

Abstract
Coarctation of the aorta (CoA) is defined as a congenital stenosis of the thoracic aorta and is one of the most common congenital cardiovascular diseases. Despite successful surgical treatment for CoA, arterial abnormalities, including refractory hypertension, aortic aneurysm, and proatherogenic phenotypic changes, frequently affect patients’ quality of life. Emerging evidence from morphological and molecular biological investigations suggest that the area of CoA is characterized by phenotypic modulation of smooth muscle cells, intimal thickening, and impaired elastic fiber formation. These changes extend to the pre-and post-stenotic aorta and impair arterial elasticity. The aim of this review is to present current findings on the pathology and molecular mechanisms of vascular remodeling due to CoA. In particular, we will discuss the association between CoA and the ductus arteriosus since the most common site for the stenosis is in the proximity of the ductus arteriosus.
Keywords: Congenital heart disease, Coarctation of the aorta, Remodeling, Ductus arteriosus
Introduction
Early diagnosis and appropriate management of coarctation of the aorta (CoA) have resulted in a low mortality rate associated with the repair of CoA. It has been suggested that patients with CoA are at increased risk of myocardial infarction because systemic hypertension and premature atherosclerosis tend to develop early in life even after successful surgical treatment [1, 2]. The increased cardiovascular risk in this patient group is the result of both frequent relapses of hypertension and proatherogenic abnormalities that are present even in the absence of arterial hypertension [3, 4]. Recent findings have demonstrated phenotypic modulation of smooth muscle cells, accumulation of excessive collagen, and inborn impaired arterial elasticity in patients with CoA. This accumulating body of evidence suggests that CoA is a systemic vascular disease rather than a simple mechanical obstruction that can be resolved through surgical intervention alone. Here, we review the historical background and current understanding of the pathology and molecular mechanisms of CoA.
Historical and clinical features
Identification of coarctation of the aorta
“Coarctation” derives from the Latin term coarctatio, which literally means drawing together to make tight [5, 6]. CoA, therefore, is defined as a narrowing or obstruction of the aortic arch. The first description of this condition is generally attributed to Johann Freidrich Meckel, a Prussian anatomist who demonstrated the case of an 18-year-old female to the Royal Academy of Sciences of Berlin in 1750. The postmortem examination of the young woman revealed that her aorta was extremely narrow, with a diameter that was less than half that of her pulmonary artery [6]. A second account of CoA was written in 1791 by M. Paris, the prosecutor of the amphitheater at the Hotel-Dieu in Paris, who described the case of a 50-year-old woman as follows [6]: “The part of the aorta which is beyond the arch, between the ligamentum arteriosum and the first inferior intercostal, was so narrowed it had at most the thickness of a goosequill. The part of the vessel which was above the constriction was slightly dilated. Neither of these early cases revealed any cause, either in the aorta or in its vicinity, to which this extraordinary condition could be attributed.”
Epidemiology
Coarctation of the aorta is the fifth or sixth most common congenital heart defect, accounting for 5–8% of neonates with congenital heart diseases [7, 8]. The incidence of CoA has been reported to one per 2500 births [7, 9] and the ratio of males to females with the condition to be between 1.27:1 and 1.74:1 [7, 9]. In these studies, CoA was considered to be hemodynamically dominant in complex congenital heart defects with a left-to-right shunt.
Morphology and hemodynamics
Coarctation of the aorta was originally classified as either infantile coarctation, characterized by a long narrow segment, or adult coarctation, characterized by a sharp localized constriction [10]. However, this classification does not account for the frequent finding of adult-type coarctation in newborn infants. Elzenga and Gittenberger-de Groot proposed that the name tubular hypoplasia be used for the type characterized by a long narrow segment, while the term coarctation be reserved for the type characterized by a sharp localized constriction [10]. In a typical case, CoA is located near the entrance of the ductus arteriosus. Although the relationship between CoA and the ductus arteriosus is sometimes difficult to determine when the ductus arteriosus is closed and ligamentous [6], CoA is currently classified into three types based on this relationship: preductal, paraductal, and postductal. Preductal CoA is most frequently encountered in infants and young children, while postductal CoA, on the other hand, is rare in patients under 5 years of age and can coexist with an open duct [11].
Narrowing of the aortic isthmus without other significant intracardiac anomalies is defined as simple CoA. Some patients with CoA have additional congenital heart defects [6], of which the two most common concurrent defects are perimembranous-type ventricular septal defects and postero-inferior overriding of the aortic valve. Other malformations which have been reported to be associated with CoA are left ventricular outlet tract stenosis, bicuspid aortic valve, and mitral valve malformations [12, 13]. CoA can also occur with a double-outlet right ventricle (also known as Taussig-Bing anomaly) and with transposition when there is a restrictive subaortic infundibulum [14].
After birth, depending on the severity of coarctation and the presence of associated intra-cardiac lesions, CoA causes mild systolic hypertension, severe congestive heart failure, and shock. In isolated CoA, left ventricular pressure overload rapidly occurs upon closure of the ductus arteriosus, resulting in left ventricular dysfunction (Fig. 1a). In complex CoA, the ventricular septal defect increases left ventricular end-diastolic volume and ventricular preload, which leads to an increase in left ventricular end-diastolic pressure and the subsequent development of pulmonary venous and arterial hypertension (Fig. 1b).
Fig. 1.

Hemodynamics of simple coarctation of the aorta (CoA) and complex CoA. a Hemodynamics of simple CoA. Closure of the ductus arteriosus leads to left ventricular pressure overload, resulting in left ventricular dysfunction. b In complex CoA, blood flow from the left ventricle to the right ventricle through the ventricular septal defect causes left ventricular volume overload. Subsequent increase in left ventricular endo-diastolic pressure leads to pulmonary venous and arterial hypertension
Treatment and prognosis
Patients with CoA have a poor prognosis if they do not receive surgical or catheter interventions. Campbell et al. examined the natural history of CoA and demonstrated that the median age of death for unrepaired CoA is 31 years, with 76% of deaths attributable to complications of aortic coarctation (25.5% cardiac failure, 21% aortic rupture, 18% bacterial endocarditis, 11.5% intracranial hemorrhage) [15].
Crafoord and Nylin performed the first surgical intervention in 1944, about 200 years after the first description of CoA [16]. These surgeons resected the narrowed segment and re-attached the transected ends using an end-to-end anastomosis. The Blalock–Park procedure was reported in that same year [17]. Following the introduction of prosthetic patch aortoplasty in 1961 [18], the subclavian flap aortoplasty procedure was reported in 1966 by Waldhausen and Nahrwold as a strategy to lower the high rates of restenosis [19]. At that time, mortality among patients with CoA after repair surgery at the Mayo Clinic was almost 30% over a follow-up period of 30 years (1946–1981) [20]. Similarly, Pedersen et al. reported that the estimated mortality after surgery in Denmark from 1965 to 1985 was almost 23% [21]. Since recoarctation is the most frequent complication after surgical repair, patients with CoA frequently underwent multiple operations to correct stenosis and other complications. For small children with/without hypoplasia of the isthmus or transverse arch, an extended end-to-end anastomosis using a broader longitudinal incision across the proximal aorta improved the outcome and is currently preferred because of low mortality rates and low rates of restenosis [22, 23].
In the early 1980s, the technique of balloon angioplasty for CoA was developed as an alternative to repeat surgery [24, 25]. This technique was rapidly introduced into routine clinical practice, and in the intervening years it has become an established option for patients with CoA. However, balloon angioplasty is associated with its own set of complications, including re-coarctation, aneurysm, and aortic dissection [26]; consequently, it is often preferred for recurrent CoA rather than native CoA. In the subsequent decade, endovascular expandable stents were introduced to replace balloon dilatation for both native CoA and recurrent CoA, although the use of endovascular stents in small children remains controversial [23]. The primary argument for this change was that endovascular stents support the integrity of the vessel wall and permit a more controlled tear, thereby minimizing tear extension and subsequent dissection or aneurysm formation [26].
Due to advancements such as these in the treatment and management of CoA, the mortality of patients with this condition has declined. Peres et al. compared surgery and balloon angioplasty as treatment and management strategies in 100 patients with simple CoA between 1987 and 2008 in Portugal [27]. The immediate mortality was 2% in the surgical group but 0% in the balloon angioplasty group; likewise, there was no late mortality in either the surgical or balloon angioplasty group during a mean follow-up period of 7.2 ± 5.4 years [27]. Burch et al. reported that the immediate and late mortality rates in 167 patients with simple CoA who underwent surgical repair between 1996 and 2006 were 0.6 and 1.2%, respectively [28].
Etiology
Hemodynamic theory
Various developmental aberrations have been proposed as explanations for abnormalities of the aortic arch. One of the morphological abnormalities proposed as an explanation for CoA concerns fetal blood flow [29, 30]. Rudolph et al. described three features of CoA in support of this hypothesis, namely, the localized shelf, the associated intracardiac defects, and the hypoplastic aortic arch with a reduced anterograde aortic flow. Indeed, hypoplasia of the left heart has been associated with decreased anterograde flow through the developing left ventricle in the fetus [31, 32]. A recent report also supports the hemodynamic theory not only in tubular hypoplasia of the aortic arch but also in simple CoA. Van den Boom et al. analyzed four cases of monochorionic diamniotic pregnancies with twin–twin transfusion syndrome and found an increased risk of simple CoA in the donor twin [33]. Thus, the pattern of blood flow in the fetal circulation influences embryogenesis; in particular, a reduction in the volume of blood passing through the ascending aorta during the fetal period in CoA with ventricular septal defect, left ventricular outflow obstruction, and tubular hypoplasia of the transverse arch leads postnatally to the development of CoA.
Ductus tissue theory
In 1855, Skoda speculated that the constriction of the aorta is related to the closure of the ductus arteriosus extending into the walls of the aorta [34], a proposal which is referred to as the Skodiac hypothesis [5]. In a normal junction with no coarctation, the extension of the ductus into the aortic wall is minimal. When coarctation is present, however, there is a variable but greatly increased quantity of ductus tissue in the aorta, frequently in the form of rings that almost completely surround the vessel [35]. Russell et al. reported that the ductal tissue forming the circumferential sling extends distally beyond the major coarctation shelf in simple CoA and complex CoA [35], leading these authors to suggest that the ductus tissue theory may be applicable to both simple CoA and complex CoA.
It is well known that the ductus arteriosus is dilated by prostaglandin E1 [36–39]. Several studies have demonstrated that CoA is relieved following prostaglandin E1 infusion, leading to the suggestion that in such cases the ductus arteriosus functionally extends into the aortic wall. Callahan et al. reported a case of a 9-day-old male with CoA accompanied by ventricular septal defect who responded to a prostaglandin E1 infusion [40]. Liberman et al. reported similar results from three cases of infants presenting with critical CoA that included simple CoA and complex CoA [41, 42]. The age of these patients ranged from 2 days to 7 weeks, and all patients were born after uneventful pregnancies lasting at least 36 weeks. In all these patients, prostaglandin E1 was administrated after closure of the ductus arteriosus and did not cause reopening of the ductus arteriosus. Since vascular remodeling progresses significantly after the anatomical closure of the ductus arteriosus [36–39, 43–46], it is reasonable to conclude that prostaglandin E1 did not open the ductus arteriosus in these patients. These results support the concept that CoA is caused by excessive distribution of the tissue of the ductus arteriosus.
Histology
Elastic fiber formation
The concept that CoA is related to an abnormal extension of the tissue unique to the ductus arteriosus is largely based on histological findings on elastic fiber formation [10, 47, 48]. Elastic fibers are differentially formed between the aorta and the ductus arteriosus [44]. The normal thoracic aorta is characterized by an aortic media with regular elastic lamellae alternating with smooth muscle cell layers [10]. Although the aorta and ductus arteriosus are connected and exposed to essentially the same hemodynamics, the ductus arteriosus has structural properties similar to those of muscular arteries rather than those of elastic arteries [39, 44]. The ductus arteriosus exhibits disassembly and fragmentation of the internal elastic lamina and sparse elastic fibers in the middle layer, in contrast to its connecting arteries, i.e., the main pulmonary trunk and the descending aorta. This impaired elastogenesis is a hallmark of the vascular remodeling of the ductus arteriosus in late gestation [44, 49–51].
Elzenga and Gittenberger-de Groot examined the extension of ductal tissue into the aortic media in 45 CoA specimens that had no intracardiac abnormality [10]. The range of the patients ranged from 2 weeks to 40 years. The ductal tissue in the coarctation shelf was represented by loosely arranged spindle cells without elastic fibers, as visualized using an elastic tissue stain. Ductal tissue bordered the aortic lumen for more than half of the total aortic circumference in all patients despite the existence of patent ductus arteriosus. In transverse sections of normal aortas, on the other hand, ductal tissue did not extend beyond one-third of the total circumference of the aorta at the level of the ductus arteriosus. These histological properties were also found in CoA combined with tubular hypoplasia and/or intracardiac abnormality [5, 52]. As show in Fig. 2, impaired elastic fiber formation occurs in the ductus arteriosus and extends to the proximal portion of the ascending aorta, which is considered to be CoA. Kim et al. reported that the percentage of ductal tissue relative to the whole luminal circumference was 69 ± 24 and 61 ± 21% in coarctation segments and transitional zones, respectively [52]. In their study, the extension of ductal tissue was also observed in preterm-delivered babies.
Fig. 2.
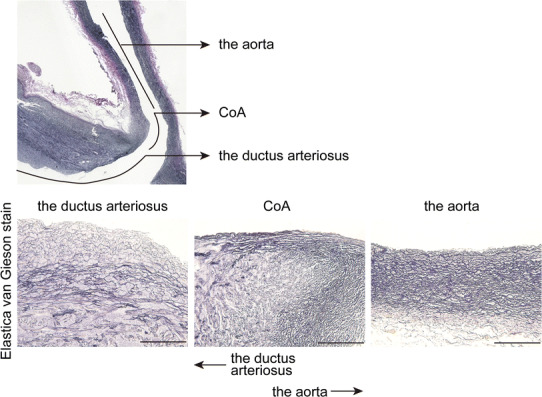
Elastic fiber formation of CoA and the ductus arteriosus. Upper panel Elastic fiber formation in the tissue of CoA with hypoplastic left heart syndrome (1-month-old). Lower panel Magnified images of upper panel. Elastic fiber formation was impaired in the ductal side and elastic fibers were abundantly formed in the aortic side in CoA tissue. Scale bars 100 μm. Elastic fibers were visualized by elastica–van Gieson stain as previously described [44]
Iwaki et al. recently demonstrated the three-dimensional extent of ductal tissue in resected human coarctation segments using synchrotron radiation-based X-ray phase contrast tomography, based on the Talbot grating interferometer X-ray imaging system at the SPring-8 synchrotron radiation facility (Hyōgo Prefecture, Japan) [53] (Fig. 3). These images show the circular or partial distribution of ductal media in aortic tissues as low-density areas (indicated in blue), which correspond to impaired elastic fiber formation, and that ductal tissues spread more distally on the inner curvature than on the greater curvature [53].
Fig. 3.
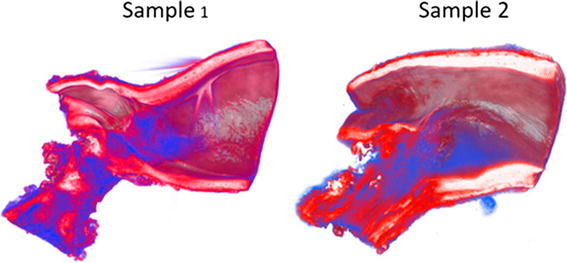
Three-dimensional extent of ductal tissue with three-dimensional reconstruction of phase-contrast tomography images. Synchrotron radiation-based X-ray phase contrast tomography based on the Talbot grating interferometer at the SPring-8 synchrotron radiation facility shows the circular or partial distribution of ductal media in aortic tissues as low-density areas (indicated in blue), which correspond to impaired elastic fiber formation
Reprinted from Iwaki et al. [53] copyright (2016), with permission from Elsevier (License Number: 3986330814990)
We also recently found that the prostaglandin E receptor EP4, which is known to be a predominant prostanoid receptor in the ductus arteriosus, is abundantly expressed in human coarctation segments [44]. In the process of elastic fiber formation, soluble tropoelastins are deposited on microfibrils, following which lysyl oxidase cross-links tropoelastins to confer elastic properties to elastic fibers. Our study demonstrated that prostaglandin E-EP4 signaling inhibits elastogenesis in the ductus arteriosus by degrading lysyl oxidase proteins [44]. Similarly, we observed lower lysyl oxidase expression in human coarctation segments, as well as abundant EP4 expression (Fig. 4a, b). These data further support the concept of ductal extension into the aortic wall in CoA.
Fig. 4.
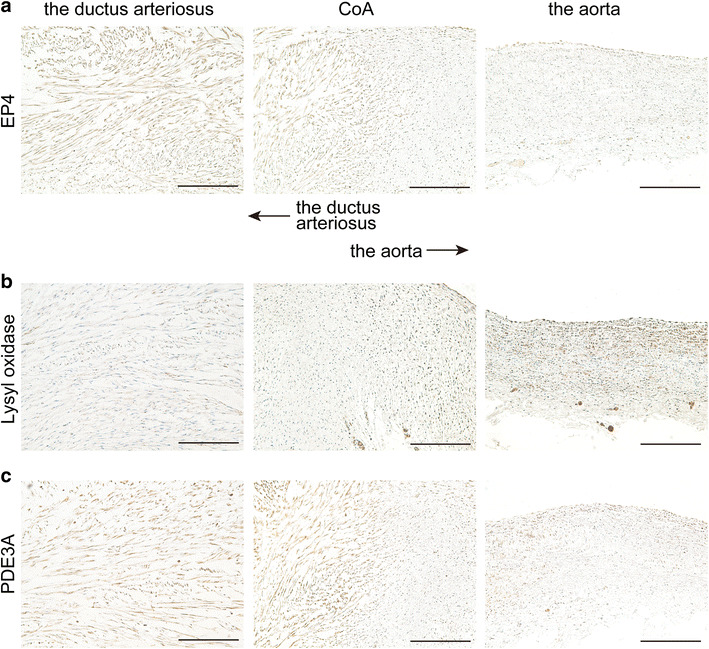
Protein expressions in CoA and the ductus arteriosus. Immunohistochemistry for prostaglandin E receptor EP4 (EP4; a), lysyl oxidase (b), and phosphodiesterase 3A (PDE3A; c) in the tissue of CoA with hypoplastic left heart syndrome (1-month-old patient). The expressions of EP4 and PDE3A were higher in the ductus arteriosus and ductal side of CoA, and lower in the aorta and aortic side of CoA. Lysyl oxidase was abundantly expressed in the aorta and aortic side of CoA, but not in the ductus arteriosus and ductal side of CoA. Immunohistochemical analysis was performed as previously described [44, 63]. Serial sections of Fig. 2 were used for immunohistochemistry. Scale bars 100 μm
Impaired elastic fiber formation is also known to cause aortic aneurysm [38, 54, 55], which was the most common cause of death in patients with CoA before the introduction of surgical repair. Thus, abnormally extended ductal tissue may be the cause of the increased risk of aneurysm formation in CoA patients through the mechanism of impaired elastic fiber formation.
Intimal thickening
The aortic intima consists of a simple flattened endothelium that directly covers the internal elastic lamina. Intimal thickening is one of the prominent phenotypic changes of the ductus arteriosus [36–39, 46] and has also been observed in coarctation segments [10, 52, 56] (Fig. 2). In the ductus arteriosus, intimal thickening is characterized by an area of subendothelial deposition of extracellular matrix, the disassembly of the internal elastic lamina and loss of elastic fiber in the medial layer, and the migration of undifferentiated medial smooth muscle cells into the subendothelial space [36–39, 46, 57, 58].
Jimenez et al. examined ten cases of CoA with no intracardiac lesions or prostaglandin E1 infusion in patients ranging in age from 15 days to 4 years [56]. Widened subendothelial regions and collagen deposition in this area were demonstrated. The prostaglandin E receptor EP4 and its downstream signaling have been found to mediate hyaluronan-induced intimal thickening of the ductus arteriosus [36–39, 45, 46]. Although deposition of hyaluronan in the subendothelial area of CoA was not examined in the study by Jimenez et al., it is most likely that prostaglandin E promotes the intimal thickening in coarctation segment during late gestation.
In addition to widened subendothelial spaces, fragmentation of the internal elastic lamina has also been observed in specimens of CoA [44]. Proliferative smooth muscle cells have not been observed in the intimal thickening of coarctation segments. Endothelial cells have been observed to be aligned at the border of the lumen with no proliferation [56], suggesting that smooth muscle cell migration contributes to the formation of intimal thickening in CoA.
Secondary intimal proliferation
Elzenga and Gittenberger-de Groot observed secondary intimal proliferation in coarctation segments of older patients with CoA in both ductal and aortic tissue, and based on these observations they precisely describe the difference between intimal thickening and secondary intimal proliferation [10]. According to these authors, secondary intimal proliferation consists of loose fibrous tissue where smooth muscle cells and elastic fibers are scattered. In contrast, intimal thickening in the ductus arteriosus and ductal tissue in coarctation segments exhibit more abundant fine elastic fibers. This pathological architecture has been shown to narrow the residual lumen of the CoA and has been observed from around 1 year of age, becoming prominent in the teenage years [10]. This secondary intimal proliferation is thought to be the result of flow disturbance at the point of stenosis [59].
Molecular mechanism and functions
Smooth muscle phenotype
Smooth muscle cell differentiation has been shown to occur earlier in the fetal ductus arteriosus than in adjacent arteries [60–62]. The resulting mature-phenotype smooth muscle cells in the ductus arteriosus are believed to participate in the immediate closure of the ductus arteriosus after birth. Following observations of the extension of ductus tissue into the aortic wall in CoA, Kim et al. performed immunohistochemical analysis of tissues of 15 patients with CoA either with or without intracardiac anomalies [52]. Most patients were less than 2 months old. These authors found that the expression levels of the myosin heavy chain isoforms SM1 and SM2, markers for differentiated smooth muscle cells, were lower in the intima of the coarctation segments than in that of the ductus arteriosus [52]. The dedifferentiated smooth muscle cell marker SMemb was abundantly expressed in both the intima of the coarctation segments and in the ductus arteriosus. In the medial layer, expression levels of SM1 and SM2 were similar in coarctation segments and the ductus arteriosus. These data suggest that in the medial layer, the expression of the smooth muscle cell phenotype in CoA is similar to that in the ductus arteriosus, while in the intimal layer the expression of the dedifferentiated smooth muscle cell phenotype in CoA is greater than that in the ductus arteriosus.
Jimenez et al. examined the smooth muscle phenotype in a set of older CoA patients with a mean age of 15.8 ± 12.2 months, focusing on analyzing the smooth muscle cell phenotype in normal aortas, in moderately stenotic areas of CoA, and in the coarcted shelf [56]. In the medial layers, immunoreactivity with smooth muscle-myosin heavy chain (SM-MHC) and high-molecular-weight caldesmon (h-caldesmon), both differentiated smooth muscle cell markers, was observed in moderately stenotic areas and the coarcted shelf as well as in the normal aorta. In the intima, the faint expression of SM-MHC and h-caldesmon was observed in the moderately stenotic areas. In the intima of moderately stenotic areas, electron microscopic analysis also confirmed the dedifferentiated smooth muscle cell phenotype, which was characterized by a dramatic increase in rough endoplasmic reticulum. In the coarcted shelf, in contrast, the strong expression of differentiated markers was observed in the intima. These data suggest that, in older populations, the smooth muscle cells in the intima of CoA and in that of the ductus arteriosus are dedifferentiated, while the smooth muscle cells in the coarcted shelf, the most stenotic part of CoA, are redifferentiated.
The ductus arteriosus has greater contractile ability, and we previously demonstrated that phosphodiesterase type 3 (PDE3) is highly expressed in the ductal tissues and that inhibition of PDE3 maintains the ductal opening, but not dilation of the adjacent arteries [63]. Expression of PDE3 was highly expressed both in the ductus arteriosus and CoA compared to the aorta (Fig. 4c).
Apoptosis
Regulated cell death, known as apoptosis, has been demonstrated in various developmental vascular remodeling processes, including the closure of the ductus arteriosus. In the ductus arteriosus, extensive apoptosis is observed in areas of cytolytic necrosis and, to a lesser extent, in the intima in this region [62, 64]. Kim et al. performed apoptosis assays using tissues from coarctation sites and the ductus arteriosus and frequently found terminal dUTP nick end labeling (TUNEL)-positive cell deaths in the intima and media of both coarctation segments and the ductus arteriosus but none in normal aortas and transitional zones [52]. These results further support the concept that abnormal extension of ductal tissue into the aorta plays a crucial role in the pathogenesis of CoA.
Neural crest perturbation
Subpopulations of cardiac neural crest cells differentiate into vascular smooth muscle cells that consist of the ascending aorta, aortic arch, and head vessels [65, 66]. Since CoA is often associated with the bicuspid aortic valve, these abnormalities of the left ventricular outflow tract are suggested to developmentally and genetically relate to a disorder of the neural crest [13]. In accordance with this concept, Jain et al. used mice with primary and secondary cardiac neural crest deficiencies to demonstrate that perturbation of the neural crest contributes to abnormal semilunar valves and CoA [67]. In their study, loss of Pax3, which is expressed in the premigratory neural crest, led to thickened aortic valve leaflets and increased extracellular matrix deposition in the aortic smooth muscle layer. In addition, the authors generated mice with disrupted Notch signaling in the second heart field by Cre-inducible expression of a truncated form of the mastermind-like protein DNMAML using Mef2c-AHF-Cre. These mice also exhibited aortic valve abnormalities and thickened aortic walls [67].
The ductus arteriosus is also derived largely from neural crest cells. In humans, patent ductus arteriosus (PDA) is associated with DNA-binding mutations in the gene encoding the neural crest-enriched transcription factor TFAP2β [68]. In mice, Tfap2β mRNA was found to be strongly expressed in smooth muscle cells of mouse ductus arteriosus at embryonic day 13. However, Tfap2β was not expressed in the ascending aorta [68]. In addition, in the above-mentioned mice with primary and secondary cardiac neural crest deficiencies, abnormality of the ductus arteriosus was not demonstrated [67]. These data suggest that subpopulations of neural crest cells may be differentially involved in CoA and ductal tissues.
Gridlock/HEY2 mutation
Mutant zebrafish homozygous for gridlock have no circulation to the posterior trunk and tail because of a localized block to caudal blood flow at the base of the dorsal aorta, the region where the two anterior lateral dorsal aortae merge to form the single midline dorsal aorta [69–71]. Peterson et al. further demonstrated that inducing vascular endothelial growth factor is sufficient to suppress the gridlock phenotype, including aortic abnormality in zebrafish [72]. These results led to further study of gridlock as a candidate gene for CoA.
Surprisingly, however, knock-out of the mouse gridlock gene did not result in CoA but instead in other cardiac malformations, including ventricular septal defect [73–75]. In humans, Reamon-Buettner et al. analyzed the sequences encoding the bHLH domain of the human HEY2, a homolog of gridlock, in 52 explanted hearts and concluded that mutations in these domains are rare in complex congenital heart disease [76]. Although the gridlock mutation contributes to the CoA phenotype in zebrafish, the role of the gene in human CoA remains unclear.
Genetics
Coarctation of the aorta has a high rate of sibling recurrence, as do other congenital heart diseases [69]. Tagariello et al. recently examined 83 cases of non-syndromic CoA and found two missense mutations in exons 8 and 9 of TBL1Y [77]. TBL1Y is similar to its gonosomal homolog, TBL1X, and its autosomal homolog, TBLR1. Both genes are part of co-repressor machineries and are required for transcriptional activation by transcription factors that involve CtBP1/2, which contributes to Notch signaling. It has also been reported that Alagille syndrome is associated with CoA [78] and is caused by mutations in human Jagged1, which encodes a ligand for Notch1 [79]. These findings suggest an association of Notch signaling with CoA.
Quintero-Rivera et al. recently reported the association of the MATR3 mutation with CoA and PDA [80]. MATR3 encodes the nuclear matrix protein Matrin 3, and mouse Matr3 is highly expressed in the neural crest. These authors generated mice in which the 3′ portion of Matr3 was disrupted and found that heterozygotes of the mice exhibited incompletely penetrant bicuspid aortic valve, CoA, and PDA phenotypes that are similar to those in the human proband [80].
A high prevalence of CoA (10–20%) in Turner syndrome has been noted [81, 82]. CoA is more prevalent in subjects with 45,X than in those with mosaic monosomy X [82]. In addition, familial bicuspid aortic valve, occasionally with aortic coarctation, has been shown to be a channelopathy caused by mutations in the potassium channel gene KCNJ2 [83]. A recent study has demonstrated that microdeletion of the centromeric gene, MCTP2 (multiple C2-domains with two transmembrane regions 2) is associated with CoA and hypoplastic left heart syndrome. Alteration of Mctp2 gene expression in Xenopus laevis embryos resulted in the failure of proper left ventricular outflow tract development [84].
Hemodynamics
Accumulating evidence of vasoreactivity and endothelial function in patients with CoA suggests that CoA is not a simple mechanical obstruction that can be cured surgically. Sehested et al. measured isometric tension using pre- and post-stenotic segments of CoA in response to potassium, noradrenaline, and prostaglandin F2α. These CoA samples were obtained during surgical repair from patients ranging in age from 1 month to 38 years. The isometric force induced by all contractile agents was significantly lower in the pre-stenotic proximal segments than in the post-stenotic distal segments [85]. This study showed significantly more collagen and less smooth muscle mass in pre-stenotic segments than in post-stenotic segments, both in the arch and distal to the ligamentum arteriosum in the normal aorta. Elastic fiber formation did not differ between pre-stenotic and post-stenotic segments. These results are consistent with a rabbit model of CoA in which increased gene expression for collagen types I and III has been demonstrated in the aorta proximal to the coarctation site [86].
More recently, echocardiographic studies have suggested that distensibility of the pre-stenotic aorta is lower in preoperative simple CoA patients than in normal control subjects and that this impairment of distensibility is not improved during a 3-year follow-up period after surgical repair [87, 88]. Taken together, these data suggest that CoA is not only a localized anatomical problem of the stenotic segment but also an inborn systemic vascular disease of the pre-coarctational arteries. Impaired distensibility and response to contractile agents of the pre-stenotic segment of CoA may contribute to long-term cardiovascular morbidity and mortality.
Other studies have documented early vascular wall changes, including impaired flow-mediated vasodilation and increased carotid intimal and medial thickness, in children after successful coarctation repair [1, 3, 4]. The authors of these studies concluded that arterial hypertension and a resting pressure gradient are the major contributing factors to early atherosclerotic development. However, further studies are needed to determine whether this impairment in endothelial function and atherosclerotic change in the carotid artery are simply due to a residual pressure gradient.
Conclusions
It is widely accepted that CoA consists of ductal tissue extending into the aortic wall. Recent findings have revealed that smooth muscle cells in the intima of CoA stenotic segments dedifferentiate at an early stage and redifferentiate in older populations. In addition to this localized phenotypic modulation of the aorta, vascular distensibility is decreased and collagen content is increased in precoarctational aortas, even in preoperative patients with simple CoA that does not exhibit apparent gross pathology in precoarctational segments. Although it is not clear whether a gridlock mutation is associated with human CoA, genetic studies suggest that CoA is associated with the regulation of Notch signaling, which contributes to the development of the aortic arch. Further research into the cellular mechanisms involved in CoA could have great implications for ensuring that longer-term morbidity, secondary to early-onset hypertension and atherosclerotic progression, is minimized.
Acknowledgements
We thank Ryo Ishiwata (Cardiovascular Research Institute, Yokohama City University, Japan) for assistance with graphical illustrations of congenital heart disease.
Compliance with ethical standards
Conflict of interest
Utako Yokoyama has received a speaker honorarium from Eisai Co., Ltd., Japan Blood Products Organization and Mitsubishi Tanabe Pharma Corporation. Yasuhiro Ichikawa, Susumu Minamisawa, and Yoshihiro Ishikawa declare that they have no conflict of interest.
Ethical approval
Human CoA tissue was obtained from Yokohama City University Hospital at the time of corrective operations. The study was approved by the human subject committees at Yokohama City University (B100107034). A sample was obtained after receiving written informed parental consent.
Funding
This study has been supported in part by JSPS (UY, H1605358, 15H05761, 26670506; SM, 26293249, 26670096, 23136516; YI, 24390200, 25670131), MEXT (YI, 22136009), NEDO (YI, 60890021), NCVC (YI, 22-2-3), AMED (YI, 66890005, 66890011, 66890001, 66890023), the Vehicle Racing Commemorative Foundation (UY and SM), and the MEXT fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (UY, 15638648).
Contributor Information
Utako Yokoyama, Phone: +81-45-7872575, Email: utako@yokohama-cu.ac.jp.
Yoshihiro Ishikawa, Phone: +81-45-7872575, Email: yishikaw@med.yokohama-cu.ac.jp.
References
- 1.Meyer AA, Joharchi MS, Kundt G, Schuff-Werner P, Steinhoff G, Kienast W. Predicting the risk of early atherosclerotic disease development in children after repair of aortic coarctation. Eur Heart J. 2005;26:617–622. doi: 10.1093/eurheartj/ehi037. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, Stefanescu Schmidt A. Treatment of hypertension in coarctation of the aorta. Curr Treat Options Cardiovasc Med. 2016;18:40. doi: 10.1007/s11936-016-0462-x. [DOI] [PubMed] [Google Scholar]
- 3.Gardiner HM, Celermajer DS, Sorensen KE, Georgakopoulos D, Robinson J, Thomas O, et al. Arterial reactivity is significantly impaired in normotensive young adults after successful repair of aortic coarctation in childhood. Circulation. 1994;89:1745–1750. doi: 10.1161/01.CIR.89.4.1745. [DOI] [PubMed] [Google Scholar]
- 4.Brili S, Tousoulis D, Antoniades C, Aggeli C, Roubelakis A, Papathanasiu S, et al. Evidence of vascular dysfunction in young patients with successfully repaired coarctation of aorta. Atherosclerosis. 2005;182:97–103. doi: 10.1016/j.atherosclerosis.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 5.Ho SY, Anderson RH. Coarctation, tubular hypoplasia, and the ductus arteriosus. Histological study of 35 specimens. Br Heart J. 1979;41:268–274. doi: 10.1136/hrt.41.3.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson RH. Pediatric cardiology. 3. Philadelphia: Chuchill Livingstone Elsevier; 2009. pp. 945–966. [Google Scholar]
- 7.Samanek M, Voriskova M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999;20:411–417. doi: 10.1007/s002469900502. [DOI] [PubMed] [Google Scholar]
- 8.Izukawa T, Mulholland HC, Rowe RD, Cook DH, Bloom KR, Trusler GA, et al. Structural heart disease in the newborn. Changing profile: comparison of 1975 with 1965. Arch Dis Child. 1979;54:281–285. doi: 10.1136/adc.54.4.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenny D, Hijazi ZM. Coarctation of the aorta: from fetal life to adulthood. Cardiol J. 2011;18:487–495. doi: 10.5603/CJ.2011.0003. [DOI] [PubMed] [Google Scholar]
- 10.Elzenga NJ, Gittenberger-de Groot AC. Localised coarctation of the aorta. An age dependent spectrum. Br Heart J. 1983;49:317–323. doi: 10.1136/hrt.49.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho SY, Micheal LY, Anderson RH. Echocardiology in congenital heart disease. London: Imperial College Press; 2008. [Google Scholar]
- 12.Anderson RH, Lenox CC, Zuberbuhler JR. Morphology of ventricular septal defect associated with coarctation of aorta. Br Heart J. 1983;50:176–181. doi: 10.1136/hrt.50.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kappetein AP, Gittenberger-de Groot AC, Zwinderman AH, Rohmer J, Poelmann RE, Huysmans HA. The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. J Thorac Cardiovasc Surg. 1991;102:830–836. [PubMed] [Google Scholar]
- 14.Sadow SH, Synhorst DP, Pappas G. Taussig-Bing anomaly and coarctation of the aorta in infancy: surgical options. Pediatr Cardiol. 1985;6:83–89. doi: 10.1007/BF02282743. [DOI] [PubMed] [Google Scholar]
- 15.Campbell M. Natural history of coarctation of the aorta. Br Heart J. 1970;32:633–640. doi: 10.1136/hrt.32.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crafoord C, Nylin G. Congenital coarctation of the aorta and its surgical treatment. J Thorac Surg. 1945;14:347. [Google Scholar]
- 17.Blalock A, Park EA. The surgical treatment of experimental coarctation (atresia) of the aorta. Ann Surg. 1944;119:445–456. doi: 10.1097/00000658-194403000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vossschulte K. Surgical correction of coarctation of the aorta by an “isthmusplastic” operation. Thorax. 1961;16:338–345. doi: 10.1136/thx.16.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waldhausen JA, Nahrwold DL. Repair of coarctation of the aorta with a subclavian flap. J Thorac Cardiovasc Surg. 1966;51:532–533. [PubMed] [Google Scholar]
- 20.Cohen M, Fuster V, Steele PM, Driscoll D, McGoon DC. Coarctation of the aorta. Long-term follow-up and prediction of outcome after surgical correction. Circulation. 1989;80:840–845. doi: 10.1161/01.CIR.80.4.840. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen TA, Munk K, Andersen NH, Lundorf E, Pedersen EB, Hjortdal VE, et al. High long-term morbidity in repaired aortic coarctation: weak association with residual arch obstruction. Congenit Heart Dis. 2011;6:573–582. doi: 10.1111/j.1747-0803.2011.00575.x. [DOI] [PubMed] [Google Scholar]
- 22.van Heurn LW, Wong CM, Spiegelhalter DJ, Sorensen K, de Leval MR, Stark J, et al. Surgical treatment of aortic coarctation in infants younger than three months: 1985 to 1990. Success of extended end-to-end arch aortoplasty. J Thorac Cardiovasc Surg. 1994;1994(107):74–85. [PubMed] [Google Scholar]
- 23.Torok RD, Campbell MJ, Fleming GA, Hill KD. Coarctation of the aorta: management from infancy to adulthood. World J Cardiol. 2015;7:765–775. doi: 10.4330/wjc.v7.i11.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singer MI, Rowen M, Dorsey TJ. Transluminal aortic balloon angioplasty for coarctation of the aorta in the newborn. Am Heart J. 1982;103:131–132. doi: 10.1016/0002-8703(82)90539-7. [DOI] [PubMed] [Google Scholar]
- 25.Lock JE, Niemi T, Burke BA, Einzig S, Castaneda-Zuniga WR. Transcutaneous angioplasty of experimental aortic coarctation. Circulation. 1982;66:1280–1286. doi: 10.1161/01.CIR.66.6.1280. [DOI] [PubMed] [Google Scholar]
- 26.Padua LM, Garcia LC, Rubira CJ, de Oliveira Carvalho PE. Stent placement versus surgery for coarctation of the thoracic aorta. Cochrane Database Syst Rev. 2012;5:CD008204. doi: 10.1002/14651858.CD008204.pub2. [DOI] [PubMed] [Google Scholar]
- 27.Peres A, Martins JD, Parames F, Gil R, Matias C, Franco J, et al. Isolated aortic coarctation: experience in 100 consecutive patients. Rev Port Cardiol. 2010;29:23–35. [PubMed] [Google Scholar]
- 28.Burch PT, Cowley CG, Holubkov R, Null D, Lambert LM, Kouretas PC, et al. Coarctation repair in neonates and young infants: is small size or low weight still a risk factor? J Thorac Cardiovasc Surg. 2009;138:547–552. doi: 10.1016/j.jtcvs.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 29.Rudolph AM, Heymann MA, Spitznas U. Hemodynamic considerations in the development of narrowing of the aorta. Am J Cardiol. 1972;30:514–525. doi: 10.1016/0002-9149(72)90042-2. [DOI] [PubMed] [Google Scholar]
- 30.Hutchins GM. Coarctation of the aorta explained as a branch-point of the ductus arteriosus. Am J Pathol. 1971;63:203–214. [PMC free article] [PubMed] [Google Scholar]
- 31.Grossfeld PD. The genetics of hypoplastic left heart syndrome. Cardiol Young. 1999;9:627–632. doi: 10.1017/S1047951100005722. [DOI] [PubMed] [Google Scholar]
- 32.Gardiner HM. Response of the fetal heart to changes in load: from hyperplasia to heart failure. Heart. 2005;91:871–873. doi: 10.1136/hrt.2004.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Boom J, Battin M, Hornung T. Twin-twin transfusion syndrome, coarctation of the aorta and hypoplastic aortic arch: a case series report. J Paediatr Child Health. 2010;46:76–79. doi: 10.1111/j.1440-1754.2009.01641.x. [DOI] [PubMed] [Google Scholar]
- 34.Skoda J (1855) Protokoll der Sektions-Sitzung fur Physiologie und Pathologie, am 19. okt. Z kaiserlichkoniglichen Gesellschft der Aerzte zu Wien 710–722
- 35.Russell GA, Berry PJ, Watterson K, Dhasmana JP, Wisheart JD. Patterns of ductal tissue in coarctation of the aorta in the first three months of life. J Thorac Cardiovasc Surg. 1991;102:596–601. [PubMed] [Google Scholar]
- 36.Yokoyama U, Minamisawa S, Quan H, Ghatak S, Akaike T, Segi-Nishida E, et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J Clin Invest. 2006;116:3026–3034. doi: 10.1172/JCI28639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokoyama U, Minamisawa S, Ishikawa Y. Regulation of vascular tone and remodeling of the ductus arteriosus. J Smooth Muscle Res (Nihon Heikatsukin Gakkai kikanshi) 2010;46:77–87. doi: 10.1540/jsmr.46.77. [DOI] [PubMed] [Google Scholar]
- 38.Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]
- 39.Yokoyama U. Prostaglandin E-mediated molecular mechanisms driving remodeling of the ductus arteriosus. Pediatr Int. 2015;57:820–827. doi: 10.1111/ped.12769. [DOI] [PubMed] [Google Scholar]
- 40.Callahan PF, Quivers ES, Bradley LM, Sell JE, Martin GR. Echocardiographic evidence for a ductal tissue sling causing discrete coarctation of the aorta in the neonate: case report. Pediatr Cardiol. 1998;19:182–184. doi: 10.1007/s002469900276. [DOI] [PubMed] [Google Scholar]
- 41.Liberman L, Gersony WM, Flynn PA, Lamberti JJ, Cooper RS, Stare TJ. Effectiveness of prostaglandin E1 in relieving obstruction in coarctation of the aorta without opening the ductus arteriosus. Pediatr Cardiol. 2004;25:49–52. doi: 10.1007/s00246-003-0549-5. [DOI] [PubMed] [Google Scholar]
- 42.Hascoet JM, Didier F, Monin P, Vert P. Efficiency of prostaglandin E1 in a tiny baby with coarctation of the aorta and ligated ductus arteriosus. Acta Paediatr. 1992;81:938–940. doi: 10.1111/j.1651-2227.1992.tb12142.x. [DOI] [PubMed] [Google Scholar]
- 43.Smith GC. The pharmacology of the ductus arteriosus. Pharmacol Rev. 1998;50:35–58. [PubMed] [Google Scholar]
- 44.Yokoyama U, Minamisawa S, Shioda A, Ishiwata R, Jin MH, Masuda M, et al. Prostaglandin E2 inhibits elastogenesis in the ductus arteriosus via EP4 signaling. Circulation. 2014;129:487–496. doi: 10.1161/CIRCULATIONAHA.113.004726. [DOI] [PubMed] [Google Scholar]
- 45.Yokoyama U, Minamisawa S, Quan H, Akaike T, Suzuki S, Jin M, et al. Prostaglandin E2-activated Epac promotes neointimal formation of the rat ductus arteriosus by a process distinct from that of cAMP-dependent protein kinase A. J Biol Chem. 2008;283:28702–28709. doi: 10.1074/jbc.M804223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokoyama U, Minamisawa S, Katayama A, Tang T, Suzuki S, Iwatsubo K, et al. Differential regulation of vascular tone and remodeling via stimulation of type 2 and type 6 adenylyl cyclases in the ductus arteriosus. Circ Res. 2010;106:1882–1892. doi: 10.1161/CIRCRESAHA.109.214924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brom AG. Narrowing of the aortic isthmus and enlargement of the mind. J Thorac Cardiovasc Surg. 1965;50:166–180. [PubMed] [Google Scholar]
- 48.Rosenberg HS. Coarctation of the aorta: morphology and pathogenetic considerations. Perspect Pediatr Pathol. 1973;1:339–368. [PubMed] [Google Scholar]
- 49.Toda T, Tsuda N, Takagi T, Nishimori I, Leszczynski D, Kummerow F. Ultrastructure of developing human ductus arteriosus. J Anat. 1980;131:25–37. [PMC free article] [PubMed] [Google Scholar]
- 50.Ho SY, Anderson RH. Anatomical closure of the ductus arteriosus: a study in 35 specimens. J Anat. 1979;128:829–836. [PMC free article] [PubMed] [Google Scholar]
- 51.Jager BV, Wollenman OJ. An anatomical study of the closure of the ductus arteriosus. Am J Pathol. 1942;18:595–613. [PMC free article] [PubMed] [Google Scholar]
- 52.Kim JE, Kim EK, Kim WH, Shim GH, Kim HS, Park JD, et al. Abnormally extended ductal tissue into the aorta is indicated by similar histopathology and shared apoptosis in patients with coarctation. Int J Cardiol. 2010;145:177–182. doi: 10.1016/j.ijcard.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 53.Iwaki R, Matsuhisa H, Hoshino M, Oshima Y. Three-dimensional evaluation of ductal tissue in coarctation of the aorta using X-ray phase-contrast tomography. J Thorac Cardiovasc Surg. 2016;152:1454–1456. doi: 10.1016/j.jtcvs.2016.05.053. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama U, Ishiwata R, Jin MH, Kato Y, Suzuki O, Jin H, et al. Inhibition of EP4 signaling attenuates aortic aneurysm formation. PLoS One. 2012;7:e36724. doi: 10.1371/journal.pone.0036724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo DC, Papke CL, He R, Milewicz DM. Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:339–352. doi: 10.1196/annals.1383.013. [DOI] [PubMed] [Google Scholar]
- 56.Jimenez M, Daret D, Choussat A, Bonnet J. Immunohistological and ultrastructural analysis of the intimal thickening in coarctation of human aorta. Cardiovasc Res. 1999;41:737–745. doi: 10.1016/S0008-6363(98)00287-9. [DOI] [PubMed] [Google Scholar]
- 57.De Reeder EG, Girard N, Poelmann RE, Van Munsteren JC, Patterson DF, Gittenberger-De Groot AC. Hyaluronic acid accumulation and endothelial cell detachment in intimal thickening of the vessel wall. The normal and genetically defective ductus arteriosus. Am J Pathol. 1988;132:574–585. [PMC free article] [PubMed] [Google Scholar]
- 58.Rabinovitch M. Cell-extracellular matrix interactions in the ductus arteriosus and perinatal pulmonary circulation. Semin Perinatol. 1996;20:531–541. doi: 10.1016/S0146-0005(96)80067-X. [DOI] [PubMed] [Google Scholar]
- 59.Edwards JE, Christensen NA, et al. Pathologic considerations of coarctation of the aorta. Proc Staff Meet Mayo Clin. 1948;23:324–332. [PubMed] [Google Scholar]
- 60.Kim HS, Aikawa M, Kimura K, Kuro-o M, Nakahara K, Suzuki T, et al. Ductus arteriosus. Advanced differentiation of smooth muscle cells demonstrated by myosin heavy chain isoform expression in rabbits. Circulation. 1993;88:1804–1810. doi: 10.1161/01.CIR.88.4.1804. [DOI] [PubMed] [Google Scholar]
- 61.Jin MH, Yokoyama U, Sato Y, Shioda A, Jiao Q, Ishikawa Y, et al. DNA microarray profiling identified a new role of growth hormone in vascular remodeling of rat ductus arteriosus. J Physiol Sci. 2011;61:167–179. doi: 10.1007/s12576-011-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slomp J, Gittenberger-de Groot AC, Glukhova MA, Conny van Munsteren J, Kockx MM, Schwartz SM, et al. Differentiation, dedifferentiation, and apoptosis of smooth muscle cells during the development of the human ductus arteriosus. Arterioscler Thromb Vasc Biol. 1997;17:1003–1009. doi: 10.1161/01.ATV.17.5.1003. [DOI] [PubMed] [Google Scholar]
- 63.Ichikawa Y, Yokoyama U, Iwamoto M, Oshikawa J, Okumura S, Sato M, et al. Inhibition of phosphodiesterase type 3 dilates the rat ductus arteriosus without inducing intimal thicknening. Circ J. 2012;76:2456–2464. doi: 10.1253/circj.CJ-12-0215. [DOI] [PubMed] [Google Scholar]
- 64.Goldbarg S, Quinn T, Waleh N, Roman C, Liu BM, Mauray F, et al. Effects of hypoxia, hypoglycemia, and muscle shortening on cell death in the sheep ductus arteriosus. Pediatr Res. 2003;54:204–211. doi: 10.1203/01.PDR.0000072519.61060.E5. [DOI] [PubMed] [Google Scholar]
- 65.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, et al. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–715. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 67.Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, et al. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. doi: 10.1172/JCI44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivey KN, Sutcliffe D, Richardson J, Clyman RI, Garcia JA, Srivastava D. Transcriptional regulation during development of the ductus arteriosus. Circ Res. 2008;103:388–395. doi: 10.1161/CIRCRESAHA.108.180661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong TP, Rosenberg M, Mohideen MA, Weinstein B, Fishman MC. Gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science. 2000;287:1820–1824. doi: 10.1126/science.287.5459.1820. [DOI] [PubMed] [Google Scholar]
- 70.Zhong TP, Childs S, Leu JP, Fishman MC. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414:216–220. doi: 10.1038/35102599. [DOI] [PubMed] [Google Scholar]
- 71.Weinstein BM, Stemple DL, Driever W, Fishman MC. Gridlock, a localized heritable vascular patterning defect in the zebrafish. Nat Med. 1995;1:1143–1147. doi: 10.1038/nm1195-1143. [DOI] [PubMed] [Google Scholar]
- 72.Peterson RT, Shaw SY, Peterson TA, Milan DJ, Zhong TP, Schreiber SL, et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22:595–599. doi: 10.1038/nbt963. [DOI] [PubMed] [Google Scholar]
- 73.Sakata Y, Kamei CN, Nakagami H, Bronson R, Liao JK, Chin MT. Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci USA. 2002;99:16197–16202. doi: 10.1073/pnas.252648999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gessler M, Knobeloch KP, Helisch A, Amann K, Schumacher N, Rohde E, et al. Mouse gridlock: no aortic coarctation or deficiency, but fatal cardiac defects in Hey2 -/- mice. Curr Biol. 2002;12:1601–1604. doi: 10.1016/S0960-9822(02)01150-8. [DOI] [PubMed] [Google Scholar]
- 75.Donovan J, Kordylewska A, Jan YN, Utset MF. Tetralogy of fallot and other congenital heart defects in Hey2 mutant mice. Curr Biol. 2002;12:1605–1610. doi: 10.1016/S0960-9822(02)01149-1. [DOI] [PubMed] [Google Scholar]
- 76.Reamon-Buettner SM, Borlak J. HEY2 mutations in malformed hearts. Hum Mutat. 2006;27:118. doi: 10.1002/humu.9390. [DOI] [PubMed] [Google Scholar]
- 77.Tagariello A, Breuer C, Birkner Y, Schmidt S, Koch AM, Cesnjevar R, et al. Functional null mutations in the gonosomal homologue gene TBL1Y are associated with non-syndromic coarctation of the aorta. Curr Mol Med. 2012;12:199–205. doi: 10.2174/156652412798889027. [DOI] [PubMed] [Google Scholar]
- 78.Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. doi: 10.1161/01.CIR.0000121361.01862.A4. [DOI] [PubMed] [Google Scholar]
- 79.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 80.Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, et al. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. 2015;24:2375–2389. doi: 10.1093/hmg/ddv004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hasegawa T, Ogata T, Hasegawa Y, Honda M, Nagai T, Fukushima Y, et al. Coarctation of the aorta and renal hypoplasia in a boy with Turner/Noonan surface anomalies and a 46, XY karyotype: a clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata. Hum Genet. 1996;97:564–567. doi: 10.1007/BF02281861. [DOI] [PubMed] [Google Scholar]
- 82.Gotzsche CO, Krag-Olsen B, Nielsen J, Sorensen KE, Kristensen BO. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child. 1994;71:433–436. doi: 10.1136/adc.71.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663–668. doi: 10.1086/342360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lalani SR, Ware SM, Wang X, Zapata G, Tian Q, Franco LM, et al. MCTP2 is a dosage-sensitive gene required for cardiac outflow tract development. Hum Mol Genet. 2013;22:4339–4348. doi: 10.1093/hmg/ddt283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sehested J, Baandrup U, Mikkelsen E. Different reactivity and structure of the prestenotic and poststenotic aorta in human coarctation. Implications for baroreceptor function. Circulation. 1982;65:1060–1065. doi: 10.1161/01.CIR.65.6.1060. [DOI] [PubMed] [Google Scholar]
- 86.Xu C, Zarins CK, Bassiouny HS, Briggs WH, Reardon C, Glagov S. Differential transmural distribution of gene expression for collagen types I and III proximal to aortic coarctation in the rabbit. J Vasc Res. 2000;37:170–182. doi: 10.1159/000025728. [DOI] [PubMed] [Google Scholar]
- 87.Kuhn A, Baumgartner D, Baumgartner C, Horer J, Schreiber C, Hess J, et al. Impaired elastic properties of the ascending aorta persist within the first 3 years after neonatal coarctation repair. Pediatr Cardiol. 2009;30:46–51. doi: 10.1007/s00246-008-9280-6. [DOI] [PubMed] [Google Scholar]
- 88.Vogt M, Kuhn A, Baumgartner D, Baumgartner C, Busch R, Kostolny M, et al. Impaired elastic properties of the ascending aorta in newborns before and early after successful coarctation repair: proof of a systemic vascular disease of the prestenotic arteries? Circulation. 2005;111:3269–3273. doi: 10.1161/CIRCULATIONAHA.104.529792. [DOI] [PubMed] [Google Scholar]