

Abstract
Eccentric contractions (ECC) induce myofibrillar collapse, edema, and inflammation in muscle cells. Although apoptosis of myonuclei following ECC is activated during the inflammatory phase, the apoptosis response of the regenerative phase remains to be elucidated. The aim of the present study was to determine the inflammatory and regenerative phase of the apoptosis responses induced by ECC. In anesthetized rats, the tibialis anterior muscles were subjected to ECC repeated 40 times, evoked by surface electric stimulation (100 Hz, 10 V) with mechanical muscle stretch. Apoptosis was examined in the control group and in groups 1, 3, 7, and 14 days after ECC (each group, n = 4–6). Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL)-positive myonuclei were assessed by further labeling with dystrophin staining and DAPI. The expression of proteins related to apoptosis (Bcl-2 and Bax) was examined by Western blot assay. At 1 and 3 days, focal edema and necrotic myofibers invaded by mononuclear phagocytes were present, whereas regenerated myofibers with central nuclei were detected at 7 and 14 days. The occurrence of TUNEL-positive myonuclei increased significantly at 7 (7.0 ± 1.5%) and 14 days (5.6 ± 0.6%) compared with control (0.9 ± 0.5%). Further we found that myonuclear apoptosis was restricted to the subsarcolemmal space at 7 and 14 days and markedly absent from the central nucleus. The Bax/Bcl-2 ratio was significantly higher at 3 (4.5 ± 0.9) and 7 days (3.4 ± 0.5) after ECC. In conclusion, myofiber apoptotic responses following ECC are present not only in the inflammatory phase but also persist during the regenerative phase.
Keywords: Muscle damage, Bcl-2, Bax
Introduction
It is well known that repetitive eccentric muscle contractions (ECC) induce muscle damage. ECC-induced muscle damage leads to myofiber necrosis accompanied by ultrastructural collapse, edema, and inflammation of myofibers [6, 16, 17, 46]. These inflammatory reactions achieve their peak responses 3 days after ECC, and subsequently the muscle fiber shifts to the process of regeneration in ~7 days [6, 20, 21]. The elimination of damaged myofibers by necrosis constitutes one of a series of responses in the process of myofiber regeneration. It is also known that a damaged or dysfunctional cell or myofiber is removed by a genetic control through apoptosis [23, 31]. In fact, apoptotic induction is observed with necrosis in myofibers due to exercise [7, 12, 13, 30, 36, 42]. Previous studies demonstrated that apoptosis plays an important role in controlling the cell differentiation processes of skeletal muscle [8, 27]. For example, caspase-3 activation of apoptosis-induced factor is required for skeletal muscle differentiation [19, 25]. MyoD, a muscle-specific transcription factor, promotes differentiation of satellite cells. Recently, Asakura et al. [8] reported that MyoD plays an essential role in apoptosis during proliferation and differentiation phases. Therefore apoptosis may play an important role for controlling the cell differentiation processes of skeletal muscle. Based on these studies, we hypothesized that the apoptosis response induced by ECC would be activated in the regeneration phase as well as the inflammation phase. The purpose of the present investigation, therefore, was to test this hypothesis by examining the time course of the apoptotic response from the early inflammation phase through the regeneration phase following ECC.
In addition, endurance training causes apoptosis to a muscular capillary (i.e., endothelial cell), but little is known about whether ECC contraction with significant damage induces endothelial cell apoptosis. We also examined apoptosis induction in endothelial cells during inflammation and regeneration process.
Materials and methods
Animals
Male Wistar rats (n = 27, Japan SLC) 12 weeks of age were used in this study. Rats were maintained on a 12:12-h light-dark cycle and received food and water ad libitum. All experiments were conducted under the guidelines established by the Physiological Society of Japan and were approved by University of Electro-Communications Institutional Animal Care and Use Committee.
Eccentric contractions protocol
During eccentric exercise and all surgical procedures, the rats were anesthetized with intraperitoneal injection of pentobarbital sodium (70 mg kg−1 i.p.), and supplemental doses of anesthesia were administered as needed. The right tibialis anterior (TA) muscle was stimulated electrically via a surface electrode (10 V stimulation, 100 Hz frequency, 700 ms stimulation period, i.e., 70 pulses). In preliminary experiments and our previous study [26], we confirmed that maximum muscle tension was achieved by electrical stimulation of a surface electrode (100 Hz, <10 V). In the resting condition before ECC, the right foot was attached to the clamp unit and the plate was connected to the electromotor system (Model RU-72, NEC Medical Systems). The right ankle joint was maintained at 50° as the initial angle, and during electrostimulation of the TA muscle, the electromotor was rotated at an angular velocity of 260° s−1 to 180° of the ankle joint, which lengthened the dorsiflexor muscle group. The muscle tension generated during ECC was monitored using a strain gauge that was incorporated into the plate fixing the foot. The strain gauge was calibrated using precision calibration weights that spanned the expected range of strains. The right TA muscle was subjected to 40 repeated ECCs. Each rat was assigned randomly to one of five groups and examined at control (n = 5) or 1 (n = 6), 3 (n = 4), 7 (n = 6), or 14 (n = 6) days post-ECC.
Histological evaluation for muscle damage
At 1, 3, 7, and 14 days after ECC, the TA muscles of both legs were carefully dissected and the mid-belly region cut transversely to the long axis of the muscle. The tissue blocks were frozen rapidly in isopentane cooled by liquid nitrogen. Transverse sections of 10 μm were made with a cryostat (Leica, CM1510) at −20°C and stained with hematoxylin–eosin (HE) to examine the histological features of muscle damage. To avoid sampling bias, each section was subsampled at six different regions: (1) anterior-medial, (2) anterior-central, (3) anterior-lateral, (4) posterior-medial, (5) posterior-central, and (6) posterior-lateral; each of these fields was analyzed in all muscles. Muscle fiber damage was determined by a point counting method using a 30 × 40 mounted grid (i.e., 1,200 points total; one square = 18 × 18 μm) on microscopic fields. Damaged myofibers were defined as those with infiltration of inflammatory cells, pale staining of the cytoplasm, swollen appearance, or multiple central nuclei (Fig. 1A, B). Muscle fiber damage was expressed as a percentage of counted grid squares.
Fig. 1.
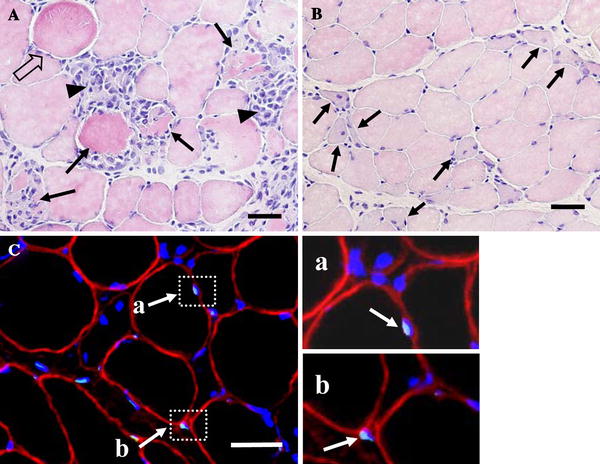
a Hematoxylin and eosin staining 3 days after ECC. Damaged myofibers were defined as those inflammatory cells with swollen (outlined arrows), swollen with infiltration (arrows), and infiltration (arrow-heads) appearance. b The multiple central nuclei can be detected (arrows) after 7 days of ECC. c TUNEL-positive nuclei were found both inside (a) and outside (b) myofibers. Muscle transverse sections were immunofluorescent-stained with dystrophin antibody to identify the sarcolemma (red), fluorescein-mediated TUNEL assay was performed to identify apoptotic nuclei (light blue), and all nuclei were labeled by DAPI staining (blue). TUNEL-positive nuclei positioned inside the dystrophin stain (a) were identified as myofiber nuclei, whereas other nuclei located outside (b) were counted as endothelial or interstitial cell nuclei. Bar 50 μm
Identification of myofiber apoptosis
Double labeling with the TUNEL assay was performed after dystrophin labeling. The sections were air dried at room temperature, fixed in 4% paraformaldhyde for 15 min, and washed twice with phosphate-buffered saline (PBS, 10 min each). The tissue was blocked by 1% bovine serum albumin (BSA) in PBS for 30 min at room temperature. After washing in PBS, sections were incubated with an anti-dystrophin mouse–monoclonal antibody (1:30, VP-D 505, Vector Laboratories) for 1 h at room temperature, followed by an anti-mouse IgG rhodamine-conjugated antibody fragment incubation for 1 h at 37°C (1:100, R0270, Dako).
After dystrophin labeling, apoptotic nuclei were assessed from muscle cross-sections via a TUNEL assay. Apoptotic nuclei were identified by a fluorometric TUNEL detection kit (MEBSTAIN Apoptosis Kit Direct, Co. 8445, Medical & Biological Laboratories) according to the manufacturers’ instructions for both muscle cross-sections. Briefly, tissue sections were incubated with a fluorescein-conjugated TUNEL reaction. Negative control experiments were performed by omitting the TdT enzyme in the TUNEL reaction mixture on the tissue sections. After dystrophin and TUNEL labeling, the muscle sections were mounted with 4′6′-diamidino-2-phenylindole (DAPI; H-1200, Vector Laboratories). TUNEL- and DAPI-positive nuclei and dystrophin staining were captured under a fluorescence microscope (Eclipse E800, Nikon). The number of TUNEL and DAPI-positive nuclei was counted from six random, nonoverlapping fields at an objective magnification of ×10. Only the TUNEL-positive nuclei inside the dystrophin were quantified as myofiber apoptotic nuclei (Fig. 1C). The TUNEL labeling was quantified as the number of myofiber TUNEL-positive nuclei per total myofiber nuclei.
Identification of endothelial apoptosis
For endothelial apoptosis, serial cross-sections were stained using TUNEL and alkaline phosphatase-dipeptidyl peptidase IV (AP-DPPIV) stain [29], respectively. A small amount of substrate solution specific for DPPIV was poured onto the sections, which were maintained at 4°C overnight. The sections were rinsed with PBS and treated with the substrate solution specific for AP at 37°C for 60 min. The solution for demonstrating DPPIV consisted of glycyl-l-proline-4-methoxy-beta-naphthyamine, which is a substrate for DPPIV, dissolved in 1 ml of N,N-dimethylformamide, and fast blue B dissolved in 0.1 M acetate buffer (pH 7.4). The solution for AP consists of naphthol AS-MAX phosphate, which is a substrate for AP, dissolved in N,N-dimethylformamide, and variamine blue salt RT dissolved in 0.1 M Tris–HCl buffer (pH 9.2).
Western blot analysis for Bcl-2 and Bax
Protein expressions of Bcl-2 and Bax were determined in TA muscles after ECC. The samples were homogenized in homogenizing buffer (50 mM Tris, 1 mM EDTA, PMSF) and centrifuged at 8,000g for 15 min (4°C). Soluble protein was heated for 5 min at 95°C, loaded on each lane of a 12% polyacrylamide gel, and separated by routine SDS-PAGE for 50 min (20 V) at room temperature. The gels were blotted to PVDF membranes (Millipore) by semi-dry assay. The membranes were blocked in 10% skim milk in PBS with 0.05% Tween-20 (PBS-T) at room temperature for 1 h and probed with anti-Bcl-2 mouse monoclonal antibody (1:500, sc-7382, Santa Cruz Biotechnology) and anti-Bax rabbit polyclonal antibody (1:500, sc-493, Santa Cruz Biotechnology) diluted in PBS-T.
All primary antibody incubations were at room temperature for 1.5 h. Membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody, AP1254 for anti-mouse IgG antibody, and AP758 for anti-rabbit IgG antibody, and the signals were developed by ECL plus Western Blotting Detection System (RPN2132, Amersham Biosciences). The resulting bands were quantified as OD × band area by CS analyzer (ATTO Corporation). The size of the immuno-detected proteins was verified by using standard molecular-weight markers (Bio-Rad Laboratories).
Statistical analyses
Statistical analyses consisted of a one-way ANOVA with Dunnett post-hoc testing using Prism version 4.0 (GraphPad software). For all statistical tests, the 0.05 and 0.01 levels of confidence were accepted for statistical significance. All values are reported as mean ± SE.
Results
Muscle fiber damage
Histological examination of control muscles revealed no visible evidence of muscle tissue inflammation. In contrast, typical histological lesions appeared in the exercised muscle fiber after around 1–3 days (Fig. 2, Table 1). One day after ECC, damaged muscle fiber showed the swollen condition (2.4 ± 2.1% of total fiber area). At 3 days after ECC, focal edema and necrotic fiber invaded by mononuclear phagocytes were recognized and the extent of the damaged fiber area was significantly increased (23.4 ± 6.2% of total fiber area, p < 0.01 vs. control). A regeneration process was observed at 7 and 14 days after ECC, characterized by a small-diameter fiber with multiple central nuclei (7 days: 4.5 ± 1.3%, 14 days: 3.7 ± 1.4% of total fiber area, p < 0.05 vs. control).
Fig. 2.
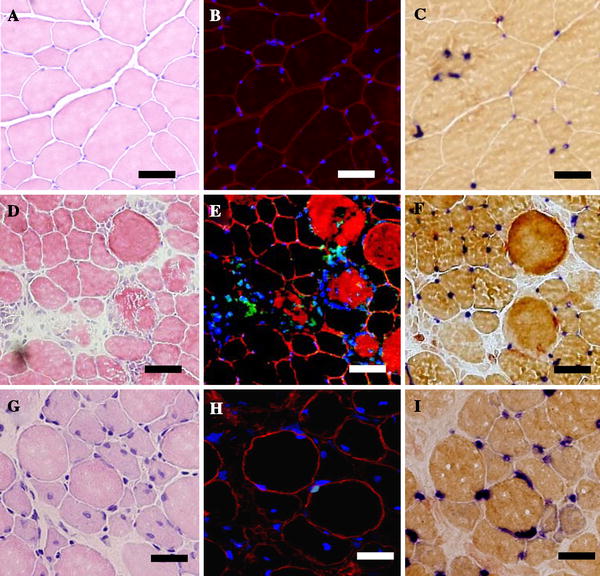
Serial transverse sections from control (a–c), 3 days (d–f), and 7 days (g–i) after ECC in TA muscle. H&E staining (a, d, g) was performed to examine the histological features of muscle damage, triple histochemical staining (b, e, h) for myofiber apoptotic nuclei was used as shown in Fig. 1, and AP-DPPIV staining was performed to identify endothelial cells (c, f, i). Bar 50 μm
Table 1.
Percentage of inflammation and regeneration muscle fiber after ECC
| Control | 1 day | 3 days | 7 days | 14 days | |
|---|---|---|---|---|---|
| Inflammation fiber (%) | 0.0 ± 0.0 | 2.4 ± 2.2 | 23.4 ± 6.2** | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Regeneration fiber (%) | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.5 ± 1.3* | 3.7 ± 1.4* |
Values are mean ± SE
* p < 0.05, ** p < 0.01, significant difference from control
Myofiber apoptosis and dystrophin
The TUNEL assay and dystrophin staining to investigate the expression of myofiber apoptosis are presented in Fig. 1. Loss of dystrophin was not observed in the control and 1 day after ECC. Myofiber TUNEL-positive nuclei were marginally detectable in control and 1 day after ECC (control: 0.9 ± 0.5%, 1 day: 2.5 ± 1.1%, Fig. 3). In contrast, intense staining of TUNEL-positive nuclei and loss of dystrophin were observed 3 days after ECC (Fig. 2). Because of sarcolemmal degeneration after 3 days of ECC, we were unable to quantify myofiber apoptosis at this time point. In the regeneration phase (7 and 14 days after ECC), loss of dystrophin was not observed, however, regenerated myofibers with central nuclei were detected. TUNEL-positive myonuclei were significantly increased at 7 (7.0 ± 1.5%, p < 0.01) and 14 days (5.6 ± 0.6%, p < 0.05) compared with control.
Fig. 3.
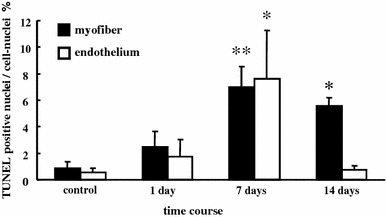
Percentage of myofiber and endothelial cell nuclei identified as apoptotic in control and post-ECC muscles at 1, 7, and 14 days. Values are mean ± SE. *p < 0.05, **p < 0.01, significant difference from control
Figure 4 shows the longitudinal section of TA muscle at 7 and 14 days after ECC by TUNEL and dystrophin staining. We found that myonuclear apoptosis was located in the subsarcolemmal region rather than the central nucleus at 7 and 14 days.
Fig. 4.
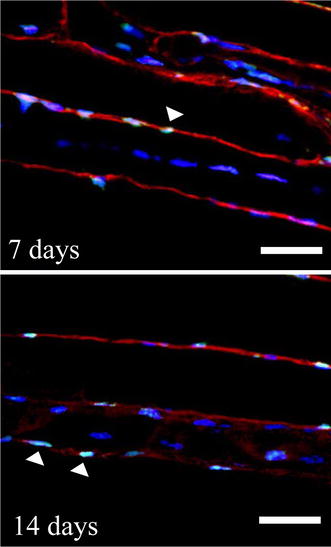
Longitudinal sections taken 7 and 14 days after ECC in TA muscle. All nuclei were stained with DAPI (blue), and nuclei with internal dystrophin stain (red) were defined as myofiber nuclei. TUNEL-positive myofiber nuclei (arrow) were located in the subsarcolemma, but central nuclei were not detected. Bar 50 μm
Endothelial apoptosis
Using serial sections by TUNEL and AP-DPPIV assay, we evaluated vascular endothelial cell apoptosis (Fig. 2). We observed TUNEL-positive endothelial nuclei at 7 days (7.6 ± 3.6%, p < 0.05 vs. control) after ECC. The myonuclear apoptosis remained higher than control at 14 days after ECC, whereas endothelial nuclear apoptosis had returned to basal levels.
Expression of apoptotic proteins
We performed Western blotting to determine the pro-apoptotic (Bax) and anti-apoptotic (Bcl-2) muscle protein content (Fig. 5). Bcl-2 protein significantly decreased in muscles at 1, 3, and 7 days after ECC compared with control (1 day: −35%, 3 days: −60%, 7 days: −56%, p < 0.05, Fig. 5b). Also, there was a tendency toward an increase in Bax protein content at 3 days (88%) and 7 days (51%). Thus the Bax/Bcl-2 ratio was significantly higher at 3 (4.5 ± 0.9) and 7 (3.4 ± 0.5) days after ECC (Fig. 5c).
Fig. 5.
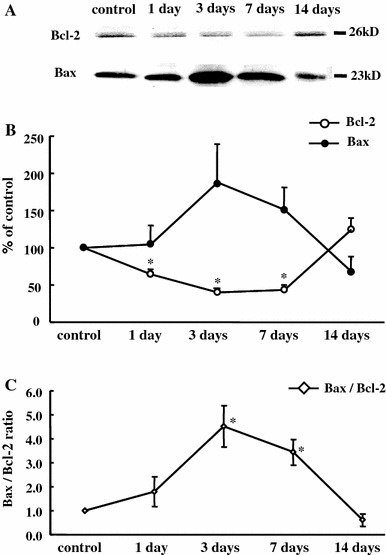
Immunoblot analysis of the protein expression of Bcl-2 and Bax. Inset displays representative images for control and ECC muscles (a). Quantification by densitometry of Bcl-2 protein was significantly decreased 1, 3, and 7 days after ECC compared with control (b). Values are expressed as the percentage of the mean values of the control. Bax/Bcl-2 ratio was significantly higher at 3 and 7 days after ECC (c). *p < 0.05, significant difference from control
Discussion
The present investigation has demonstrated for the first time that, following ECC, myofiber apoptosis is not restricted solely to the inflammation phase, but also occurs during the regeneration phase. Furthermore, this regeneration phase myonuclear apoptosis did not occur in central nuclei but rather was restricted to the subsarcolemmal region.
Myofiber apoptosis during the inflammation phase
After ECC, inflammatory responses progress over the first 3 days following insult, and damaged cells undergo phagocytosis [10, 32]. Previous studies [9, 15, 24, 33] have shown that many genes related to inflammation are expressed in skeletal muscle following high-intensity ECC. Urso et al. [48] reported that gene expression related to apoptotic pathways was approximately 9% for a total expressed gene at 8 h after ECC in human skeletal muscle. In the present investigation, we attempted to determine the time course and spatial resolution of ECC-induced myofiber apoptosis histologically. One day following ECC some apoptotic nuclei could be identified within myofibers. After 3 days, most damaged myofibers were accompanied by collapse of a plasma membrane structure that precluded identification of myofiber nuclei at that time. However, many TUNEL-positive nuclei were observed within damaged myofibers that lacked sarcolemmal dystrophin. It is generally accepted that dystrophic muscle fibers are disassembled by necrotic pathways, but several studies have suggested that acute muscle fiber degeneration occurs by apoptotic pathways [1, 41, 42, 47]. Biral and co-workers [12] also reported that TUNEL-positive nuclei were detected in fibers deprived of dystrophin or a sarcoglycan sarcolemmal staining following 6 and 24 h of ECC. This latter paper considered that the loss of several membrane skeletal proteins after ECC could lead to fiber death by apoptosis or necrosis.
Myonuclear apoptosis during the regeneration phase
We have provided original evidence that myonuclear apoptosis occurs in the regeneration phase following ECC. These findings suggest the participation of apoptosis in the processes of myofiber regeneration. The myocyte is a multinucleated cell, and it is necessary to keep a fixed muscle nuclei domain. Taking into consideration physiological significance, apoptosis in the regeneration phase is not targeting damaged myofiber for removal. We predict that apoptosis plays a major role in regulating nuclear localization and domains during myofiber regeneration. It is well known that the number of nuclei is a causative factor for the regulation of muscle fiber size [18, 22]. For example, in atrophying fibers, apoptosis contributes to the elimination of myonuclei and/or satellite cells [4, 5, 14, 43, 44]. A similar mechanism may contribute to the spatial definition of muscle nuclear domains during myofiber regeneration.
As shown in the myofiber longitudinal sections in Fig. 4, myonuclear apoptosis was localized to the subsarcolemma rather than the central cytoplasm at 7 and 14 days after ECC. If a central nucleus represents a neogenic nucleus from the satellite cell, it is possible that an existing myofiber nucleus located in the sarcolemma leads to preferential apoptosis. Muscle satellite cells are responsible for repair as they function as muscle stem cells [2, 11]. The myogenic regulatory factors such as MyoD, Myf5, and myogenin play an essential role in regulating the satellite cell activation program. In the ECC animal model employed by Okada et al. [35], which was similar to the present investigation, it was reported that the MyoD protein expression level was increased markedly from 3 to 7 days after ECC. Also, it is known that myotoxic-induced muscle damage causes high MyoD expression after 5–21 days [40]. Interestingly, Asakura et al. [8] have demonstrated that MyoD regulates not only differentiation and proliferation of satellite cells but also the apoptosis response during the regenerating phase. In the present investigation, we found a very different quantitative and temporal profile of expression of Bax and Bcl-2 between 7 and 14 days post-ECC. Though apoptosis was induced at 14 days, the Bax/Bcl-2 expression ratio declined to control levels. This profile suggests participation of other pro-apoptosis genes in the post-ECC response. Indeed, there is evidence that MyoD alters the expression of many genes involved in apoptosis [8].
Apoptosis response in endothelial cells
This study found that apoptosis occurs not only in myofibers but also in capillary endothelial cells after ECC. These findings were unexpected because we have shown that ECC does not induce necrosis of endothelial cells [26]. Little is currently known about exercise-induced capillary endothelial apoptosis. There is one report that examined apoptosis after running in mice [37]. That previous study showed that acute spontaneous running increased the number of apoptotic nuclei in endothelial cells.
There is evidence that both reactive oxygen species (ROS) [28, 34] and intracellular [Ca2+] ([Ca2+]i) dysregulation may participate in ECC-induced muscle damage [3, 45]. Specifically, high [Ca2+]i serves to stimulate calpain activity and consequently promotes neutrophil activation [39]. Activated neutrophils are potential sources of free radicals and ROS production, which subsequently damage proteins and DNA [38]. Thus, ROS caused by neutrophilic activity acts on endothelial cells from inside the vascular lumen and may induce endothelial cell apoptosis.
In conclusion, we have discovered myofiber and endothelial cell apoptosis throughout the 14-day inflammation-regeneration period following ECC in male rat skeletal muscle. Future investigations might usefully explore the specific myonuclear domains during apoptosis found in the regeneration phase following ECC-induced damage.
Acknowledgments
We gratefully acknowledge Dr. D.C. Poole at Kansas State University for helpful comments on the manuscript and Dr. S. Yamada at the University of Tokyo for excellent technical support.
References
- 1.Abmayr S, Crawford RW, Chamberlain JS. Characterization of ARC, apoptosis repressor interacting with CARD, in normal and dystrophin-deficient skeletal muscle. Hum Mol Genet. 2004;13:213–221. doi: 10.1093/hmg/ddh018. [DOI] [PubMed] [Google Scholar]
- 2.Adams GR. Satellite cell proliferation and skeletal muscle hypertrophy. Appl Physiol Nutr Metab. 2006;31:782–790. doi: 10.1139/H06-053. [DOI] [PubMed] [Google Scholar]
- 3.Allen DG, Whitehead NP, Yeung EW. Mechanisms of stretch-induced muscle damage in normal and dystrophic muscle: role of ionic changes. J Physiol. 2005;567:723–735. doi: 10.1113/jphysiol.2005.091694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen DL, Linderman JK, Roy RR, Bigbee AJ, Grindeland RE, Mukku V, Edgerton VR. Apoptosis: a mechanism contributing to remodeling of skeletal muscle in response to hindlimb unweighting. Am J Physiol. 1997;273:C579–C587. doi: 10.1152/ajpcell.1997.273.2.C579. [DOI] [PubMed] [Google Scholar]
- 5.Alway SE, Siu PM. Nuclear apoptosis contributes to sarcopenia. Exerc Sport Sci Rev. 2008;36:51–57. doi: 10.1097/JES.0b013e318168e9dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong RB. Mechanisms of exercise-induced delayed onset muscular soreness: a brief review. Med Sci Sports Exerc. 1984;16:529–538. [PubMed] [Google Scholar]
- 7.Arslan S, Erdem S, Sivri A, Hascelik Z, Tan E. Exercise-induced apoptosis of rat skeletal muscle and the effect of meloxicam. Rheumatol Int. 2002;21:133–136. doi: 10.1007/s00296-001-0156-9. [DOI] [PubMed] [Google Scholar]
- 8.Asakura A, Hirai H, Kablar B, Morita S, Ishibashi J, Piras BA, Christ AJ, Verma M, Vineretsky KA, Rudnicki MA. Increased survival of muscle stem cells lacking the MyoD gene after transplantation into regenerating skeletal muscle. Proc Natl Acad Sci USA. 2007;104:16552–16557. doi: 10.1073/pnas.0708145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL. Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. Am J Physiol Cell Physiol. 2004;286:C355–C364. doi: 10.1152/ajpcell.00211.2003. [DOI] [PubMed] [Google Scholar]
- 10.Best TM, Hunter KD. Muscle injury and repair. Phys Med Rehabil Clin N Am. 2000;11:251–266. [PubMed] [Google Scholar]
- 11.Bhagavati S. Stem cell based therapy for skeletal muscle diseases. Curr Stem Cell Res Ther. 2008;3:219–228. doi: 10.2174/157488808785740343. [DOI] [PubMed] [Google Scholar]
- 12.Biral D, Jakubiec-Puka A, Ciechomska I, Sandri M, Rossini K, Carraro U, Betto R. Loss of dystrophin and some dystrophin-associated proteins with concomitant signs of apoptosis in rat leg muscle overworked in extension. Acta Neuropathol (Berl) 2000;100:618–626. doi: 10.1007/s004010000231. [DOI] [PubMed] [Google Scholar]
- 13.Boffi FM, Cittar J, Balskus G, Muriel M, Desmaras E. Training-induced apoptosis in skeletal muscle. Equine Vet J. 2002;34(Suppl):275–278. doi: 10.1111/j.2042-3306.2002.tb05432.x. [DOI] [PubMed] [Google Scholar]
- 14.Bruusgaard JC, Gundersen K. In vivo time-lapse microscopy reveals no loss of murine myonuclei during weeks of muscle atrophy. J Clin Invest. 2008;118:1450–1457. doi: 10.1172/JCI34022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen YW, Hubal MJ, Hoffman EP, Thompson PD, Clarkson PM. Molecular responses of human muscle to eccentric exercise. J Appl Physiol. 2003;95:2485–2494. doi: 10.1152/japplphysiol.01161.2002. [DOI] [PubMed] [Google Scholar]
- 16.Clarkson PM, Hubal MJ. Exercise-induced muscle damage in humans. Am J Phys Med Rehabil. 2002;81:S52–S69. doi: 10.1097/00002060-200211001-00007. [DOI] [PubMed] [Google Scholar]
- 17.Clarkson PM, Sayers SP. Etiology of exercise-induced muscle damage. Can J Appl Physiol. 1999;24:234–248. doi: 10.1139/h99-020. [DOI] [PubMed] [Google Scholar]
- 18.Favier FB, Benoit H, Freyssenet D. Cellular and molecular events controlling skeletal muscle mass in response to altered use. Pflugers Arch. 2008;456:587–600. doi: 10.1007/s00424-007-0423-z. [DOI] [PubMed] [Google Scholar]
- 19.Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA. 2002;99:11025–11030. doi: 10.1073/pnas.162172899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friden J. Changes in human skeletal muscle induced by long-term eccentric exercise. Cell Tissue Res. 1984;236:365–372. doi: 10.1007/BF00214240. [DOI] [PubMed] [Google Scholar]
- 21.Friden J, Lieber RL. Segmental muscle fiber lesions after repetitive eccentric contractions. Cell Tissue Res. 1998;293:165–171. doi: 10.1007/s004410051108. [DOI] [PubMed] [Google Scholar]
- 22.Gallegly JC, Turesky NA, Strotman BA, Gurley CM, Peterson CA, Dupont-Versteegden EE. Satellite cell regulation of muscle mass is altered at old age. J Appl Physiol. 2004;97:1082–1090. doi: 10.1152/japplphysiol.00006.2004. [DOI] [PubMed] [Google Scholar]
- 23.Henriquez M, Armisen R, Stutzin A, Quest AF. Cell death by necrosis, a regulated way to go. Curr Mol Med. 2008;8:187–206. doi: 10.2174/156652408784221289. [DOI] [PubMed] [Google Scholar]
- 24.Hubal MJ, Chen TC, Thompson PD, Clarkson PM. Inflammatory gene changes associated with the repeated-bout effect. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1628–R1637. doi: 10.1152/ajpregu.00853.2007. [DOI] [PubMed] [Google Scholar]
- 25.Hunter AL, Zhang J, Chen SC, Si X, Wong B, Ekhterae D, Luo H, Granville DJ. Apoptosis repressor with caspase recruitment domain (ARC) inhibits myogenic differentiation. FEBS Lett. 2007;581:879–884. doi: 10.1016/j.febslet.2007.01.050. [DOI] [PubMed] [Google Scholar]
- 26.Kano Y, Sampei K, Matsudo H. Time course of capillary structure changes in rat skeletal muscle following strenuous eccentric exercise. Acta Physiol Scand. 2004;180:291–299. doi: 10.1111/j.0001-6772.2003.01250.x. [DOI] [PubMed] [Google Scholar]
- 27.King KL, Cidlowski JA. Cell cycle and apoptosis: common pathways to life and death. J Cell Biochem. 1995;58:175–180. doi: 10.1002/jcb.240580206. [DOI] [PubMed] [Google Scholar]
- 28.Kon M, Tanabe K, Lee H, Kimura F, Akimoto T, Kono I. Eccentric muscle contractions induce greater oxidative stress than concentric contractions in skeletal muscle. Appl Physiol Nutr Metab. 2007;32:273–281. doi: 10.1139/H06-115. [DOI] [PubMed] [Google Scholar]
- 29.Koyama T, Xie Z, Gao M, Suzuki J, Batra S. Adaptive changes in the capillary network in the left ventricle of rat heart. Jpn J Physiol. 1998;48:229–241. doi: 10.2170/jjphysiol.48.229. [DOI] [PubMed] [Google Scholar]
- 30.Lim JH, Kim DY, Bang MS. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle Nerve. 2004;30:456–462. doi: 10.1002/mus.20139. [DOI] [PubMed] [Google Scholar]
- 31.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Lowe DA, Warren GL, Ingalls CP, Boorstein DB, Armstrong RB. Muscle function and protein metabolism after initiation of eccentric contraction-induced injury. J Appl Physiol. 1995;79:1260–1270. doi: 10.1152/jappl.1995.79.4.1260. [DOI] [PubMed] [Google Scholar]
- 33.Mahoney DJ, Safdar A, Parise G, Melov S, Fu M, MacNeil L, Kaczor J, Payne ET, Tarnopolsky MA. Gene expression profiling in human skeletal muscle during recovery from eccentric exercise. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1901–R1910. doi: 10.1152/ajpregu.00847.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruhashi Y, Kitaoka K, Yoshiki Y, Nakamura R, Okano A, Nakamura K, Tsuyama T, Shima Y, Tomita K. ROS scavenging activity and muscle damage prevention in eccentric exercise in rats. J Physiol Sci. 2007;57:211–216. doi: 10.2170/physiolsci.RP013006. [DOI] [PubMed] [Google Scholar]
- 35.Okada A, Ono Y, Nagatomi R, Kishimoto KN, Itoi E. Decreased muscle atrophy F-box (MAFbx) expression in regenerating muscle after muscle-damaging exercise. Muscle Nerve. 2008;38:1246–1253. doi: 10.1002/mus.21110. [DOI] [PubMed] [Google Scholar]
- 36.Podhorska-Okolow M, Dziegiel P, Murawska-Cialowicz E, Krajewska B, Ciesielska U, Jethon Z, Zabel M. Exercise-induced apoptosis in renal tubular cells of the rat. Folia Morphol (Warsz) 2004;63:213–216. [PubMed] [Google Scholar]
- 37.Podhorska-Okolow M, Sandri M, Zampieri S, Brun B, Rossini K, Carraro U. Apoptosis of myofibres and satellite cells: exercise-induced damage in skeletal muscle of the mouse. Neuropathol Appl Neurobiol. 1998;24:518–531. doi: 10.1046/j.1365-2990.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- 38.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raj DA, Booker TS, Belcastro AN. Striated muscle calcium-stimulated cysteine protease (calpain-like) activity promotes myeloperoxidase activity with exercise. Pflugers Arch. 1998;435:804–809. doi: 10.1007/s004240050587. [DOI] [PubMed] [Google Scholar]
- 40.Richard-Bulteau H, Serrurier B, Crassous B, Banzet S, Peinnequin A, Bigard X, Koulmann N. Recovery of skeletal muscle mass after extensive injury: positive effects of increased contractile activity. Am J Physiol Cell Physiol. 2008;294:C467–C476. doi: 10.1152/ajpcell.00355.2007. [DOI] [PubMed] [Google Scholar]
- 41.Sandri M, Minetti C, Pedemonte M, Carraro U. Apoptotic myonuclei in human Duchenne muscular dystrophy. Lab Invest. 1998;78:1005–1016. [PubMed] [Google Scholar]
- 42.Sandri M, Podhorska-Okolow M, Geromel V, Rizzi C, Arslan P, Franceschi C, Carraro U. Exercise induces myonuclear ubiquitination and apoptosis in dystrophin-deficient muscle of mice. J Neuropathol Exp Neurol. 1997;56:45–57. doi: 10.1097/00005072-199701000-00005. [DOI] [PubMed] [Google Scholar]
- 43.Siu PM, Pistilli EE, Butler DC, Alway SE. Aging influences cellular and molecular responses of apoptosis to skeletal muscle unloading. Am J Physiol Cell Physiol. 2005;288:C338–C349. doi: 10.1152/ajpcell.00239.2004. [DOI] [PubMed] [Google Scholar]
- 44.Smith HK, Maxwell L, Martyn JA, Bass JJ. Nuclear DNA fragmentation and morphological alterations in adult rabbit skeletal muscle after short-term immobilization. Cell Tissue Res. 2000;302:235–241. doi: 10.1007/s004410000280. [DOI] [PubMed] [Google Scholar]
- 45.Sonobe T, Inagaki T, Poole DC, Kano Y. Intracellular calcium accumulation following eccentric contractions in rat skeletal muscle in vivo: role of stretch-activated channels. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1329–R1337. doi: 10.1152/ajpregu.00815.2007. [DOI] [PubMed] [Google Scholar]
- 46.Stupka N, Tarnopolsky MA, Yardley NJ, Phillips SM. Cellular adaptation to repeated eccentric exercise-induced muscle damage. J Appl Physiol. 2001;91:1669–1678. doi: 10.1152/jappl.2001.91.4.1669. [DOI] [PubMed] [Google Scholar]
- 47.Tews DS, Goebel HH. DNA-fragmentation and expression of apoptosis-related proteins in muscular dystrophies. Neuropathol Appl Neurobiol. 1997;23:331–338. doi: 10.1111/j.1365-2990.1997.tb01304.x. [DOI] [PubMed] [Google Scholar]
- 48.Urso ML, Clarkson PM, Hittel D, Hoffman EP, Thompson PD. Changes in ubiquitin proteasome pathway gene expression in skeletal muscle with exercise and statins. Arterioscler Thromb Vasc Biol. 2005;25:2560–2566. doi: 10.1161/01.ATV.0000190608.28704.71. [DOI] [PubMed] [Google Scholar]