VRAC current. Intracellular loading of the STZ-diabetic myocytes with phosphatidylinositol 3,4,5-trisphosphate (PIP3), but not phosphatidylinositol 4,5-bisphosphate (PIP2), also reversed the attenuation of VRAC current. Furthermore, treatment of the normal cells with wortmannin, a phosphatidylinositol 3-kinase (PI3K) inhibitor, suppressed the development of VRAC current. We postulate that an impairment PI3K-PIP3 pathway, which may be insulin-dependent, is responsible for the attenuation of VRAC currents in STZ-diabetic myocytes.
Keywords: Chloride channels, Heart, Diabetes mellitus, Patch clamp, Cell volume regulation
Introduction
It is well-known that diabetes mellitus is one of the strongest risk factors for cardiovascular disease. In addition to an increased incidence of coronary artery disease, cardiac dysfunction is a frequently reported complication of clinical diabetes mellitus [1]. One of the major complications leading to cardiac dysfunction is the development of diabetic cardiomyopathy, which may be associated with increased risk of sudden cardiac death [1] in the absence of major cardiac risk factors (i.e. atherosclerosis, hyperlipidaemia, hypertension or heart failure).
Experimental studies showed that streptozotocin (STZ)-induced diabetes in animals is associated with a variety of cardiac defects including disturbed heart rhythm, prolonged time-course of cardiac muscle contraction [2], changes in the surface electrocardiogram including T wave abnormalities and QT prolongation [3], and action potential prolongation [4]. Abnormalities of cardiac ion channels and transporters have also been reported in diabetic cardiomyocytes; i.e. impairments of transient outward K+ current [4–6], slow delayed rectifier K+ current (I Ks) [7], steady-state outward K+ current (I ss) [6, 8], L-type Ca2+ current [6, 8] and Na+–K+ pump [9]. No attenuation was reported in the inward rectifier K+ current (I K1) and rapid delayed rectifier K+ current (I Kr) [7]. Thus, many studies have been done to examine the diabetes-related alterations of cardiac cation channels. In contrast, the properties of anion channels in diabetic myocytes are still unknown.
One of the important physiological roles of Cl− channels is to regulate the cell-volume, and the cell-volume regulation is an essential cellular function coupled to a variety of physiological processes, such as cell proliferation, differentiation and migration, in most animal cell types [10]. The cells can re-adjust their volume after transient osmotic swelling by a mechanism known as regulatory volume decrease (RVD) which involves an activity of Cl− channels. It has been reported that the survival of dorsal root ganglion (DRG) neurons in hypotonic culture medium was decreased in diabetic mice [11]. To our knowledge, however, there is no information on RVD of cardiac cells derived from diabetic animals.
RVD is accomplished mainly by KCl efflux induced by parallel activation of K+ and Cl− channels in cardiac cells [12]. Among several types of cardiac Cl− channels, one of the most important channels in cell-volume regulation is volume-regulated outwardly rectifying anion channel (VRAC) [10, 12]. In the present study, we examined VRAC current and RVD in single ventricular cells derived from insulin-deficient diabetic mice, which were prepared by a single intraperitoneal (i.p.) injection of STZ. In these ventricular cells, we found loss of RVD, which was presumably caused by diabetes-related suppression of VRAC current. Our results show also that insulin exerts a restorative effect on the impaired VRAC current in diabetic cardiac cells, probably via the signaling pathway involving phosphoinositide 3-kinases.
Material and methods
Animal models
Saga University Animal Care and Use Committee approved the use and treatment of all animals used in the experiments described here. The investigation conforms also with the Guiding Principles of the Physiological Society of Japan. Insulin-deficient diabetes was induced in C57BL/6J mice (8 weeks male) by a single i.p. injection of streptozotocin (STZ; Sigma, St. Louis, MO; 2.5 mg per body-weight (BW(g)), which was dissolved in 0.1 M sodium citrate buffer (pH 4.5) just before injection, as described previously [13]. Age-matched normal control mice received saline alone. Fasting blood glucose level was evaluated 3 days later. A supplemental STZ injection (1.25 mg per BW(g), i.p.) was given if the fasting blood glucose was below 150 mg dl−1. All diabetic (blood glucose level after 8 h fasting >150 mg dl−1) and normal mice were maintained with the same diet and water ad libitum for a total of 2 weeks. Each blood sample was taken from the tail vein, and its glucose level was measured with Freestyle Flash Blood Monitoring System (Nipro. Osaka, Japan). In some experiments the mice were pretreated with 3-O-methylglucose (3-OMG; Sigma, 2.5 mg per BW(g) i.p.) before STZ injection, to prevent STZ’s β cell toxicity and thus to prevent the development of diabetes mellitus.
Cell preparation
Single ventricular myocytes were isolated from mouse hearts using an enzymatic dispersion technique. Briefly, mice were anaesthetized with sodium pentobarbitone (50 mg ml−1, i.p.). The chest was opened, and the heart was rapidly removed and perfused, by using a modified Langendorff technique, with a physiological saline solution (PSS, see ‘Solutions and drugs’) warmed to 37°C to wash out blood, and then with a nominally Ca2+-free PSS until the heart ceased to beat, and finally with Ca2+-free solution containing 0.1% collagenase (CLSII, Worthington, Lakewood, NJ, USA) and 1.0% bovine serum albumin (BSA) for 20–30 min. The ventricles were removed and cell dissociation was achieved by gentle mechanical agitation of the tissue in high-K+, low-Cl− storage (modified KB) solution (see ‘Solutions and drugs’) and the dissociated cells were stored in a refrigerator (4°C) for later use (within 8 h). Only rod-shaped myocytes with clear cross-striations and no blebs were used in the experiments. Some cells were incubated with insulin or wortmannin before electrophysiological measurements. They were stored at 37°C for 5–8 h in the modified Tyrode solution (see ‘Solutions and drugs’) containing 100 nM insulin (Sigma) or 100 nM wortmannin (Sigma), with 2 mg ml−1 BSA, penicillin-streptomycin solution (P-4458, Sigma, final concentration of 100 units ml−1 penicillin and 100 μg ml−1 streptomycin), and 15 μg ml−1 phenol red for pH monitor [14].
Electrophysiological techniques
The tight-seal whole-cell patch-clamp technique was used to record whole-cell currents. Patch pipettes (1.5 mm O.D. borosilicate glass electrodes) had a tip resistance of 1–3 MΩ when filled with pipette solution. Voltage-clamp recordings were performed using patch-clamp amplifiers (Axopatch 200B; Axon Instruments, Foster City, CA, USA) and membrane currents were filtered at a frequency of 2.5 kHz, and sampled at 5 kHz with a Digidata 1322A and pCLAMP 9.2 software (Axon Instruments). A 3 M KCl-agar bridge between the bath and the Ag–AgCl reference electrode was used to minimize changes in liquid junction potential. Unless otherwise stated, current recordings were made by applying voltage pulses of 400 ms duration to various potentials (between −100 and +100 mV in +20 mV steps) from a holding potential of −40 mV every 2 s. When necessary, the current density was calculated by membrane capacitance, which was obtained using pCLAMP 9.2 software. Usually, 5 min was allowed for adequate cell dialysis after membrane rupture, before beginning of the voltage clamp protocol. All experiments were performed at 36.5 ± 0.5°C.
Videomicroscopy and image analysis
The method to measure the cell area on the videomicroscopic image was almost the same as described previously [12, 15]. Briefly, microscopic images were recorded with a CCD video camera (CS3330; Tokyo Electronic, Tokyo, Japan), and fed into a computer (DELL Dimension 4100; Dell Computer Corporation, Round Rock, TX, USA) through an 8-bit frame grabber (LG-3; Scion, Frederick, MD, USA). Number of the pixels included within the captured cell-image was detected by custom-made macro programs on Scion-Image software ver. 4.03 (Scion). It should be noted that the cell area is only a rough index of the cell volume. The validity of this method to estimate the cell volume has been discussed in the previous paper [15].
Solutions and drugs
The PSS for cell preparation contained [16]: 126 NaCl, 10 glucose, 4.4 KCl, 5.0 MgCl2, 1.5 CaCl2, 20 taurine, 5.0 creatine, 5.0 sodium pyruvate, 1.0 NaHPO4, 10 Hepes; pH 7.4 adjust with NaOH; 300 mOsm with mannitol. Ca2+-free PSS was prepared by simply omitting CaCl2 from the PSS. The modified KB solution for cell storage contained [16]: 70 potassium glutamate, 20 KCl, 1.0 MgCl2, 10 KH2PO4, 10 taurine, 10 EGTA, 10 glucose, 0.1% albumin, 10 β-hydroxybutyric acid and 10 Hepes; pH 7.2 with KOH; 300 mOsm with mannitol. The modified Tyrode solution contained [16]: NaCl 140, KCl 5.4, MgCl2 0.5, CaCl2 1.8, NaH2PO4 0.33, glucose 5.5 and Hepes 5, pH 7.4 with NaOH; 300 mOsm with mannitol. In electrophysiological studies, all bath and pipette solutions were chosen to maximize recording of Cl− currents and to reduce possible contamination with cation currents and Ca2+ dependent currents. For VRAC currents recording, the standard (isotonic) extracellular solutions contained [16]: 80 NaCl, 20 TEA-Cl, 1.0 MgCl2, 0.5 CaCl2, 0.5 BaCl2, 0.5 CdCl2, 0.01 GdCl3, 5.5 glucose, 10 HEPES, 0.01 nicardipine; [Cl−]o = 105 mM; pH 7.4 with CsOH: 310 mOsm with mannitol. Hypotonic bath solutions were prepared by reducing the amount of mannitol in the standard bath solution to achieve 200 mOsm. Thus, both hypotonic and isotonic solutions had a constant ionic strength. For measurements of cell area, bath solutions were prepared by subtracting 50 mM NaCl from the modified Tyrode solution, and the osmolarity was adjusted to 200 and 300 mOsm with mannitol for hypotonic and isotonic solution, respectively. The standard intracellular pipette solution for VRAC current, contained [16]: 105 NMDG, 90 l-aspartic acid, 40 TEA-Cl, 5 NaCl, 5 MgATP, 0.1 Tris–GTP, 5 EGTA, 5 Hepes; pH 7.3 adjusted with NMDG; total [Cl−]i = 45 mM; 290 mOsm using mannitol. For the experiments in which phosphatidylinositol 4,5-bisphosphate (PIP2) or phosphatidylinositol 3,4,5-trisphosphate (PIP3) was used, PIP2 dic8 (Echelon Biosciences, Salt Lake City, UT, USA) [17] or PIP3 dic8 (Echelon Biosciences) [18] was directly dissolved in the control pipette solution at a concentration of 10 μM. The osmolarity of all solutions was measured using freezing point depression osmometers (Model OM-801; Vogel, Giessen, Germany).
Data analysis
Data are expressed as mean ± SEM; N and n indicate the number of animals and cells, respectively. Statistical comparisons were performed either by one-way ANOVA (analysis of variance) with a post hoc test (Scheffé’s multiple comparison test) for group data, or by paired or unpaired Student’s t-test when only two groups were compared. Statistical comparisons of the time course of changes in cell area between two groups were made according to two-way repeated measures ANOVA. A two tailed probability of <0.05 is taken to indicate statistical significance.
Results
We first measured body weight and fasting blood glucose levels of STZ-treated mice (N = 9) before and 2 weeks after injection of STZ. STZ-treated mice exhibited frequent urination and water drinking, and their mean body weight decreased from 23.3 ± 0.8 to 21.4 ± 1.0 g after the injection (Fig. 1a). Fasting blood glucose level before injection of STZ was less than 150 mg dl−1, and the level after injection was higher than 300 mg dl−1 (Fig. 1b). These results are consistent with previous studies using acute diabetic model of STZ-induced insulin-deficiency [19]. Hearts of STZ-induced chronic diabetic rats were reported to be hypertrophied; i.e. the heart-to-body weight ratio significantly exceeded that of age-matched normal rats [5]. However, as shown in Fig. 1c, the heart-to-body weight ratio (HW BW−1) did not seem to be altered in our short-term diabetic mice: 4.50 ± 0.05 mg g−1 (N = 5) for age-matched normal control mice versus 4.71 ± 0.25 mg g−1 (N = 6) for STZ-diabetic mice. In addition, measurements of cell capacitance, which is an index of cell size, showed no significant difference between myocytes from normal and those from STZ-diabetic mice (Fig. 1d). Though, in most previous studies, STZ-diabetic mice have been used after a period of more than 4 weeks after STZ injection, Shimoni and his co-workers [20] reported that high-dose STZ treatment altered the properties of several ion currents in rat ventricular myocytes within 4–6 days, the alterations being similar to those observed 7 weeks after a low dose treatment. We used the ventricular myocytes which were isolated from mice 2 weeks after STZ injection.
Fig. 1.
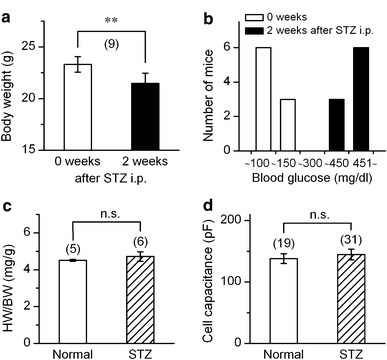
General characteristics of STZ-induced mice. a Mean data showing body-weight of mice measured before (0 week) and 2 weeks after STZ treatment. **Significantly different with P < 0.01 according to a paired t-test. b Distribution of blood glucose level (mg dl−1) among the normal control mice (0 week) and those treated with STZ (2 weeks). c and d comparison of the ratio of heart-weight and body-weight (c), and the cell capacitance of ventricular myocyte (d). Cells were obtained from the mice 2 weeks after STZ treatment (STZ) or from the age-matched un-treated mice (normal). Number in parentheses indicates number of animals (a and c) or number of cells examined (d). n.s., no significant difference
In the initial series of experiments, we compared the change in the cell volume during hypotonic challenge between the cells from STZ-diabetic mice and those from age-matched normal mice. The relative cell area on captured video image was used as an index of cell volume (see methods). Figure 2a shows the time-course of the changes in relative cell area during hypotonic challenge in myocytes from normal mice. Upon switching the bathing solution from isotonic to hypotonic solution, the cell area increased, as expected. But the increase peaked about 5 min after the initiation of hypotonic perfusion, and the cell area decreased thereafter. This spontaneous recovery of cell volume following the hypotonic cell swelling has been shown in several types of cardiac cells [12], and is termed regulatory volume decrease (RVD).
Fig. 2.
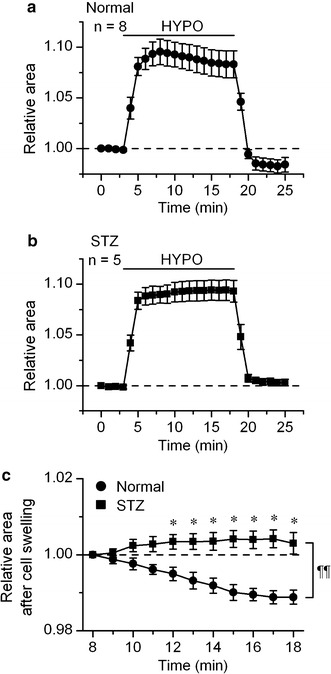
Hypotonicity-induced swelling of ventricular myocyte from normal and STZ-diabetic mice. a and b, time course of change of the relative cell area observed in the myocytes of normal (a) and STZ-diabetic (b) mice exposed to hypotonic (HYPO) solutions. The cells were initially bathed in isotonic solution, and then hypotonic (HYPO) solution was applied during the period indicated by bar. The values are expressed as relative to those obtained before application of hypotonic solution. c Shows an expanded illustration of a part of a (filled circle) and b (filled square). The values are expressed as relative to those obtained 5 min after application of hypotonic solution. *Significantly larger than the time-matched control data with P < 0.05 according to an unpaired t-test. A comparison of curves with repeated measures ANOVA yielded P < 0.01 (¶¶)
The cells from STZ-diabetic mice also inflated in response to external hypotonicity (Fig. 2b) but there was little RVD in these cells (Fig. 2b, c). Thus the cell area attained 15 min after application of hypotonic solution, as relative to the value seen 5 min after hypotonic solution, was 0.989 ± 0.002 (n = 8) and 1.003 ± 0.003 (n = 5) in cells from normal and STZ-diabetic mice, respectively. Re-application of isotonic solution decreased the cell area in both types of hypotonically swollen cells, as expected (Fig. 2a, b). In cells from normal mice, the level of cell area attained in the reapplied isotonic solution was smaller than the initial level, showing excessive cell shrinkage (Fig. 2a). This undershoot of the relative cell area below the initial level is attributed to a loss of solutes from the intracellular medium during RVD [10, 12]. In contrast, there was no clear undershoot of cell area in the diabetic cells which showed little RVD (Fig. 2b).
In our previous study, Cl− efflux through volume-regulated outwardly rectifying anion channel (VRAC) was implicated in the RVD in ventricular myocytes [12]. Therefore, we examined VRAC currents in ventricular myocytes from normal and STZ-diabetic mice. Figure 3a shows the time-course of changes in the amplitude of whole-cell currents at +60 (filled circle) and −60 mV (circle) in normal cells exposed to hypotonic (HYPO) solution. The current amplitude gradually increased during hypotonic perfusion, and decreased upon resumption of isotonic (ISO) solution. The hypotonicity-induced current displayed a very weak time-dependent inactivation at positive potentials (Fig. 3c). Its current–voltage (I–V) relationship showed a moderate outward rectification with the reversal potentials (E rev) of −19.8 ± 1.28 mV (n = 11), of which value being close to the predicted Cl− equilibrium potential (−21 mV) under the present condition (Fig. 3e). These properties are consistent with previous reports for native VRAC currents in mouse cardiac cells [21, 22]. The VRAC currents developed also in the cells derived from STZ-diabetic mice (Fig. 3b, d–f), but their amplitude in these cell was much smaller than that of VRAC currents in normal control cells (Fig. 3f). The mean amplitude of the VRAC current (defined as the difference current) at +100 mV measured 9 min after the beginning of hypotonic challenge was 4.31 ± 0.53 pA pF−1 (n = 11) in normal cells and 1.49 ± 0.16 pA pF−1 (n = 7) in diabetic cells (Fig. 3e).
Fig. 3.
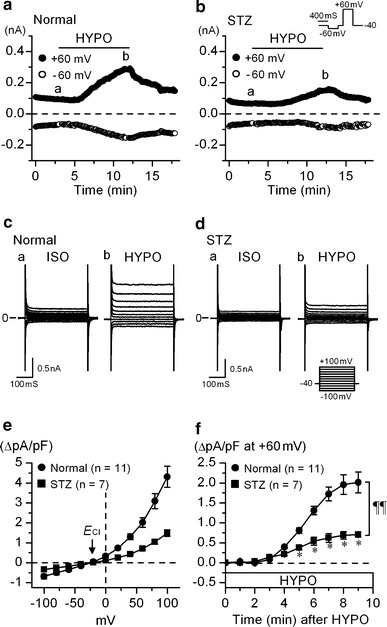
Activation of whole cell currents by hypotonic solutions in ventricular myocyte from normal and STZ-diabetic mice. a, b Time course of activation of VRAC currents at +60 mV (filled circle) and −60 mV (circle) observed in the myocytes of normal (a) and STZ-diabetic (b) mice exposed to hypotonic (HYPO) solutions. [Cl−]o/[Cl−]i ratio was constantly 105 mM/45 mM with which the predicted equilibrium potential (E Cl) was −21 mV. The cells were initially bathed in isotonic solution (ISO), and then hypotonic (HYPO) solution was applied during the period indicated by bar. The pulse protocol is shown in the upper part of b. c and d Recordings of membrane currents in myocytes from normal (c) and STZ-diabetic mice (d) in isotonic and hypotonic solutions, respectively. Currents were recorded by applying 400 ms voltage-clamp steps to membrane potentials between −100 and +100 mV in +20 mV steps from a holding potential of −40 mV every 6 s, at the time points (a and b) indicated in a and b. e The mean I–V relationships of the difference current between the current in HYPO and that in ISO (a, b in c and d), obtained in myocyte from normal (filled circle) and STZ-diabetic (filled square) mice. Arrow indicates the predicted Cl− equilibrium potential. f Mean time course of activation of VRAC current at +60 mV after hypotonic solutions, in myocytes from normal (filled circle) and STZ-diabetic mice (filled square). In this plot, the current level at the beginning of hypotonic perfusion (0 min) was set to be 0. Abscissa, time after exposure to hypotonic solution. *Significantly smaller than the control value at matched time point with P < 0.05 according to an unpaired t-test. A comparison of curves with repeated measures ANOVA yielded P < 0.01 (¶¶)
We considered that the suppression of VRAC current accompanied by loss of RVD observed in the cells derived from STZ-treated mice was caused by the STZ-induced diabetic state of animals, but not by the effects of this drug other than the toxicity on β cells, if any. This view was tested in a series of experiments in which the mice were pretreated with 3-O-methylglucose (3-OMG; 2.5 mg per BW(g) i.p.) before STZ injection, to prevent STZ’s β cell toxicity and thus to prevent the development of diabetes [23]. In five mice treated in this way, no mice showed frequent urination and water drinking, body weight loss or an increase of fasting blood glucose level (less than 123 mg dl−1) for at least 2 weeks after STZ injection, indicating that pretreatment with 3-OMG protected the onset of diabetes in mice, in agreement with previous studies [23].
Further, change in VRAC currents were examined in myocytes from mice treated with 3-OMG and STZ. Figure 4a show that the amplitude of VRAC current at +100 mV (4.20 ± 0.59 pA pF−1) observed in such myocytes was significantly larger than that observed in myocytes from the mice treated with STZ alone. Furthermore, the above value was not significantly different from the control value (4.31 ± 0.53 pA pF−1) obtained in normal mice. Treatment with 3-OMG alone without STZ did not induce any significant change in the amplitude of VRAC current at +100 mV (4.57 ± 0.64 pA pF−1). Thus the development of diabetes appeared to be an important causal factor for the attenuation of VRAC current. In accordance with the above finding, RVD was not impaired in myocytes from the mice treated with 3-OMG and STZ (Fig. 4b). The cell area attained 15 min after application of hypotonic solution, as relative to the value seen 5 min after hypotonic solution, was 0.986 ± 0.003 (n = 4), of which value being comparable to the value obtained in normal control cells (0.989 ± 0.002; n = 8, see Fig. 2c).
Fig. 4.
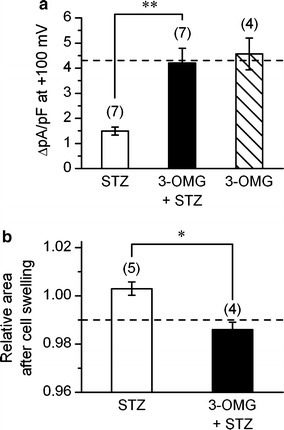
Prevention by 3-OMG of STZ-induced suppression of cardiac VRAC current. a and b Mean density of VRAC current (difference current) measured at +100 mV (a) and cell area (b), observed after 15 min of hypotonic perfusion. The currents and cell area were measured in the myocytes from STZ-diabetic mice (STZ), and those from the mice pretreated with 3-OMG before STZ injection (3-OMG + STZ). In a, the density of VRAC current observed in cells from the mice treated with 3-OMG alone is also shown. **Significantly different with P < 0.01, according to one-way ANOVA with post-hoc test In b, the values are expressed as relative to those obtained 5 min after application of hypotonic solution. Dashed line in a and b indicates the control level derived from the data in Fig. 3e (filled circle) and Fig. 2c (filled circle), respectively. *Significantly different with P < 0.05 according to an unpaired t-test. Number in parentheses indicates number of cells examined
The injection of STZ to mice results in a definite decrease in the plasma insulin level [24]. Therefore, the observed attenuation of VRAC current could causally be related to the insulin-deficiency. We examined whether the insulin treatment could restore the attenuated VRAC currents. Ventricular myocytes from normal and STZ-diabetic mice were incubated with insulin (100 nM) for 5–8 h [25]. The basal background current (I bb), which was recorded in isotonic solution before hypotonic challenge (e.g. Fig. 3c(a)), was little influenced by the insulin treatment in normal or diabetic cells. Thus the magnitude of I bb at +100 mV was 2.85 ± 0.25 pA pF−1 (n = 6) and 2.60 ± 0.24 pA pF−1 (n = 5) in normal and insulin-treated normal cells, respectively. It was 2.30 ± 0.23 pA pF−1 (n = 4) and 2.29 ± 0.04 pA pF−1 (n = 4) in non-treated diabetic and insulin-treated diabetic cells, respectively. As with the VRAC current, its magnitude was not significantly influenced by insulin in normal cells (Fig. 5b). However, the attenuated VRAC current in cells from STZ-diabetic mice appeared to be normalized by the insulin treatment. The magnitude of VRAC current in the insulin-treated diabetic cells was similar to that in normal cells (Fig. 5b). Since a short-term treatment (less than 15 min) with 100 nM insulin did not affect VRAC current in cells from STZ-diabetic mice (data not shown), the restoration of VRAC current observed above likely involved a slow intracellular metabolic process.
Fig. 5.
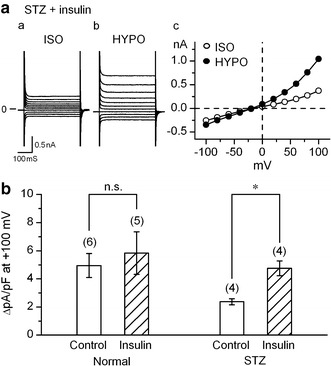
Effects of insulin on VRAC currents in ventricular myocytes from STZ-diabetic mice. a recordings of whole cell currents in myocytes from STZ-diabetic mice, obtained after ~6 h exposure to 100 nM insulin. Currents were recorded in isotonic (a, ISO) and hypotonic (b, HYPO) solutions with the same voltage steps as in Fig. 3d, and c shows their I–V relationships. b Magnitude of VRAC currents at +100 mV observed in the cells derived from normal (Normal) and STZ-diabetic mice (STZ) with and without insulin treatment. The cells were incubated at 37°C for 5–8 h, in the modified Tyrode solution without (Control) or with 100 nM insulin. *Significantly larger than control with P < 0.05, according to one-way ANOVA with post-hoc test. Number in parentheses indicates number of cells examined
The binding of insulin to its receptor (IR) results in changes in the activity of various enzymes and exerts effects on transcription and/or translation of various gene products, which in turn results in phosphorylation of the insulin receptor substrates (IRSs) [26]. Especially, phosphatidylinositol 3-kinase (PI3K) plays an essential role in the regulation of intracellular metabolism by insulin [26]. Insulin stimulated PI3K activity in cardiac cells [27], and in STZ-induced diabetic rats there was a significant decrease of PI3K activity in the vagus nerve [28]. Furthermore, recent reports showed that PI3K inhibitors attenuate VRAC currents in rabbit adult ventricular cells [29, 30]. Based on these findings, we hypothesized that PI3K activity was suppressed in the cardiac cells from STZ-diabetic mice due to insulin-deficiency, which in turn led to inhibition of VRAC current, and that insulin treatment could restore the attenuated PI3K activity.
To test the above hypothesis, we examined VRAC current in the cells from STZ-diabetic mice dialyzed with phosphatidylinositol 4,5-bisphosphate (PIP2) or phosphatidylinositol 3,4,5-trisphosphate (PIP3), considering that PI3K converts PIP2 to PIP3 and that PIP3 is an important mediator of many PI3K-related cellular responses [31]. These compounds were dissolved in the pipette solution at a concentration of 10 μM, and this concentration is comparable to the concentrations that have been used to demonstrate their effects on PIP2- or PIP3-regulated transporters and channels [17, 18, 32]. The original current traces obtained in PIP2- or PIP3-loaded myocytes exposed to isotonic and hypotonic solutions are displayed in Fig. 6a, b, and the mean I–V relationships of VRAC current and the mean time course of activation of VRAC current observed in these cells are shown in Fig. 6c, d, respectively. VRAC current in PIP3-loaded cells was significantly larger than that in PIP2-loaded cells (Fig. 6d). PIP2- and PIP3-loading appeared to have little or no influence on the kinetics of VRAC current (Fig. 6a, b versus Fig. 3c, d).
Fig. 6.

Effects of PIP3 loading on VRAC currents in ventricular myocytes from STZ-diabetic mice. a and b, Recordings of membrane currents in myocytes from STZ-diabetic mice in isotonic (ISO) and hypotonic (HYPO) solutions. The cells were loaded with PIP2 (a) or PIP3 (b). Voltage pulses were applied as in Fig. 3c and d. c Mean I–V relationships of VRAC current in ventricular myocyte from STZ-diabetic mice dialyzed with PIP2 (circle, n = 5) or PIP3 (filled circle, n = 5). In this series of experiments, pipette solution contained 10 μM of PIP2 or PIP3, and the current recordings were begun ~ 30 min after the membrane rupture, for more complete cell dialysis. d Mean time course of activation of VRAC current at +60 mV after hypotonic solutions, in STZ-diabetic myocyte dialyzed with PIP2 (circle) and PIP3 (filled circle). In this plot, the current level at the beginning of hypotonic perfusion (0 min) was set to be 0. Abscissa, time after exposure to hypotonic solution. *Significantly smaller than the control value at matched time point with P < 0.05 according to an unpaired t-test. A comparison of curves with repeated measures ANOVA yielded P < 0.01 (¶¶). e Mean density of VRAC current at +100 mV obtained under different conditions. Currents were measured in cells from normal and STZ-diabetic mice, with intracellular PIP2 or PIP3, respectively. f Mean density of VRAC current at +100 mV in normal (Normal) and STZ-diabetic (STZ) myocytes incubated with wortmannin (100 nM) for 5–8 h. In e and f, dashed line indicates the control level derived from the data in Fig. 3e (filled circle). * and **Significantly smaller than control with P < 0.05 and 0.01, respectively, according to one-way ANOVA with post-hoc test. Number in parentheses indicates number of cells examined
VRAC current was measured also in non-diabetic control cells dialyzed with PIP2 or PIP3. The amplitude of VRAC current in PIP3-loaded diabetic cells was similar to that in PIP3- or PIP2-loaded normal (non-diabetic) cells (Fig. 6e) and also to that in non-loaded normal cells (dashed line in Fig. 6e). In contrast, VRAC current in PIP2-loaded diabetic cells was significantly smaller than that in normal control cells (Fig. 6e), and its magnitude was similar to that observed in diabetic cells without PIP-loading (see Fig. 4a). Thus, PIP3-loading appeared to restore the attenuated VRAC current in the cells from STZ-diabetic mice, but PIP2-loading, not. Further, we attempted to see whether PI3K activity was involved in the VRAC activation, using wortmannin, a PI3K inhibitor. As shown in Fig. 6f, the development of VRAC current was strongly inhibited by application of wortmannin (100 nM) in normal (non-diabetic) cells, suggesting a crucial role of this enzyme in the VRAC activation. Wortmannin appeared to exert no further effect on the attenuated VRAC current in diabetic cells. Thus, the magnitude of VRAC current in wortmannin-treated STZ-diabetic cells (Fig. 6f) was comparable to that in non-treated diabetic cells (cf. Fig. 4a or Fig. 5b). These results are in line with our idea that suppression of PI3K activity played a key role in the inhibition of VRAC current observed in the cells from STZ-diabetic mice, and may support our recent proposal that PIP3 is an important mediator in the activation of VRAC current in mouse ventricular cells [33].
Discussion
We report here, for the first time, the recording of cardiac anion currents in an animal model of insulin-deficient diabetes. In single ventricular cells from STZ-induced diabetic mice, activation of VRAC current was suppressed (Fig. 3) and RVD was prevented (Fig. 2). Since RVD of cardiac cells strongly depends on the activation of VRAC current [12], the loss of RVD in the cells from STZ-diabetic mice is likely due to suppression of VRAC current. The current suppression does not appear to be due to any direct pharmacological effect of STZ on the VRAC current, because the cells derived from the animals pretreated with 3-OMG, which was expected to prevent the β cell toxicity of STZ, exhibited little suppression of VRAC current (Fig. 4).
Thus, it is likely that the attenuation of VRAC current was due to a chronic effect of diabetic state on the cellular metabolism. Since STZ treatment definitely induces insulin-deficiency, it seems reasonable to hypothesize that plasma insulin level played a key role in the development of an impairment of the membrane current. In accordance with this view, VRAC current was restored in diabetic cells by incubating these cells with insulin (100 nM) for 5–9 h (Fig. 5). Similar restorative effect of insulin on cardiac membrane current has been shown in the previous studies [25, 34], in which incubation of the myocytes from diabetic rats with insulin completely normalized the impaired Ito. Our results can be taken to indicate that plasma insulin is required to maintain VRAC current in mouse ventricular cells. If this were the case, it is possible that incubation of the isolated normal cells in insulin-free medium gradually causes an attenuation of their VRAC current. This point remains to be clarified.
If the chronic insulin-deficiency is an important factor for the impairment of VRAC current, what does the insulin-deficiency cause in the cells in relation to the current system? It is established that insulin stimulates PI3K activity in many types of vertebrate cells including cardiac cells [27]. Cai and Helke [28] reported that there was a significant decrease in PI3K activity without changes in the protein expression of p85 subunit of PI3K in the vagus nerve of STZ-induced diabetic rats. Similar results were shown in cardiac cells from alloxan-induced insulin-deficient diabetic rats [35]. Decrease of the phosphorylation of Akt, a downstream reaction of insulin-PI3K signaling, was shown in STZ-induced diabetic mouse heart [36]. On the other hand, evidence is accumulating that PI3K plays a role in cell swelling and related cellular responses. Cell swelling has been shown to activate PI3K in several cell types (for review see [37]), and osmotic activation of VRAC current was shown to depend on PI3K in rabbit ventricular myocytes [29, 30] and cultured smooth muscle cells [38]. In the present study, dialyzing the diabetic cells with PIP3, but not PIP2, restored VRAC current (Fig. 6). Furthermore, treatment with wortmannin, a PI3K inhibitor, strongly inhibited the development of VRAC current in cells from normal mice (Fig. 6f). These findings can be taken to indicate that the PIP3 concentration in diabetic cells was low due to low PI3K activity, resulting in an inhibition of VRAC current.
Based on the above considerations, we postulate that insulin-deficiency results in an inhibition of PI3K activity in cardiac cells, which leads to a decrease in the intracellular concentration of PIP3, while the decrease of PIP3 interferes with the activation of VRAC current via an unknown mechanism. However, the VRAC current system may not exclusively be regulated by the insulin-PI3K-PIP3 pathway. In ventricular myocyte of STZ-diabetic rat, the restoration of I to and I ss by insulin was blocked by an inhibition of the mitogen-activated protein (MAP) kinase-dependent pathway [25] and the cytoskeletal function [39]. Since the inhibitors of MAP kinase [40] and cytoskeleton (actin polymerization) [41] have been reported to inhibit the activation of VRAC current, these signaling pathways also might concern with the restoration of VRAC current by insulin observed in the present study. This issue as well as the precise role of PIP3 in the activation process of VRAC current must be investigated in future experiments.
STZ is an islet cell toxin used most commonly to induce type-1 (insulin-deficient) diabetes in experimental animal models. The metabolic features evoked by STZ treatment include not only a markedly reduced plasma insulin level but also hyperglycemia, hypertriglyceridemia, and ketosis [27]. Our finding that incubation of diabetic cells with insulin restored the impaired VRAC current may not necessarily indicate that the impairment was not at all related to the above diabetic complications other than insulin-deficiency. Further studies are necessary to clarify this point. As with hyperglycemia, it is noteworthy that high glucose rather increases the PI3K activity in cultured glomerular mesangial cells [42] and cultured cardiac fibroblasts [16].
Clinical and experimental studies support the existence of a specific cardiomyopathy associated with diabetes mellitus. The diabetic cardiomyopathy frequently exhibits left ventricular systolic and diastolic dysfunction and left ventricular hypertrophy [27]. Cl− channels may contribute to cardiac ischemic preconditioning and the adaptive remodeling of the heart during myocardial hypertrophy and heart failure [43], and alterations of cell volume are thought to be the key events during cell proliferation and apoptotic cell death [44]. It is possible that the attenuation of VRAC current and associated loss of RVD are somehow related to development of cardiac hypertrophy associated with diabetes. Further studies are necessary to elucidate the pathophysiological roles of VRAC current in this animal model as well as to clarify whether the reduction of VRAC activity is a universal phenomenon in the heart of diabetic animals including non-insulin-dependent diabetes model.
Acknowledgments
This research was partly supported by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture (No. 17500276 to T.E., and Nos. 18790158 and 20500366 to S.Y.).
References
- 1.McNally PG, Lawrence IG, Panerai RB, Weston PJ, Thurston H. Sudden death in type 1 diabetes. Diabetes Obes Metab. 1999;1:151–158. doi: 10.1046/j.1463-1326.1999.00025.x. [DOI] [PubMed] [Google Scholar]
- 2.Cesario DA, Brar R, Shivkumar K. Alterations in ion channel physiology in diabetic cardiomyopathy. Endocrinol Metab Clin North Am. 2006;35:601–610. doi: 10.1016/j.ecl.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Shehadeh A, Regan TJ. Cardiac consequences of diabetes mellitus. Clin Cardiol. 1995;18:301–305. doi: 10.1002/clc.4960180604. [DOI] [PubMed] [Google Scholar]
- 4.Jourdon P, Feuvray D. Calcium and potassium currents in ventricular myocytes isolated from diabetic rats. J Physiol. 1993;470:411–429. doi: 10.1113/jphysiol.1993.sp019866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nobe S, Aomine M, Arita M, Ito S, Takaki R. Chronic diabetes mellitus prolongs action potential duration of rat ventricular muscles: circumstantial evidence for impaired Ca2+ channel. Cardiovasc Res. 1990;24:381–389. doi: 10.1093/cvr/24.5.381. [DOI] [PubMed] [Google Scholar]
- 6.Shimoni Y. Hormonal control of cardiac ion channels and transporters. Prog Biophys Mol Biol. 1999;72:67–108. doi: 10.1016/S0079-6107(99)00005-X. [DOI] [PubMed] [Google Scholar]
- 7.Lengyel C, Virag L, Biro T, Jost N, Magyar J, Biliczki P, Kocsis E, Skoumal R, Nanasi PP, Toth M, Kecskemeti V, Papp JG, Varro A. Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc Res. 2007;73:512–520. doi: 10.1016/j.cardiores.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Wang DW, Kiyosue T, Shigematsu S, Arita M. Abnormalities of K+ and Ca2+ currents in ventricular myocytes from rats with chronic diabetes. Am J Physiol. 1995;269:H1288–H1296. doi: 10.1152/ajpheart.1995.269.4.H1288. [DOI] [PubMed] [Google Scholar]
- 9.Hansen PS, Clarke RJ, Buhagiar KA, Hamilton E, Garcia A, White C, Rasmussen HH. Alloxan-induced diabetes reduces sarcolemmal Na+-K+ pump function in rabbit ventricular myocytes. Am J Physiol Cell Physiol. 2007;292:C1070–C1087. doi: 10.1152/ajpcell.00288.2006. [DOI] [PubMed] [Google Scholar]
- 10.Okada Y. Cell volume-sensitive chloride channels. Contrib Nephrol. 1998;123:21–33. doi: 10.1159/000059920. [DOI] [PubMed] [Google Scholar]
- 11.Sango K, Horie H, Takano M, Inoue S, Takenaka T. Diabetes-induced reduction of neuronal survival in hypotonic environments in culture. Brain Res Bull. 1994;34:365–368. doi: 10.1016/0361-9230(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto S, Ishihara K, Ehara T, Shioya T. Cell-volume regulation by swelling-activated chloride current in guinea-pig ventricular myocytes. Jpn J Physiol. 2004;54:31–38. doi: 10.2170/jjphysiol.54.31. [DOI] [PubMed] [Google Scholar]
- 13.Ceylan-Isik AF, LaCour KH, Ren J. Gender disparity of streptozotocin-induced intrinsic contractile dysfunction in murine ventricular myocytes: role of chronic activation of Akt. Clin Exp Pharmacol Physiol. 2006;33:102–108. doi: 10.1111/j.1440-1681.2006.04331.x. [DOI] [PubMed] [Google Scholar]
- 14.Shioya T. A simple technique for isolating healthy heart cells from mouse models. J Physiol Sci. 2007;57:327–335. doi: 10.2170/physiolsci.RP010107. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto S, Ehara T, Shioya T. Changes in cell volume induced by activation of the cyclic AMP-dependent chloride channel in guinea-pig cardiac myocytes. Jpn J Physiol. 2001;51:31–41. doi: 10.2170/jjphysiol.51.31. [DOI] [PubMed] [Google Scholar]
- 16.Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2008;294:H2078–H2087. doi: 10.1152/ajpheart.01363.2007. [DOI] [PubMed] [Google Scholar]
- 17.Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res. 2007;73:531–538. doi: 10.1016/j.cardiores.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 18.Brady JD, Rich ED, Martens JR, Karpen JW, Varnum MD, Brown RL. Interplay between PIP3 and calmodulin regulation of olfactory cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 2006;103:15635–15640. doi: 10.1073/pnas.0603344103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, Kang YJ. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–1697. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 20.Shimoni Y, Firek L, Severson D, Giles W. Short-term diabetes alters K+ currents in rat ventricular myocytes. Circ Res. 1994;74:620–628. doi: 10.1161/01.res.74.4.620. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto-Mizuma S, Wang GX, Liu LL, Schegg K, Hatton WJ, Duan D, Horowitz TL, Lamb FS, Hume JR. Altered properties of volume-sensitive osmolyte and anion channels (VSOACs) and membrane protein expression in cardiac and smooth muscle myocytes from Clcn3−/- mice. J Physiol. 2004;557:439–456. doi: 10.1113/jphysiol.2003.059261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong W, Xu H, Shimizu T, Morishima S, Tanabe S, Tachibe T, Uchida S, Sasaki S, Okada Y. ClC-3-independent, PKC-dependent activity of volume-sensitive Cl channel in mouse ventricular cardiomyocytes. Cell Physiol Biochem. 2004;14:213–224. doi: 10.1159/000080330. [DOI] [PubMed] [Google Scholar]
- 23.Ganda OP, Rossini AA, Like AA. Studies on streptozotocin diabetes. Diabetes. 1976;25:595–603. doi: 10.2337/diabetes.25.7.595. [DOI] [PubMed] [Google Scholar]
- 24.Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- 25.Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. J Physiol. 1998;507(Pt 2):485–496. doi: 10.1111/j.1469-7793.1998.485bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitehead JP, Clark SF, Urso B, James DE. Signalling through the insulin receptor. Curr Opin Cell Biol. 2000;12:222–228. doi: 10.1016/S0955-0674(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 27.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- 28.Cai F, Helke CJ. Abnormal PI3 kinase/Akt signal pathway in vagal afferent neurons and vagus nerve of streptozotocin-diabetic rats. Brain Res Mol Brain Res. 2003;110:234–244. doi: 10.1016/S0169-328X(02)00652-6. [DOI] [PubMed] [Google Scholar]
- 29.Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3 K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–251. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl- current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 32.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001;2001:RE19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 33.Ichishima K, Yamamoto S, Ehara T. α1-adrenergic receptor-mediated depletion of phosphatidylinositol 4, 5-bisphosphate inhibits activation of volume-regulated chloride current in mouse ventricular myocytes. J Physiol Sci. 2008;58:S80. doi: 10.1111/j.1476-5381.2010.00896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Z, Patel KP, Rozanski GJ. Metabolic basis of decreased transient outward K+ current in ventricular myocytes from diabetic rats. Am J Physiol. 1996;271:H2190–H2196. doi: 10.1152/ajpheart.1996.271.5.H2190. [DOI] [PubMed] [Google Scholar]
- 35.De Luca JP, Garnache AK, Rulfs J, Miller TB., Jr Wortmannin inhibits insulin-stimulated activation of protein phosphatase 1 in rat cardiomyocytes. Am J Physiol. 1999;276:H1520–H1526. doi: 10.1152/ajpheart.1999.276.5.H1520. [DOI] [PubMed] [Google Scholar]
- 36.Bilim O, Takeishi Y, Kitahara T, Arimoto T, Niizeki T, Sasaki T, Goto K, Kubota I. Diacylglycerol kinase zeta inhibits myocardial atrophy and restores cardiac dysfunction in streptozotocin-induced diabetes mellitus. Cardiovasc Diabetol. 2008;7:2. doi: 10.1186/1475-2840-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jakab M, Furst J, Gschwentner M, Botta G, Garavaglia ML, Bazzini C, Rodighiero S, Meyer G, Eichmueller S, Woll E, Chwatal S, Ritter M, Paulmichl M. Mechanisms sensing and modulating signals arising from cell swelling. Cell Physiol Biochem. 2002;12:235–258. doi: 10.1159/000067895. [DOI] [PubMed] [Google Scholar]
- 38.Wang GX, McCrudden C, Dai YP, Horowitz B, Hume JR, Yamboliev IA. Hypotonic activation of volume-sensitive outwardly rectifying chloride channels in cultured PASMCs is modulated by SGK. Am J Physiol Heart Circ Physiol. 2004;287:H533–H544. doi: 10.1152/ajpheart.00228.2003. [DOI] [PubMed] [Google Scholar]
- 39.Shimoni Y, Rattner JB. Type 1 diabetes leads to cytoskeleton changes that are reflected in insulin action on rat cardiac K+ currents. Am J Physiol Endocrinol Metab. 2001;281:E575–E585. doi: 10.1152/ajpendo.2001.281.3.E575. [DOI] [PubMed] [Google Scholar]
- 40.Crepel V, Panenka W, Kelly ME, MacVicar BA. Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J Neurosci. 1998;18:1196–1206. doi: 10.1523/JNEUROSCI.18-04-01196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fatherazi S, Izutsu KT, Wellner RB, Belton CM. Hypotonically activated chloride current in HSG cells. J Membr Biol. 1994;142:181–193. doi: 10.1007/BF00234940. [DOI] [PubMed] [Google Scholar]
- 42.Sheu ML, Ho FM, Chao KF, Kuo ML, Liu SH. Activation of phosphoinositide 3-kinase in response to high glucose leads to regulation of reactive oxygen species-related nuclear factor-kappaB activation and cyclooxygenase-2 expression in mesangial cells. Mol Pharmacol. 2004;66:187–196. doi: 10.1124/mol.66.1.187. [DOI] [PubMed] [Google Scholar]
- 43.Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, Ye L, Hume JR. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26:265–278. doi: 10.1111/j.1745-7254.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 44.Lang F, Foller M, Lang K, Lang P, Ritter M, Vereninov A, Szabo I, Huber SM, Gulbins E. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 2007;428:209–225. doi: 10.1016/S0076-6879(07)28011-5. [DOI] [PubMed] [Google Scholar]