

Abstract
Arrhythmogenic effects of slowed action potential conduction produced by the gap junction and sodium-channel inhibitor heptanol (0.1–2 mM) were explored in Langendorff-perfused mouse hearts. Monophasic action potential recordings showed that 2 mM heptanol induced ventricular tachycardia in the absence of triggered activity arising from early or after-depolarizations during regular 8 Hz pacing and programmed electrical stimulation (PES). It also increased activation latencies and ventricular effective refractory periods (VERPs), but did not alter action potential duration (APD), thereby reducing local critical intervals for re-excitation given by APD90 − VERP. Bipolar electrogram recordings showed that 2 mM heptanol increased electrogram duration (EGD) and ratios of EGDs obtained at the longest to those obtained at the shortest S1S2 intervals studied during PES, suggesting increased dispersion of conduction velocities. These findings show, for the first time in the mouse heart, that slowed conduction induces reversible arrhythmogenic effects despite repolarization abnormalities expected to reduce arrhythmogenicity.
Keywords: Heptanol, Mice, Ventricular arrhythmia, Slowed conduction
Introduction
Normal cardiac excitation depends on an orderly sequence of electrical activation and recovery in successive myocardial regions. Disruption of this pattern can arise from re-excitation of previously activated tissue, and result in ventricular tachyarrhythmia. Mouse hearts have been used to study arrhythmogenesis, because they enable the use of genetic modification to study the consequences of ion-channel abnormalities [1]. The latter can be used to produce action potential abnormalities in repolarization and conduction.
Mouse models of re-entrant ventricular arrhythmogenesis can be divided into three groups. First, repolarization abnormalities alone are sufficient to induce arrhythmogenesis in hypokalaemic hearts, Scn5a +/Δ hearts containing a gain-of-function mutation in the α-subunit of the sodium channel, and KCNE1 −/− hearts with deletion of the coding sequence for the β-subunit of the slow delayed rectifier potassium (I Ks) channel. In these hearts action potential duration (commonly measured at 90% repolarization, APD90) is prolonged [2–4], transmural repolarization gradients are altered [2–4], and critical intervals for re-excitation given by APD90 − ventricular effective refractory periods (VERP) are increased [3, 5, 6], predisposing the hearts to triggered and re-entrant arrhythmias.
Second, repolarization and conduction abnormalities occur together in Cx43 +/− hearts with cardiac-specific inactivation of the gap junction protein connexin 43, resulting in both shortened APDs and slowed conduction, predisposing the hearts to re-entrant arrhythmias [7]. In mXinα +/− hearts with a deficiency of Xinα, a protein located in gap junctions, prolonged APDs and slowed conduction are observed [8], predisposing the hearts to triggered and re-entrant arrhythmias. In Scn5a +/− hearts with a targeted disruption of the gene coding for the α-subunit of the sodium channel, shortened APDs, altered repolarization gradients [9], and slowed conduction [10] are observed, predisposing the hearts to re-entrant arrhythmias.
A third possible case might involve conduction abnormalities only, but such a situation has not yet been demonstrated in mouse hearts, despite their extensive use in modelling arrhythmogenesis. In this work, therefore, experiments were conducted to examine whether slowed conduction alone can induce arrhythmogenic effects in the absence of the repolarization abnormalities described above in Langendorff-perfused mouse hearts. Conduction velocity depends on sodium current activation and gap junction conduction [11]. Heptanol at concentrations up to 1–2 mM specifically inhibits gap junctions [12, 13], and at ≥2 mM also inhibits sodium channels [12, 14]. It was therefore an appropriate agent to slow conduction [15–17].
Heptanol at 2 mM had significant arrhythmogenic effects under conditions of both regular 8 Hz pacing and programmed electrical stimulation (PES) [18, 19]. These were accompanied by increases in activation latencies, suggesting slowed conduction. The repolarization changes observed here were in a direction expected to reduce rather than increase arrhythmogenicity. Thus, they took the form of increased VERPs in an absence of alterations in APDs. These findings therefore demonstrate, for the first time in the whole mouse heart, that reduced conduction velocity alone was sufficient to cause arrhythmogenesis.
Methods
Experimental animals
Wild-type mice of the 129 genetic background were kept in an animal house facility at room temperature (21 ± 1°C), subject to a 12:12 h light/dark cycle. Sterile rodent chow and drinking water were available at all times. Male and female mice aged between 5 and 7 months were used for the experiments. All procedures carried out were in accordance with the UK Animals (Scientific Procedures) Act 1986.
Preparation of Langendorff-perfused mouse hearts
Intact Langendorff-perfused mouse hearts were used for this study. Techniques used for the preparation of whole hearts for Langendorff perfusion have been described elsewhere [20]. Briefly, mice were killed by cervical dislocation in accordance with Sections 1(c) and 2 of Schedule 1 of the UK Animals (Scientific Procedures) Act 1986. The hearts were rapidly excised and submerged in ice-cold bicarbonate-buffered, Krebs–Henseleit solution (mM: NaCl 119, NaHCO3 25, KCl 4, KH2PO4 1.2, MgCl2 1, CaCl2 1.8, glucose 10, and sodium pyruvate 2, pH 7.4) bubbled with 95% O2–5% CO2 [21]. A small section of aorta (3–4 mm) was identified and cannulated with a tailor-made 21-gauge cannula that had been prefilled with ice-cold buffer. The aorta was secured on to the cannula by use of a micro-aneurysm clip (Harvard Apparatus, Kent, UK). The heart was then quickly transferred and attached to the perfusion system. The perfusate was passed through 200 and 5 μm filters, then warmed to 37°C by use of a water jacket and circulator. Perfusion was started in a retrograde manner through the aorta at 2–2.5 ml min−1 by use of a peristaltic pump (model 505S; Watson–Marlow Bredel Pumps, Cornwall, UK). In this way, the heart was perfused with the Krebs–Henseleit solution passing through the aorta, into the coronary ostia, and thence into the coronary arteries, reaching the right atrium. After the start of perfusion, hearts that regained a pink colouration and spontaneous rhythmic contractions were studied further. The remaining 10% of hearts did not and were therefore discarded. Hearts were perfused with Krebs–Henseleit solution for a further 20 min before experimentation, to minimize any residual effects of endogenous catecholamine release.
Electrophysiology of perfused hearts
Hearts were extrinsically stimulated using paired platinum electrodes (1 mm interpole distance) gently positioned at the right ventricular epicardium. After the start of perfusion, hearts were paced at 8 Hz, by use of square-wave pulses 2 ms in duration, with a stimulation voltage set to thrice the threshold of excitation (Grass S48 stimulator; Grass-Telefactor, Berkshire, UK), enabling direct comparison with previous mouse studies of arrhythmogenesis [5, 22].
Determination of time taken for solutions to reach the perfusion cannula
Coloured solution was added to a colourless Krebs–Henseleit solution. The time taken for the discarded solution to become coloured was noted. Three repeats were performed and the mean of the durations was calculated.
Monophasic action potential recordings
Monophasic action potentials (MAPs) were recorded from the left ventricular epicardium using a MAP electrode (Linton Instruments, Kent, UK). Endocardial MAPs were recorded from the left ventricle with a custom-made MAP electrode, prepared from two strands of 0.25 mm Teflon-coated silver wire (99.99% purity; Advent Research Materials, Oxfordshire, UK). The tips of the endocardial MAP electrode had previously been galvanically-chlorided to eliminate DC offset. The electrode was introduced through a small access window made by removal of a small portion of the apex, to gain access to the left ventricular endocardium, as has previously been performed [2]. The positions of the stimulating and recording electrodes were kept constant, in the middle of the ventricular epicardium, and with a fixed inter-electrode distance of 3 mm.
All recordings were performed using a baseline cycle length (BCL) of 125 ms (8 Hz) to exclude rate-dependent differences in action potential duration (APD). MAPs were pre-amplified using a NL100AK head stage, amplified with a NL 104A amplifier, and band-pass filtered between 0.5 and 1 kHz using a NL125/6 filter (Neurolog, Hertfordshire, UK) and then digitized (1401plus MKII; Cambridge Electronic Design, Cambridgeshire, UK) at 5 kHz. MAP waveforms were analysed using Spike2 software (Cambridge Electronic Design) and those not matching the following stringent criteria for MAP signals [23] were rejected: MAPs must have a stable baseline and MAP morphology, fast upstroke without inflection or negative spikes, and rapid first phase of repolarization. Zero percent repolarization was measured at the peak of the MAP and 100% repolarization was taken as the minimum value of the membrane potential recorded [2, 23–25].
Programmed electrical stimulation
Programmed electrical stimulation (PES), previously adapted from clinical practice [18, 19], was performed. The PES procedure consisted of eight S1 pacing stimuli separated by a BCL of 125 ms, followed by an S2 extrastimulus. The S1S2 interval was initially 125 ms. With subsequent cycles this interval was reduced by 1 ms until the final cycle in which the S2 extrastimulus either provoked arrhythmic activity or failed to initiate a MAP. The latter indicated that ventricular effective refractory period (VERP) had been reached.
Bipolar electrogram recordings
Bipolar electrograms (BEGs) were recorded from the left ventricular epicardium using paired platinum recording electrodes (1 mm interpole spacing). Application of paced electrogram fractionation analysis (PEFA) [18, 26] to BEG recordings yielded conduction curves in which latencies of peaks and troughs were expressed as functions of S1S2 intervals. The point at which latency of the first peak starts to increase to 5% above its initial value was designated as the critical S1S2 interval. Electrogram duration (EGD) was calculated from the difference between the latencies of the first and last peaks or troughs. The EGD ratio was calculated by dividing the EGD obtained at the shortest S1S2 interval by the EGD obtained at the longest S1S2 interval.
Heptanol solutions
Heptanol (Sigma, Dorset, UK; density 0.82 g ml−1) is a lipophilic agent which is soluble in water up to approximately 9 mM (Merck Index, New Jersey, USA). Heptanol was added directly to Krebs–Henseleit solution to produce final concentrations between 0.1 and 2 mM.
Measurement of electrophysiological data
The following data were obtained from the experimental records:
activation latency, defined as the time difference between the stimulus and the peak of the MAP;
conduction velocity, defined as the ratio of the inter-electrode distance to the activation latency; as the latter distance was kept constant, the conduction velocities were inversely proportional to the corresponding activation latencies;
APDx, the time difference between the peak of the MAP and x = 30, 50, 70, and 90% repolarization;
local and transmural critical intervals, given by epicardial APD90 − epicardial VERP and (endocardial latency + endocardial APD90) − (epicardial latency + epicardial VERP) [5], respectively; and
the wavelength of excitation—the product of VERP and conduction velocity.
Statistical analysis
All values were expressed as mean ± standard error of the mean (SEM). Different experimental groups were compared by one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference test and Student’s t test, as appropriate. Categorical data were compared by use of Fisher’s exact test (two-tailed). P < 0.05 was considered statistically significant.
Results
Introduction of 2 mM heptanol produces a gradual increase in activation latency without altering action potential time course
The initial experiments were performed on hearts extrinsically paced at 8 Hz, which is close to the mouse heart rate observed in vivo [27], before and after introduction of 2 mM heptanol. Figure 1 shows epicardial MAP traces from a representative heart at different time points. MAPs with similar waveforms were observed before the introduction of heptanol (Fig. 1A). Each MAP had a stable baseline, fast upstroke without inflection or negative spikes, and a rapid first phase of repolarization [23]. MAP waveforms remained consistent at all times after introduction of heptanol. Both activation latencies and action potential durations at 90% repolarization (APD90) were unaltered immediately after introduction of heptanol (Fig. 1B). In contrast, activation latency increased from 13.1 to 29.5 ms 120 s after introduction of heptanol (Fig. 1C). This was consistent with slowed conduction. Furthermore, APD90 was 44.9 ms before and 46.1 ms 120 s after introduction of heptanol, suggesting that the time course of repolarization was not significantly altered.
Fig. 1.
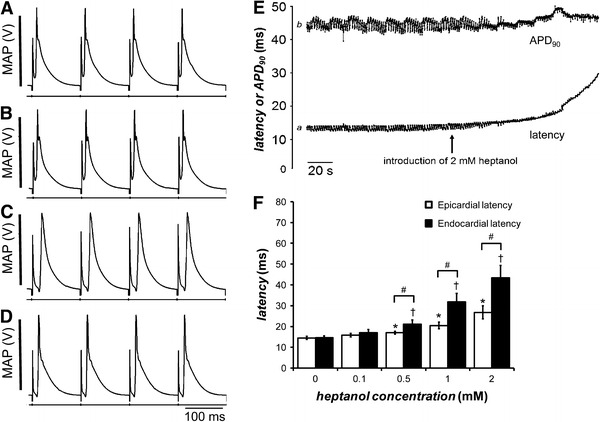
Traces showing monophasic action potentials (MAPs) recorded from the epicardium of a representative heart during regular 8 Hz pacing at different time points (A–D). MAPs recorded 120 s before the introduction of 2 mM heptanol (A). MAPs recorded immediately after introduction of 2 mM heptanol (B). MAPs recorded 120 s after introduction of 2 mM heptanol (C). MAPs recorded 900 s after withdrawal of 2 mM heptanol (D). Activation latency (trace a) and APD90 (trace b) plotted as a function of time (E). The time at which 2 mM heptanol was introduced is indicated by an arrow. Before introduction of 2 mM heptanol, activation latency was 13.1 ms. 120 s after introduction of 2 mM heptanol, this was increased to 29.5 ms. In contrast, APD90 was 44.9 ms before and 46.1 ms after introduction of heptanol. Epicardial activation latencies (white bars), obtained before (n = 8) and 120 s after introduction of 0.1 (n = 8), 0.5 (n = 7), 1 (n = 9), and 2 mM (n = 7) heptanol during regular 8 Hz pacing (F). Endocardial activation latencies (black bars) before (n = 9) and 120 s after introduction of 0.1 (n = 5), 0.5 (n = 10), 1 (n = 5), and 2 mM (n = 5) heptanol, obtained during regular 8 Hz pacing. Asterisks indicate the epicardial activation latencies that were significantly increased by heptanol (ANOVA, P < 0.05). Daggers indicate the endocardial activation latencies that were significantly increased by heptanol (ANOVA, P < 0.05). Hashes indicate that endocardial activation latencies were significantly larger than the corresponding epicardial activation latencies at a given heptanol concentration (ANOVA, P < 0.05)
To investigate the reversibility of the effects of heptanol, the heptanol-containing solution was changed to one consisting of Krebs–Henseleit solution alone. Activation latency recovered to 21.2 ms, with APD90 remaining at 45.8 ms 15 min after this replacement (Fig 1D). Figure 1E shows an example of the changes in activation latency (trace a) and APD90 (trace b) over time. The arrow indicates the point at which heptanol was introduced, after which there was a smooth, progressive increase in activation latency but little change in APD90. The action of heptanol was time-dependent, continuously increasing without reaching a stable value during the time it was possible to investigate its effects. Thus, activation latency progressively increased until complete conduction block occurred in approximately 60% of the hearts at 539 ± 107 s (n = 13) after treatment with the highest (2 mM) concentration of heptanol. It was therefore necessary to specify a consistent time point at which activation latency was assessed to enable the effects of heptanol to be compared between experiments. We observed that 120 s after introduction of heptanol was sufficient to demonstrate the changes in conduction, and therefore consistently used this time point for every experiment. Thus there was a 56% slowing of conduction but only a 3% change in APD90 120 s after introduction of heptanol. Figure 1F also shows both epicardial (n = 8; white bars) and endocardial (n = 9; black bars) activation latencies before and after introduction of heptanol at concentrations between 0.1 and 2 mM. The mean epicardial activation latency was 14.5 ± 0.8 ms before the introduction of heptanol. It increased with increasing concentrations of heptanol from 0.1 to 2 mM, reaching a value of 26.8 ± 3.1 ms at 2 mM heptanol (asterisks, ANOVA, P < 0.05). The mean endocardial activation latency was 14.6 ± 0.9 ms before introduction of heptanol. It increased with increasing concentrations of heptanol from 0.1 to 2 mM, reaching a value of 43.3 ± 6.0 ms at 2 mM heptanol (daggers, ANOVA, P < 0.05). Finally, activation latencies at the epicardium were not significantly different from those at the endocardium before and after introduction of 0.1 mM heptanol (ANOVA, P > 0.05). In contrast, endocardial activation latencies were significantly larger than the corresponding epicardial activation latencies after introduction of heptanol at concentrations between 0.5 and 2 mM heptanol (hashes, ANOVA, P < 0.05). These results suggest that action potential conduction to the endocardium was slower than that to the epicardium. Taken together, these findings associate increased arrhythmogenicity induced by heptanol directly with slowed conduction.
These findings show that 2 mM heptanol selectively slowed conduction without significantly affecting the time courses of action potential repolarization. They extend to the mouse ventricle previous results demonstrating slowed conduction from the rat ventricular papillary muscle [16], the sheep Purkinje fibre [28], and the rabbit ventricle [15].
Introduction of 2 mM heptanol results in ventricular tachycardia in the absence of triggered activity during regular 8 Hz pacing
Having established that 2 mM heptanol slowed conduction in the heart, we next systematically sought spontaneous ventricular arrhythmogenicity before and after introduction of heptanol at the concentrations investigated during regular 8 Hz stimulation. An incident of ventricular tachycardia (VT) was defined as the occurrence of a succession of five or more action potentials at intervals closer than the basic cycle length (BCL). Hearts were studied for similar durations of exposure to different solutions, which were 600 ± 50, 630 ± 105, 630 ± 55, 673 ± 185, and 420 ± 92 s for 0 mM (n = 13), 0.1 mM (n = 13), 0.5 mM (n = 13), 1 mM (n = 13), and 2 mM (n = 13) heptanol, respectively. Exposure to 2 mM heptanol was for a shorter period because its effects were observed sooner after application.
Epicardial MAPs followed directly after their preceding extrinsic stimuli, with consistent waveforms and no evidence of triggered activity arising from either early and delayed after-depolarizations or VT before introduction of heptanol (Fig. 2A). There was an increase in the proportion of hearts for which VT was observed (Fig. 2B), from 0 out of 13 to 5 out of 13, when the concentration of heptanol was increased from 0.1 to 2 mM. Figure 2C summarises the incidences of spontaneous VT observed at each heptanol concentration, revealing that it was significantly arrhythmogenic only at 2 mM (asterisk, Fisher’s exact test, P < 0.05).
Fig. 2.
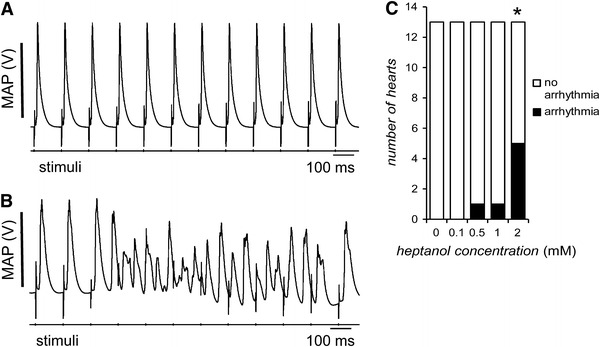
Representative traces of MAPs recorded during regular 8 Hz pacing. Before introduction of heptanol, a typical regular rhythm with each MAP occurring directly after its preceding extrinsic stimulus can be observed (A). In contrast, after introduction of 2 mM heptanol, VT can be observed (B). Incidences of VT over the first 10 min after introduction of 0, 0.1, 0.5, 1, and 2 mM heptanol (C). The asterisk indicates that 2 mM heptanol induced significant arrhythmogenic effects (Fisher’s exact test, P < 0.05)
Programmed electrical stimulation induces ventricular tachycardia in hearts treated with 2 mM heptanol
Subsequent experiments applied programmed electrical stimulation (PES), which has been used clinically in electrophysiological studies [18, 19]. The PES procedure consisted of eight S1 pacing stimuli separated by a BCL of 125 ms, followed by an S2 extrastimulus. The S1S2 interval was initially 125 ms. With subsequent cycles this interval was reduced by 1 ms, until the final cycle in which the S2 extrastimulus either provoked arrhythmic activity or failed to initiate an MAP. The latter indicated that the ventricular effective refractory period (VERP) had been reached. Because heptanol induced time-dependent effects that did not reach a steady state, arrhythmogenicity was assessed and ventricular effective refractory periods (VERPs) were measured at the same standardized time point as for measurement of activation latency.
PES consistently failed to provoke VT before the introduction of heptanol (Fig. 3A; S2 extrastimuli indicated by arrows). There was an increase in the proportion of hearts for which provoked VT was observed (Fig. 3B; S2 extrastimulus indicated by an arrow), from 0 out of 7 to 3 out of 5, when the concentration of heptanol was increased from 0.1 to 2 mM. The heptanol-containing solution was then changed to one consisting of Krebs–Henseleit solution alone, after which PES did not induce VT in any heart. Figure 3C summarises the incidences of provoked VT observed at each heptanol concentration, revealing that it was significantly arrhythmogenic only at 2 mM (asterisk, Fisher’s exact test, P < 0.05).
Fig. 3.
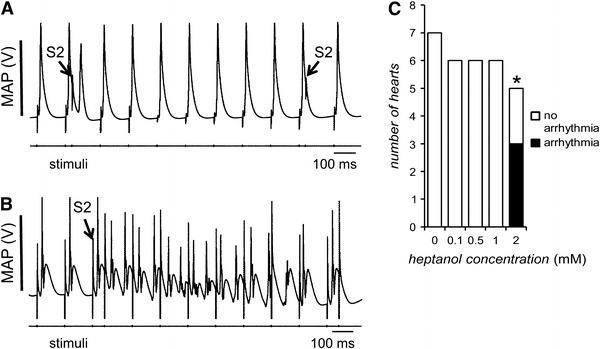
Representative MAP recordings from programmed electrical stimulation (PES). Before introduction of heptanol, the S2 extrastimulus failed to induce ventricular tachycardia (VT) (A). In contrast, 120 s after introduction of 2 mM heptanol, the S2 extrastimulus induced VT (B). Incidences of VT in hearts subject to PES before and 120 s after introduction of 0.1, 0.5, 1, and 2 mM heptanol (C). The asterisk indicates that 2 mM heptanol induced significant arrhythmogenic effects (Fisher’s exact test, P < 0.05)
Heptanol does not alter the duration of epicardial or endocardial action potentials
We next assessed the time courses of action potential repolarization, represented by action potential duration at x = 30, 50, 70, and 90% repolarization (APDx) at the epicardium and endocardium, before and after introduction of heptanol during 8 Hz pacing. Transmural repolarization gradients were determined by the difference (endocardial APD90 − epicardial APD90) (ΔAPD90). Epicardial APD90, endocardial APD90, and ΔAPD90 values obtained before and after introduction of heptanol at all the concentrations investigated are summarised in Tables 1, 2, and 3, respectively. These tables indicate that heptanol at any concentration did not significantly alter any of these properties (ANOVA, P > 0.05). These results therefore reveal that introduction of heptanol did not alter the time courses of repolarization at either the epicardium or the endocardium, and accordingly left the transmural repolarization gradients unaltered.
Table 1.
Epicardial APDx ± SEM values before and after introduction of heptanol at concentrations between 0.1 and 2 mM
| [Heptanol] (mM) | n | APD30 (ms) | APD50 (ms) | APD70 (ms) | APD90 (ms) |
|---|---|---|---|---|---|
| 0 | 6 | 5.55 ± 0.97 | 9.11 ± 1.25 | 16.37 ± 1.62 | 37.72 ± 1.30 |
| 0.1 | 6 | 6.66 ± 0.95 | 12.87 ± 0.92 | 20.76 ± 1.23 | 40.43 ± 2.92 |
| 0.5 | 6 | 6.07 ± 1.31 | 12.41 ± 1.58 | 21.82 ± 2.30 | 40.81 ± 2.70 |
| 1 | 5 | 5.27 ± 0.16 | 9.09 ± 0.67 | 18.61 ± 0.41 | 37.32 ± 0.68 |
| 2 | 6 | 6.04 ± 1.11 | 12.21 ± 1.33 | 20.44 ± 1.46 | 41.23 ± 2.57 |
None of the properties was significantly altered by heptanol (ANOVA, P > 0.05)
Table 2.
Endocardial APDx ± SEM values before and after introduction of heptanol at concentrations between 0.1 and 2 mM
| [Heptanol] (mM) | n | APD30 (ms) | APD50 (ms) | APD70 (ms) | APD90 (ms) |
|---|---|---|---|---|---|
| 0 | 6 | 11.22 ± 2.37 | 16.67 ± 3.49 | 25.28 ± 4.72 | 49.48 ± 3.50 |
| 0.1 | 6 | 8.24 ± 1.19 | 14.38 ± 1.66 | 26.40 ± 2.05 | 46.88 ± 4.24 |
| 0.5 | 6 | 7.21 ± 1.18 | 13.10 ± 1.18 | 24.47 ± 2.85 | 46.85 ± 3.85 |
| 1 | 5 | 6.83 ± 1.33 | 12.22 ± 2.85 | 21.34 ± 3.87 | 41.72 ± 2.52 |
| 2 | 6 | 9.44 ± 1.57 | 16.78 ± 2.24 | 29.55 ± 2.42 | 51.58 ± 2.02 |
None of the properties was significantly altered by heptanol (ANOVA, P > 0.05)
Table 3.
Endocardial − epicardial APDx, expressed as ΔAPDx, ±SEM values before and after introduction of heptanol at concentrations between 0.1 and 2 mM
| [Heptanol] (mM) | n | ΔAPD30 (ms) | ΔAPD50 (ms) | ΔAPD70 (ms) | ΔAPD90 (ms) |
|---|---|---|---|---|---|
| 0 | 6 | 5.67 ± 2.56 | 7.57 ± 3.71 | 8.91 ± 4.99 | 11.76 ± 3.73 |
| 0.1 | 6 | 1.58 ± 1.53 | 1.51 ± 1.90 | 5.65 ± 2.39 | 6.44 ± 5.15 |
| 0.5 | 6 | 1.14 ± 1.76 | 0.69 ± 1.97 | 2.65 ± 3.66 | 6.04 ± 4.70 |
| 1 | 5 | 1.56 ± 1.34 | 3.13 ± 2.93 | 2.73 ± 3.89 | 4.40 ± 2.61 |
| 2 | 6 | 3.40 ± 1.93 | 4.57 ± 2.61 | 9.11 ± 2.83 | 10.35 ± 3.27 |
None of the properties was significantly altered by heptanol (ANOVA, P > 0.05)
Heptanol induces a concentration-dependent increase in VERP
Values of VERP were determined by use of the same PES procedure described above both before and after introduction of heptanol at the concentrations investigated (Fig. 4A). The mean VERP was 43.0 ± 3.3 ms before introduction of heptanol (n = 20). It increased when the concentration of heptanol was increased from 0.1 to 2 mM, reaching a value of 66.8 ± 5.8 ms at 2 mM heptanol (ANOVA, P < 0.05). These results indicate that introduction of heptanol increased the VERP, from which a decrease, rather than increase, in arrhythmogenicity would be predicted.
Fig. 4.
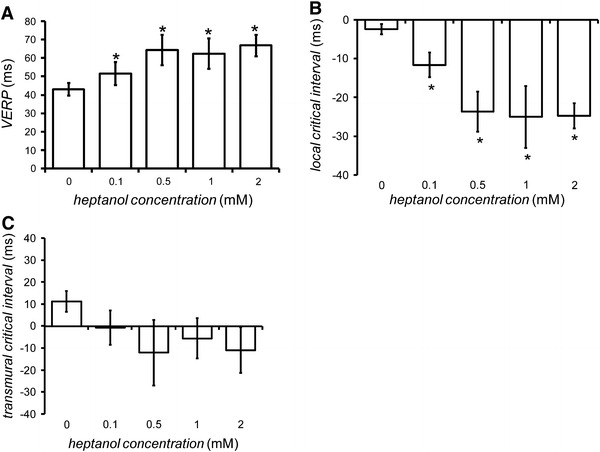
Ventricular effective refractory period (VERP) obtained during PES, before (n = 20) and 120 s after introduction of 0.1 mM (n = 5), 0.5 mM (n = 5), 1 mM (n = 6), and 2 mM (n = 5) heptanol (A). Asterisks indicate values that were significantly increased by heptanol (ANOVA, P < 0.05). Local critical interval, given by epicardial APD90 − epicardial VERP, obtained before (n = 6) and after introduction of 0.1 mM (n = 5), 0.5 mM (n = 5), 1 mM (n = 5) and 2 mM (n = 5) heptanol (B). This was reduced by heptanol at all the concentrations investigated (asterisk; ANOVA, P < 0.05). Transmural critical interval, given by (endocardial latency + endocardial APD90) − (epicardial latency + epicardial VERP), obtained before (n = 6) and after introduction of 0.1 mM (n = 5), 0.5 mM (n = 5), 1 mM (n = 5) and 2 mM (n = 5) heptanol (C). This was not altered by heptanol at any concentration (Student’s t test, P > 0.05)
Heptanol induces a concentration-dependent decrease in the local critical interval but does not alter the transmural critical interval
The critical intervals reflect the time periods that permit re-excitation before full action potential repolarization and can involve the APD90 exceeding VERP either in the local epicardial region or across the myocardial wall [5]. Our experiments assessed these values before and after introduction of heptanol. The local critical interval was −2.4 ± 1.3 ms before introduction of heptanol (n = 6; Fig. 4B). It decreased when the concentration of heptanol was increased from 0.1 to 2 mM, reaching a value of −24.8 ± 3.3 ms at 2 mM heptanol (ANOVA, P < 0.05). The transmural critical interval was 11.3 ± 4.8 ms before introduction of the heptanol (n = 6; Fig. 4C). However, this was not significantly altered by heptanol at any concentration (Student’s t test, P > 0.05). These results indicate that heptanol treatment resulted in reduced critical intervals for local re-excitation, which would be compatible with a decrease rather than an increase in arrhythmogenicity. Furthermore, heptanol treatment resulted in unchanged critical intervals for transmural re-excitation, from which no change, rather than an increase, in arrhythmogenicity would be predicted. The arrhythmogenic effects of heptanol were thus associated with slowed conduction in the presence of unchanged APD90 and reduced critical intervals.
Heptanol at 2 mM increases activation latency and reduces conduction velocity, VERP/latency ratio, and excitation wavelength
The activation latency was determined at the same time point as the VERP during the penultimate cycle of PES (Fig. 5A). This was 15.1 ± 0.7 ms before introduction of heptanol (n = 20) and was not altered by 0.1 mM heptanol (n = 5, ANOVA, P > 0.05). In contrast, it increased when the concentration of heptanol was increased from 0.1 to 2 mM, reaching a value of 37.7 ± 5.7 ms at 2 mM heptanol (ANOVA, P < 0.05). Note that there was variability in the time at which the S2 extrastimulus was applied in the final PES cycle. This led to differences in the epicardial activation latencies obtained during PES shown in this figure and those obtained during regular 8 Hz pacing shown in Fig. 1F. The ratio of VERP to activation latency was then determined (Fig. 5B). This was 2.9 ± 0.3 ms before introduction of heptanol (n = 20), and was not significantly altered by heptanol at concentrations between 0.1 and 1 mM heptanol (Student’s t test, P > 0.05). In contrast, it was reduced by 2 mM heptanol (n = 5), taking a value of 1.9 ± 0.3 ms (Student’s t test, P < 0.05).
Fig. 5.
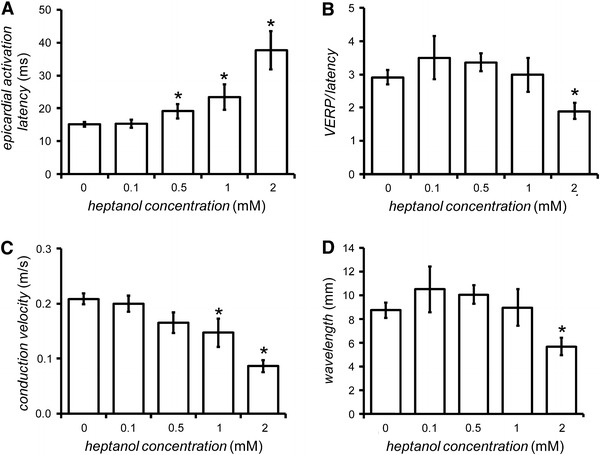
Epicardial activation latency (A) and conduction velocity (CV) (B) obtained in response to S1 stimulation, VERP/latency ratio (C) and wavelength of excitation (VERP × CV) (D), obtained during PES, before (n = 20) and 120 s after introduction of 0.1 mM (n = 5), 0.5 mM (n = 5), 1 mM (n = 6), and 2 mM (n = 5) heptanol. Asterisks indicate values that were significantly altered by heptanol (ANOVA, P < 0.05)
Conduction velocity was then determined (Fig. 5C). The conduction velocity was 0.21 ± 0.01 m/s before introduction of heptanol (n = 20) and was not altered by heptanol at concentrations between 0.1 and 0.5 mM (n = 5 or 6; ANOVA, P > 0.05). In contrast, it decreased when the concentration of heptanol was increased from 1 to 2 mM, reaching a value of 0.09 ± 0.01 m/s at 2 mM heptanol (n = 5, ANOVA, P < 0.05). The wavelength was 8.7 ± 0.6 mm before introduction of heptanol (n = 20) (Fig. 5D). It was not significantly altered by heptanol at concentrations between 0.1 and 1 mM (ANOVA, P > 0.05). In contrast, it was reduced by 2 mM heptanol (n = 5), taking a value of 5.7 ± 0.5 mm (ANOVA, P < 0.05).
These results indicate that 2 mM heptanol reduced conduction velocity more than it increased VERP, leading to a decrease in wavelength. This therefore explains the increase in arrhythmogenicity observed.
Introduction of 2 mM heptanol increases electrogram duration in response to both regular and extrasystolic stimulation
The final experiments recorded bipolar electrograms (BEGs) before and after introduction of 2 mM heptanol during PES. Figure 6A, B show representative BEG recordings from the last two cycles of PES before and 120 s after introduction of 2 mM heptanol, respectively. These BEGs contain multiple peaks and troughs resulting from activation of individual paths with different conduction velocities within the myocardium that was in close proximity to the recording electrode [29]. These deflections show progressively greater latencies and separations from each other with the application of successively more premature stimuli. Paced electrogram fractionation analysis (PEFA) of such data enables quantification of this asynchronous activation that provides an indication of re-entrant arrhythmogenic tendency [18, 21, 26, 30]. In the penultimate cycle, S1 stimulus and S2 extrastimulus artifacts followed by their resulting electrograms can be seen. In the final cycle, the S2 extrastimulus did not evoke an electrogram, indicating that refractoriness had been reached. The same number of deflections was observed for all the BEGs before and after introduction of heptanol (n = 5).
Fig. 6.
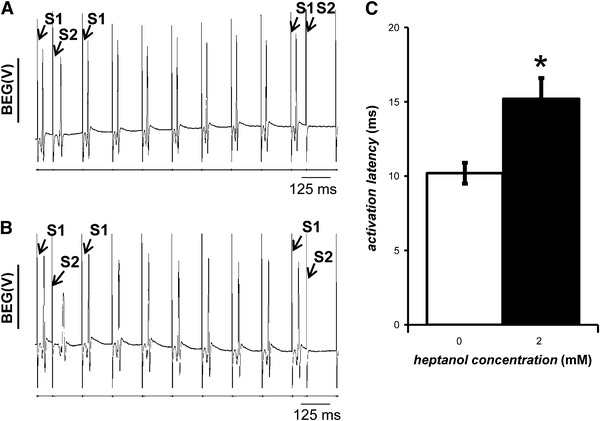
Representative bipolar electrogram (BEG) recordings during programmed electrical stimulation (PES) before (A) and 120 s after (B) introduction of 2 mM heptanol. Activation latencies from BEG recordings obtained before (clear bar; n = 5) and 120 s after (black bar; n = 5) introduction of 2 mM heptanol (C). The asterisk indicates these were significantly increased by 2 mM heptanol (ANOVA, P < 0.05)
The activation latencies from BEG recordings, defined as the time difference between the stimulus and the largest deflection of the resulting waveform, obtained in response to regular S1 stimulation, were determined before and 120 s after introduction of heptanol (Fig. 6C). The activation latency was 10.2 ± 0.7 ms before introduction of heptanol (n = 5). This was significantly increased by 2 mM heptanol (n = 5), taking a value of 15.2 ± 1.4 ms (asterisk, ANOVA, P < 0.05), thus corroborating the findings from MAP recordings.
Figure 7A, B show BEGs obtained during the penultimate cycle of PES before and after introduction of 2 mM heptanol, respectively. The latencies of the successive peaks and troughs of the BEGs obtained in response to S1 stimulation were designated a 0, b 0, c 0 and d 0, and those obtained in response to S2 stimulation were designated a, b, c and d. Application of PEFA yielded series of conduction curves in which the latencies a, b, c and d were plotted against the S1S2 intervals explored. Figure 7C, D show typical examples of such curves obtained before and after introduction of 2 mM heptanol, respectively. The vertical spread of data points was wider at short S1S2 intervals than the vertical spread at long S1S2 intervals in all the conduction curves obtained before introduction of heptanol (Fig. 7C). The spreads at a given S1S2 interval were more marked after introduction of 2 mM heptanol (Fig. 7D), indicating slowed conduction.
Fig. 7.
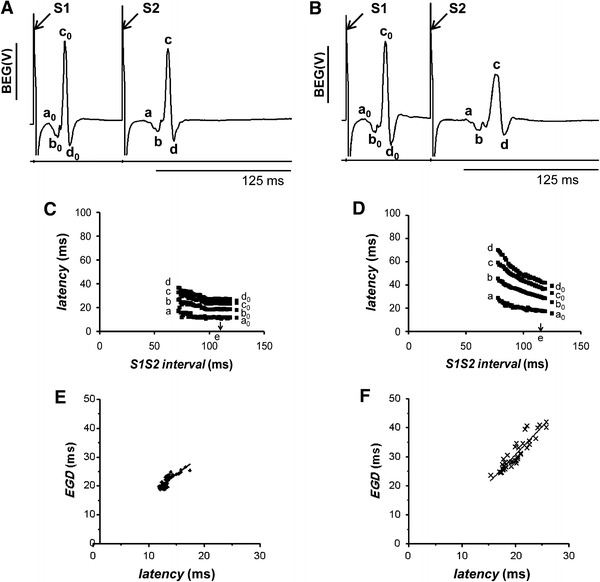
Representative bipolar electrogram (BEG) recordings taken in the penultimate cycle of the programmed electrical stimulation (PES) procedure after the final regular stimulus (S1) and the extrastimulus (S2), before (A) and 120 s after (B) introduction of 2 mM heptanol. The latencies of the successive peaks and troughs of the BEGs obtained in response to S2 stimulation were designated a, b, c, and d respectively. Those latencies obtained in response to S1 stimulation were designated a 0, b 0, c 0, and d 0. Application of paced electrogram fractionation analysis (PEFA) to BEG recordings obtained before (C) and after (D) introduction of 2 mM heptanol, yielding series of conduction curves. The critical S1S2 interval (arrow, point e) was defined as the S1S2 interval at which a 0 increased by 5%. The electrogram duration (EGD) was calculated from the difference between the latencies of the first and last peaks or troughs. The EGD ratio was defined as (a − d)/(a 0 − d 0). Activation latency plotted against EGD before (E) and after (F) introduction of 2 mM heptanol
The EGD was calculated from the difference between the latencies of the first and last peaks or troughs. It was then plotted as a function of the latency of the first deflection, which is denoted a. Figure 7E, F show typical examples of these plots obtained before and after introduction of 2 mM heptanol, respectively. It can be seen that as the latency increased, EGD also increased in both cases. EGDs therefore provided an indication of the degree of slowed conduction. Linear regression analysis of EGD and activation latency by least-squares fitting was then performed. In each heart, the correlation coefficient was statistically significant both before and after introduction of 2 mM heptanol (Student’s t test, P < 0.05). The means of the gradients of the linear functions for all the hearts were then determined. The gradients obtained both before (1.0 ± 0.2, n = 5) and after introduction of heptanol (0.8 ± 0.2, n = 5) were significantly greater than zero (Student’s t test, P < 0.05), suggesting significant relationships between latency and EGD. However, they were not significantly different from each other (Student’s t test, P > 0.05), suggesting that the relationship between latency and EGD was not altered by heptanol.
The EGD ratio was given by (a − d)/(a 0 − d 0). The critical S1S2 interval (point e, indicated by an arrow in Fig. 7D) was defined as the S1S2 interval at which a 0 increased by 5% [21]. The EGD ratio was 1.3 ± 0.04 before introduction of 2 mM heptanol (n = 5). This was significantly increased by 2 mM heptanol to 1.8 ± 0.2 (n = 5; ANOVA, P < 0.05), indicating increased dispersion of conduction velocities. However, the critical S1S2 intervals obtained before (107.3 ± 5.9 ms, n = 5) and after introduction of 2 mM heptanol (115.1 ± 2.1 ms, n = 5) were statistically indistinguishable from each other (ANOVA, P > 0.05). The EGD ratios were plotted against critical S1S2 intervals to stratify arrhythmogenic risk, as previously performed elsewhere [18, 21, 26]. Figure 8 shows such a plot for our experimental system. Diamonds represent untreated hearts and crosses represent hearts treated with 2 mM heptanol. This plot clearly indicates that increased EGD ratios were obtained for hearts treated with 2 mM heptanol without changes in the critical S1S2 intervals in the resulting scattergram.
Fig. 8.
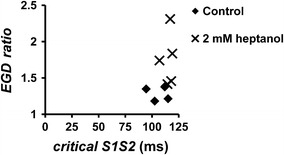
A scattergram of EGD ratios obtained before (n = 5) and after (n = 5) introduction of 2 mM heptanol plotted against critical S1S2 intervals
These results therefore indicate that 2 mM heptanol increased activation latencies, consistent with slowed conduction, and increased EGDs and EGD ratios, consistent with increased dispersion of conduction velocities. Finally, we demonstrated, for the first time in the mouse heart, that EGD directly correlated with activation latency and therefore with conduction velocity. Taken together, all the above results indicate that slowed conduction alone exerted reversible arrhythmogenic effects despite repolarization abnormalities expected to reduce arrhythmogenicity in the mouse heart.
Discussion
Heptanol is a known inhibitor of two determinants of conduction velocity [11], sodium current (I Na) activation and gap-junction conduction between myocytes [12, 14]. Slowed conduction is a possible arrhythmogenic mechanism in pathological conditions such as myocardial ischaemia [31], ventricular hypertrophy [32], heart failure [33], and the Brugada syndrome [10]. This study therefore investigated whether slowed conduction can induce arrhythmogenic effects in the absence of the remaining repolarization abnormalities previously also implicated in arrhythmogenesis. This was achieved by applying heptanol to Langendorff-perfused mouse hearts, which have previously been used to study the consequences of genetic modification on ion-channel abnormalities [1]. Electrical stimulation took the form of both regular pacing at 8 Hz, which is close to the mouse heart rate observed in vivo [27], and programmed electrical stimulation (PES) [18, 19]. The stimulating electrode was placed at the right ventricle rather than the right atrium, in order to study the specific effects of heptanol on the ventricle.
Electrical recordings were obtained by use of both monophasic action potential (MAP) and bipolar electrogram (BEG) techniques, and were used to assess changes in arrhythmogenicity, activation latencies, action potential durations (APDs) and ventricular effective refractory periods (VERPs). There are several justifications for use of the MAP technique to study cardiac electrophysiological properties. First, it enables electrical recordings to be made in the intact, perfused heart. Second, APDs measured by MAPs closely replicate the durations of the intracellular action potential [23, 34, 35], despite the fact that MAP electrodes record electrical activity from groups of cells rather than single cells. Third, this technique has proved sufficiently sensitive in mouse hearts to detect alterations in both APDs and activation latencies after pharmacological and genetic manipulation [2–4, 6, 9, 10], although it does not provide information on the upstroke velocity of the cellular action potential. Fourth, the VERP values obtained during PES were accurate to the nearest millisecond, because the S1S2 interval was shortened successively by 1 ms until an action potential was not elicited by the S2 extrastimulus.
Both epicardial and endocardial MAPs were recorded in order to study the transmural differences in the effects produced by heptanol. It was necessary to remove a small portion of the apex of the hearts to gain access to and record MAPs from the endocardium. When we recorded endocardial MAPs from the site where the apex was cut, prolonged activation latencies were observed that were indicative of conduction abnormalities resulting from tissue injury [36, 37]. However, for the experiments we placed the endocardial MAP electrode away from the apex and the injury site. The resulting endocardial MAPs had stable and reproducible waveforms for prolonged periods, and activation latencies that were not significantly different from those recorded from the epicardium before introduction of heptanol.
Heptanol increases ventricular arrhythmogenicity, slows conduction, and increases the dispersion of conduction velocities
Introduction of 2 mM heptanol significantly increased the proportions of hearts in which spontaneous and provoked ventricular tachycardia (VT) were observed during regular pacing and PES, respectively. These arrhythmogenic phenomena were accompanied by increases in activation latencies of MAPs and BEGs, consistent with slowed conduction. For both MAP and BEG findings the changes in activation latency were in agreement. Furthermore, conduction of action potentials to the endocardium was slower than that to the epicardium after introduction of heptanol at concentrations >0.5 mM. These results contrast with previous findings that heptanol slows conduction to the epicardium and endocardium to similar extents in the rabbit ventricle [15], consistent with heptanol inducing more major alterations in the detailed pathways taken by the observed excitation in mouse hearts. These results are in agreement with reports that heptanol slows conduction in sheep Purkinje fibres [28], papillary muscle of the rat ventricle [16], and rabbit ventricle [15]. They extend previous findings that heptanol at 0.05 mM increases arrhythmogenicity but at 0.1–1 mM reduces arrhythmogenicity in the ischaemic rat heart [16]. Our results also complement reports associating slowed conduction with increased arrhythmogenicity in mXinα +/− [8], Scn5a +/− [10, 19], and Cx43 +/− hearts [7, 38, 39].
The arrhythmogenesis observed in heptanol-treated hearts was further analysed by application of paced electrogram fractionation analysis (PEFA) [18, 26] to the BEG recordings obtained during PES. PEFA plotted the latency of each peak and trough against S1S2 intervals for each heart, yielding a family of conduction curves. Electrogram durations (EGDs) and conduction latencies were derived from these curves. Increases in the EGD ratios reflect abnormal patterns of myocardial activation that resulted in increases in the dispersion of conduction velocities. Increases in both the EGDs and the ratios of EGDs obtained at the longest S1S2 interval to those obtained at the shortest S1S2 interval were observed for heptanol-treated hearts. Our results were, therefore, similar to those also indicative of increases in EGD ratios in KCNE1 −/− [21], Scn5a +/Δ [20], and Scn5a +/− [10] hearts. Furthermore, we demonstrated for the first time that there was a significant and positive linear relationship between latency and EGDs that was not altered by heptanol. EGDs therefore directly reflected the degree of slowed conduction.
Heptanol induces repolarization abnormalities that are expected to reduce rather than increase arrhythmogenicity
The observed arrhythmogenic phenomena occurred despite repolarization abnormalities that were expected to reduce rather than increase arrhythmogenicity in the mouse heart. First, epicardial or endocardial APDx values at x = 30, 50, 70, and 90% repolarization were not significantly altered at all of the heptanol concentrations investigated during 8-Hz pacing. Therefore, ΔAPD90, given by endocardial APD90 − epicardial APD90, was also unaltered. Thus, increased arrhythmogenicity after introduction of heptanol did not involve local or transmural changes in APDs [40]. Out results contrast with those from previous mouse studies that have associated increased arrhythmogenicity with alterations in the time courses of action potential repolarization. Increased APDs, reflected in the APD90 values, have been observed for hypokalaemic [2] and mXinα +/− [8] hearts. This predisposed them to early after-depolarizations and triggered activity. In contrast, increased arrhythmogenicity in Cx43 +/− hearts was associated with reduced APDs reflected in the APD50, APD70, and APD90 values. This predisposed them to re-entrant activity [7]. However, their decreases in APD were attributed to ion-channel remodelling that resulted in increased levels of I sus and I K1 rather than decreases in gap junction conductance per se. Furthermore, increased arrhythmogenicity in heptanol-treated rabbit hearts was associated with both reduced APDs, reflected in the activation recovery intervals, and slowed conduction [15]. Therefore, it was not then possible to study the contributions of slowed conduction to arrhythmogenesis in the absence of alterations in repolarization time in the rabbit heart. This was possible using mouse hearts in our study.
This study confirmed that action potential features in mouse hearts differ from those of other species, for example rabbit hearts, in showing shorter time-courses and an absence of plateau components. This has been attributed to different ion-current contributions to their action potentials. Thus, larger calcium (I Ca) and rapid and slow delayed rectifier potassium (I Kr and I Ks, respectively) relative to their transient outward current (I to) contributions have been established for the latter species [41, 42]. Such differences would, in turn, be compatible with different effects on APDs produced by the inhibitory actions of heptanol on sodium channels and gap junctions. The experiments here thus demonstrated that heptanol did not alter APDs in mouse hearts, in contrast with reducing APDs in rabbit hearts [15]. Nevertheless this empirical observation enabled the demonstration that ventricular arrhythmias could result from acute reductions in conduction velocity without alterations in action potential recovery time course, even with increased refractory periods. Finally, these results contrast with those of the previous studies using hypokalaemic [2], KCNE1 −/− [3], and Scn5a +/Δ [4] and Scn5a +/− [9] hearts, for which alterations in the transmural gradients of repolarization were observed.
Second, heptanol produced concentration-dependent increases in VERP, reaching significance at 0.1 mM. This resulted in decreases in the local critical intervals given by APD90 − VERP. This would predict a decrease rather than an increase in arrhythmogenicity. Furthermore, the transmural critical interval given by (endocardial latency + endocardial APD90) − (epicardial latency + epicardial VERP) was unaltered. This would predict no change in arrhythmogenicity. These critical intervals reflect the time periods permitting re-excitation during the course of action potential repolarization [5]. The heptanol-treated hearts therefore contrast with hypokalaemic [5], KCNE1 −/− [3], and Scn5a +/Δ [6] hearts, because increased critical intervals in these models were associated with arrhythmogenesis.
Our findings show, for the first time in the mouse heart, that slowed conduction alone is sufficient to produce ventricular arrhythmogenesis, despite repolarization abnormalities expected to reduce rather than increase arrhythmogenicity, and may do so by reducing the wavelength of excitation.
Acknowledgments
This work was supported by the Wellcome Trust, the Medical Research Council, the British Heart Foundation, the Helen Kirkland Trust, Papworth Hospital, UK, and Xention Discovery. GT was supported by a Wellcome Trust Vacation Scholarship, Trinity Hall, Cambridge, a Biotechnology and Biological Sciences Research Council CASE Studentship, and Xention Discovery. SSH was supported by a Medical Research Council Capacity Building Studentship.
Footnotes
G. Tse and S. S. Hothi contributed equally to this paper.
References
- 1.Sabir IN, Killeen MJ, Grace AA, Huang CL. Ventricular arrhythmogenesis: insights from murine models. Prog Biophys Mol Biol. 2008;98:208–218. doi: 10.1016/j.pbiomolbio.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Killeen MJ, Thomas G, Gurung IS, Goddard CA, Fraser JA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL. Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol (Oxf) 2007;189:33–46. doi: 10.1111/j.1748-1716.2006.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hothi SS, Thomas G, Killeen MJ, Grace AA, Huang CL. Empirical correlation of triggered activity and spatial and temporal re-entrant substrates with arrhythmogenicity in a murine model for Jervell and Lange-Nielsen syndrome. Pflugers Arch. 2009;458:819–835. doi: 10.1007/s00424-009-0671-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CL. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Δ murine hearts modelling long QT syndrome 3. J Physiol. 2007;578:69–84. doi: 10.1113/jphysiol.2006.117945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang CL. The contribution of refractoriness to arrhythmic substrate in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. 2007;454:209–222. doi: 10.1007/s00424-007-0217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hothi SS, Booth SW, Sabir IN, Killeen MJ, Simpson F, Zhang Y, Grace AA, Huang CL. Arrhythmogenic substrate and its modification by nicorandil in a murine model of long QT type 3 syndrome. Prog Biophys Mol Biol. 2008;98:267–280. doi: 10.1016/j.pbiomolbio.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Danik SB, Rosner G, Lader J, Gutstein DE, Fishman GI, Morley GE. Electrical remodeling contributes to complex tachyarrhythmias in connexin-43-deficient mouse hearts. FASEB J. 2008;22:1204–1212. doi: 10.1096/fj.07-8974com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin CI, Cheng CP, Loh YX, Gustafson-Wagner EA, Lai YJ, Chen YC, Lin JJ (2005) Electrophysiological characteristics of ventricular myocytes of Xinα-deficient mice. In: Proceedings of the 12th World Congress on Heart Disease. Advances in Heart Disease, Vancouver
- 9.Martin CA, Zhang Y, Grace AA, Huang CL. Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada syndrome. J Cardiovasc Electrophysiol. 2010;21:1153–1159. doi: 10.1111/j.1540-8167.2010.01767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stokoe KS, Balasubramaniam R, Goddard CA, Colledge WH, Grace AA, Huang CL. Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/− murine hearts modelling the Brugada syndrome. J Physiol. 2007;581:255–275. doi: 10.1113/jphysiol.2007.128785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 12.Christ GJ, Spektor M, Brink PR, Barr L. Further evidence for the selective disruption of intercellular communication by heptanol. Am J Physiol. 1999;276:H1911–H1917. doi: 10.1152/ajpheart.1999.276.6.H1911. [DOI] [PubMed] [Google Scholar]
- 13.Rudisuli A, Weingart R. Electrical properties of gap junction channels in guinea-pig ventricular cell pairs revealed by exposure to heptanol. Pflügers Arch. 1989;415:12–21. doi: 10.1007/BF00373136. [DOI] [PubMed] [Google Scholar]
- 14.Nelson WL, Makielski JC. Block of sodium current by heptanol in voltage-clamped canine cardiac Purkinje cells. Circ Res. 1991;68:977–983. doi: 10.1161/01.res.68.4.977. [DOI] [PubMed] [Google Scholar]
- 15.Keevil VL, Huang CL, Chau PL, Sayeed RA, Vandenberg JI. The effect of heptanol on the electrical and contractile function of the isolated, perfused rabbit heart. Pflügers Arch. 2000;440:275–282. doi: 10.1007/s004240000264. [DOI] [PubMed] [Google Scholar]
- 16.Chen BP, Mao HJ, Fan FY, Bruce IC, Xia Q. Delayed uncoupling contributes to the protective effect of heptanol against ischaemia in the rat isolated heart. Clin Exp Pharmacol Physiol. 2005;32:655–662. doi: 10.1111/j.0305-1870.2005.04246.x. [DOI] [PubMed] [Google Scholar]
- 17.Sims JJ, Schoff KL, Loeb JM, Wiegert NA. Regional gap junction inhibition increases defibrillation thresholds. Am J Physiol Heart Circ Physiol. 2003;285:H10–H16. doi: 10.1152/ajpheart.01074.2002. [DOI] [PubMed] [Google Scholar]
- 18.Saumarez RC, Grace AA. Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res. 2000;47:11–22. doi: 10.1016/S0008-6363(00)00096-1. [DOI] [PubMed] [Google Scholar]
- 19.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CL. Paced electrogram fractionation analysis of arrhythmogenic tendency in ΔKPQ Scn5a mice. J Cardiovasc Electrophysiol. 2005;16:1329–1340. doi: 10.1111/j.1540-8167.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- 21.Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CL. Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol. 2003;552:535–546. doi: 10.1113/jphysiol.2003.048249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hothi SS, Gurung I, Heathcote J, Zhang Y, Booth S, Skepper J, Grace AA, Huang CL. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflügers Arch. 2008;457:253–270. doi: 10.1007/s00424-008-0508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knollmann BC, Katchman AN, Franz MR. Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol. 2001;12:1286–1294. doi: 10.1046/j.1540-8167.2001.01286.x. [DOI] [PubMed] [Google Scholar]
- 24.Gussak I, Chaitman BR, Kopecky SL, Nerbonne JM. Rapid ventricular repolarization in rodents: electrocardiographic manifestations, molecular mechanisms, and clinical insights. J Electrocardiol. 2000;33:159–170. doi: 10.1016/S0022-0736(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 25.Fabritz L, Kirchhof P, Franz MR, Eckardt L, Mönnig G, Milberg P, Breithardt G, Haverkamp W. Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol. 2003;98:25–32. doi: 10.1007/s00395-003-0386-y. [DOI] [PubMed] [Google Scholar]
- 26.Saumarez RC, Chojnowska L, Derksen R, Pytkowski M, Sterlinski M, Huang CL, Sadoul N, Hauer RN, Ruzyłło W, Grace AA. Sudden death in noncoronary heart disease is associated with delayed paced ventricular activation. Circulation. 2003;107:2595–2600. doi: 10.1161/01.CIR.0000068342.96569.A1. [DOI] [PubMed] [Google Scholar]
- 27.Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci USA. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jalife J, Sicouri S, Delmar M, Michaels DC. Electrical uncoupling and impulse propagation in isolated sheep Purkinje fibers. Am J Physiol. 1989;257:H179–H189. doi: 10.1152/ajpheart.1989.257.1.H179. [DOI] [PubMed] [Google Scholar]
- 29.Saumarez RC, Camm AJ, Panagos A, Gill JS, Stewart JT, de Belder MA, Simpson IA, McKenna WJ. Ventricular fibrillation in hypertrophic cardiomyopathy is associated with increased fractionation of paced right ventricular electrograms. Circulation. 1992;86:467–474. doi: 10.1161/01.cir.86.2.467. [DOI] [PubMed] [Google Scholar]
- 30.Saumarez RC, Heald S, Gill J, Slade AK, de Belder M, Walczak F, Rowland E, Ward DE, Camm AJ. Primary ventricular fibrillation is associated with increased paced right ventricular electrogram fractionation. Circulation. 1995;92:2565–2571. doi: 10.1161/01.cir.92.9.2565. [DOI] [PubMed] [Google Scholar]
- 31.Stables CL, Curtis MJ. Development and characterization of a mouse in vitro model of ischaemia-induced ventricular fibrillation. Cardiovasc Res. 2009;83:397–404. doi: 10.1093/cvr/cvp068. [DOI] [PubMed] [Google Scholar]
- 32.Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–H2692. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boulaksil M, Winckels SK, Engelen MA, Stein M, van Veen TA, Jansen JA, Linnenbank AC, Bierhuizen MF, Groenewegen WA, van Oosterhout MF, Kirkels JH, de Jonge N, Varró A, Vos MA, de Bakker JM, van Rijen HV. Heterogeneous connexin-43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur J Heart Fail. 2010;12:913–921. doi: 10.1093/eurjhf/hfq092. [DOI] [PubMed] [Google Scholar]
- 34.Franz MR, Burkhoff D, Spurgeon H, Weisfeldt ML, Lakatta EG. In vitro validation of a new cardiac catheter technique for recording monophasic action potentials. Eur Heart J. 1986;7:34–41. doi: 10.1093/oxfordjournals.eurheartj.a061954. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman BF, Cranefield PF, Lepeschkin E, Surawicz B, Herrlich HC. Comparison of cardiac monophasic action potentials recorded by intracellular and suction electrodes. Am J Physiol. 1959;196:1297–1301. doi: 10.1152/ajplegacy.1959.196.6.1297. [DOI] [PubMed] [Google Scholar]
- 36.Franz MR, Flaherty JT, Platia EV, Bulkley BH, Weisfeldt ML. Localization of regional myocardial ischemia by recording of monophasic action potentials. Circulation. 1984;69:593–604. doi: 10.1161/01.CIR.69.3.593. [DOI] [PubMed] [Google Scholar]
- 37.Donaldson RM, Taggart P, Swanton H, Fox K, Noble D, Rickards AF. Intracardiac electrode detection of early ischaemia in man. Br Heart J. 1983;50:213–221. doi: 10.1136/hrt.50.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin-43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antzelevitch C, Fish J. Electrical heterogeneity within the ventricular wall. Basic Res Cardiol. 2001;96:517–527. doi: 10.1007/s003950170002. [DOI] [PubMed] [Google Scholar]
- 41.Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008;118:2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Killeen MJ, Thomas G, Sabir IN, Grace AA, Huang CL. Mouse models of ventricular arrhythmias. Acta Physiol (Oxf) 2008;192:455–469. doi: 10.1111/j.1748-1716.2007.01822.x. [DOI] [PubMed] [Google Scholar]