

Abstract
Endothelial dysfunction has been implicated in the pathogenesis of atherosclerosis. Salvia miltiorrhiza (danshen) is a traditional Chinese medicine that has been effectively used to treat cardiovascular disease. Cryptotanshinone (CTS), a major lipophilic compound isolated from S. miltiorrhiza, has been reported to possess cardioprotective effects. However, the anti-atherogenic effects of CTS, particularly on tumor necrosis factor-α (TNF-α)-induced endothelial cell activation, are still unclear. This study aimed to determine the effect of CTS on TNF-α-induced increased endothelial permeability, monocyte adhesion, soluble intercellular adhesion molecule 1 (sICAM-1), soluble vascular cell adhesion molecule 1 (sVCAM-1), monocyte chemoattractant protein 1 (MCP-1) and impaired nitric oxide production in human umbilical vein endothelial cells (HUVECs), all of which are early events occurring in atherogenesis. We showed that CTS significantly suppressed TNF-α-induced increased endothelial permeability, monocyte adhesion, sICAM-1, sVCAM-1 and MCP-1, and restored nitric oxide production. These observations suggest that CTS possesses anti-inflammatory properties and could be a promising treatment for the prevention of cytokine-induced early atherogenesis.
Keywords: Cryptotanshinone, Nitric oxide, Soluble cellular adhesion molecule, TNF-α, Monocyte adhesion, Chemokine
Introduction
Atherosclerosis is a chronic inflammatory disease characterized by narrowing of the blood vessel walls, resulting from the development of atherosclerotic plaque in the intima. Endothelial dysfunction is now widely accepted as one of the underlying mechanisms by which atherogenesis is initiated. Increased endothelial permeability allows low-density lipoprotein (LDL) to migrate through the endothelium and become oxidized in the intima. Oxidized LDL (ox-LDL), a primary trigger of atherogenesis, in turn activates monocytes to secrete a variety of soluble factors, including pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukins, which are considered as the secondary triggers of atherogenesis [1]. TNF-α helps to perpetuate the inflammatory process by causing an upregulation of cellular adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), promoting the adherence of monocytes to the endothelium and a subsequent migration of monocytes across the endothelium [2]. Interestingly, monocyte migration can occur through both the paracellular and transcellular pathways, although the latter process is rare [3].
Previous studies have shown that the expression of ICAM-1 and VCAM-1 was elevated in atherosclerotic lesions isolated from both human and animal models [4, 5]. In addition, increased level of soluble form of adhesion molecules in the plasma was found to correlate with the occurrence of cardiovascular diseases including atherosclerosis [5]. Besides, chemokines such as monocyte chemoattractant protein 1 (MCP-1) bind to their receptor CCR2 and govern the recruitment and infiltration of monocytes to the atherosclerotic lesions. TNF-α has also been reported to reduce nitric oxide (NO) bioavailability, a key factor in endothelial dysfunction, which is primarily due to the inhibition of endothelial nitric oxide synthase (eNOS). The downregulation of NO production has been reported to cause destabilization of the endothelial barrier, enhance expression of cellular adhesion molecules and chemokines, and increase leukocyte adhesion [6]. Therefore, the restoration of NO production can be used as a therapeutic strategy to prevent the inflammatory process that occurs in the early stages of atherosclerosis.
Salvia miltiorrhiza (danshen), a traditional Chinese medicine, has beneficial effects in the circulatory system, and is widely used in China and other Asian countries as a therapeutic agent in the management of cardiovascular diseases such as stroke, heart attack and angina pectoris. S. miltiorrhiza has been found to promote blood flow, reduce cerebral infarction, and to possess anti-atherosclerotic, antihypertensive and anti-inflammatory properties [7]. Cryptotanshinone (CTS), a major tanshinone compound derived from the dried root of S. miltiorrhiza, has been reported to possess vasculoprotective [8] and anti-inflammatory effects [9, 10] and inhibit the development of atherosclerotic lesions [11]. Our group previously reported that CTS attenuated oxLDL-induced pre-lesional events of atherosclerosis in human umbilical vein endothelial cells (HUVECs) [12]. Similarly, a recent study has demonstrated that CTS attenuated oxLDL-induced atherogenic events through lectin-like oxLDL receptor 1 (LOX-1) pathway [13]. However, there are no reports on how CTS improves endothelial dysfunction induced by TNF-α, a secondary stimulus of atherogenesis, which sustains the inflammatory phenotype.
Therefore, the aim of this study was to examine the effect of CTS on TNF-α-induced increases in endothelial permeability, upregulation of sICAM-1 and sVCAM-1 protein secretion, increases in monocyte adhesion and production of MCP-1, and attenuation of NO bioavailability. We suggest that CTS exhibits anti-inflammatory properties and thus might prevent the early atherogenic events induced by TNF-α.
Materials and methods
Materials
CTS with a purity of 95.8 %, as determined by high-performance liquid chromatography (HPLC), was purchased from ChromaDex (Irvine, CA, USA). CTS was dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 10 mM and stored at −20 °C. M200 medium and low-serum growth supplement (LSGS) kits were purchased from Cascade Biologics (Portland, OR, USA). Antibiotics (5000 U/mL penicillin and 5000 μg/mL streptomycin), amphotericin B (0.25 μg/mL), fetal bovine serum (FBS), Roswell Park Memorial Institute medium (RPMI)-1640 and Dulbecco’s modified Eagle's medium (DMEM) were purchased from HyClone Laboratories (Logan, UT, USA). 2′,7′-bis-(2-Carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM), bovine serum albumin (BSA) and trypsin/ethylenediaminetetraacetic acid (EDTA) solution were purchased from Sigma-Aldrich (St. Louis, MO, USA). Human recombinant tumour necrosis factor-α (TNF-α) was purchased from Calbiochem (Nottingham, UK).
Cell culture
HUVECs were purchased from Cascade Biologics and grown in collagen type I-coated flasks. The cells were nourished with M200 medium supplemented with LSGS kit containing 2 % FBS, hydrocortisone, human epidermal growth factor, basic fibroblast growth factor and heparin. Cells from passages 2–6 were used in all the experiments. The media was changed every 2 days until the cells reached 80 % confluence. U937 monocytic cells were purchased from American Type Culture Collection (ATCC) and cultured in RPMI-1640 supplemented with 10 % FBS, 4.5 g/L glucose, sodium pyruvate (1 mmol/L), l-glutamine (2 mmol/L), amphotericin B, streptomycin (50 μg/mL) and penicillin (50 U/mL). The media was changed every two days. The cells were maintained at a density of 5 × 105–2 × 106 cells/mL and used for monocyte adhesion assay.
In vitro vascular permeability assay
The permeability of the HUVEC monolayer upon exposure to TNF-α and CTS was determined using an in vitro vascular permeability assay kit (EMD Millipore, Billerica, MA, USA). The assay was carried out according to the manufacturer’s protocol, with some modifications. Briefly, HUVECs were grown onto collagen-coated cell culture inserts for 72 h. CTS was added concurrently with TNF-α (10 ng/mL) and incubated for 6 h, after which all the treatments were replaced with 100 µL of fluorescein isothiocyanate (FITC)-dextran (dilution 1:50) and incubated for 5 min to allow the tracer to permeate across the monolayer. The media was collected from the bottom wells, and the fluorescence intensities were measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence microplate reader (Infinite 200; Tecan Group, Männedorf, Switzerland). Results were expressed as permeability index (%) using the formula reported by Maruo et al. [14].
U937 monocyte adhesion assay
Quantitative monocyte adhesion assay was performed as described previously [15]. U937 cells were labeled with 4 µM BCECF-AM/RPMI and incubated for 30 min at 37 °C in 5 % CO2. The cells were washed twice with 0.5 % BSA/PBS to remove excess dye after labeling. U937 monocytes were then resuspended in M200 media at a cell density of 1 × 106 cells/mL.
HUVECs were grown to confluence in 96-well plates. TNF-α and CTS were added concurrently to HUVECs for 6 h. All the treatments were then removed, and 100 µL of BCECF-labeled monocytes was added and incubated for 1 h at 37 °C in 5 % CO2 to allow the binding process to occur. Then, each well was washed twice with assay buffer (0.5 % BSA in DMEM without phenol red, with Mg2+ and Ca2+) and once with PBS. The adhered monocytes were lysed with 100 µL of 0.2 % Triton X-100/PBS. Fluorescence intensities were measured at 485-nm excitation and 530 nm emission using a fluorescence microplate reader (Tecan Infinite 200). The results were expressed as a percentage compared to the control.
Concentration of soluble cellular adhesion molecules
The concentrations of sICAM-1 and sVCAM-1 were quantified using a human sICAM-1 enzyme-linked immunosorbent assay (ELISA) kit and a human sVCAM-1 ELISA kit (Bender MedSystems, Vienna, Austria), respectively. HUVECs were grown to confluence in 24-well plates and incubated with TNF-α (10 ng/mL) in the presence or absence of various concentrations of CTS (1, 2.4, 5, 10 and 20 µM) for 6 h. The supernatant was collected and stored at −20 °C for batch measurement of the sICAM-1 and sVCAM-1 levels, according to the manufacturer’s protocol. The absorbance for each well was measured using a microplate reader (Tecan Infinite 200) at a wavelength of 450 nm and a reference wavelength of 620 nm. Results were expressed as a percentage compared to control.
Concentration of chemokine monocyte chemoattractant protein 1 (MCP-1)
To evaluate the effect of CTS on TNF-α-induced increases in MCP-1 secretion, a human MCP-1 ELISA kit (Becton Dickinson, East Rutherford, NJ, USA) was used. HUVECs were seeded onto 24-well plates and grown until confluent. HUVECs were incubated with TNF-α (10 ng/mL) in the presence or absence of CTS for 6 h. Supernatant was collected and stored at −20 °C for batch measurement according to the manufacturer’s protocol. The optical density for each well was read using a microplate reader (Tecan Infinite 200) at a wavelength of 450 nm and corrected with a reference wavelength of 570 nm. Results were expressed as a percentage compared to normal control.
Measurement of nitric oxide production
Total concentrations of nitrite (NO2 −) and nitrate (NO3 −) in cell culture supernatant were measured using a nitrate/nitrite fluorometric assay kit (Cayman Chemical Co., Ann Arbor, MI, USA). HUVECs were seeded in 24-well plates and grown until confluent. TNF-α and CTS were added concurrently to HUVECs for 6 h. Supernatant was collected for the measurement of total NO level, according to the manufacturer’s protocol. The fluorescence intensities were measured using a fluorescence microplate reader (Tecan Infinite 200) with an excitation and an emission wavelengths of 360 and 430 nm, respectively. Results are expressed as a percentage compared to normal control.
Statistical analysis
All the data are expressed as the mean ± the standard error of the mean (SEM) and analyzed using one-way analysis of variance (ANOVA) with a post hoc analysis (Dunnett's test) for pairwise comparison. Statistical significance was defined as p < 0.05.
Results
CTS protects against endothelial barrier disruption induced by TNF-α
The paracellular permeability of HUVECs to FITC-dextran was examined using an in vitro vascular permeability assay. As shown in Fig. 1, the incubation of HUVECs with TNF-α (10 ng/mL) for 6 h showed an increase in permeability index, from a basal permeability level of 2.75 ± 0.83 to 71.4 ± 7.57 %. The administration of CTS at 2.5 µM resulted in significant suppression of the permeability index, to 35.2 ± 7.96 % (p < 0.05). Maximal inhibition of the permeability index was achieved with 5 µM CTS, achieving a reduction to 27.75 ± 7.1 % (p < 0.05). CTS at a dose of 20 µM failed to produce significant suppression of endothelial hyperpermeability. A previous study by our group showed that application of CTS 1–20 µM for 24 h did not reduce HUVEC viability [12]. These results demonstrate that CTS protects against endothelial hyperpermeability elicited by TNF-α, without causing cell death.
Fig. 1.
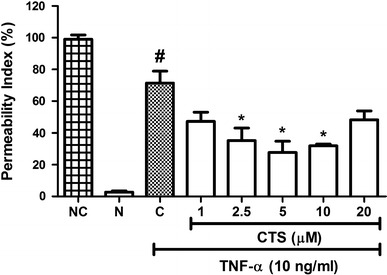
Effect of CTS on TNF-α-induced increases in endothelial permeability. CTS was incubated concurrently with TNF-α for 6 h. Values indicate the permeability index. NC no cell, N non-induced, C control. Values are expressed as the mean ± SEM from three independent experiments. Hash (#) indicates significant difference from the non-induced group, p < 0.05. Asterisk (*) indicates significant difference from control, p < 0.05
CTS suppresses TNF-α-induced increase in U937 monocyte adhesion
Upon exposure to TNF-α, endothelial cells express a number of cell surface adhesion molecules, such as ICAM-1 and VCAM-1, which facilitate circulating monocytes to adhere to the endothelium before migrate to the intima through diapedesis. Quantitative monocyte adhesion assay, therefore, may reflect the surface expression of these cellular adhesion molecules under different treatment conditions. TNF-α (10 ng/mL) caused a significant increase in adhesion of U937 monocytic cells, to 217.8 ± 14.34 % of the control (Fig. 2). CTS at doses of 1, 2.5, 5, 10 and 20 µM reduced monocyte attachment to 176.4 ± 2.62, 167.8 ± 5.11, 150.3 ± 8.21, 130.3 ± 6.47 and 227.4 ± 4.60 %, respectively (p < 0.05; Fig. 2), and thus a dose-dependent effect of CTS was observed. As such, CTS reduced the number of monocytes attached to HUVECs, which might be due to the reduction in the surface expression of cellular adhesion molecules.
Fig. 2.
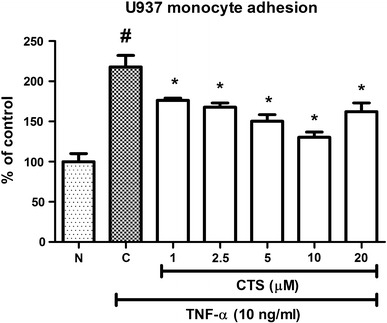
Effect of CTS on TNF-α-induced increase in U937 monocytic cells adhesion to HUVECs. N non-induced, C control. Values are expressed as the mean ± SEM from three independent experiments. Hash (#) indicates significant difference from non-induced group, p < 0.05. Asterisk (*) indicates significant different from the control, p < 0.05
CTS reduces sICAM-1 and sVCAM-1 levels in TNF-α-treated HUVECs
Because the soluble forms of cellular adhesion molecules are a biomarker for cardiovascular diseases, we were interested in examining the effect of CTS on TNF-α-induced increase in sICAM-1 and sVCAM-1. TNF-α (10 ng/mL) significantly increased the concentration of sICAM-1, to 275.8 ± 6.50 % of the control (Fig. 3a), and significantly (p < 0.05) inhibited the TNF-α-induced increase in sICAM-1 protein secretion in a dose-dependent manner (Fig. 3a). A dose of 20 μM CTS, the highest concentration used in this study, suppressed the sICAM-1 level to 143.7 ± 1.37 % of the control. Moreover, TNF-α caused a significant elevation in sVCAM-1 production, to 580.7 ± 36.59 % of the control (Fig. 3b). The increased expression of sVCAM-1 was reduced by CTS in a dose-dependent manner (2.5, 5, 10 and 20 μM), to 495.7 ± 13.72 %, 456.9 ± 13.34 %, 409.7 ± 11.73 % and 359.1 ± 9.16 %, respectively (p < 0.05; Fig. 3b). However, 1 μM CTS did not significantly reduce the TNF-α-induced higher sVCAM-1 level (568.5 ± 15.63 %, p > 0.05). Taken together, these results show that CTS inhibits the increased production of sICAM-1 and sVCAM-1 stimulated by TNF-α.
Fig. 3.
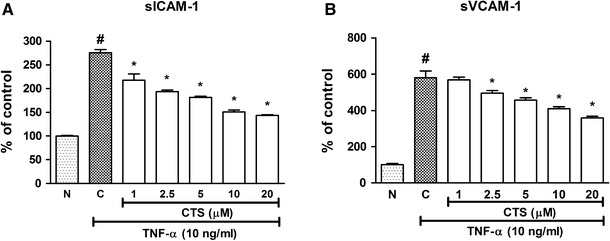
Effect of CTS on the TNF-α-induced sICAM-1 and sVCAM-1 protein secretions in HUVECs. CTS was incubated concurrently with TNF-α for 6 h. a sICAM-1 expression; b sVCAM-1 expression. Values are expressed as the mean ± SEM from three independent experiments. Hash (#) indicates significant difference from the non-induced group, p < 0.05. Asterisk (*) indicates significant difference from the control, p < 0.05. N non-induced, C control
CTS decreases TNF-α-induced increase in MCP-1 level
It is well known that MCP-1 plays an important role in mediating monocyte adhesion. To investigate the protective effect of CTS against increased monocyte adhesion caused by TNF-α, the production of MCP-1 was studied using an ELISA assay. MCP-1 production was stimulated by TNF-α to 1283 ± 85.88 % of the control (Fig. 4). CTS applied at concentrations of 1 μM and 2.5 µM reduced MCP-1 levels to 1130 ± 65.115 and 1089 ± 10.04 %, respectively, but these data were not statistically different from those in the TNF-α-treated group (Fig. 4). CTS at the higher concentrations of 10 and 20 μM showed a significant inhibitory effect on the MCP-1 level (729.3 ± 37.48 and 613.1 ± 64.36 %, respectively; p < 0.05; Fig. 4). A dose–response effect was observed. These results indicate that CTS attenuates the secretion of MCP-1 stimulated by TNF-α.
Fig. 4.
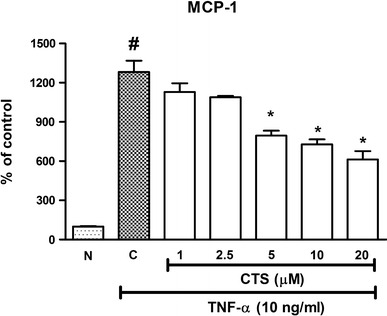
Effect of CTS on TNF-α-induced increase in MCP-1 levels in HUVECs. CTS was concurrently incubated with TNF-α for 6 h. N non-induced, C control. Values are expressed as the mean ± SEM from three independent experiments. Hash (#) indicates significant difference from the non-induced group, p < 0.05. Asterisk (*) indicates significant difference from the control, p < 0.05
CTS restores TNF-α-impaired NO bioavailability
An alteration in the baseline NO level, either increasing or decreasing NO production, was found to cause endothelial dysfunction. TNF-α administered at 10 ng/mL for 6 h reduced baseline NO to 24.17 ± 3.54 % of the control (Fig. 5). CTS at all tested doses (1, 2.5, 5, 10 and 20 μM) significantly increased NO from the reduced levels elicited by TNF-α, in a dose-dependent manner, with near-restoration of baseline NO level at a CTS dose of 20 μM (97.3 ± 5.91 % of control, p < 0.05; Fig. 5). Thus, CTS prevented TNF-α-induced endothelial dysfunction by restoring the attenuated NO production.
Fig. 5.
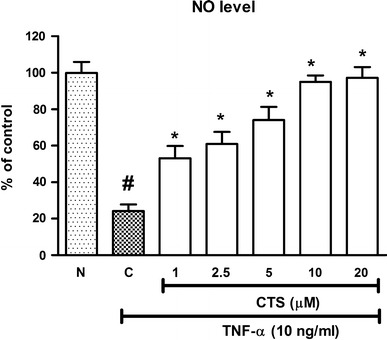
Effect of CTS on TNF-α-attenuated NO production in HUVECs. N non-induced, C control. All values are expressed as the mean ± SEM from three independent experiments. Hash (#) indicates significant difference from the non-induced group, p < 0.05. Asterisk (*) indicates significant difference from the control, p < 0.05
Discussion
The anti-atherosclerotic effects of CTS in vivo have been reported previously [13], and the suppressive effects of CTS on ox-LDL-induced early events of atherosclerosis in vitro were demonstrated by our group as well. In this study, we extended our previous work to examine how CTS protects against endothelial dysfunction elicited by TNF-α, a secondary trigger in early atherogenesis. It is worth mentioning that, to date, no studies have investigated the effects of CTS on TNF-α-induced endothelial barrier disruption, increased monocyte adhesion and increased production of MCP-1, sICAM-1 and sVCAM-1. In the current study, we showed that CTS inhibited TNF-α-induced increased permeability, increased monocyte adhesion and elevated production of sICAM-1 and sVCAM-1, as well as MCP-1. In addition, CTS restored TNF-α-attenuated NO bioavailability.
While we had previously reported the suppressive effect of CTS on ox-LDL-induced enhanced permeability of HUVECs [12], the question of whether CTS would improve TNF-α-impaired endothelial barrier integrity remained to be investigated. In the present study, we suggest that CTS protects against TNF-α-induced barrier disruption by suppressing the passage of FITC-dextran across the endothelial monolayer. The two bioactive compounds of Salvia miltiorrhiza, danshensu and salvianolic acid B, have also been shown to reduce the permeability of BSA in TNF-α-treated HUVECs [16], and our data strongly suggest that CTS contributes to the barrier protective effect of S. miltiorrhiza as well.
Researchers have shown that CTS undergoes dehydrogenation and is rapidly converted into its metabolites, including tanshinone IIA [17]. The maximum plasma concentration (C max) of CTS in rats after single oral administration of 100 mg/kg of cryptotanshinone was approximately 303 nM [18]. Although the concentrations used in this study (1, 2.5, 5, 10 and 20 µM) are much higher than the concentration found in rat plasma, we cannot exclude the possibility that higher doses are needed, as in vitro experiments are usually confined to short incubation periods such as 24 h. Chronic treatment of CTS in animal models up to several weeks or months may result in the accumulation of CTS and its metabolites in the plasma. A recent study found that oral administration of 15 mg/kg of CTS for 22 weeks inhibited atherosclerotic plaque formation in apolipoprotein E-deficient mice through a reduction in the expression of lectin-like oxLDL receptor 1 (LOX-1) and generation of reactive oxygen species (ROS) [13]. These data indicate that chronic relatively low-dose treatment (15 mg/kg) is required for CTS to exert an anti-atherogenic effect in animal models. For in vitro experiments, these authors used doses (2.5, 5 and 10 µM) of CTS similar to those in the present study to treat ox-LDL-stimulated HUVECs. These doses were effective in suppressing oxLDL-induced increased monocyte adhesion and increased expression of both ICAM-1 and VCAM-1. In line with this earlier study, we demonstrated a protective effect of CTS against endothelial dysfunction through inhibition of increased monocyte adherence and increased production of sICAM-1 and sVCAM-1 elicited by TNF-α.
The cleavage of cellular adhesion molecules by sheddase is an event that occurs during endothelial activation, and as a consequence, circulating adhesion molecule levels are elevated in the plasma. Increased levels of sICAM-1 and sVCAM-1 are significantly correlated with the angiographic severity of atherosclerosis and plaque instability [19]. Therefore, the soluble form of adhesion molecules such as sICAM-1 and sVCAM-1 serves as an important biomarker for endothelial activation and cardiovascular risk factors in clinical practice. In this study, the reduction of sICAM-1 and sVCAM-1 production by CTS provides evidence to support its use as a potential cardioprotective agent.
Increased expression of MCP-1 can be found in atherosclerotic plaque, which facilitates the accumulation of monocytes in the lesion [20]. Previous studies have demonstrated that leukocyte–endothelial interaction stimulates the release of MCP-1 from endothelial cells, resulting in enhanced expression of surface adhesion molecules, which then further promotes the firm adhesion of leukocytes to endothelial cells [21]. Thus, it is clear that leukocyte–endothelial interaction is driven by overlapping inflammatory events that occur in parallel rather than sequentially. CTS has been shown to reduce TNF-α-increased ICAM-1 and VCAM-1 levels [9], but whether this leads to the suppression of monocyte adhesion has yet to be explored. Our data show that CTS decreased TNF-α-induced monocyte adhesion on HUVECs and MCP-1 production in a concentration-dependent manner. Therefore, we suggest that CTS reduces the attachment of monocytes to the endothelium and that this may occur through the inhibition of MCP-1 expression, in addition to the suppressed ICAM-1 and VCAM-1 expression.
NO is a pleiotropic molecule produced at low levels by endothelial cells. Apart from its role as an important signaling molecule in neurotransmission, NO also regulates vascular tone, platelet activity and vascular smooth muscle cell proliferation. NO is central to endothelial dysfunction in early atherogenesis, where overproduction or reduced synthesis of NO is regarded as harmful to normal vascular function. Previous studies have reported that NO exerts its anti-atherogenic effect through the inhibition of MCP-1 [22], cellular adhesion molecule expression, platelet aggregation and leukocyte adhesion [23]. A decrease in NO production or activity causes enhanced permeability of endothelial cells and monocyte attachment to the endothelium, the latter of which is mediated through elevated expression of surface adhesion molecules. Therefore, NO donors, such as nicorandil and sydnonimines, or drugs that can increase the bioavailability of NO, such as statins, angiotensin-converting enzyme inhibitors and l-arginine, are used as therapeutic agents in clinical practice to prevent atherosclerosis [24]. Nitric oxide synthases (NOS) are enzymes that catalyze the conversion of l-arginine to NO, and their expression levels and activities are important for NO production. There are three types of NOS: endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS). Among these, eNOS and iNOS are commonly present in endothelial cells. Importantly, iNOS is expressed in response to inflammatory stimuli such as cytokines, and stimulates excess production of NO during the inflammatory response [25]. TNF-α has been found to up-regulate the expression of iNOS, leading to increased NO production, regardless of the cell type [26]. Surprisingly, our data showed that HUVECs exposed to 10 ng/mL of TNF-α for 6 h had lower NO levels than those in an unstimulated group. It is important to highlight that data published on TNF-α-induced NO level and iNOS expression remain controversial. It appears that TNF-α-induced iNOS expression is influenced by various experimental conditions, including cell type used, concentration and exposure time of TNF-α, and oxygen availability [27, 28]. It is possible, therefore, that under our experimental conditions, iNOS expression was not altered by TNF-α. We found that CTS restored TNF-α-impaired NO level, from 24.17 ± 3.54 % to 95.03 ± 3.56 % of the control. However, the mechanism by which NO regulates CTS-attenuated increased permeability, monocyte adhesion, and sICAM-1, sVCAM-1 and MCP-1 production remains to be elucidated. An earlier study demonstrated that the restoration of NO bioavailability by CTS may involve upregulation of eNOS [29].
In conclusion, here we report that CTS exhibits anti-inflammatory effects through suppression of TNF-α-induced early atherogenic events in HUVECs. In addition, CTS protects against endothelial dysfunction through the restoration of NO production that has been reduced by TNF-α. Our data enrich the current scientific findings on CTS and support the use of S. miltiorrhiza in treating cardiovascular diseases such as atherosclerosis. However, future studies are needed to elucidate the signaling pathways that underlie the anti-atherogenic effect of CTS.
Acknowledgments
This research project was funded by the Fundamental Research Grant Scheme (Project No. 04/04/10/845FR) from the Ministry of Education, Malaysia.
Compliance with ethical standards
Conflict of interest
The authors have declared that there is no conflict of interest.
References
- 1.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 2.Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto K, et al. Live-cell visualization of the trans-cellular mode of monocyte transmigration across the vascular endothelium, and its relationship with endothelial PECAM-1. J Physiol Sci. 2012;62:63–69. doi: 10.1007/s12576-011-0181-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Huo Y. Adhesion molecules and atherosclerosis, in atherosclerosis: molecular and cellular mechanisms. In: George SJ, Johnson J, editors. Atherosclerosis: molecular and cellular mechanisms. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2010. [Google Scholar]
- 6.Funk SD, Yurdagul A, Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. 2012;2012:569654. doi: 10.1155/2012/569654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin TH, Hsieh CL. Pharmacological effects of Salvia miltiorrhiza (Danshen) on cerebral infarction. Chin Med. 2010;5:22. doi: 10.1186/1749-8546-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams JD, et al. Preclinical and clinical examinations of Salvia miltiorrhiza and its tanshinones in ischemic conditions. Chin Med. 2006;1:3. doi: 10.1186/1749-8546-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin YC, et al. Cryptotanshinone, a lipophilic compound of Salvia miltiorrriza root, inhibits TNF-alpha-induced expression of adhesion molecules in HUVEC and attenuates rat myocardial ischemia/reperfusion injury in vivo. Eur J Pharmacol. 2009;614:91–97. doi: 10.1016/j.ejphar.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 10.Tang S, et al. Cryptotanshinone suppressed inflammatory cytokines secretion in RAW264.7 macrophages through inhibition of the NF-kappaB and MAPK signaling pathways. Inflammation. 2011;34:111–118. doi: 10.1007/s10753-010-9214-3. [DOI] [PubMed] [Google Scholar]
- 11.Suh SJ, et al. Cryptotanshinone from Salvia miltiorrhiza BUNGE has an inhibitory effect on TNF-alpha-induced matrix metalloproteinase-9 production and HASMC migration via down-regulated NF-kappaB and AP-1. Biochem Pharmacol. 2006;72:1680–1689. doi: 10.1016/j.bcp.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Ang KP, et al. Cryptotanshinone attenuates in vitro oxLDL-induced pre-lesional atherosclerotic events. Planta Med. 2011;77:1782–1787. doi: 10.1055/s-0030-1271119. [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, et al. Cryptotanshinone, an orally bioactive herbal compound from Danshen, attenuates atherosclerosis in apolipoprotein E-deficient mice: role of lectin-like oxidized LDL receptor-1 (LOX-1) Br J Pharmacol. 2015;172(23):5661–5675. doi: 10.1111/bph.13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maruo N, et al. IL-6 increases endothelial permeability in vitro. Endocrinology. 1992;131:710–714. doi: 10.1210/endo.131.2.1639018. [DOI] [PubMed] [Google Scholar]
- 15.Schleser S, Ringseis R, Eder K. Conjugated linoleic acids have no effect on TNF alpha-induced adhesion molecule expression, U937 monocyte adhesion, and chemokine release in human aortic endothelial cells. Atherosclerosis. 2006;186:337–344. doi: 10.1016/j.atherosclerosis.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Ding M, et al. Aqueous extract of Salvia miltiorrhiza attenuates increased endothelial permeability induced by tumor necrosis factor-alpha. Int Immunopharmacol. 2005;5:1641–1651. doi: 10.1016/j.intimp.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Wei Y, et al. Metabolism of tanshinone IIA, cryptotanshinone and tanshinone I from radix salvia miltiorrhiza in zebrafish. Molecules. 2012;17:8617–8632. doi: 10.3390/molecules17078617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, et al. A mechanistic study of the intestinal absorption of cryptotanshinone, the major active constituent of salvia miltiorrhiza. J Pharmacol Exp Ther. 2006;317:1285–1294. doi: 10.1124/jpet.105.100701. [DOI] [PubMed] [Google Scholar]
- 19.Ribeiro F, et al. Endothelial function and atherosclerosis: circulatory markers with clinical usefulness. Rev Port Cardiol. 2009;28:1121–1151. [PubMed] [Google Scholar]
- 20.Yla-Herttuala S, et al. Expression of monocyte chemoattractant protein 1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci U S A. 1991;88:5252–5256. doi: 10.1073/pnas.88.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukacs NW, et al. Production of chemokines, interleukin-8 and monocyte chemoattractant protein-1, during monocyte: endothelial cell interactions. Blood. 1995;86:2767–2773. [PubMed] [Google Scholar]
- 22.Tsao PS, et al. Nitric oxide regulates monocyte chemotactic protein-1. Circulation. 1997;96:934–940. doi: 10.1161/01.CIR.96.3.934. [DOI] [PubMed] [Google Scholar]
- 23.Napoli C, et al. Nitric oxide and atherosclerosis: an update. Nitric Oxide. 2006;15:265–279. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Herman AG, Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur Heart J. 2005;26:1945–1955. doi: 10.1093/eurheartj/ehi333. [DOI] [PubMed] [Google Scholar]
- 25.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(829–837):837a–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanders DB, et al. Comparison of tumor necrosis factor-alpha effect on the expression of iNOS in macrophage and cardiac myocytes. Perfusion. 2001;16:67–74. doi: 10.1177/026765910101600110. [DOI] [PubMed] [Google Scholar]
- 27.Paz Y, et al. Effect of tumor necrosis factor-alpha on endothelial and inducible nitric oxide synthase messenger ribonucleic acid expression and nitric oxide synthesis in ischemic and nonischemic isolated rat heart. J Am Coll Cardiol. 2003;42:1299–1305. doi: 10.1016/S0735-1097(03)00992-6. [DOI] [PubMed] [Google Scholar]
- 28.Zulueta JJ, et al. Modulation of inducible nitric oxide synthase by hypoxia in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 2002;26:22–30. doi: 10.1165/ajrcmb.26.1.4510. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Z, et al. Cryptotanshinone inhibits endothelin-1 expression and stimulates nitric oxide production in human vascular endothelial cells. Biochim Biophys Acta. 2006;1760:1–9. doi: 10.1016/j.bbagen.2005.09.009. [DOI] [PubMed] [Google Scholar]