

Abstract
Skeletal muscle is one of the most abundant and highly plastic tissues. The ubiquitin–proteasome system (UPS) is recognised as a major intracellular protein degradation system, and its function is important for muscle homeostasis and health. Although UPS plays an essential role in protein degradation during muscle atrophy, leading to the loss of muscle mass and strength, its deficit negatively impacts muscle homeostasis and leads to the occurrence of several pathological phenotypes. A growing number of studies have linked UPS impairment not only to matured muscle fibre degeneration and weakness, but also to muscle stem cells and deficiency in regeneration. Emerging evidence suggests possible links between abnormal UPS regulation and several types of muscle diseases. Therefore, understanding of the role of UPS in skeletal muscle may provide novel therapeutic insights to counteract muscle wasting, and various muscle diseases. In this review, we focussed on the role of proteasomes in skeletal muscle and its regeneration, including a brief explanation of the structure of proteasomes. In addition, we summarised the recent findings on several diseases and elaborated on how the UPS is related to their pathological states.
Keywords: Ubiquitin proteasome system, Muscle homeostasis, Muscle stem cell, Myopathy, Muscular dystrophy, Cachexia, Amyotrophic lateral sclerosis
Introduction
The skeletal muscle mass accounts for approximately 40% of the total human body weight, making it the largest tissue mass present in the body [1]. Maintaining muscle homeostasis is essential for preserving the body’s integrity and activities of daily living, and thus, muscle loss or impairment is associated with several diseases, which ultimately leads to a poor quality of life. Skeletal muscle is highly plastic tissue, and its mass can change dynamically. Muscle atrophy is caused by an imbalance in proteostasis; during muscle atrophy, protein degradation overwhelms protein synthesis, leading to loss of muscle mass and muscle weakness. Paradoxically, in general, proteolysis is critical for preventing cellular dysfunction and the progression of diseases, causing complexity of proteolysis in skeletal muscle.
Perhaps the ubiquitin–proteasome system (UPS) is the most well-known cellular proteolytic system, which is responsible for degrading majority of the misfolded or defective cellular proteins [2]. Most proteins undergo degradation by being the target of the 26S proteasome through covalent attachment of a multi-ubiquitin chain. The ubiquitination of proteins involves the action of the E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin–protein ligases. These tagged proteins are then recognised by the 26S proteasome, consisting of a central barrel-shaped 20S core associated with two 19S regulatory subunits [3, 4]. The latter subunits recognise and bind to the ubiquitinated proteins and initiate the adenosine triphosphate (ATP)-dependent degradation process within the catalytic core [3]. Using such a mechanism above, the UPS performs substrate-specific proteolysis.
Although there remains a lot to understand how the UPS recognises misfolded or defective proteins, disruption of the UPS is associated with pathological states, highlighting the importance of this system in cellular and whole-body homeostasis. Accordingly, several reports have demonstrated a relationship between proteasome system and lifespan. Proteasome activity has been reported to decrease with age in the brain [5], liver [6, 7], heart [8], and skeletal muscles [9], causing age-associated deteriorations. On the other hand, the genetical activation of the proteasome in yeast and Caenorhabditis elegans show protective effect against cellular aging and prolonged lifespan [10, 11].
It has also been shown that the overexpression of proteasome subunits in yeast and Caenorhabditis elegans results in an increase in the proteasome activity and leads to a prolonged lifespan [10, 11]. Conversely, transgenic mice constitutively expressing the β5t subunit of the proteasome showed reduced proteasome activity and a shorter lifespan [12]. Moreover, the loss of function of the Rpn11 subunit of the proteasome results in decreased proteasome activity, accumulation of ubiquitinated proteins, and a shortened lifespan [13]. These previous studies suggest that continuous clearance of misfolded proteins mediated by the UPS is necessary for healthy aging. We recently reported that skeletal muscle-specific reduction in proteasome activity is associated with a shortened lifespan in mouse models [14, 15]. Thus, the UPS in skeletal muscle, which comprises the largest mass in the body, may have a strong impact on aging, longevity, and whole-body homeostasis.
Proteolysis in skeletal muscle has dual nature: proteolytic pathways play a substantial physiological role in muscle atrophy, while their inhibition also promotes muscle dysfunction and weakness [16]. Indeed, contrary to the protective role of the UPS in maintaining cellular functions, many studies also have implicated that the UPS promotes muscle wasting, myofibre degeneration, and muscle weakness. Skeletal muscles function as a storage place for amino acids, and it might be one reason for the disuse atrophy. These contradictions result in variable effects of inhibitions of UPS in atrophies. Therefore, better understanding of the pathogenic role of this proteolytic system in skeletal muscle may provide novel therapeutic insights to counteract the UPS-associated diseases.
In this manuscript, we reviewed published important articles about UPS in the sequence of molecules, myotubes, skeletal muscles, until muscle atrophy in the patients. We focussed on the role of the UPS in skeletal muscle, by outlining the results of our recent studies on proteasomes in skeletal muscle and muscle stem cells. We further summarised recent findings on several diseases, and elaborated on how the UPS is related to their pathological states.
Structure of the 26S proteasome
Proteolysis by the UPS is mainly performed through a series of complex structures. The rapid degradation of ubiquitinated proteins by the 26S proteasome involves multiple enzymatic and non-enzymatic steps. The 26S proteasome is a multi-catalytic protease localised both in the nucleus and cytoplasm. As shown in Fig. 1, it is composed of one proteolytically active cylinder-shaped particle (the 20S proteasome), and two ATPase-containing complexes (known as the 19S cap complexes) [3, 4, 17]. The 19S cap complexes unfold the ubiquitin-conjugated proteins, allowing them to enter into the 20S core particle. The 20S core is composed of inner α-rings and outer β-rings, each of them has seven structurally similar subunits, respectively; α1–7 and β1–7 [18]. In particular, β1, β2, and β5 subunits display caspase-like, trypsin-like, and chymotrypsin-like proteasome activity, respectively [19, 20]. Also, these β subunits contain immunoproteasomes, in which β1, β2, β5 subunits are replaced with β1i (LMP2), β2i (MECL1), β5i (LMP7) subunits, respectively. Recently, an association between the immunoproteasome and disease has been reported and is described in Sect. 4.2.1. The 19S regulatory particles are composed of at least 18 subunits [3, 4]. Two components form the 19S proteasome, the lid and the base. The lid, which is responsible for the recognition of the polyubiquitin signal [21, 22], is composed of nine subunits, Rpn3, Rpn5-9, Rpn11-12, and Rpn15. The base is composed of the two largest subunits of the proteasome, Rpn1 (PSMD2) and Rpn2 (PSMD1), the ubiquitin receptor Rpn13, Rpn10 (PSMD4), and six ATPases, Rpt1–Rpt6 (PSMC2, PSMC1, PSMC4, PSMC6, PSMC3, PSMC5, respectively). These subunits form a large family of proteins with a highly conserved ATPase domain [23]. Rpt3, also known as PSMC4, is an essential subunit of the 26S proteasome [23] and is required for the degradation of most proteasomal substrates. The ubiquitin receptor Rpn10 attaches to Rpn1, although this association is stabilised by presence of Rpn2 [24]. Rpn2 acts as a receptor of the ubiquitin receptor Rpn13 [25–27].
Fig. 1.
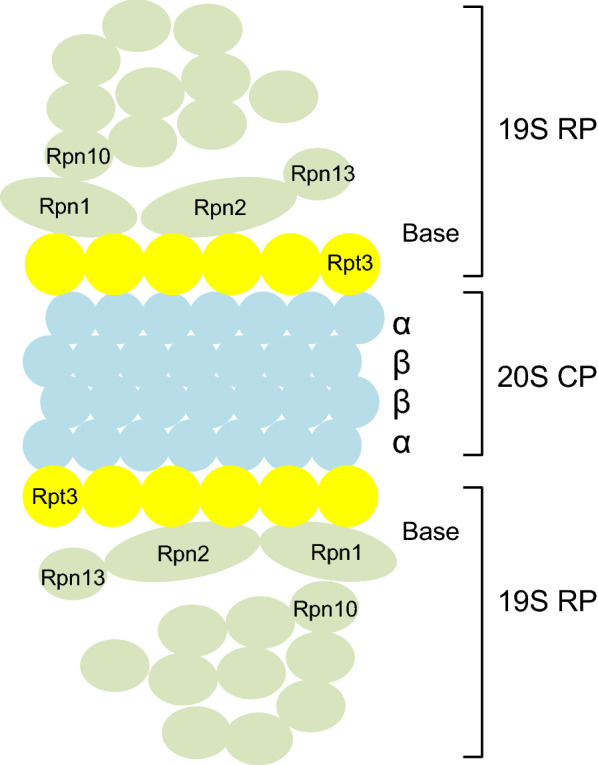
The 26S proteasome is composed of one proteolytically active cylinder-shaped particle (the 20S proteasome) and ATPase-containing complexes (the 19S cap complexes). The 19S cap complex unfolds ubiquitin-conjugated proteins to allow their entry into the 20S cylindrical particle. The 19S regulatory particles on the ends of the 20S proteasome are composed of at least 18 subunits. The base contains the two largest subunits of the proteasome, Rpn1 and Rpn2, the ubiquitin receptor Rpn10 and Rpn13, and the six ATPases, Rpt1-Rpt6. 19S RP, 19S regulatory particle. 20S CP, 20S core particle
Protein degradation in the skeletal muscle
E3 ubiquitin ligases and UPS in skeletal muscle
Muscle atrophy is a serious problem that limits the daily activities and reduces the quality of life. Regulation of the skeletal muscle mass is highly dependent on both the protein synthesis and protein degradation processes. Proteolysis in skeletal muscle is closely regulated by ubiquitin ligases. In skeletal muscle, Cullin-RING ubiquitin ligases comprise the largest known category of ubiquitin ligases as shown in Table 1. Cullin-RING ubiquitin ligases regulate various cellular processes, including multiple aspects of the cell proliferation, transcription, signal transduction, and development [28]. Especially, the muscle-specific E3 ubiquitin ligases, such as the muscle RING finger 1 (MuRF1) and the muscle atrophy F-Box (Atrogin-1/MAFbx), are involved in the regulation of protein degradation in skeletal muscle [29–31]. The expression of these two ubiquitin ligases has been shown to markedly increase in skeletal muscle atrophy [32, 33]. Studies have suggested that titin, Myosin Heavy Chains, Myosin Light Chain-1/2 as the substrates of MuRF1 and elongation initiation factor 3 subunit f (eIF3-f), MyoD, and myogenin as the substrates of Atrogin-1/MAFbx [34]. Although their specific substrate in vivo is yet to be elucidated, MuRF1 or Atrogin-1/MAFbx knockout mice are resistant to muscle atrophy induced by denervation [32], suggesting that these two genes are important regulators of muscle atrophy.
Table 1.
Ubiquitin ligases related with UPS in skeletal muscles modified from Ref. [82]
| Ubiquitin ligases | Target or affected proteins | Refs. |
|---|---|---|
| MuRF1 (TRIM63) | Sarcomeric proteins, myosin-binding protein (MYBPC1), troponin 3, telethonin | [83–86] |
| MuRF3 (TRIM 54) | Sarcomeric proteins, filamin | [83, 87] |
| TRIM32 | Actin, desmin | [88] |
| MUSA1 | – | [37] |
| SMART | – | [37, 38] |
| Nedd4 | MTMR4, FGFR1, Notch 1 | [89, 90] |
| TRAF6 | Ubc13, K63-linked ubiquitination | [128–130] |
| Cullin adaptors | ||
| Atrogin-1/MAFbx | Actin, titin, calsarcin-1, MYHBPC3 | [91, 92] |
| Cbl-b | Insulin receptor substrate 1 (IRS-1) | [35, 36] |
| KLHL40 | Filament protein | [95] |
| KBTBD13 | Z-disc proteins | [96] |
| KLHL41 | Nebulin, nebulin-related anchoring protein | [97–99] |
| KLHL20 | Autophagy-related protein 13 | [100, 101] |
MuRF1: muscle RING finger 1; MuRF3: muscle RING finger 3; TRAF6: tumour necrosis factor receptor-associated factor 6; Cbl-b: Casitas B-lineage lymphoma proto-oncogene-b; KLHL40: Kelch-like protein 40; KLHL41: Kelch-like protein 41; KLHL20: Kelch-like protein 20
In addition to MuRF1 and Atrogin-1/MAFbx, Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b) is also a known E3-ligase related to muscle atrophy. Unloading or spaceflight promotes the expression of Cbl-b; which targets IRS-1, an intermediate of IGF-I signalling which induces protein synthesis in muscle. Thus, it seems Cbl-b, at least partly, works as mechanosensing-mediated muscle atrophy [35, 36]. There are E3-ligases associated with denervation-induced muscle atrophy: muscle ubiquitin ligase of SCF complex atrophy-1 (MUSA1) and specific for muscle atrophy and regulated by transcription (SMART) [37, 38]. Moreover, Hughes et al. recently reported that an E3-ligase, F-box and leucine-rich protein 22 (Fbxl22) mediates neural inactivity-induced muscle atrophy [39]. Knockdown of Fbxl22 in denervated muscle resulted in significant muscle sparing. The expression of ubiquitin ligase is regulated by a transcription factor, Forkhead box O (FoxO) [30]. Akt phosphorylates FoxOs, thereby resulting in their export from the nucleus to the cytoplasm. On the other hand, when the Akt pathway is attenuated by models of muscle atrophy, FoxOs are imported to the nucleus and induce the expression of ubiquitin ligases. Although most of the E3-ligases remain to be explored in the context of muscle atrophy, these findings indicate that the UPS plays a substantial role in muscle atrophies.
UPS dysfunction causes skeletal muscle atrophy
The UPS degrades most of the long- and short-lived normal as well as abnormal intracellular proteins [40]. Especially in the muscle, most of the myofibrillar proteins are degraded through the UPS [41, 42]. We generated muscle-specific Rpt3-knockout mice (Mlc1f-Cre;Rpt3f/f) to better understand the role of the proteasomal system in the skeletal muscle tissue. The proteasomal subunit Rpt3 deletion significantly decreases protease activities and increases ubiquitinated proteins [14]. The muscle-specific deletion of Rpt3 resulted in reduced physical activity, and a decrease in the force production in mice, accompanied with the accumulation of abnormal proteins [14]. In addition, in muscle-specific Rpt3-deficient mice, muscle weight divided by body weight was significantly smaller in the gastrocnemius and tibialis anterior muscles, which are predominantly type II fibres, but not in the soleus muscle, which is predominantly type I fibres [14]. Previous studies have reported that type II glycolytic muscle fibres are more susceptible to muscle wasting conditions than are type I oxidative fibres [43, 44]. Since genetic induction of PGC-1α shows resistance to atrophy [45], the pro-atrophic response in type II fibres may be partly due to a low content of PGC1α. Interestingly, the proteasome-deficient mice showed premature death [14], where it further shows the importance of skeletal muscle homeostasis maintained by the UPS.
Crosstalk with autophagy system in muscle-specific proteasome dysfunction
Another degradation process, which is known as autophagy is the natural regulatory cellular mechanism that is mainly involved in the removal of unnecessary or dysfunctional components from the cell [46]. Autophagic process initiates by forming a flat membrane cistern that envelops a portion of cytoplasm, eventually forming a closed double-membrane vesicle, which is known as the autophagosome. The autophagosome further fuses with the lysosome where its cargo components are degraded. Although autophagy is marginally activated in basal conditions, main factor responsible for the activation is the nutrient starvation [46, 47]. It has also been shown that mice deficient in Atg3, Atg5, or Atg7, which are involved in autophagy, respectively, appear almost normal at birth but die on the first day of birth [48–50].
Autophagy is also shown to be essential for the maintenance of the skeletal muscle homeostasis. With regard to the skeletal muscle, the excessive activation of autophagy promotes muscle wasting [51–53]. Conversely, the muscle-specific deletion of a crucial autophagy gene, Atg7, results in profound muscle atrophy and age-dependent decrease in the generation of muscle force [54]. Furthermore, when mTORC1 was constitutively activated in skeletal muscle by TSC1-knockout, autophagy is inhibited, and results in late onset severe myopathy [55]. Thus, autophagy plays an important role in muscle plasticity and homeostasis.
Since the UPS and autophagy pathways are differently oriented, these two proteolysis systems were viewed as independent [56, 57]. However, there are emerging evidences suggesting the occurrence of a crosstalk between the autophagy and proteasomal pathways in the skeletal muscle. Although autophagy had been thought as a non-specific degradation system, it was found to degrade ubiquitinated proteins [58]. Moreover, muscle-specific autophagy dysfunction induces an increase in ubiquitinated proteins during denervation [54], which would be compensatory upregulation of UPS. Our previous study [14] also suggests the autophagy–UPS complementary relationship. Proteasome-deficient mice exhibited the activation of autophagy. Briefly, the protein levels of LC3II, a standard marker for autophagosomes, and ubiquitin-binding p62 were found to be increased in the Rpt3−/− gastrocnemius muscle. Moreover, the levels of Beclin-1 and Atg5 that are involved in the formation of the isolation membrane, and LC3I, which is involved in the initiation of autophagosome formation, were increased in the Rpt3−/− muscle. Therefore, the autophagy pathway seems to be enhanced in the UPS-deficient muscle. Previous studies also have demonstrated that inhibition of the UPS induces autophagy in vivo and in vitro [59, 60]. Taken together, the UPS and autophagy systems, at least in part, compensatory maintain myocellular homeostasis and integrity.
UPS in muscle stem cells and regeneration
The adult muscle stem cells (also known as satellite cells) are required for the regeneration of the adult skeletal muscle [61]. Responding to muscle damage, satellite cells are rapidly activated and start to proliferate. Three days after the cardiotoxin injection-induced muscle damage, when the satellite cells are under the process of muscle regeneration, we found that both chymotrypsin-like and trypsin-like proteasome activities were increased. Using satellite cell-specific proteasome-deficient mice (Pax7-CreERT2; Rpt3f/f), we demonstrated that proteasome dysfunction impaired satellite cell ability to proliferate, survive, and differentiate, resulting in defective muscle regeneration [62] (Fig. 2). These findings indicate that the UPS activity in satellite cell is associated with muscle regeneration, especially in the early phase [62]. Consistently, in a previous study, proteasome activity was found to be strongly correlated with proliferating cell nuclear antigen protein levels, suggesting that proteasomes play a key role in satellite cell proliferation [63]. Therefore, the findings suggest that the enhancement of the proteasome system is important for satellite cell proliferation and normal muscle regeneration.
Fig. 2.
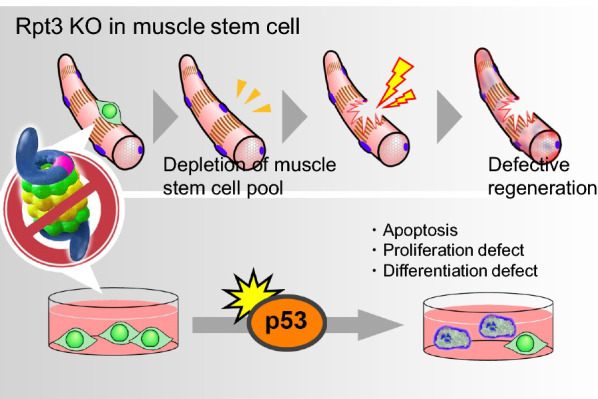
Proteasome dysfunction in Rpt3-deficient muscle stem cells impaired their ability to proliferate, survive and differentiate, resulting in defective muscle regeneration. We also found that proteasome inactivation by Rpt3 deficiency in primary myoblasts inhibits cell proliferation and induces apoptosis. Further, proteasome dysfunction conferred by satellite cell-specific Rpt3-knockout induces p53 activation
Previous studies have also reported that the UPS is associated with the process of myogenic differentiation of satellite cells, which begins with cell cycle arrest and ends with the fusion of individual cells to form multinucleated myotubes [64–66]. In addition, it has also been shown that the inhibition or knockdown of the proteasome can block the fusion of myoblasts and thereby inhibit the process of differentiation [65, 66].
A lot of uncertainty still remains, regarding the relationship between the UPS and muscle regeneration. One possible candidate to explain the relationship would be p53, which regulates apoptosis and cell survival, and promotes muscle differentiation in myoblasts [67–71]. A recent study reported that the tight control of p53 levels in myoblasts regulates the balance between the differentiation process and the return to the quiescence stage [72], which indicates the importance of the p53 during the process of myogenic differentiation. Interestingly, p53 was upregulated in Rpt3-deficient primary satellite cells and the inhibition of p53 expression by siRNA in those cells reduced cell death [62]. These data indicate that Rpt3 in satellite cells is important for cell survival via p53. There is still ambiguity with regard to muscle regeneration and the UPS. Although further work needs to be carried out to understand the UPS, recent studies have revealed a link between the UPS and various diseases discussed the following section.
Proteasome system and related diseases
As the UPS is important for the maintenance of the homeostasis in skeletal muscle, dysfunctions in the system may lead to pathological conditions such as muscular dystrophy, myositis, cachexia and amyotrophic lateral sclerosis.
Diseases with disorganisation of skeletal muscle itself
Muscular dystrophy: aberrant developmental process
Dystrophin, a 427-kDa skeletal muscle protein, links the interior of the myofibres to the extracellular matrix. Mutations in the dystrophin gene are linked to a severe form of muscular dystrophy known as Duchenne muscular dystrophy (DMD) or a mild form known as Becker muscular dystrophy (BMD) [73]. Dystrophin missense mutations cause a wide range of severe phenotypical features in DMD patients. When these missense mutations are stably expressed in mammalian myoblasts, dystrophin proteins are unstable and the protein levels are decreased by proteasomal degradation, but still mutated dystrophin functions to some extent [74]. Continuous administration of the proteasomal inhibitor MG-132 effectively rescues the plasma membrane localisation and the amount of dystrophin in skeletal muscle fibres in dystrophin-deficient model mice [75]. Proteasomal inhibitors have been shown to reduce muscle membrane damage, as revealed by vital staining of the diaphragm and gastrocnemius muscle isolated from treated mice with dystrophin deficiency [75]. Bortezomib, which is a proteasome inhibitor, restores dystrophin and dystrophin–glycoprotein complex at the sarcolemma, which improves the dystrophic phenotype [76]. Compared with control animals, muscular dystrophy dogs treated with bortezomib had lower amount of connective tissue deposition and inflammation as evidenced by muscle histology, collagen morphometry, and ultrastructural microscopy [77].
Congenital muscular dystrophy, which is caused by mutations in the gene encoding the laminin α2 chain, is a severe and incapacitating disease [78]. Proteasome activity is increased in laminin α2 chain-deficient muscle, and treatment with MG-132 reduces muscle pathology in laminin α2 chain-deficient mice [78]. In addition, bortezomib reduces proteasome activity in congenital muscular dystrophy type 1A myoblasts and myotubes [78]. Hence, proteasome inhibition might be useful in patients lacking the laminin α2 chain as a supportive treatment [79]. In case of dysferlinopathy, it was reported that proteasomal inhibition restores biological function of missense mutated dysferlin in patient-derived cells, but the paper was retracted [80]. The effect of proteasome inhibition in muscular dystrophy is yet to be elucidated.
Overall, proteasome inhibition might block the degradation of the mutant dystrophin and recover the function to some extent.
Disease related to mutants in the proteins of UPS system: abnormal maintenance of muscle structures
Protein aggregate myopathies are characterised by protein accumulation in myofibres. These myofibres contain inclusions of sarcomere proteins including myosin and myosin-associated proteins with aberrantly distributed microtubules [81]. Since sarcomere proteins are degraded by UPS, UPS dysfunction might be central to the development of these diseases. It is important to understand how genetic mutations of UPS impact on sarcomere integrity.
Several disease-causing mutations have been found in ubiquitin E3-ligase and its adaptors (Table1) [82]. The genes tripartite motif-containing 63 and 54 (TRIM63 and TRIM54) encoding MuRF1 and MuRF3, respectively, are RING ubiquitin ligases involved in UPS. Both MuRFs are microtubule-associated proteins located in the M bands and Z discs of the sarcomere [83]. These E3 ubiquitin ligases play a role in the degradation of sarcomeric proteins, myogenesis, and stabilisation of microtubules. The targets of MuRF1 include myosin-binding protein (MYBPC1), troponin 3, and telethonin, which cause distal arthrogryposis-1B myopathy [84], dilated cardiomyopathy-2A [85], and limb–girdle muscular dystrophy (LGMD)-7 [86], respectively. MuRF3 targets filamin, and MuRF3 mutations are associated with distal myopathy, myofibrillar myopathy, and restrictive cardiomyopathy-5 [87]. Another TRIM protein, TRIM32, targets actin, and desmin. A mutation in TRIM32 results in LGMD, nemaline myopathy, or myofibrillar myopathy [88]. The ubiquitin ligase Nedd4 participates in denervation-induced muscle atrophy in mice [89], and a relationship between myotonic dystrophy type 2 and NEDD4 has also been reported in human [90]. Atrogin-1/MAFbx is the Cullin adaptor of UPS. Atrogin-1 targets actin and disconnects actin, titin, and calsarcin-1 [91]. Atrogin-1 also targets MYHBPC3, and mutation in the gene results in dilated cardiomyopathy [92].
The Kelch-like family members, the adaptor proteins of E3-ligase, are mutated in nemaline myopathy [93]. Mutations in the Kelch family affect Cullin 3 (CUL3) interaction, which in turn affects the ubiquitination and degradation of protein substrates targeted by CUL3 protein complex [94]. Several genes among the Kelch-like family members are involved in nemaline myopathy; Kelch-like protein 40 (KLHL40) decreases thin filament protein stability [95], KBTBD13 disorganises the Z-disc [96], and Kelch-like protein 41 (KLHL41) impacts on nebulin degradation [97, 98]. KLHL41 degrades nebulin-related anchoring protein, and this degradation is dysregulated in nemaline myopathy [99]. Especially, the CUL3 and KLHL20 complex coordinates the amount of autophagy-related protein 13 (ATG13) in prolonged starvation and controls autophagy [100, 101].
Overall, mutations in several E3-ligases can be the cause of muscle diseases. Ubiquitin ligases could mediate the interplay between autophagy and ubiquitin proteasomal degradation.
Cachexia: reduced stress tolerance of the skeletal muscle
Cachexia reflects muscle wasting syndromes associated with several chronic diseases, such as cancer, diabetes, chronic obstructive pulmonary disease, congestive heart failure and chronic kidney disease (CKD) [102]. Body weight loss, muscle wasting, adipose tissue depletion and abnormal metabolism are among the characteristics of cachexia [103]. While the underlying mechanisms are complex, elevated angiotensin II (Ang II) levels are frequently found in patients with cachexia, and treatment with angiotensin-converting enzyme inhibitor prevents weight loss [104]. Diaphragm muscle biopsies from 22 critically ill patients under mechanical ventilation appeared approximately 25% smaller in myofibre diameter, and this reduced their contractile force by one-half or more [105]. This protein degradation is mediated by proteolytic pathways, including proteasome and lysosomes [103]. The expression levels of muscle-specific ubiquitin ligases, such as Atrogin-1/MAFbx, MuRF1/TRIM63, SMART [38], have been accepted as molecular markers of enhanced proteasome-dependent proteolysis in cancer-related cachexia demonstrated in several types of experimental models [106]. Small-molecule inhibiting MuRF1 attenuates skeletal muscle atrophy in cardiac cachexia mouse model [107]. Sestrin1, which is a stress-inducible metabolic regulator, preserves muscle mass and force in atrophy condition mouse models, including sarcopenia by blunting FoxO-dependent atrogenes [108]. On the other hand, specific proteasome inhibitors such as bortezomib do not improve the muscle phenotype in cancer-associated cachexia [109]. Others have reported unchanged levels of the UPS pathway in cancer patients [110, 111].
Depending on the type of cachexia pathology, there could be different protein degradation pathways involved. Interventional studies using cachexic models are needed to elucidate the role of UPS in the cachexic state.
Diseases characterised with inflammations
Nakajo–Nishimura syndrome
Proteasome-associated auto-inflammatory syndromes was firstly described in 1939 in patients presenting with recurrent fever beginning in early childhood, accompanied by nodular erythema, rash, and joint contractures [112]. Since then, several syndromes, such as the chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE) syndrome, the Nakajo–Nishimura syndrome (NNS), Joint contractures/Muscle atrophy/microcytic anaemia/the Panniculitis-induced lipodystrophy (JMP) syndrome, and the Japanese auto-inflammatory syndrome with lipodystrophy, have been used to categorise patients with diseases within the same spectrum, including myositis [113, 114]. As mentioned in Sect. 2, an association between the immunoproteasome and disease has also been reported. Independent studies identified mutations in the immunoproteasome subunit β5i gene that result in a sustained inflammatory response [113, 115]. In response to cytokines, DNA damage, or oxidative stress, cells selectively upregulate the expression of the immunoproteasome. Immunoproteasome accelerates the proteolysis of specific peptide substrates and allows for the facilitated degradation of oxidant damaged proteins, which may accumulate during inflammation. Immunoproteasome-mediated proteolysis generates immunogenic epitopes presented by MHC class I molecules [116].
Immunoproteasomes are also critical for skeletal muscle differentiation of myoblasts [117]. The oxidative pathway is dysregulated in β5i-mutated iPS cell-induced myeloid cells [118]. In a DMD animal model, inhibition of the immunoproteasome was reported to ameliorate cardiomyopathy in mdx mice, reducing the fibrosis [119, 120]. The mechanism of β5i mutations in skeletal muscles remains to be elucidated. These studies are enhancing the importance of analysing about immunoproteasome in the disease mechanism of NNS and muscular dystrophy.
Sporadic inclusion body myositis
Sporadic inclusion body myositis (sIBM) is the most common form of inflammatory myopathy in individuals aged 50 years or more [121, 122]. Muscle weakness apparent in the quadriceps, wrist flexors, and finger flexors are the typical clinical findings of sIBM [123]. A muscle biopsy would typically reveal endomysial inflammation, invasion of mononuclear cells into non-necrotic fibres, and rimmed vacuoles. These pathological hallmarks indicate that both inflammation and degeneration contribute to the pathology. As such, sIBM is considered to be caused by protein unfolding/misfolding combined with the formation of inclusion bodies [124]. Inhibition of the elimination of ubiquitinated misfolded/unfolded proteins by proteasome results in cellular accumulation of protein aggregates found in sIBM-affected muscle fibres [14]. Amyloid beta (Aβ) and phosphorylated tau (p-tau) can be found in these aggregates. The effect of Aβ precursor protein (APP) overexpression on proteasome function and the influence of proteasome inhibition on aggresome formation was examined using cultured human muscle fibres [125]. In sIBM-affected muscle biopsies, the 26S proteasome subunits have been immune-detected in the β-tubulin-associated aggresomes, which also contained Aβ, p-tau, ubiquitin, and heat shock protein 70 (HSP70). Cultured muscle fibres have been observed to overexpress APP and display diminished proteasomal proteolytic activity, and the addition of a proteasome inhibitor strikingly increases aggresome formation apparently. The formation of inclusion bodies might be followed by abnormal intracellular accumulation of unfolded proteins [126]. As mentioned in Sect. 3.3, the crosstalk between proteasome and autophagy is important. Recently, the role of chaperone-mediated autophagy in the aetiology of sIBM was investigated [127]. Several ubiquitin proteasome genes modulate autophagy. For example, Beclin-1, an essential gene for activation of autophagy, is ubiquitinated by TNF receptor-associated factor 6 (TRAF6) ligase [128]. In addition, TRAF6 has been reported to be involved in muscle atrophy in several cases [128–130].
Dysfunction of UPS, which results in an enhanced autophagic machinery, may be the cause of muscle atrophy in sIBM. Accordingly, in contrast to the protective effect of proteasome inhibitor in muscular dystrophy, proteasome dysfunction may play a role in the accumulation of misfolded, potentially cytotoxic proteins in sIBM myofibres.
Motor neuron disease: indirect cause of muscle atrophy
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterised by the fatal progressive loss of upper and lower motor neurons. In particular, the accumulation of ubiquitinated inclusions containing ALS-causing gene products is a common feature in most familial ALS models; it is also a pathological hallmark of sporadic ALS, indicating that the failure to eliminate detrimental proteins is linked to the pathogenesis of both sporadic and familial ALS. The involvement of UPS dysfunction is strongly suggested by the presence of ubiquitinated inclusions such as skein-like and round hyaline inclusions [131]. The proteasome system is dysregulated in ALS, and the accumulation of superoxide dismutase 1 (SOD1) deposits can be found in the spinal cord of experimental animals and human autopsy cases [132–134]. The continuous expression of mutant SOD1 decreases proteasome activity, and primary cultured embryonic motor neurons are vulnerable to proteasome inhibitor [134]. Proteasome subunit Rpt3 conditional knockout mouse in a motor neuron-specific system showed locomotor dysfunction accompanied by progressive motor neuron loss and gliosis [135]. Promoting proteasomal degradation could be a therapeutic strategy for ALS [136].
Mutation causing ALS can also affect the function of UPS. Ubiquilin-2 and p62, two disease-causing genes in ALS, are mainly related to the protein aggregation and degradation pathways; therefore, mutations in the ubiquilin-2 and p62 genes can cause ALS related disorders [137–139]. Recently, exome sequencing of the ALS-FTD family identified the CCNF (encoding cyclin-F) gene as a novel gene associated with ALS [140]. Cyclin-F is the part of Skp1-Cul1-F-box (SCF) E3 ubiquitin-ligase complex that enables proteasome degradation [141]. Cyclin-F binds to valosin-containing protein (VCP), which is also reported to be mutated in ALS [142]. The ATPase activity of VCP promotes cytoplasmic aggregation of TAR DNA-binding protein 43 (TDP-43), which is commonly observed in degenerating neurons in ALS patients [142]. The inhibition of proteasome in motor neuron can be the cause of aggregation of SOD1 or TDP-43, which would be involved in ALS pathomechanism.
Overall, in case of motor neuron disease, UPS has a role to eliminate pathologically aggregated proteins rather than preserving the amounts of functional molecules as is in the case of structural proteins in muscular dystrophy. The inhibition of proteasome in motor neuron can be the cause of aggregation of SOD1 or TDP-43, which would be involved in ALS pathomechanism.
Conclusions
In skeletal muscle, a functional decline due to atrophy is regulated by proteolysis. This nature of the tissue makes the relationship between proteolysis and skeletal muscle complicated. Indeed, the UPS inhibition often leads muscle atrophy and deficit in regeneration, while preserving skeletal muscle in some conditions or diseases. The proteasome inhibitors and ubiquitin ligases are important regulators of the proteolytic system and may be potential therapeutic targets. Further studies are needed to understand how the UPS regulates the dynamics of proteostasis in skeletal muscle, and how its aberrance/dysfunction induces the pathological state. The study considerably enhances our understanding of the UPS regulation/maintenance of skeletal muscle. It will thus be of immense importance to further elucidate the mechanisms behind proteasome-mediated proteolysis, which will ultimately allow us to develop therapeutic intervention for the related diseases.
Acknowledgements
Not applicable.
Authors’ contributions
YK, KY and NS wrote, reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Grant-in-Aid for Scientific Research KAKENHI (15H05667, 16H05318, 18K07519, 18K17857, and 20H04078). This work was also supported by the Suzuken Memorial Foundation.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yasuo Kitajima, Email: kitajima-y@kumamoto-u.ac.jp.
Naoki Suzuki, Email: naoki@med.tohoku.ac.jp.
References
- 1.Kwee BJ, Mooney DJ. Biomaterials for skeletal muscle tissue engineering. Curr Opin Biotechnol. 2017;47:16–22. doi: 10.1016/j.copbio.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 3.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 4.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 5.Zeng BY, Medhurst AD, Jackson M, Rose S, Jenner P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech Ageing Dev. 2005;126:760–766. doi: 10.1016/j.mad.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Dasuri K, Nguyen A, Zhang L, Fernandez-Kim OS, Bruce-Keller AJ, Blalock BA, Cabo RD, Keller JN. Comparison of rat liver and brain proteasomes for oxidative stress-induced inactivation: influence of ageing and dietary restriction. Free Radic Res. 2009;43:28–36. doi: 10.1080/10715760802534812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayashi T, Goto S. Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mech Ageing Dev. 1998;102:55–66. doi: 10.1016/s0047-6374(98)00011-6. [DOI] [PubMed] [Google Scholar]
- 8.Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- 9.Ferrington DA, Husom AD, Thompson LV. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Thorpe J, Dohmen JR, Li F, Keller JN. Ump1 extends yeast lifespan and enhances viability during oxidative stress: central role for the proteasome? Free Radic Biol Med. 2006;40:120–126. doi: 10.1016/j.freeradbiomed.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 11.Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489:263–268. doi: 10.1038/nature11315. [DOI] [PubMed] [Google Scholar]
- 12.Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S, Tanaka K, Kasahara M. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol. 2012;180:963–972. doi: 10.1016/j.ajpath.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, Tanaka K, Miura M. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol. 2009;29:1095–1106. doi: 10.1128/MCB.01227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitajima Y, Tashiro Y, Suzuki N, Warita H, Kato M, Tateyama M, Ando R, Izumi R, Yamazaki M, Abe M, Sakimura K, Ito H, Urushitani M, Nagatomi R, Takahashi R, Aoki M. Proteasome dysfunction induces muscle growth defects and protein aggregation. J Cell Sci. 2014;127:5204–5217. doi: 10.1242/jcs.150961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitajima Y, Suzuki N, Yoshioka K, Izumi R, Tateyama M, Tashiro Y, Takahashi R, Aoki M, Ono Y. Inducible Rpt3, a proteasome component, knockout in adult skeletal muscle results in muscle atrophy. Front Cell Dev Biol. 2020 doi: 10.3389/fcell.2020.00859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandri M, Coletto L, Grumati P, Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013;126:5325–5333. doi: 10.1242/jcs.114041. [DOI] [PubMed] [Google Scholar]
- 17.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dick TP, Nussbaum AK, Deeg M, Heinemeyer W, Groll M, Schirle M, Keilholz W, Stevanovic S, Wolf DH, Huber R, Rammensee HG, Schild H. Contribution of proteasomal beta-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J Biol Chem. 1998;273:25637–25646. doi: 10.1074/jbc.273.40.25637. [DOI] [PubMed] [Google Scholar]
- 20.Kisselev AF, Akopian TN, Castillo V, Goldberg AL. Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol Cell. 1999;4:395–402. doi: 10.1016/s1097-2765(00)80341-x. [DOI] [PubMed] [Google Scholar]
- 21.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- 22.van Nocker S, Sadis S, Rubin DM, Glickman M, Fu H, Coux O, Wefes I, Finley D, Vierstra RD. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol Cell Biol. 1996;16:6020–6028. doi: 10.1128/mcb.16.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakao Y, Kawai T, Takeuchi O, Copeland NG, Gilbert DJ, Jenkins NA, Takeda K, Akira S. Mouse proteasomal ATPases Psmc3 and Psmc4: genomic organization and gene targeting. Genomics. 2000;67:1–7. doi: 10.1006/geno.2000.6231. [DOI] [PubMed] [Google Scholar]
- 24.Rosenzweig R, Bronner V, Zhang D, Fushman D, Glickman MH. Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J Biol Chem. 2012;287:14659–14671. doi: 10.1074/jbc.M111.316323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, Groll M. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–552. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamazaki J, Iemura S, Natsume T, Yashiroda H, Tanaka K, Murata S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J. 2006;25:4524–4536. doi: 10.1038/sj.emboj.7601338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, Cohen RE. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol. 2006;8:994–1002. doi: 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- 28.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 29.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 30.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 32.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 33.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foletta VC, White LJ, Larsen AE, Leger B, Russell AP. The role and regulation of MAFbx/atrogin-1 and MuRF1 in skeletal muscle atrophy. Pflugers Arch. 2011;461:325–335. doi: 10.1007/s00424-010-0919-9. [DOI] [PubMed] [Google Scholar]
- 35.Nikawa T, Ishidoh K, Hirasaka K, Ishihara I, Ikemoto M, Kano M, Kominami E, Nonaka I, Ogawa T, Adams GR, Baldwin KM, Yasui N, Kishi K, Takeda S. Skeletal muscle gene expression in space-flown rats. FASEB J. 2004;18:522–524. doi: 10.1096/fj.03-0419fje. [DOI] [PubMed] [Google Scholar]
- 36.Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, Ohno A, Okumura Y, Nonaka I, Yasutomo K, Baldwin KM, Kominami E, Higashibata A, Nagano K, Tanaka K, Yasui N, Mills EM, Takeda S, Nikawa T. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol Cell Biol. 2009;29:4798–4811. doi: 10.1128/MCB.01347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H, Sandri M. BMP signaling controls muscle mass. Nat Genet. 2013;45:1309–1318. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- 38.Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hughes DC, Baehr LM, Driscoll JR, Lynch SA, Waddell DS, Bodine SC (2020) Identification and characterization of Fbxl22, a novel skeletal muscle atrophy-promoting E3 ubiquitin ligase. bioRxiv: 2020.2004.2024.059659 [DOI] [PubMed]
- 40.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 41.Attaix D, Aurousseau E, Combaret L, Kee A, Larbaud D, Ralliere C, Souweine B, Taillandier D, Tilignac T. Ubiquitin-proteasome-dependent proteolysis in skeletal muscle. Reprod Nutr Dev. 1998;38:153–165. doi: 10.1051/rnd:19980202. [DOI] [PubMed] [Google Scholar]
- 42.Hasselgren PO, Wray C, Mammen J. Molecular regulation of muscle cachexia: it may be more than the proteasome. Biochem Biophys Res Commun. 2002;290:1–10. doi: 10.1006/bbrc.2001.5849. [DOI] [PubMed] [Google Scholar]
- 43.Li JB, Goldberg AL. Effects of food deprivation on protein synthesis and degradation in rat skeletal muscles. Am J Physiol. 1976;231:441–448. doi: 10.1152/ajplegacy.1976.231.2.441. [DOI] [PubMed] [Google Scholar]
- 44.Matsakas A, Patel K. Skeletal muscle fibre plasticity in response to selected environmental and physiological stimuli. Histol Histopathol. 2009;24:611–629. doi: 10.14670/HH-24.611. [DOI] [PubMed] [Google Scholar]
- 45.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 47.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 49.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 50.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev. 2005;19:1715–1722. doi: 10.1101/gad.1318305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 54.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 55.Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, Ruegg MA. Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 2013;17:731–744. doi: 10.1016/j.cmet.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 56.Ciechanover A, Finley D, Varshavsky A. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell. 1984;37:57–66. doi: 10.1016/0092-8674(84)90300-3. [DOI] [PubMed] [Google Scholar]
- 57.Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–190. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 58.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 59.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 60.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 61.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitajima Y, Suzuki N, Nunomiya A, Osana S, Yoshioka K, Tashiro Y, Takahashi R, Ono Y, Aoki M, Nagatomi R. The ubiquitin-proteasome system is indispensable for the maintenance of muscle stem cells. Stem Cell Reports. 2018;11:1523–1538. doi: 10.1016/j.stemcr.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duguez S, Bihan MC, Gouttefangeas D, Feasson L, Freyssenet D. Myogenic and nonmyogenic cells differentially express proteinases, Hsc/Hsp70, and BAG-1 during skeletal muscle regeneration. Am J Physiol Endocrinol Metab. 2003;285:E206–215. doi: 10.1152/ajpendo.00331.2002. [DOI] [PubMed] [Google Scholar]
- 64.Abu Hatoum O, Gross-Mesilaty S, Breitschopf K, Hoffman A, Gonen H, Ciechanover A, Bengal E. Degradation of myogenic transcription factor MyoD by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol Cell Biol. 1998;18:5670–5677. doi: 10.1128/mcb.18.10.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gardrat F, Montel V, Raymond J, Azanza JL. Proteasome and myogenesis. Mol Biol Rep. 1997;24:77–81. doi: 10.1023/a:1006877214153. [DOI] [PubMed] [Google Scholar]
- 66.Kim SS, Rhee S, Lee KH, Kim JH, Kim HS, Kang MS, Chung CH. Inhibitors of the proteasome block the myogenic differentiation of rat L6 myoblasts. FEBS Lett. 1998;433:47–50. doi: 10.1016/s0014-5793(98)00883-7. [DOI] [PubMed] [Google Scholar]
- 67.Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 68.Porrello A, Cerone MA, Coen S, Gurtner A, Fontemaggi G, Cimino L, Piaggio G, Sacchi A, Soddu S. p53 regulates myogenesis by triggering the differentiation activity of pRb. J Cell Biol. 2000;151:1295–1304. doi: 10.1083/jcb.151.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soddu S, Blandino G, Scardigli R, Coen S, Marchetti A, Rizzo MG, Bossi G, Cimino L, Crescenzi M, Sacchi A. Interference with p53 protein inhibits hematopoietic and muscle differentiation. J Cell Biol. 1996;134:193–204. doi: 10.1083/jcb.134.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tamir Y, Bengal E. p53 protein is activated during muscle differentiation and participates with MyoD in the transcription of muscle creatine kinase gene. Oncogene. 1998;17:347–356. doi: 10.1038/sj.onc.1201929. [DOI] [PubMed] [Google Scholar]
- 71.Weintraub H, Hauschka S, Tapscott SJ. The MCK enhancer contains a p53 responsive element. Proc Natl Acad Sci USA. 1991;88:4570–4571. doi: 10.1073/pnas.88.11.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flamini V, Ghadiali RS, Antczak P, Rothwell A, Turnbull JE, Pisconti A. The satellite cell niche regulates the balance between myoblast differentiation and self-renewal via p53. Stem Cell Rep. 2018;10:970–983. doi: 10.1016/j.stemcr.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15:373–386. doi: 10.1038/s41582-019-0203-3. [DOI] [PubMed] [Google Scholar]
- 74.Talsness DM, Belanto JJ, Ervasti JM. Disease-proportional proteasomal degradation of missense dystrophins. Proc Natl Acad Sci USA. 2015;112:12414–12419. doi: 10.1073/pnas.1508755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonuccelli G, Sotgia F, Schubert W, Park DS, Frank PG, Woodman SE, Insabato L, Cammer M, Minetti C, Lisanti MP. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol. 2003;163:1663–1675. doi: 10.1016/S0002-9440(10)63523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gazzerro E, Assereto S, Bonetto A, Sotgia F, Scarfi S, Pistorio A, Bonuccelli G, Cilli M, Bruno C, Zara F, Lisanti MP, Minetti C. Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. Am J Pathol. 2010;176:1863–1877. doi: 10.2353/ajpath.2010.090468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Araujo KP, Bonuccelli G, Duarte CN, Gaiad TP, Moreira DF, Feder D, Belizario JE, Miglino MA, Lisanti MP, Ambrosio CE. Bortezomib (PS-341) treatment decreases inflammation and partially rescues the expression of the dystrophin-glycoprotein complex in GRMD dogs. PLoS ONE. 2013;8:e61367. doi: 10.1371/journal.pone.0061367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korner Z, Fontes-Oliveira CC, Holmberg J, Carmignac V, Durbeej M. Bortezomib partially improves laminin alpha2 chain-deficient muscular dystrophy. Am J Pathol. 2014;184:1518–1528. doi: 10.1016/j.ajpath.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 79.Korner Z, Durbeej M. Bortezomib does not reduce muscular dystrophy in the dy2J/dy2J mouse model of laminin alpha2 chain-deficient muscular dystrophy. PLoS ONE. 2016;11:e0146471. doi: 10.1371/journal.pone.0146471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Azakir BA, Di Fulvio S, Kinter J, Sinnreich M. Proteasomal inhibition restores biological function of mis-sense mutated dysferlin in patient-derived muscle cells. J Biol Chem. 2017;292:12542. doi: 10.1074/jbc.A111.329078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Olive M, Abdul-Hussein S, Oldfors A, Gonzalez-Costello J, van der Ven PF, Furst DO, Gonzalez L, Moreno D, Torrejon-Escribano B, Alio J, Pou A, Ferrer I, Tajsharghi H. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum Mol Genet. 2015;24:3638–3650. doi: 10.1093/hmg/ddv108. [DOI] [PubMed] [Google Scholar]
- 82.Hnia K, Clausen T, Moog-Lutz C. Shaping striated muscles with ubiquitin proteasome system in health and disease. Trends Mol Med. 2019;25:760–774. doi: 10.1016/j.molmed.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 83.McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol. 2002;157:125–136. doi: 10.1083/jcb.200108089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–1095. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moreira ES, Wiltshire TJ, Faulkner G, Nilforoushan A, Vainzof M, Suzuki OT, Valle G, Reeves R, Zatz M, Passos-Bueno MR, Jenne DE. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24:163–166. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 87.Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, Richardson JA, Bassel-Duby R, Olson EN. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest. 2007;117:2486–2495. doi: 10.1172/JCI32827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol. 2012;198:575–589. doi: 10.1083/jcb.201110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagpal P, Plant PJ, Correa J, Bain A, Takeda M, Kawabe H, Rotin D, Bain JR, Batt JA. The ubiquitin ligase Nedd4-1 participates in denervation-induced skeletal muscle atrophy in mice. PLoS ONE. 2012;7:e46427. doi: 10.1371/journal.pone.0046427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Screen M, Jonson PH, Raheem O, Palmio J, Laaksonen R, Lehtimaki T, Sirito M, Krahe R, Hackman P, Udd B. Abnormal splicing of NEDD4 in myotonic dystrophy type 2: possible link to statin adverse reactions. Am J Pathol. 2014;184:2322–2332. doi: 10.1016/j.ajpath.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mearini G, Gedicke C, Schlossarek S, Witt CC, Kramer E, Cao P, Gomes MD, Lecker SH, Labeit S, Willis MS, Eschenhagen T, Carrier L. Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res. 2010;85:357–366. doi: 10.1093/cvr/cvp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bilodeau PA, Coyne ES, Wing SS. The ubiquitin proteasome system in atrophying skeletal muscle: roles and regulation. Am J Physiol Cell Physiol. 2016;311:C392–403. doi: 10.1152/ajpcell.00125.2016. [DOI] [PubMed] [Google Scholar]
- 94.Gupta VA, Beggs AH. Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet Muscle. 2014;4:11. doi: 10.1186/2044-5040-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garg A, O'Rourke J, Long C, Doering J, Ravenscroft G, Bezprozvannaya S, Nelson BR, Beetz N, Li L, Chen S, Laing NG, Grange RW, Bassel-Duby R, Olson EN. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J Clin Invest. 2014;124:3529–3539. doi: 10.1172/JCI74994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sambuughin N, Yau KS, Olive M, Duff RM, Bayarsaikhan M, Lu S, Gonzalez-Mera L, Sivadorai P, Nowak KJ, Ravenscroft G, Mastaglia FL, North KN, Ilkovski B, Kremer H, Lammens M, van Engelen BG, Fabian V, Lamont P, Davis MR, Laing NG, Goldfarb LG. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet. 2010;87:842–847. doi: 10.1016/j.ajhg.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramirez-Martinez A, Cenik BK, Bezprozvannaya S, Chen B, Bassel-Duby R, Liu N, Olson EN. KLHL41 stabilizes skeletal muscle sarcomeres by nonproteolytic ubiquitination. Elife. 2017;6:12. doi: 10.7554/eLife.26439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, Ogata K, Hsu C, Clarke NF, Darras BT, Farrar MA, Hashem A, Manton ND, Muntoni F, North KN, Sandaradura SA, Nishino I, Hayashi YK, Sewry CA, Thompson EM, Yau KS, Brownstein CA, Yu TW, Allcock RJ, Davis MR, Wallgren-Pettersson C, Matsumoto N, Alkuraya FS, Laing NG, Beggs AH. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet. 2013;93:1108–1117. doi: 10.1016/j.ajhg.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jirka C, Pak JH, Grosgogeat CA, Marchetii MM, Gupta VA. Dysregulation of NRAP degradation by KLHL41 contributes to pathophysiology in Nemaline Myopathy. Hum Mol Genet. 2019;28(15):2549–2560. doi: 10.1093/hmg/ddz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, Chen RH. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61:84–97. doi: 10.1016/j.molcel.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 101.Yuan WC, Lee YR, Lin SY, Chang LY, Tan YP, Hung CC, Kuo JC, Liu CH, Lin MY, Xu M, Chen ZJ, Chen RH. K33-linked polyubiquitination of Coronin 7 by Cul3-KLHL20 ubiquitin E3 ligase regulates protein trafficking. Mol Cell. 2014;54:586–600. doi: 10.1016/j.molcel.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 102.Yoshida T, Delafontaine P. Mechanisms of cachexia in chronic disease states. Am J Med Sci. 2015;350:250–256. doi: 10.1097/MAJ.0000000000000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Penna F, Ballaro R, Beltra M, De Lucia S, Garcia Castillo L, Costelli P. The skeletal muscle as an active player against cancer cachexia. Front Physiol. 2019;10:41. doi: 10.3389/fphys.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, Yusuf S. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet. 2003;361:1077–1083. doi: 10.1016/S0140-6736(03)12892-9. [DOI] [PubMed] [Google Scholar]
- 105.Hooijman PE, Beishuizen A, Witt CC, de Waard MC, Girbes AR, Spoelstra-de Man AM, Niessen HW, Manders E, van Hees HW, van den Brom CE, Silderhuis V, Lawlor MW, Labeit S, Stienen GJ, Hartemink KJ, Paul MA, Heunks LM, Ottenheijm CA. Diaphragm muscle fiber weakness and ubiquitin-proteasome activation in critically ill patients. Am J Respir Crit Care Med. 2015;191:1126–1138. doi: 10.1164/rccm.201412-2214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Argiles JM, Busquets S, Stemmler B, Lopez-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. 2014;14:754–762. doi: 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 107.Adams V, Bowen TS, Werner S, Barthel P, Amberger C, Konzer A, Graumann J, Sehr P, Lewis J, Provaznik J, Benes V, Buttner P, Gasch A, Mangner N, Witt CC, Labeit D, Linke A, Labeit S. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J Cachexia Sarcopenia Muscle. 2019;10:1102–1115. doi: 10.1002/jcsm.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Segales J, Perdiguero E, Serrano AL, Sousa-Victor P, Ortet L, Jardi M, Budanov AV, Garcia-Prat L, Sandri M, Thomson DM, Karin M, Hee Lee J, Munoz-Canoves P. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat Commun. 2020;11:189. doi: 10.1038/s41467-019-13832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Penna F, Busquets S, Argiles JM. Experimental cancer cachexia: evolving strategies for getting closer to the human scenario. Semin Cell Dev Biol. 2016;54:20–27. doi: 10.1016/j.semcdb.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 110.Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O. Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr. 2013;98:1485–1492. doi: 10.3945/ajcn.113.063859. [DOI] [PubMed] [Google Scholar]
- 111.Op den Kamp CM, Langen RC, Minnaard R, Kelders MC, Snepvangers FJ, Hesselink MK, Dingemans AC, Schols AM. Pre-cachexia in patients with stages I-III non-small cell lung cancer: systemic inflammation and functional impairment without activation of skeletal muscle ubiquitin proteasome system. Lung Cancer. 2012;76:112–117. doi: 10.1016/j.lungcan.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 112.McDermott A, Jacks J, Kessler M, Emanuel PD, Gao L. Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management. Int J Dermatol. 2015;54:121–129. doi: 10.1111/ijd.12695. [DOI] [PubMed] [Google Scholar]
- 113.Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martinez de Villarreal L, dos Santos HG, Garg A. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866–872. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, Almeida de Jesus A, Pelletier M, Tsai WL, Remmers EF, Kardava L, Hill S, Kim H, Lachmann HJ, Megarbane A, Chae JJ, Brady J, Castillo RD, Brown D, Casano AV, Gao L, Chapelle D, Huang Y, Stone D, Chen Y, Sotzny F, Lee CC, Kastner DL, Torrelo A, Zlotogorski A, Moir S, Gadina M, McCoy P, Wesley R, Rother KI, Hildebrand PW, Brogan P, Kruger E, Aksentijevich I, Goldbach-Mansky R. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015;125:4196–4211. doi: 10.1172/JCI81260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, Ichinose K, Nakamura H, Tsujino A, Kawakami A, Matsunaka M, Kasagi S, Kawano S, Kumagai S, Ohmura K, Mimori T, Hirano M, Ueno S, Tanaka K, Tanaka M, Toyoshima I, Sugino H, Yamakawa A, Tanaka K, Niikawa N, Furukawa F, Murata S, Eguchi K, Ida H, Yoshiura K. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci USA. 2011;108:14914–14919. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ohmura K. Nakajo-Nishimura syndrome and related proteasome-associated autoinflammatory syndromes. J Inflamm Res. 2019;12:259–265. doi: 10.2147/JIR.S194098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cui Z, Hwang SM, Gomes AV. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol Cell Biol. 2014;34:96–109. doi: 10.1128/MCB.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Honda-Ozaki F, Terashima M, Niwa A, Saiki N, Kawasaki Y, Ito H, Hotta A, Nagahashi A, Igura K, Asaka I, Li HL, Yanagimachi M, Furukawa F, Kanazawa N, Nakahata T, Saito MK. Pluripotent stem cell model of Nakajo-Nishimura syndrome untangles proinflammatory pathways mediated by oxidative stress. Stem Cell Rep. 2018;10:1835–1850. doi: 10.1016/j.stemcr.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Farini A, Gowran A, Bella P, Sitzia C, Scopece A, Castiglioni E, Rovina D, Nigro P, Villa C, Fortunato F, Comi GP, Milano G, Pompilio G, Torrente Y. Fibrosis rescue improves cardiac function in Dystrophin-deficient mice and Duchenne patient-specific cardiomyocytes by immunoproteasome modulation. Am J Pathol. 2019;189:339–353. doi: 10.1016/j.ajpath.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 120.Farini A, Sitzia C, Cassani B, Cassinelli L, Rigoni R, Colleoni F, Fusco N, Gatti S, Bella P, Villa C, Napolitano F, Maiavacca R, Bosari S, Villa A, Torrente Y. Therapeutic potential of immunoproteasome inhibition in duchenne muscular dystrophy. Mol Ther. 2016;24:1898–1912. doi: 10.1038/mt.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol. 2019;15:257–272. doi: 10.1038/s41584-019-0186-x. [DOI] [PubMed] [Google Scholar]
- 122.Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JM, Mammen AL. Classification and management of adult inflammatory myopathies. Lancet Neurol. 2018;17:816–828. doi: 10.1016/S1474-4422(18)30254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Needham M, Mastaglia FL. Sporadic inclusion body myositis: a review of recent clinical advances and current approaches to diagnosis and treatment. Clin Neurophysiol. 2016;127:1764–1773. doi: 10.1016/j.clinph.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 124.Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009;19:493–506. doi: 10.1111/j.1750-3639.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW, Askanas V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol. 2005;167:517–526. doi: 10.1016/s0002-9440(10)62994-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 127.Cacciottolo M, Nogalska A, D'Agostino C, Engel WK, Askanas V. Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol Appl Neurobiol. 2013;39:750–761. doi: 10.1111/nan.12038. [DOI] [PubMed] [Google Scholar]
- 128.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191:1395–1411. doi: 10.1083/jcb.201006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, Kumar A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol. 2012;32:1248–1259. doi: 10.1128/MCB.06351-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun H, Gong Y, Qiu J, Chen Y, Ding F, Zhao Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int J Mol Sci. 2014;15:11126–11141. doi: 10.3390/ijms150611126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maurel C, Dangoumau A, Marouillat S, Brulard C, Chami A, Hergesheimer R, Corcia P, Blasco H, Andres CR, Vourc'h P. Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: a link to neurodegeneration. Mol Neurobiol. 2018;55:6480–6499. doi: 10.1007/s12035-017-0856-0. [DOI] [PubMed] [Google Scholar]
- 132.Cheroni C, Peviani M, Cascio P, Debiasi S, Monti C, Bendotti C. Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol Dis. 2005;18:509–522. doi: 10.1016/j.nbd.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 133.Kato S, Saeki Y, Aoki M, Nagai M, Ishigaki A, Itoyama Y, Kato M, Asayama K, Awaya A, Hirano A, Ohama E. Histological evidence of redox system breakdown caused by superoxide dismutase 1 (SOD1) aggregation is common to SOD1-mutated motor neurons in humans and animal models. Acta Neuropathol. 2004;107:149–158. doi: 10.1007/s00401-003-0791-1. [DOI] [PubMed] [Google Scholar]
- 134.Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- 135.Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H, Sakimura K, Ito H, Takahashi R. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Urushitani M, Kurisu J, Tateno M, Hatakeyama S, Nakayama K, Kato S, Takahashi R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J Neurochem. 2004;90:231–244. doi: 10.1111/j.1471-4159.2004.02486.x. [DOI] [PubMed] [Google Scholar]
- 137.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 139.Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, Gabelle A, Didic M, De Septenville A, Millecamps S, Lenglet T, Latouche M, Kabashi E, Campion D, Hannequin D, Hardy J, Brice A, French C. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013;70:1403–1410. doi: 10.1001/jamaneurol.2013.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, Zhang KY, Farrawell N, Vance C, Hu X, Chesi A, Leblond CS, Lee A, Rayner SL, Sundaramoorthy V, Dobson-Stone C, Molloy MP, van Blitterswijk M, Dickson DW, Petersen RC, Graff-Radford NR, Boeve BF, Murray ME, Pottier C, Don E, Winnick C, McCann EP, Hogan A, Daoud H, Levert A, Dion PA, Mitsui J, Ishiura H, Takahashi Y, Goto J, Kost J, Gellera C, Gkazi AS, Miller J, Stockton J, Brooks WS, Boundy K, Polak M, Munoz-Blanco JL, Esteban-Perez J, Rabano A, Hardiman O, Morrison KE, Ticozzi N, Silani V, de Belleroche J, Glass JD, Kwok JB, Guillemin GJ, Chung RS, Tsuji S, Brown RH, Jr, Garcia-Redondo A, Rademakers R, Landers JE, Gitler AD, Rouleau GA, Cole NJ, Yerbury JJ, Atkin JD, Shaw CE, Nicholson GA, Blair IP. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7:11253. doi: 10.1038/ncomms11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.D'Angiolella V, Esencay M, Pagano M. A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013;23:135–140. doi: 10.1016/j.tcb.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu Y, Nakagawa T, Morohoshi A, Nakagawa M, Ishida N, Suzuki N, Aoki M, Nakayama K. Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum Mol Genet. 2019;28:3486–3497. doi: 10.1093/hmg/ddz119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.