

Abstract
The β-adrenergic signaling pathways and antioxidant defence mechanisms play important roles in maintaining proper heart function. Here, we examined the effect of chronic normobaric hypoxia (CNH, 10% O2, 3 weeks) on myocardial β-adrenergic signaling and selected components of the antioxidant system in spontaneously hypertensive rats (SHR) and in a conplastic SHR-mtBN strain characterized by the selective replacement of the mitochondrial genome of SHR with that of the more ischemia–resistant Brown Norway strain. Our investigations revealed some intriguing differences between the two strains at the level of β-adrenergic receptors (β-ARs), activity of adenylyl cyclase (AC) and monoamine oxidase A (MAO-A), as well as distinct changes after CNH exposure. The β2-AR/β1-AR ratio was significantly higher in SHR-mtBN than in SHR, apparently due to increased expression of β2-ARs. Adaptation to hypoxia elevated β2-ARs in SHR and decreased the total number of β-ARs in SHR-mtBN. In parallel, the ability of isoprenaline to stimulate AC activity was found to be higher in SHR-mtBN than that in SHR. Interestingly, the activity of MAO-A was notably lower in SHR-mtBN than in SHR, and it was markedly elevated in both strains after exposure to hypoxia. In addition to that, CNH markedly enhanced the expression of catalase and aldehyde dehydrogenase-2 in both strains, and decreased the expression of Cu/Zn superoxide dismutase in SHR. Adaptation to CNH intensified oxidative stress to a similar extent in both strains and elevated the IL-10/TNF-α ratio in SHR-mtBN only. These data indicate that alterations in the mitochondrial genome can result in peculiar changes in myocardial β-adrenergic signaling, MAO-A activity and antioxidant defence and may, thus, affect the adaptive responses to hypoxia.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-017-0546-8) contains supplementary material, which is available to authorized users.
Keywords: SHR, Mitochondrial genome, Myocardium, β-adrenergic receptors, Adenylyl cyclase, Monoamine oxidase A, Antioxidant defence, Chronic hypoxia
Introduction
The spontaneously hypertensive rat (SHR) is one of the most commonly used animal models in cardiovascular research. This strain, which harbors a deletion variant of the Cd36 gene, is predisposed to the development of hypertension and cardiac hypertrophy in adulthood [1]. The hearts of SHR exhibit higher vulnerability to ischemia/reperfusion (I/R) injury and susceptibility to ventricular arrhythmias when compared to normotensive rats [2–4]. Interestingly, despite their reduced ischemic tolerance, ischemic preconditioning was found to provide beneficial antiarrhythmic effects in these animals [5]. We have previously documented that transgenic rescue of defective Cd36 in SHR rat leads to smaller infarct size induced by coronary artery occlusion [6]. In addition, transgenic SHR-Cd36 rats were shown to express a higher number of myocardial β-adrenergic receptors (β-ARs) and displayed increased adenylyl cyclase (AC) activity [7]. These data imply that transgenic expression of an apparently unrelated gene may strongly affect β-adrenergic signaling and myocardial resistance to IR injury.
β-ARs and their signaling machinery are known to play a key role in regulating myocardial function [8]. There are numerous indications that β-AR-mediated signaling may also participate in the development of preconditioning-induced ischemic tolerance [9–11]. Adaptation to chronic hypoxia, which may provide potent cardioprotection, can also be associated with changes in cardiac β-ARs and AC signaling. Down-regulation of β-ARs and desensitization of AC were occasionally observed in hearts from hypoxia-adapted animals [12–16]. It is well known that hypoxia promotes the activity of the sympathetic adrenergic system [17] and that stimulation of β-ARs may enhance mitochondrial reactive oxygen species (ROS) generation in cardiomyocytes [18]. β-AR activation seems to play an essential role in the development of powerful myocardial ischemic resistance conferred by chronic hypoxic exposure [19]. However, the adaptive changes induced by different conditioning regimens or hypoxia are not limited just to β-ARs or other membrane-bound receptors and their signaling systems, but they are also tightly linked to changes in ROS production and their detoxification.
ROS, among other factors, are well-known mediators of the beneficial effects of hypoxic conditioning [20]. Whereas the appropriate rise in ROS is important for achieving suitable protective outcomes of hypoxic adaptation [21, 22], high levels of ROS can cause excessive oxidative stress in cardiomyocytes. In this respect, mitochondria have drawn a great deal of attention as a major site of ROS production and control of redox-sensitive transcription factors. At the same time, these organelles are also the major targets of the detrimental effects of ROS overproduction [23]. Interestingly, the functional properties and vulnerability of mitochondria to oxidative stress may vary somewhat between different tissues and species [24, 25]. On the other hand, increasing evidence indicates that mitochondrial DNA (mtDNA) is essential for the cell phenotype and, thus, may contribute to stress and environmental adaptability [26]. It is known that mtDNA modulates cellular bioenergetics and mitochondrial ROS generation and mtDNA sequence variation may, thus, contribute to disease susceptibility [27]. The role of mtDNA in modulating physiological plasticity and stress responses at the cellular, tissue and whole organism level can be investigated using mitochondrial replacement technology. We have shown recently that the conplastic SHR-mtBN strain characterized by the selective replacement of the mitochondrial genome with that of the more ischemia–resistant Brown Norway strain exhibited the same myocardial infarct size caused by I/R insult as the progenitor SHR. Although adaptation to chronic hypoxia improved cardiac ischemic resistance in both strains, the infarct size-limiting effect was stronger in SHR-mtBN than in SHR, and correlated with reduced sensitivity of mitochondrial permeability transition to Ca2+-induced opening [28].
To further define the potential role of mitochondrial genome in the modulation of cardiac function and resistance to I/R injury, here we focused on exploring β-AR-mediated signaling and selected components of the antioxidant defence system in LV preparations from SHR and SHR-mtBN. We monitored the levels of selected signaling molecules, antioxidant enzymes and markers of oxidative stress and inflammation in samples obtained from both normoxic and hypoxia-adapted animals. In addition, we also determined the expression and activity of monoamine oxidase A (MAO-A). MAO-A belongs among the main ROS producers in cardiac cells [29], but as yet there is a lack of information about behavior of this mitochondrial enzyme in the chronically hypoxic heart. Interestingly, replacement of the mitochondrial genome resulted in distinct changes in myocardial β-ARs and MAO-A, as well as in some components of the antioxidant system under both normal and hypoxic conditions.
Materials and methods
Materials
[3H]CGP-12177 was purchased from PerkinElmer, Inc. (Boston, MA, USA), [3H]cAMP was from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA), [α-32P]ATP was from Hartmann Analytic, GmbH (Braunschweig, Germany) and EcoLite liquid scintillation cocktail was from MP Biomedicals (Santa Ana, CA, USA). Acrylamide and bis-acrylamide were from SERVA (Heidelberg, Germany), aluminum oxide 90 (neutral, activity I) was from Merck (Darmstadt, Germany) and cOmplete protease inhibitor cocktail was from Roche Life Science (Indianapolis, IN, USA). Anti-MAO-A and anti-ALDH-2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-catalase antibody was from Abcam (Cambridge, UK) and anti-Cu/ZnSOD and anti-MnSOD antibodies were from Cayman Chemical Company (Ann Arbor, MI, USA). SuperSignal West Dura chemiluminescent substrate was from Pierce Biotechnology (Rockford, IL, USA). All other chemicals were purchased from Sigma–Aldrich (St. Louis, MO, USA).
Animal model
The SHR-mtBN conplastic strain harboring the mitochondrial genome of a highly inbred strain BN on the nuclear genetic background of SHR was created by selective replacement of a mitochondrial genome of SHR with the mitochondrial genome of BN rats as described earlier [28]. Adult male SHR and SHR-mtBN rats (280–300 g body wt) were exposed to continuous normobaric hypoxia (CNH; inspired O2 fraction 0.1) in a normobaric chamber (6 m2) equipped with hypoxic generators (Everest Summit, Hypoxico Inc., NY, USA) for 3 weeks. No reoxygenation occurred during this period. Animals were used immediately after the cessation of hypoxic exposure. The control rats were kept for the same period of time at room air. All animals were housed in a controled environment (22 ± 2 °C; 12:12 h light–dark cycle; light from 5:00 a.m. with free access to water and standard chow diet. The study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (published by the National Academy of Science, National Academy Press, Washington, DC). Experimental protocols were approved by the Animal Care and Use Committee of the Institute of Physiology, Czech Academy of Sciences.
Processing of heart tissue for biochemical analyses
Immediately after sacrifice, the hearts were rapidly excised, washed in ice-cold saline solution and the left ventricles (LV) were dissected from the right ventricles and the septum. The pieces of frozen tissue were either pulverized in liquid nitrogen and subsequently homogenized in RNAzol® RT (Molecular Research Center, Inc.) for isolation of mRNA or homogenized in TMES buffer (20 mM Tris, 3 mM MgCl2, 1 mM EDTA, 250 mM sucrose; pH 7.4) supplemented with protease inhibitors for radioligand binding assay, Western blotting, enzyme activity determination or other biochemical analyses. In the latter case, the ventricles were homogenized on ice by Ultra-Turrax blender for 30 s and subsequently by Potter–Elvehjem glass-Teflon homogenizer for 1 min. The homogenates were clarified by centrifugation at 600×g for 10 min (4 °C) in order to remove nuclei and particulate cellular debris. The resulting postnuclear supernatant was centrifuged at 50,000×g for 30 min (4 °C) to separate the membrane and cytosolic fractions. The pellet containing crude membranes was resuspended in TME buffer (20 mM Tris, 3 mM MgCl2 and 1 mM EDTA; pH 7.4). Both membrane and cytosolic fractions were aliquoted and stored at −80 °C until use.
Real time RT-PCR
RNA isolation and real time RT-PCR were performed as described previously [30] with a slight modification as follows. Briefly, tissue homogenization and total RNA isolation was performed according manufacturer’s instruction using RNAzol Reagent (Molecular Research Center, Inc.). One microgram of total RNA was loaded to reverse transcription using RevertAidTM H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) with oligo(dT) primers according to the manufacturer’s instructions. Real Time PCR analyses were, performed on Light Cycler LC 480 (Roche Applied Science, Branford, CT, USA) using Syber green Master Mix (Eurogentec SA, Seraing, Belgium). Gene-specific primers were designed to be compatible with the Roche Universal Probe Library (UPL) and are listed in Table 1. The relative levels of analyzed gene transcripts were calculated according to Pfaffl [31] using the 18S rRNA gene as a suitable reference gene (18S_F: tctagacaacaagctgcgtga; 18S_R: cctctatgggctcggatttt). This reference gene was selected from six candidates using GenEx software (MultiD Analyses AB, Göteborg, Sweden).
Table 1.
PCR primers for the real time PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| AC5 | gggagaaccagcaacagg | catctccatggcaacatgac |
| AC6 | atgagatcatcgcggacttt | gccatgtaagtgctaccgatg |
| ACO1 | ttgctgtgtctgagattgaaaag | cttgaaaacctttaaatccttgct |
| ACO2 | cgccttacagcctactggtc | ggcagaggccacatggta |
| ALDH2 | agacgtcaaagatggcatga | ttgaggatctgcatcactgg |
| CAT | cagcgaccagatgaagca | ggtcaggacatcgggtttc |
| CuZnSOD | taagaaacatggcggtcca | tggacacattggccacac |
| GSTO1 | aagcttgccagaagatgacc | ctcttcgccctaataaaactcg |
| MAOA | tggtatcatgacccagtatgga | tgtgcctgcaaagtaaatcct |
| MnSOD | tggacaaacctgagccctaa | gacccaaagtcacgcttgata |
| NRF1 | atagtcctgtctggggaaacc | tccatgcatgaactccatct |
| NRF2 | agcatgatggacttggaattg | cctccaaaggatgtcaatcaa |
| PRX3 | agaagaacctgcttgacagaca | caggggtgtggaatgaaga |
| PRX5 | gactatggccccgatcaa | aaaacacctttcttgtccttgaa |
| TXN2 | cacacagaccttgccattga | acgtccccgttcttgatg |
| TXNRD2 | gcacatggtgaagctacctaga | gctccatccacatcttctcag |
β-Adrenergic receptor binding
Myocardial β-ARs were determined by radioligand binding assay with a nonselective β-adrenergic antagonist [3H]CGP 12177 as described previously [32]. Samples of crude membranes (100 µg protein) were incubated in incubation buffer (50 mM Tris, 10 mM MgCl2 and 1 mM ascorbic acid; pH 7.4) containing increasing concentrations of [3H]CGP 12177 (0.06–4 nM) for 1 h at 37 °C in a total volume of 0.5 ml; at this time the specific binding of radioligand had reached an equilibrium. The binding reaction was terminated by addition of 3 ml of ice-cold washing buffer (50 mM Tris, 10 mM MgCl2; pH 7.4) and subsequent filtration through Whatman GF/C filters, which were presoaked with 0.3% polyethyleneimine for 1 h. The filters were then washed twice with ice-cold washing buffer and placed into scintillation vials. After addition of 4 ml EcoLite scintillation cocktail, radioactivity retained on the filters was measured by counting for 5 min. Nonspecific binding was assessed by incubating the samples with radioligand in the presence of 10 μM l-propranolol, and it represented less than 30% of the total binding. For competition experiments, samples of crude membranes were incubated with 1 nM [3H]CGP 12177 and increasing concentrations of the selective β2-AR antagonist ICI 118.551 (10−4–10−10 M). The characteristics of β-adrenergic binding sites and the proportions of β1- and β2-ARs in myocardial crude membranes were calculated using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA).
Assessment of adenylyl cyclase activity
Activity of AC was determined by measuring the conversion of [α-32P]ATP to [32P]cAMP as described previously [33]. Samples of crude myocardial membranes (20 μg protein) were incubated in the reaction mixture (in a total volume 100 μl) containing 48 mM Tris buffer (pH 8), 2 mM MgCl2, 20 μM GTP, 0.8 mg/ml BSA, 40 μM 3-isobutyl-1-methylxanthine, 5 mM potassium phosphoenolpyruvate, 3.2 U of pyruvate kinase, 100 mM NaCl, 0.1 mM cAMP and about 15,000 cpm [3H]cAMP as a tracer. For stimulation of AC, the following stimulators were used in separate experiments: 10 μM isoprenaline, 10 μM forskolin, 10 mM MnCl2 and 10 mM NaF. After 1 min preincubation, 0.4 mM ATP was added along with 2,000,000 cpm [α-32P]ATP and incubation proceeded for 20 min at 30 °C. The reaction was terminated by addition of 200 μl 0.5 M HCl and heating for 5 min at 100 °C. Samples were neutralized by 200 μl 1.5 M imidazole. Separation of cAMP produced by stimulated membrane preparations from other nucleotides was performed by filtration through alumina columns, and the detected amount of [32P]cAMP in each vial was corrected for recovery with [3H]cAMP as the internal standard.
Assessment of monoamine oxidase A activity
MAO-A activity in the LV was determined using kynuramine dihydrobromide as substrate in the presence of deprenyl (inhibitor MAO-B) as described previously [34] with a slight modification. Myocardial crude membranes (100 μg protein) were lysed by treatment with 2% Triton X-100. Resulting lysate samples (200 μl) were mixed with 0.2 μl of 1 mM deprenyl and 2.5 ml of 50 mM phosphate buffer (pH 7.4) and incubated for 60 min at 37 °C. After incubation, 30 μl of 2.19 mM kynuramine dihydrobromide were added to each reaction mixture as substrate and incubation was continued for another 30 min at 37 °C. The reaction was terminated by addition of 200 μl of 5 M perchloric acid. Samples were centrifuged at 1500×g for 10 min and 500 μl aliquots of supernatant were transferred into test tubes containing 2.5 ml of 1 M NaOH. The fluorescence of the reaction product 4-quinolinol was measured at Ex 310-nm/Em 380-nm using a Biotek Synergy HT plate reader. A standard curve of 4-quinolinol (in the range of 0.03–0.5 mM) was used to calculate MAO-A enzyme activity.
Electrophoresis and Western blotting
Individual samples of myocardial preparations were solubilized in Laemmli buffer and loaded (10–30 μg per lane) on 10 or 15% acrylamide gels for SDS-PAGE as described previously [35]. After electrophoresis, the resolved proteins were transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Buckinghamshire, UK), blocked with 5% non-fat dry milk in TBS buffer (10 mM Tris, 150 mM NaCl; pH 8.0) for 1 h and then incubated for 1.5 h at room temperature or overnight at 4 °C with relevant primary antibodies. After three 10-min washes in TBS containing 0.3% Tween 20, the membranes were incubated with secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. Immunoreactive proteins on the blots were visualized by enhanced chemiluminiscence technique according to the manufacturer’s instructions (Pierce Biotechnology, Rockford, IL, USA) and quantitatively analyzed by ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA). To correct for errors associated with sample loading and gel transfer, β-actin was used as a housekeeping protein for reliable quantification of Western blot data.
Determination of malondialdehyde
Lipid peroxidation was quantified by measuring malondialdehyde (MDA) formation. Myocardial samples (100 mg) were pulverized to a fine powder and dissolved in 500 μl of ice-cold buffer (25 mM Tris and 0.10% Triton X 100; pH). The homogenates were sonicated, centrifuged (1000×g, 10 min, 4 °C) and 100 μl samples of supernatant were taken and analyzed as described by Pilz et al. [36] with a slight modification. Briefly, 20 μl of 6 M NaOH was added to each sample, vortexed and incubated for 30 min at 60 °C. The solution was then cooled on ice and 50 μl of 35% perchloric acid was added. After centrifugation (10,000×g, 5 min, 4 °C), 100 μl of supernatant was taken and derivatization was performed using 10 μl of 5 mM 2,4-dinitrophenylhydrazine. After 10 min in the dark, the solution was analyzed using a HPLC system (Shimadzu, Kyoto, Japan) with UV detection at 310 nm (column: EC Nucleosil 100-5 C18, 4.6 mm × 125 mm heated to 30 °C; mobile phase: acetonitrile–water–acetic acid 380:620:2 (v/v/v); flow rate: 1.0 ml/min). MDA concentration was normalized to total protein content.
Determination of TNF-α, IL-6 and IL-10
Levels of TNF-α (tumor necrosis factor-α), IL-6 (interleukin-6) and IL-10 (interleukin-10) in myocardial homogenates from different experimental groups were measured using DuoSet ELISA kits (eBioscience, Vienna, Austria) according to the standard protocols described by the manufacturer. The content of cytokines is given in picograms per milligram of total protein [37].
Data analysis
Biochemical data were determined in at least three independent preparations. All results were expressed as the mean ± SEM. The Kolmogorov–Smirnov test was used to assess the normality and all parameters were distributed normally. One-way analysis of variance (ANOVA) and subsequent Student–Newman–Keuls tests were used for comparison of differences in normally distributed variables between the groups. Differences between appropriate groups were considered to be statistically significant when the p value was smaller than 0.05 (p < 0.05).
Results
All measurements were done on LV myocardial preparations obtained from normoxic rats (SHR and SHR-mtBN) as well as from those exposed to continuous normobaric hypoxia (CNH) for 3 weeks (SHR/H and SHR-mtBN/H).
β-Adrenergic receptors
The total number of β-ARs and dissociation constants of these receptors in myocardial membrane preparations from LVs were determined by using saturation binding experiments (Fig. 1a). We found that the total number of β-ARs, expressed as B max, tended to be higher (by about 15%) in SHR-mtBN than SHR, but this difference was not statistically significant. The values of B max were also affected by adaptation of rats to hypoxia. Whereas CNH induced a significant increase (by 16%) in the total number of β-ARs in SHR/H, the expression of these receptors was markedly diminished (by 26%) in SHR-mtBN/H (Table 2). Dissociation constants (K d) did not differ significantly between the strains and their values were not affected by adaptation to hypoxia. Subsequently, competition binding experiments were conducted to assess the distribution of β-AR subtypes in LV preparations (Fig. 1b). As indicated in Table 3, the proportion of β2-ARs was significantly increased (by 37%) in SHR-mtBN, compared to SHR. Exposure to hypoxia increased the proportion of β2-AR in SHR/H by 30% but did not affect the relative proportion of β-AR subtypes in SHR-mtBN/H.
Fig. 1.
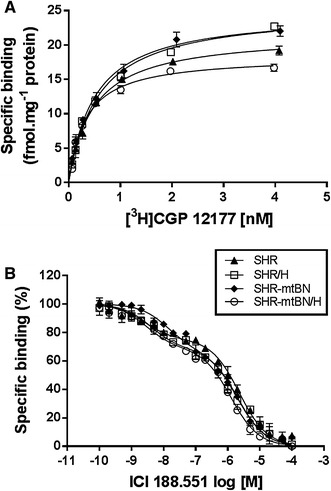
Effect of hypoxia on myocardial β-adrenergic receptors. β-ARs in LV preparations from SHR (closed triangles), SHR/H (open squares), SHR-mtBN (closed diamonds) and SHR-mtBN/H (open circles) were characterized by radioligand binding experiments. Shown are [3H]CGP 12177 saturation binding curves (a) and competitive binding curves (b) which were constructed using the β2-AR antagonist ICI 188.551. Data represent means (±SEM) of three separate experiments performed in triplicate
Table 2.
Binding characteristics of β-ARs
| SHR | SHR/H | SHR-mtBN | SHR-mtBN/H | |
|---|---|---|---|---|
| B max (fmol mg−1) | 21.71 ± 0.88 | 25.20 ± 0.38+ | 24.87 ± 0.73 | 18.37 ± 0.46§# |
| K D (nM) | 0.49 ± 0.10 | 0.58 ± 0.07 | 0.51 ± 0.03 | 0.32 ± 0.01 |
Data are means (±SEM) of three separate experiments performed in triplicates
B max maximal binding capacity, K D equilibrium dissociation constant of radioligand ([3H]CGP 12177)
+ p < 0.05 SHR vs. SHR/H; § p < 0.05 SHR/H vs. SHR-mtBN/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H
Table 3.
Distribution and properties of β-AR subtypes
| SHR | SHR/H | SHR-mtBN | SHR-mtBN/H | |
|---|---|---|---|---|
| β2 (%) | 24.03 ± 0.30 | 31.12 ± 1.42+ | 32.97 ± 2.01* | 31.15 ± 0.42 |
| K iβ2 (nM) | 0.80 ± 0.53 | 1.43 ± 0.17 | 4.80 ± 1.25 | 0.75 ± 0.21 |
| K iβ1 (µM) | 0.73 ± 0.06 | 0.77 ± 0.25 | 0.70 ± 0.07 | 0.31 ± 0.03 |
Data are means (±SEM) of three separate experiments performed in triplicates
β 2 (%) percentage of β2-ARs of total myocardial β-ARs, K i β 2 (β 1) apparent dissociation constant representing the affinity of ICI 118.551 to β2-ARs (β1-ARs)
* p < 0.05 SHR vs. SHR-mtBN; + p < 0.05 SHR vs. SHR/H
Adenylyl cyclase
Activity of AC in myocardial membrane preparations was determined by measuring cAMP production under different experimental conditions. Besides determining basal AC activity, the enzyme activity was modulated by the following stimulatory agents: isoprenaline, forskolin, MnCl2 and NaF. Isoprenaline is a β-AR agonist, forskolin or MnCl2 can stimulate the AC catalytic subunit directly and NaF elicits the enzymatic response through activation of the stimulatory G proteins [38]. Results of experiments in which AC activity was tested under different conditions are summarized in Fig. 2. Basal AC activity did not significantly differ between both strains or even after adaptation to hypoxia, but the enzyme activity was diversely influenced by different stimulators. Whereas there was no significant difference between SHR and SHR-mtBN in AC activity stimulated by forskolin or MnCl2, the enzyme activity stimulated by isoprenaline and NaF was markedly increased in SHR-mtBN (by about 35%). Adaptation of rats to hypoxia also led to specific changes in variably stimulated AC activity. Whereas CNH increased AC activity stimulated by forskolin or NaF by about 30% in preparations from SHR, there was no change in forskolin-stimulated AC activity and decrease (by 17%) in NaF-stimulated AC activity in hypoxia-adapted SHR-mtBN/H, compared to the corresponding normoxic controls. The stimulatory effects on AC activity of forskolin and NaF were lower by 10 and 14%, respectively, in SHR-mtBN/H than in SHR/H. Hypoxia did not significantly affect the ability of isoprenaline to stimulate AC activity in SHR but decreased its stimulatory effect by 13% in SHR-mtBN. Interestingly, determination of transcript levels of the dominant myocardial isoforms of AC (AC5 and AC6) revealed a decrease (by about 35%) in mRNA levels of both these isoforms in SHR-mtBN adapted to hypoxia (Suppl. Fig. S1).
Fig. 2.
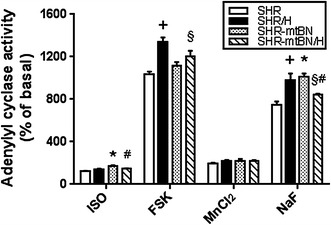
Effect of hypoxia on activity of myocardial adenylyl cyclase. AC activity in LV preparations from SHR (empty bars), SHR/H (closed bars), SHR-mtBN (dotted bars) and SHR-mtBN/H (hatched bars) was determined in the presence of the following stimulators: 10 μM isoprenaline (ISO), 10 μM forskolin (FSK), 10 mM MnCl2 and 10 mM NaF. Differently stimulated AC activity is expressed as a percentage of the corresponding basal AC activity, which did not significantly differ between different samples and was in the range of 7.9–9.2 pmol cAMP/min per mg protein in all measurements. Data represent means (±SEM) of five independent measurements performed in triplicate. *p < 0.05 SHR vs. SHR-mtBN rats; + p < 0.05 SHR vs. SHR/H rats; § p < 0.05 SHR/H vs. SHR-mtBN/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H rats
Monoamine oxidase A
Gene expression and enzyme activity of MAO-A were assessed in myocardial preparations from both normoxic and hypoxia-exposed SHR and SHR-mtBN. The mRNA level of MAO-A was significantly lower (by 30%) in SHR-mtBN than in SHR (Fig. 3a). Adaptation to hypoxia resulted in marked increase of MAO-A mRNA in both strains (by 97 and 132% in SHR and SHR-mtBN, respectively). The same trend was detected at the protein level and in the enzyme activity of MAO-A. The expression of MAO-A protein was lower by 28% in SHR-mtBN than in SHR, and CNH elevated the amount of MAO-A protein by 66 and 92% in SHR and SHR-mtBN, respectively (Fig. 3b). The enzyme activity of MAO-A in SHR-mtBN was lower by 21% than in SHR, and CNH increased the enzyme activity by 33 and 22% in SHR and SHR-mtBN, respectively (Fig. 3c).
Fig. 3.
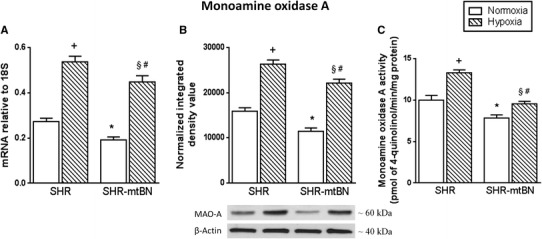
Effect of hypoxia on myocardial monoamine oxidase. Expression levels of mRNA and protein of MAO-A in LV preparations from SHR and SHR-mtBN rats kept in normoxic (empty bars) and hypoxic (hatched bars) conditions were assessed by RT-PCR and Western blotting, respectively. The amounts of specific mRNA species were normalized with 18S levels (a). The 18S levels were not significantly affected by hypoxic exposure. Representative Western blots are shown below the corresponding bar graphs depicting the relative protein expression levels (b). Specific enzyme activity of MAO-A (c) was determined spectrophotometrically by using kynuramine as substrate. Data represent means (±SEM) of at least three separate experiments. * p < 0.05 SHR vs. SHR-mtBN rats; + p < 0.05 SHR vs. SHR/H rats; § p < 0.05 SHR/H vs. SHR-mtBN/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H rats
Antioxidant defence enzymes
In the next set of experiments, we tested whether adaptation to hypoxia affects expression of selected antioxidant defence enzymes in the LV of both rat strains. CNH did not change the mRNA level of catalase (CAT) in SHR, but markedly increased (by 51%) the amount of this transcript in SHR-mtBN (Fig. 4, panel a). The expression of CAT protein was increased by 105 and 68% in hypoxia-exposed SHR/H and SHR-mtBN/H, respectively (Fig. 4, panel b). Adaptation to hypoxia elevated the level of aldehyde dehydrogenase 2 (ALDH-2) mRNA in both strains by about 40–50% (Fig. 4, panel a). The expression of ALDH-2 protein was increased by 25 and 32% in SHR/H and SHR-mtBN/H, respectively, when compared to corresponding normoxic controls (Fig. 4, panel b). In contrast to CAT and ALDH-2, mRNA and protein levels of superoxide dismutases (SODs) were decreased (Cu/ZnSOD) or remained unchanged (MnSOD) after adaptation to hypoxia (Fig. 5). The mRNA level of cytosolic Cu/ZnSOD was reduced by 12 and 16% in SHR/H and SHR-mtBN/H, respectively, but the expression of Cu/ZnSOD protein was significantly changed (a drop by 31%) just in SHR/H.
Fig. 4.
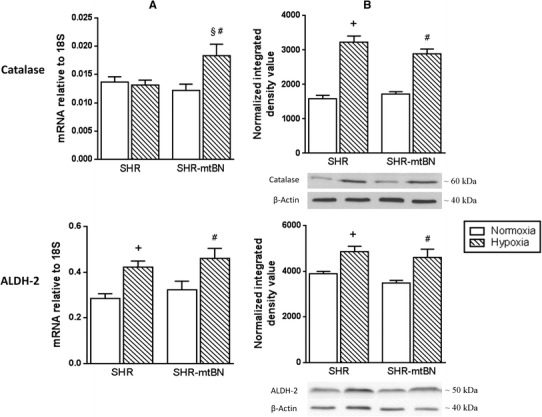
Effect of hypoxia on myocardial catalase and aldehyde dehydrogenase-2. Expression levels of mRNA and protein of CAT and ALDH-2 in LV preparations from SHR and SHR-mtBN rats kept in normoxic (empty bars) and hypoxic (hatched bars) conditions were assessed by RT-PCR and Western blotting, respectively. The amounts of specific mRNA species were normalized with 18S levels (column a). The 18S levels were not significantly affected by hypoxic exposure. Representative Western blots are shown below the corresponding bar graphs depicting the relative protein expression levels (column b). Data represent means (±SEM) of at least three separate experiments. + p < 0.05 SHR vs. SHR/H rats; § p < 0.05 SHR/H vs. SHR-mtBN/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H rats
Fig. 5.
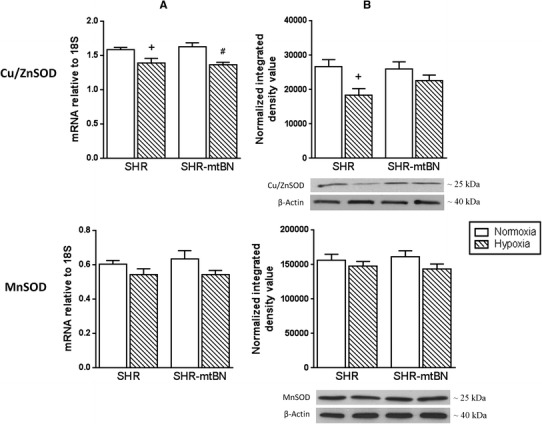
Effect of hypoxia on myocardial cytosolic copper/zinc superoxide dismutase and mitochondrial manganese superoxide dismutase. Expression levels of mRNA and protein of Cu/ZnSOD and MnSOD in LV preparations from SHR and SHR-mtBN rats kept in normoxic (empty bars) and hypoxic (hatched bars) conditions were assessed by RT-PCR and Western blotting, respectively. The amounts of specific mRNA species were normalized with 18S levels (column a). The 18S levels were not significantly affected by hypoxic exposure. Representative Western blots are shown below the corresponding bar graphs depicting the relative protein expression levels (column b). Data represent means (±SEM) of at least five separate experiments. + p < 0.05 SHR vs. SHR/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H rats
A similar downward trend was also observed in the expression of myocardial glutathione S-transferase ω1 (GTSO1), thioredoxin 2 (TXN2) and thioredoxin reductase 2 (TXNRD2) and peroxiredoxin 5 (PRX5) after adaptation to hypoxia (Suppl. Fig. S2a–c). The mRNA levels of these enzymes dropped by 19% (GSTO1 and TXN2), 18% (TXNRD2) and 16% (PRX5) in SHR/H and by 32% (TXN2), 27% (TXNRD2) and 25% (PRX5) in SHR-mtBN/H. Although hypoxia exposure did not markedly affect comparable levels of PRX3 mRNA in both rat strains, the amount of this transcript was higher (by 48%) in SHR-mtBN/H than in SHR/H (Suppl. Fig. S2c). The mRNA levels of ACO1 and ACO2 remained without significant changes (Suppl. Fig. S2d). In addition, we detected increased expression (by 31%) of nuclear respiratory factor 1 (NRF1) mRNA in SHR-mtBN/H and no change in the mRNA level of nuclear factor (erythroid-derived 2)-like 2 (NFE2L2, NRF2) (Suppl. Fig. S2e).
Markers of lipid peroxidation and inflammation
Malondialdehyde (MDA) was determined as a marker of oxidative stress-induced lipid peroxidation. Myocardial preparations from both rat strains kept under normoxic conditions did not exhibit any differences between MDA levels. Adaptation to hypoxia increased the MDA concentration by 104 and 52% in SHR/H and SHR-mtBN/H, respectively (Fig. 6a). Levels of anti-inflammatory interleukin-10 (IL-10) and pro-inflammatory tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) tended to be lower in SHR-mtBN than in SHR, and these levels tended to increase in both strains after hypoxic exposure (Suppl. Fig. 3). CNH did not significantly change the IL-10/TNF-α ratio in SHR/H and increased this ratio (by 17%) in SHR-mtBN/H (Fig. 6b).
Fig. 6.
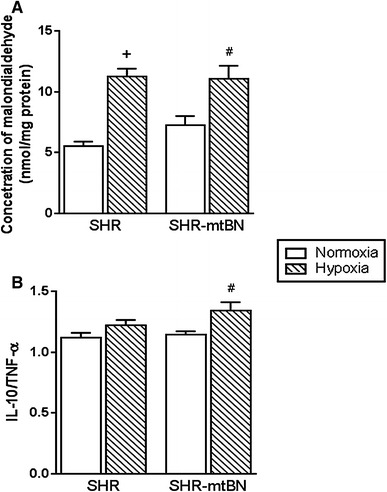
Effect of hypoxia on myocardial malondialdehyde levels and the ratio of interleukin-10/tumor necrosis factor-α. Concentrations of malondialdehyde (a) and the IL-10/TNF-α ratio (b) were assessed in LV preparations from SHR a SHR-mtBN rats kept in normoxic (empty bars) and hypoxic (hatched bars) conditions. Data represent means (±SEM) of at least five separate experiments. + p < 0.05 SHR vs. SHR/H rats; # p < 0.05 SHR-mtBN vs. SHR-mtBN/H rats
Discussion
In the present study, we sought to explore the impact of mitochondria replacement on myocardial β-adrenergic signaling and antioxidant defence in the spontaneously hypertensive rat (SHR) under normoxic and hypoxic conditions. The experiments were conducted in parallel on SHR and SHR-mtBN, i.e., the SHR-based conplastic strain carrying the mitochondrial genome from Brown Norway rats [28]. Our investigation revealed a number of distinct changes at the level of β-ARs, AC and MAO-A, as well as in some components of the antioxidant defence system.
We observed that the proportion of myocardial β2-ARs was significantly higher in SHR-mtBN than in SHR and that adaptation to hypoxia diversely affected the distribution of β-ARs in both rat strains. Whereas CNH elevated β-AR density in SHR due to increased expression of β2-AR, the total number of β-ARs in SHR-mtBN declined without a change in the proportion of β-AR subtypes. The higher β-AR density in SHR-mtBN was reflected by increased ability of isoprenaline to stimulate AC activity. However, isoprenaline-stimulated AC activity was reduced in preparations from SHR-mtBN exposed to hypoxia, which is in line with the lower expression of β-ARs and AC determined in these animals. The enzyme activity stimulated by forskolin and NaF (the agents capable of stimulating AC both directly and through G proteins or only through G proteins, respectively) was enhanced in SHR after their exposure to CNH. Interestingly, no such elevation was found in CNH-adapted SHR-mtBN. By contrast, NaF-stimulated AC activity in these rats was already increased under normoxic conditions, and it was diminished after exposure to CNH. These strain-specific differences may presumably be explained by altered ability of G proteins to regulate AC activity. Both forskolin and NaF enhanced AC activity much more strongly than isoprenaline, which is a well-known and commonly reported phenomenon [14]. The observed decline in the number β-ARs in SHR-mtBN after CNH exposure is consistent with previous findings of lower β-AR expression in rat heart following hypoxia [12, 13, 15]. As a rule, β-AR subtypes were not discerned in these studies. The only exception is a study of Mardon et al. [14], which demonstrated a selective decrease in β1-ARs caused by 5-day hypoxia. These investigators, like others [15, 16], found reduced AC activity in myocardial preparations from hypoxia-exposed rats. The reduction in NaF-stimulated AC activity observed in CNH-exposed SHR-mtBN, thus, conforms well to the previously published data. On the other hand, the CNH-induced increase in the ability of G proteins to stimulate myocardial AC activity in SHR has not been previously noted and may represent a specific feature of this rat strain. It is worth mentioning that especially β2-AR appears to be important for generating the salutary effects of preconditioning [11, 39]. Accordingly, β2-ARs were found to activate pro-survival kinases and attenuate mitochondrial dysfunction during oxidative stress [40]. The increased proportion of β2-AR in SHR-mtBN, compared to SHR, and reduction in the total number of β-ARs after adaptation to CNH may, thus, contribute to better protection of this rat strain against acute I/R injury [28].
It is well known that hypoxic stress is associated with increased production and release of catecholamines, and these compounds may be subjected to oxidative deamination catalyzed by monoamine oxidases [41]. These enzymes have recently emerged as important mitochondrial sources of oxidative stress in the cardiovascular system and MAO inhibition may apparently have a therapeutic value for treating cardiac affections of ischemic and non-ischemic origin [42]. Interestingly, the presence of MAO inhibitors during ischemic preconditioning (IPC) was shown to potentiate the IPC-induced post-ischemic functional recovery of a rat heart [43]. In the present study, we found that the expression and activity of MAO-A (the predominant cardiac isoform) was lower in SHR-mtBN than in SHR and that CNH exposure increased similarly the expression and activity of this enzyme in both strains. Nevertheless, MAO-A activity remained still significantly lower in SHR-mtBN under these conditions, which can be considered as a strain-specific feature. The observed elevation of myocardial MAO-A after hypoxic exposure may suggest the importance of MAO-A in adaptive responses to chronic hypoxia. It should be mentioned here that our finding of a relatively large increase in MAO-A activity after CNH is in contrast to the work of Maher et al. [44]. These authors did not find any change in MAO activity in a goat heart exposed to chronic hypoxia. However, this discrepancy can be explained by using different models and experimental conditions. There are some indications that the possible changes in MAO activity may depend on the duration of hypoxia. Whereas 5-day exposure to hypoxia led to decrease of MAO activity in rat liver, 21-day exposure increased the enzyme activity [45].
The harmful effects of ROS generated from different sources, including mitochondria and MAO activity, are regularly combated by a number of endogenous antioxidant defence mechanisms. The enzyme antioxidant defence system consists of different types of enzymes with different functions [46]. In the present study, we focused on selected ROS-degrading enzymes (SODs, CAT, PRXs, GSTO1) and enzymes participating in maintenance of cellular redox homeostasis (TXN2, TXNRD2, ALDH-2, ACO1 and 2). Assessment of expression levels of all these enzymes in myocardial preparations from SHR and SHR-mtBN did not reveal any significant differences between both these strains under normoxic conditions. Moreover, adaptation to hypoxia elicited very similar changes in the levels of most of these enzymes in both progenitor and conplastic SHR strains. CNH appreciably elevated the protein levels of CAT and ALDH-2, and did not change MnSOD in myocardial preparations from both strains. In contrast, CNH markedly reduced the amount of Cu/ZnSOD protein in SHR but not in SHR-mtBN. The transcript levels of mitochondrial antioxidants TXN2, TXNRD2 and PRX5 were always significantly lower in CNH-exposed animals than in corresponding normoxic controls. On the other hand, CNH did not change the mRNA levels of PRX3, ACO1 and ACO2, and decreased GSTO1 in SHR only. Interestingly, most changes associated with CNH in the expression of antioxidant enzymes in SHR and SHR-mtBN markedly differ from those reported in previous studies. In contrast to our current findings, the expression levels of Cu/ZnSOD, MnSOD, TXN2, TXNRD2, PRX5 and ACO2 were found to increase, CAT to decrease and GSTO1 not to change after adaptation of Wistar rats to the same cardioprotective regimen of hypoxia [22]. A significant increase in myocardial Cu/ZnSOD and MnSOD was also detected in Wistar rats adapted to different hypoxic conditions [47–49]. Hence, the observed discordant changes in antioxidant enzymes induced by hypoxic exposure appear to be strain-specific. The unique downregulation of Cu/ZnSOD and GSTO1 in SHR following hypoxia may partly explain the lower cardioprotective effect of CNH against acute I/R injury in these rats when compared to SHR-mtBN [28]. The effect of hypoxia on ALDH-2 expression has not yet been investigated and our finding of increased levels of ALDH-2 in both rat strains exposed to CNH supports the notion that this enzyme is highly important for reducing ischemic heart damage [50] and may follow activation of MAO-A. NRF2 is a transcription factor crucially involved in the regulation of antioxidant enzyme expression, and they can modulate the capacity of the antioxidant system at the protein level [51]. The observed increase in NRF1 in CNH-exposed SHR-mtBN might contribute to upregulation of some nuclear-encoded mitochondrial respiratory genes (such as cytochrome c oxidase (subunit VIc) and reductase, the γ subunit of ATP synthase) as NRF1 is one of the important transcription factors for structural components of the mammalian electron transport chain [52, 53]. Moreover, NRF1 has been shown to play a role in the mitochondrial DNA replication machinery and other biosynthetic and degradative pathways [54]. The elevated NRF1 expression after CNH exposure may promote mitochondrial biogenesis and, thus, increase cardiac resistance to I/R injury of SHR-mtBN/H.
Chronic hypoxia is usually accompanied by increased ROS formation [55, 56]. Therefore, we decided to assess the extent of lipid peroxidation by monitoring MDA content in myocardial preparations from rats under normoxic and hypoxic conditions. Interestingly, the MDA concentration was markedly increased in both rat strains after CNH exposure, but this increase was much more pronounced in hearts of SHR, which may reflect lower antioxidant capacity of these animals when compared to SHR-mtBN. Similar increase in myocardial MDA concentration was previously found in Wistar rats or mice after exposure to different regimens of hypoxia [37, 57–59]. These data indicate that, irrespective of animal model, ROS formation accompanied by increased lipid peroxidation is a typical sign of chronic hypoxia.
It is known that the adaptive response to hypoxia of the heart tissue can be modulated by inflammatory cytokines [60]. In the present study, we found that the amount of the principal anti-inflammatory (IL-10) and pro-inflammatory (TNF-α and IL-6) cytokines tended to be increased under hypoxic conditions. In parallel, these animals exhibited a significant increase in the IL-10/TNF-α ratio. Interestingly, these results appear rather contradictory with respect to the previously observed decrease in IL-10 and lower IL-10/TNF-α ratio in Wistar rats adapted to hypoxia [37]. We think that the disproportionate changes in the content of myocardial cytokines under hypoxic conditions can be ascribed to specific differences between individual rat strains. Moreover, the increased IL-10/TNF-α ratio in SHR-mtBN exposed to CNH can likely contribute to the lesser extent of myocardial damage caused by I/R.
The main findings obtained in the present study are schematically shown in Fig. 7. Taken together, our results indicate that the selective replacement of mitochondria of SHR with those of more ischemia–resistant Brown Norway strain can modify the functioning of myocardial AC signaling regulated by β-ARs and G proteins, as well as the expression and activity of MAO-A and some components of the antioxidant defence system. This is the first study to demonstrate relatively far-reaching consequences arising from the manipulation of the mitochondrial genome for β-adrenergic signaling and redox balance in rat heart. Concurrently, these data provide new evidence that mitochondria function not only as key ATP-producing organelles, but they may influence other aspects of normal cell functioning. Importantly, manipulating the mitochondrial genome may apparently lead to complex alterations within target cells, and this should be taken into account when planning application of such manipulations for research or therapeutic purposes.
Fig. 7.
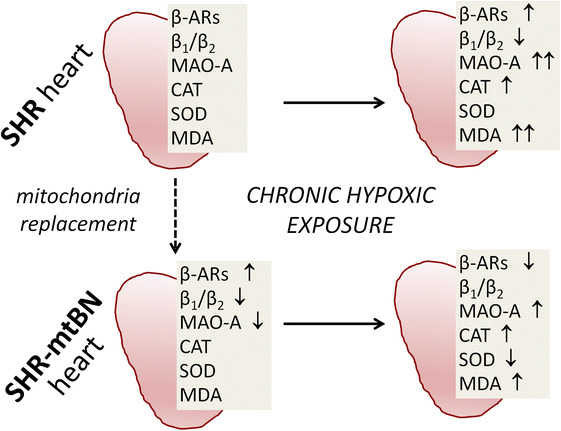
Schematic summarizing the main findings. Replacement of the mitochondrial genome of SHR with that of the more ischemia–resistant Brown Norway strain (mtBN) distinctly affects myocardial expression of β-ARs and MAO-A. Adaptation to chronic hypoxia is associated with partially dissimilar changes in β-ARs, MAO-A and some components of the antioxidant defence in the myocardium of SHR and SHR-mtBN. These differences may contribute to more efficient cardioprotection conferred by chronic hypoxia in SHR-mtBN, compared to progenitor SHR. The arrows indicate changes relative to SHR or to SHR-mtBN in the case of SHR-mtBN/H. β-ARs, β-adrenergic receptors; β1/β2, the β1-AR/β2-AR ratio; MAO-A monoamine oxidase A, CAT catalase, SOD superoxide dismutase, MDA malondialdehyde, up arrow (double up arrow) and down arrow, increase (higher increase) and decrease, respectively
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Grant 13-10267 from the Czech Science Foundation, Grant 1214214 from the Charles University Grant Agency, and by the institutional research projects no. 67985823 (Institute of Physiology, CAS) and SVV-260434/2017 (Charles University in Prague). MP was supported by Grants LL1204 (within the ERC CZ program) from the Ministry of Education, Youth and Sports and P301/12/0696 from the Czech Science Foundation.
Compliance with ethical standards
Conflict of interests
The authors have no conflict of interest to declare.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
References
- 1.Hajri T, Ibrahimi A, Coburn CT, Knapp FF, Kurtz T, Pravenec M, Abumrad NA. Defective fatty acid uptake in the spontaneously hypertensive rat is a primary determinant of altered glucose metabolism, hyperinsulinemia, and myocardial hypertrophy. J Biol Chem. 2001;276:23661–23666. doi: 10.1074/jbc.M100942200. [DOI] [PubMed] [Google Scholar]
- 2.Itter G, Jung W, Juretschke P, Schoelkens BA, Linz W. A model of chronic heart failure in spontaneous hypertensive rats (SHR) Lab Anim. 2004;38:138–148. doi: 10.1258/002367704322968812. [DOI] [PubMed] [Google Scholar]
- 3.Penna C, Pasqua T, Amelio D, Perrelli MG, Angotti C, Tullio F, Mahata SK, Tota B, Pagliaro P, Cerra MC, Angelone T. Catestatin increases the expression of anti-apoptotic and pro-angiogenetic factors in the post-ischemic hypertrophied heart of SHR. PLoS One. 2014;9:e102536. doi: 10.1371/journal.pone.0102536. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Ravingerová T, Bernátová I, Matejíková J, Ledvényiová V, Nemčeková M, Pecháňová O, Tribulová N, Slezák J. Impaired cardiac ischemic tolerance in spontaneously hypertensive rats is attenuated by adaptation to chronic and acute stress. Exp Clin Cardiol. 2011;16:e23–e29. [PMC free article] [PubMed] [Google Scholar]
- 5.Kolar F, Parratt JR. Antiarrhythmic effect of ischemic preconditioning in hearts of spontaneously hypertensive rats. Exp Clin Cardiol. 1997;2:124–128. [Google Scholar]
- 6.Neckář J, Šilhavy J, Zídek V, Landa V, Mlejnek P, Šimáková M, Seidman JG, Seidman C, Kazdová L, Klevstig M, Novák F, Vecka M, Papoušek F, Houštěk J, Drahota Z, Kurtz TW, Kolář F, Pravenec M. CD36 overexpression predisposes to arrhythmias but reduces infarct size in spontaneously hypertensive rats: gene expression profile analysis. Physiol Genom. 2012;44:173–182. doi: 10.1152/physiolgenomics.00083.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klevstig M, Manakov D, Kasparova D, Brabcova I, Papousek F, Zurmanova J, Zidek V, Silhavy J, Neckar J, Pravenec M, Kolar F, Novakova O, Novotny J. Transgenic rescue of defective Cd36 enhances myocardial adenylyl cyclase signaling in spontaneously hypertensive rats. Pflugers Arch. 2013;465:1477–1486. doi: 10.1007/s00424-013-1281-5. [DOI] [PubMed] [Google Scholar]
- 8.Florea SM, Blatter LA. Regulation of cardiac alternans by β-adrenergic signaling pathways. Am J Physiol Heart Circ Physiol. 2012;303:H1047–H1056. doi: 10.1152/ajpheart.00384.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frances C, Nazeyrollas P, Prevost A, Moreau F, Pisani J, Davani S, Kantelip JP, Millart H. Role of beta 1- and beta 2-adrenoceptor subtypes in preconditioning against myocardial dysfunction after ischemia and reperfusion. J Cardiovasc Pharmacol. 2003;41:396–405. doi: 10.1097/00005344-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Tong H, Bernstein D, Murphy E, Steenbergen C. The role of beta-adrenergic receptor signaling in cardioprotection. FASEB J. 2005;19:983–985. doi: 10.1096/fj.04-3067fje. [DOI] [PubMed] [Google Scholar]
- 11.Salie R, Moolman JA, Lochner A. The role of β-adrenergic receptors in the cardioprotective effects of beta-preconditioning (βPC) Cardiovasc Drugs Ther. 2011;25:31–46. doi: 10.1007/s10557-010-6275-3. [DOI] [PubMed] [Google Scholar]
- 12.Mader SL, Downing CL, Van Lunteren E. Effect of age and hypoxia on beta-adrenergic receptors in rat heart. J Appl Physiol. 1991;71:2094–2098. doi: 10.1152/jappl.1991.71.6.2094. [DOI] [PubMed] [Google Scholar]
- 13.Kacimi R, Richalet JP, Corsin A, Abousahl I, Crozatier B. Hypoxia-induced downregulation of beta-adrenergic receptors in rat heart. J Appl Physiol. 1992;73:1377–1382. doi: 10.1152/jappl.1992.73.4.1377. [DOI] [PubMed] [Google Scholar]
- 14.Mardon K, Merlet P, Syrota A, Mazière B. Effects of 5-day hypoxia on cardiac adrenergic neurotransmission in rats. J Appl Physiol. 1998;85:890–897. doi: 10.1152/jappl.1998.85.3.890. [DOI] [PubMed] [Google Scholar]
- 15.León-Velarde F, Bourin MC, Germack R, Mohammadi K, Crozatier B, Richalet JP. Differential alterations in cardiac adrenergic signaling in chronic hypoxia or norepinephrine infusion. Am J Physiol Regul Integr Comp Physiol. 2001;280:R274–R281. doi: 10.1152/ajpregu.2001.280.1.R274. [DOI] [PubMed] [Google Scholar]
- 16.Hrbasová M, Novotny J, Hejnová L, Kolár F, Neckár J, Svoboda P. Altered myocardial Gs protein and adenylyl cyclase signaling in rats exposed to chronic hypoxia and normoxic recovery. J Appl Physiol. 2003;94:2423–2432. doi: 10.1152/japplphysiol.00958.2002. [DOI] [PubMed] [Google Scholar]
- 17.Johnson TS, Young JB, Landsberg L. Sympathoadrenal responses to acute and chronic hypoxia in the rat. J Clin Invest. 1983;71:1263–1272. doi: 10.1172/JCI110876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A, Westerblad H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J Physiol. 2011;589:1791–1801. doi: 10.1113/jphysiol.2010.202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mallet RT, Ryou MG, Williams AG, Howard L, Downey HF. β1-Adrenergic receptor antagonism abrogates cardioprotective effects of intermittent hypoxia. Basic Res Cardiol. 2006;101:436–446. doi: 10.1007/s00395-006-0599-y. [DOI] [PubMed] [Google Scholar]
- 20.Zuo L, Roberts WJ, Tolomello RC, Goins AR. Ischemic and hypoxic preconditioning protect cardiac muscles via intracellular ROS signaling. Front Biol. 2013;8:305–311. doi: 10.1007/s11515-012-1225-z. [DOI] [Google Scholar]
- 21.Kolár F, Jezková J, Balková P, Breh J, Neckár J, Novák F, Nováková O, Tomásová H, Srbová M, Ost’ádal B, Wilhelm J, Herget J. Role of oxidative stress in PKC-delta upregulation and cardioprotection induced by chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2007;292:H224–H230. doi: 10.1152/ajpheart.00689.2006. [DOI] [PubMed] [Google Scholar]
- 22.Kasparova D, Neckar J, Dabrowska L, Novotny J, Mraz J, Kolar F, Zurmanova J. Cardioprotective and nonprotective regimens of chronic hypoxia diversely affect the myocardial antioxidant systems. Physiol Genom. 2015;47:612–620. doi: 10.1152/physiolgenomics.00058.2015. [DOI] [PubMed] [Google Scholar]
- 23.Cadenas S, Aragonés J, Landázuri MO. Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res. 2010;88:219–228. doi: 10.1093/cvr/cvq256. [DOI] [PubMed] [Google Scholar]
- 24.Mueller IA, Grim JM, Beers JM, Crockett EL, O’Brien KM. Inter-relationship between mitochondrial function and susceptibility to oxidative stress in red- and white-blooded Antarctic notothenioid fishes. J Exp Biol. 2011;214:3732–3741. doi: 10.1242/jeb.062042. [DOI] [PubMed] [Google Scholar]
- 25.Cortie CH, Hulbert AJ, Hancock SE, Mitchell TW, McAndrew D, Else PL. Of mice, pigs and humans: an analysis of mitochondrial phospholipids from mammals with very different maximal lifespans. Exp Gerontol. 2015;70:135–143. doi: 10.1016/j.exger.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Kelly RD, Rodda AE, Dickinson A, Mahmud A, Nefzger CM, Lee W, Forsythe JS, Polo JM, Trounce IA, McKenzie M, Nisbet DR, St John JC. Mitochondrial DNA haplotypes define gene expression patterns in pluripotent and differentiating embryonic stem cells. Stem Cells. 2013;31:703–716. doi: 10.1002/stem.1313. [DOI] [PubMed] [Google Scholar]
- 27.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neckář J, Svatoňová A, Weissová R, Drahota Z, Zajíčková P, Brabcová I, Kolář D, Alánová P, Vašinová J, Šilhavý J, Hlaváčková M, Tauchmannová K, Milerová M, Ošťádal B, Červenka L, Žurmanová J, Kalous M, Nováková O, Novotný J, Pravenec M, Kolář F. Selective replacement of mitochondrial DNA increases the cardioprotective effect of chronic continuous hypoxia in spontaneously hypertensive rats. Clin Sci. 2017;131:865–881. doi: 10.1042/CS20170083. [DOI] [PubMed] [Google Scholar]
- 29.Anderson EJ, Efird JT, Davies SW, O’Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, Chitwood WR, Koutlas TC, Williams JM, Rodriguez E, Kypson AP. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. 2014;3:e000713. doi: 10.1161/JAHA.113.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waskova-Arnostova P, Elsnicova B, Kasparova D, Sebesta O, Novotny J, Neckar J, Kolar F, Zurmanova J. Right-to-left ventricular differences in the expression of mitochondrial hexokinase and phosphorylation of Akt. Cell Physiol Biochem. 2013;31:66–79. doi: 10.1159/000343350. [DOI] [PubMed] [Google Scholar]
- 31.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahnova K, Kasparova D, Zurmanova J, Neckar J, Kolar F, Novotny J. β-Adrenergic signaling in rat heart is similarly affected by continuous and intermittent normobaric hypoxia. Gen Physiol Biophys. 2016;35:165–167. doi: 10.4149/gpb_2015053. [DOI] [PubMed] [Google Scholar]
- 33.Ihnatovych I, Hejnová L, Kostrnová A, Mares P, Svoboda P, Novotný J. Maturation of rat brain is accompanied by differential expression of the long and short splice variants of G(s)alpha protein: identification of cytosolic forms of G(s)alpha. J Neurochem. 2001;79:88–97. doi: 10.1046/j.1471-4159.2001.00544.x. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Ku BS, Yao HY, Lin YH, Ma X, Zhang YH, Li XJ. The effects of curcumin on depressive-like behaviors in mice. Eur J Pharmacol. 2005;518:40–46. doi: 10.1016/j.ejphar.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Novotny J, Bourová L, Kolár F, Svoboda P. Membrane-Bound and cytosolic forms of heterotrimeric G proteins in young and adult rat myocardium: influence of neonatal hypo- and hyperthyroidism. J Cell Biochem. 2001;82:215–224. doi: 10.1002/jcb.1157. [DOI] [PubMed] [Google Scholar]
- 36.Pilz J, Meineke I, Gleiter CH. Measurement of free and bound malondialdehyde in plasma by high-performance liquid chromatography as the 2,4-dinitrophenylhydrazine derivative. J Chromatogr B Biomed Sci Appl. 2000;742:315–325. doi: 10.1016/S0378-4347(00)00174-2. [DOI] [PubMed] [Google Scholar]
- 37.Chytilová A, Borchert GH, Mandíková-Alánová P, Hlaváčková M, Kopkan L, Khan MA, Imig JD, Kolář F, Neckář J. Tumour necrosis factor-α contributes to improved cardiac ischaemic tolerance in rats adapted to chronic continuous hypoxia. Acta Physiol. 2015;214:97–108. doi: 10.1111/apha.12489. [DOI] [PubMed] [Google Scholar]
- 38.Giannuzzi CE, Seidler FJ, Slotkin TA. Beta-adrenoceptor control of cardiac adenylyl cyclase during development: agonist pretreatment in the neonate uniquely causes heterologous sensitization, not desensitization. Brain Res. 1995;694:271–278. doi: 10.1016/0006-8993(95)00781-K. [DOI] [PubMed] [Google Scholar]
- 39.Mieno S, Horimoto H, Sawa Y, Watanabe F, Furuya E, Horimoto S, Kishida K, Sasaki S. Activation of beta2-adrenergic receptor plays a pivotal role in generating the protective effect of ischemic preconditioning in rat hearts. Scand Cardiovasc J. 2005;39:313–319. doi: 10.1080/14017430510009104. [DOI] [PubMed] [Google Scholar]
- 40.Fajardo G, Zhao M, Berry G, Wong LJ, Mochly-Rosen D, Bernstein D. β2-adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. J Mol Cell Cardiol. 2011;51:781–789. doi: 10.1016/j.yjmcc.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schömig A, Fischer S, Kurz T, Richardt G, Schömig E. Nonexocytotic release of endogenous noradrenaline in the ischemic and anoxic rat heart: mechanism and metabolic requirements. Circ Res. 1987;60:194–205. doi: 10.1161/01.RES.60.2.194. [DOI] [PubMed] [Google Scholar]
- 42.Kaludercic N, Carpi A, Menabò R, Di Lisa F, Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta. 2011;1813:1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dănilă MD, Privistirescu AI, Mirica SN, Sturza A, Ordodi V, Noveanu L, Duicu OM, Muntean DM. Acute inhibition of monoamine oxidase and ischemic preconditioning in isolated rat hearts: interference with postischemic functional recovery but no effect on infarct size reduction. Can J Physiol Pharmacol. 2015;93:819–825. doi: 10.1139/cjpp-2015-0103. [DOI] [PubMed] [Google Scholar]
- 44.Maher JT, Deniiston JC, Wolfe DL, Cymerman A. Mechanism of the attenuated cardiac response to beta-adrenergic stimulation in chronic hypoxia. J Appl Physiol Respir Environ Exerc Physiol. 1978;44:647–651. doi: 10.1152/jappl.1978.44.5.647. [DOI] [PubMed] [Google Scholar]
- 45.Shatemirova KK, Zelenshchikova VA, Min’ko IV. Catalytic properties of monoamine oxidases during adaptation to altitude chamber hypoxia. Kosm Biol Aviakosm Med. 1990;24:54–56. [PubMed] [Google Scholar]
- 46.Yan F, Mu Y, Yan G, Liu J, Shen J, Luo G. Antioxidant enzyme mimics with synergism. Mini Rev Med Chem. 2010;10:342–356. doi: 10.2174/138955710791330972. [DOI] [PubMed] [Google Scholar]
- 47.Nakanishi K, Tajima F, Nakamura A, Yagura S, Ookawara T, Yamashita H, Suzuki K, Taniguchi N, Ohno H. Effects of hypobaric hypoxia on antioxidant enzymes in rats. J Physiol. 1995;489(Pt 3):869–876. doi: 10.1113/jphysiol.1995.sp021099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neckár J, Borchert GH, Hlousková P, Mícová P, Nováková O, Novák F, Hroch M, Papousek F, Ost’ádal B, Kolár F. Brief daily episode of normoxia inhibits cardioprotection conferred by chronic continuous hypoxia. Role of oxidative stress and BKCa channels. Curr Pharm Des. 2013;19:6880–6889. doi: 10.2174/138161281939131127115154. [DOI] [PubMed] [Google Scholar]
- 49.Bu HM, Yang CY, Wang ML, Ma HJ, Sun H, Zhang Y. K(ATP) channels and MPTP are involved in the cardioprotection bestowed by chronic intermittent hypobaric hypoxia in the developing rat. J Physiol Sci. 2015;65:367–376. doi: 10.1007/s12576-015-0376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283:33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chau CM, Evans MJ, Scarpulla RC. Nuclear respiratory factor 1 activation sites in genes encoding the gamma-subunit of ATP synthase, eukaryotic initiation factor 2 alpha, and tyrosine aminotransferase. Specific interaction of purified NRF-1 with multiple target genes. J Biol Chem. 1992;267:6999–7006. [PubMed] [Google Scholar]
- 53.Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 54.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 55.Földes-Papp Z, Domej W, Demel U, Tilz GP. Oxidative stress caused by acute and chronic exposition to altitude. Wien Med Wochenschr. 2005;155:136–142. doi: 10.1007/s10354-005-019-3. [DOI] [PubMed] [Google Scholar]
- 56.Singh M, Thomas P, Shukla D, Tulsawani R, Saxena S, Bansal A. Effect of subchronic hypobaric hypoxia on oxidative stress in rat heart. Appl Biochem Biotechnol. 2013;169:2405–2419. doi: 10.1007/s12010-013-0141-2. [DOI] [PubMed] [Google Scholar]
- 57.Liu JN, Zhang JX, Lu G, Qiu Y, Yang D, Yin GY, Zhang XL. The effect of oxidative stress in myocardial cell injury in mice exposed to chronic intermittent hypoxia. Chin Med J. 2010;123:74–78. [PubMed] [Google Scholar]
- 58.Zhou W, Li S, Wan N, Zhang Z, Guo R, Chen B. Effects of various degrees of oxidative stress induced by intermittent hypoxia in rat myocardial tissues. Respirology. 2012;17:821–829. doi: 10.1111/j.1440-1843.2012.02157.x. [DOI] [PubMed] [Google Scholar]
- 59.Wang WY, Wan WY, Zeng YM, Chen XY, Zhang YX. Effect of Telmisartan on local cardiovascular oxidative stress in mouse under chronic intermittent hypoxia condition. Sleep Breath. 2013;17:181–187. doi: 10.1007/s11325-012-0669-3. [DOI] [PubMed] [Google Scholar]
- 60.Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47:32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.