

Abstract
The skeletal muscle is one of the important target tissues for the actions of estrogen via both nuclear and extranuclear (non-genomic) pathways. However, there is a paucity of information about the receptor (ER) involved. The aim of this study was thus to explore the ER expression in skeletal muscle, and the influence of estrogen on it, by using C2C12 myoblasts derived from mouse skeletal muscle. Significant expression of a ~66-kD protein immunoreactive to ER type α (ERα) monoclonal antibody, which was comparable to that in ovary, was detected in the whole-cell (total) and nucleus-free (nonnuclear) fractions of C2C12 myoblasts. The expression level of these ER proteins increased in several hours with treatment with 17β-estradiol (E2), which was preceded by the elevation of the ER mRNA level. This increase appeared to reflect the acceleration of de novo synthesis of ER protein, as proved by the 35S-methionine immunoprecipitation method. A similar extent of fast increase in ER expression was also induced by a membrane-impermeable, BSA-conjugated estradiol (E2-BSA). Unexpectedly, the E2-induced increases in total and nonnuclear ER were further enhanced by the classic ER antagonists tamoxifen and ICI182,780 in a wide concentration range, implying some structural difference of the involved ER from the classical one. Treatment with the ERK1/2 inhibitor, PD98059 (10 μM), or the p38 MAPK-specific inhibitor, SB203580 (10 μM), greatly inhibited the E2-induced ER increase, while the protein kinase C (PKC) activator TPA (1 μM) enhanced it. These results collectively suggest that C2C12 skeletal myoblasts express a high level of ER, a considerable part of which is extranuclear. Further, the expression of ER in these cells may be significantly upregulated by estrogen itself via increased biosynthesis linked to membrane-bound ER and downstream MAPK-mediated signaling pathways.
Electronic supplementary material
The online version of this article (doi:10.1007/s12576-009-0023-0) contains supplementary material, which is available to authorized users.
Keywords: Estrogen receptor, 17β-Estradiol, Mouse skeletal myoblast, MAPK
Introduction
The sex hormone estrogen is important for growth, differentiation, and the emergence of various cellular functions via the promotion of protein synthesis. Many estrogenic actions are genomic, being mediated via intracellular estrogen receptors (ERs) that dimerize upon estrogen binding, translocate into the nucleus, and function as transcription factors to regulate the expression of estrogen-responsive genes [1–3]. In some cell types, however, rapid estrogenic, i.e., non-genomic effects may be initiated by the binding of estrogen to membrane-associated ERs. Although the nature and identity of non-genomic ERs still remain elusive and controversial, early evidence has indicated that the classical ERs (mainly ERα) could also be integrated into or associated with the plasma membrane as a homodimer via S-palmitoylation in the ligand-binding domain of ERα [4], allowing its coupling to distinct intracellular signaling pathways [5]. As a possible alternative candidate, a novel seven transmembrane domain G-protein-coupled receptor (GPR30) has also been shown to localize on the cell membrane and mediate rapid E2 signalings [6, 7]. To date, the most convincing evidence of membrane-bound ER comes from immunohistochemistry and functional studies with a membrane-impermeable ligand, i.e., E2-BSA, which remains extracellularly and does not activate the estrogen responsive element (ERE)-dependent transcription [8–10]. Numerous studies have demonstrated that E2 and other estrogenic compounds can rapidly activate a diverse array of intracellular signaling cascades via membrane-initiated, non-genomic mechanisms. Many of these seem to involve a mechanism independent of the classic nuclear ERs and associated with G-protein coupled receptor (GPCR)-mediated signaling pathways [11–14]. For example, E2 can activate adenylyl cyclase (AC) to generate cAMP, elevate the intracellular Ca2+ concentration, activate phospholipase C (PLC) to generate inositol 1,4,5-trisphosphate and diacylglycerol, stimulate nitric oxide synthase to generate nitric oxide, and activate the extracellular signal-regulated kinase 1/2 (ERK) mitogen-activated protein kinase (MAPK) pathway [15]. In other studies of overexpressing ERα or ERβ in the reportedly ER-deficient cell line CHO-K1, it has been suggested that a certain fraction of both ERs can be expressed in the cell membrane, activation of which leads to stimulation of phospholipase C and adenylyl cyclase via G protein, or activation of MAPK/ERK transcriptional pathways [16]. These antecedents clearly indicate that the mechanism by which estrogen activates a rapid intracellular signaling differs from a classical, genomic mode of estrogen actions.
The skeletal muscle is among the important target tissues for estrogen, but the physiological role of estrogen and its receptors therein remains poorly elucidated. It has been well documented, however, that replacement therapy for decreased blood estrogen levels mitigates postmenopausal disorders, such as osteoporosis and some comorbid conditions [17]. It is also generally believed that estrogen may exert some beneficial effects on commonly observed symptoms of the elderly associated with muscle weakness, such as dyspnea, dysphagia, and dysuria. Nevertheless, the mechanism by which estrogen ameliorates muscle weakness remains largely unclear.
In this study, to elucidate the potential therapeutic value of estrogen in improving skeletal muscle functions, we started to characterize its physiological impact on them by using the C2C12 myoblast cell line as a model skeletal muscle cell. Since we previously found rapid contractile effects of E2 on frog skeletal muscle [18], we were especially interested in the nongenomic actions of estrogen. For this purpose, we explored both total and nonnuclear expression of ER in relation to its biosynthesis and associated cellular signaling cascades in mouse C2C12 myoblasts. Part of this study has been communicated to the 85th annual meeting of the Japanese Physiological Society [19].
Materials and methods
Cell culture
C2C12 murine skeletal muscle cell line [20], obtained from Dainippon Pharmaceutical Ltd., was routinely cultured in Dulbecco’s Modified Eagle’s Medium F12 (DMEM/F12) containing 10% heat-inactivated (30 min, 56°C) fetal bovine serum (FBS). Cells were maintained at 37°C in humidified 5% CO2-gassed air, and the culture medium was changed every 2 days. Phenol red-free medium was used throughout the experiment to avoid its weak estrogen-like actions.
Nucleus-free and whole-cell (nucleus-containing) preparation
Nucleus-free preparation from C2C12
A monolayer of C2C12 myoblasts was washed twice in ice-cold Dulbecco’s phosphate-buffered saline (PBS) and harvested by scraping. After homogenization in 250 μl of buffer [0.25 M sucrose/20 mM Tris/HCl (pH 7.5)/2 mM EGTA/2 mM EDTA/2 mM phenylmethylsulfonyl fluoride (PMSF)], a protease inhibitor mixture (antipain, chymostatin, elastinal, leupeptin, pepstatin A, and phosphoramidin; final concentration: 10 μM/ml), the homogenate was passed through a 28-gauge needle 20 times. The homogenate was then separated into supernatant (cytosol, organelles, and membrane) and pellet (nucleus) by centrifugation at 10,000 rpm for 10 min. The supernatant (nucleus-free fraction) was used for immunoprecipitation and SDS-PAGE.
Whole-cell (nucleus-containing) fraction from C2C12
After rinsing in PBS twice, the harvested cells were lysed in a 250-μl ice-cold solution composed of 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and a protease inhibitor cocktail (antipain, chymostatin, elastinal, leupeptin, pepstatin A, and phosphoramidin; final concentration: 10 μM/ml; Sigma). Cell debris was removed by centrifugation at 10,000 rpm for 10 min.
To assess a possible contamination of nuclear proteins, anti-lamin B antibody (Biotechnik, Heidelberg) was employed to immunoblot the nuclear protein marker lamin B (supplementary Fig. 1). The band of lamin B (66 kD) was observed in the whole cell preparation (lanes 3 and 4), but not in the nucleus-free preparation (lanes 1 and 2). These results indicate that nucleus was effectively removed from the preparation made by the needle passage.
Whole-cell (nucleus-containing) fraction from tissues and organs
Adult female mice (300 g) were anesthetized with pentobasrbital (0.3 ml). After opening the thoracic and abdominal cavities, various organs were excised and transferred into ice-cold saline. Skeletal muscle [soleus and extensor digitorum longus (EDL) muscles] was excised from legs. The obtained organs and tissues were homogenized on ice (1 ml buffer/g tissue; for composition, see above) and then centrifuged to obtain the whole-cell fraction.
Protein concentration of the sample was determined according to Bradford’s method using bovine serum albumin as a standard.
Immunoblotting
In all immunoblot experiments, equal amounts of protein were loaded in each lane of a 9% Tris-acrylamide gel and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The molecular weight of immunoreactive bands was determined by biotinylated molecular mass standards (Bio-Rad). After the electrophoresis, polyacrylamide gels were transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad), which were subsequently incubated overnight at room temperature in a 5% blocking solution consisting of 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween-20. The blotted PVDF membrane was successively incubated with primary monoclonal anti-ERα mouse antibody (Upstate Biotechnology, Lake Placid, NY), and, after fpur washes, in 0.05% Tween 20-TBS (20 mM Tris-HCl, 150 mM NaCl) for 5 min, with secondary antibodies conjugated to horseradish peroxidase (Amersham Biosciences, Piscataway, NJ) (for each diluted 1,000 times; 1 h at room temperature). The membrane was then washed three times for 5 min in T-TBS, followed by washing in TBS two times for 5 min. To quantify the immunoreactive bands, the blotted membranes were incubated with enhanced chemiluminescence reagent (Amersham) for 1 min and exposed to Hyperfilm for 15 s to 10 min, depending on signal strength, and subjected to densitometric analysis (Las 3000, Fujifilm).
Reverse transcriptase-polymerase chain reaction (RT-PCR) experiment
Total RNA was extracted from C2C12 cells using the RNeasy Mini Kit (Qiagen, Tokyo) according to the manufacturer’s instructions. RNA concentration was determined spectrophotometrically at 260 nm. The first strand DNA was synthesized by reverse transcription from total RNA (2 μg) and subsequently amplified using an RT-PCR kit according to the manufacturer’s instructions (Invitrogen). The PCR primers used for mouse ERα were (5′–3′): CCTTCTACAGGTCTAATTCTGACAATC (forward) and ACTGAAGGGTCTAGAAGGATCATATTC (backward). To quantify the expression level of ERα mRNA, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA was amplified as a control. The PCR amplification protocol consisted of 30 cycles for ERαmRNA and 19 cycles for GAPDH: denaturation at 94°C for 20 s: primer annealing at 60°C for 30 s: extension at 72°C for 1 min. PCR products were electrophoresed on a 2% agarose gel and visualized by the ethidium bromide with UV illumination.
35S-Methionine incorporation assay
After pre-incubation at 37°C for 30 min in methionine-free medium with or without the addition of E2 (10−8 M), C2C12 myoblasts (106 cells/3.5 mm dish) were labeled with 35S-methionine in 2 ml of DMEM/F12 medium devoid of methionine for the time periods indicated in the figures. The cell lysate was incubated with the anti-ERα antibody at 4°C overnight followed by the addition of protein G-Sepharose. After shaking at 4°C for 1 h, the beads were washed extensively. The resultant immunoprecipitate (IP) was electrophoresed on a 9% acrylamide SDS-PAGE, and immunoreactive bands were visualized by UV exposure on an X-ray film that was subjected to densitometric analysis (FLA 2000).
Cell proliferation
Sub-confluent C2C12 cells cultured in a six-well culture plate were deprived of serum for 2 days and consecutively cultured in DMEM/F12 medium supplemented with various concentrations of serum (1 and 10%) in the presence and absence of E2 (10 nM). Cells were dissociated by trypsin treatment every 24 h after the start of E2 incubation, and their number was counted by using a Buerger-Tuerk plate.
Chemicals
DMEM/F12 and fetal bovine serum were purchased from Gibco BRL. 17β-Estradiol and tamoxifen, PKC activator, TPA (12-o-tetradecanoil-phorbol 13-acetate), PD98,059 and SB 203,580 from Sigma (St Louis, MO), and ICI 192,780 from Wako. 17β-Estradiol, tamoxifen, ICI182,780, TPA, PD98,059, and SB203,580 were dissolved in dimethyl sulfoxide (DMSO) and 17β-estradiol BSA in water. The supplier of E2-BSA was SIGMA, and to prepare each concentration of E2-BSA, we followed the manufacturer's instructions.
Results
Tissue distribution of ER protein in adult mouse
At the beginning of this study, we investigated the in vivo distribution of ER protein. Several tissues and organs were isolated from the adult mouse and subjected to Western blot analysis with a specific monoclonal ERα antibody. As summarized in the histogram of Fig. 1a, the expression of ER protein at the whole-cell (total) level, which was detected as a 66-kDa protein (upper panel in Fig. 1a), was highest in the uterus, followed by kidney, cardiac muscle, lung, adipose tissue, skeletal muscle, and ovary (lower panel in Fig. 1a). The level of total ER expression in skeletal muscle was comparable to that in ovary, one of the major target organs of estrogen, as well as in C2C12 myoblasts, an established cell line from mouse skeletal muscle. Furthermore, when the nucleus-free (non-nuclear) ER fraction was prepared by the needle method (see “Materials and methods”), about half of the total ER was found to be non-nuclear (Fig. 1b).
Fig. 1.
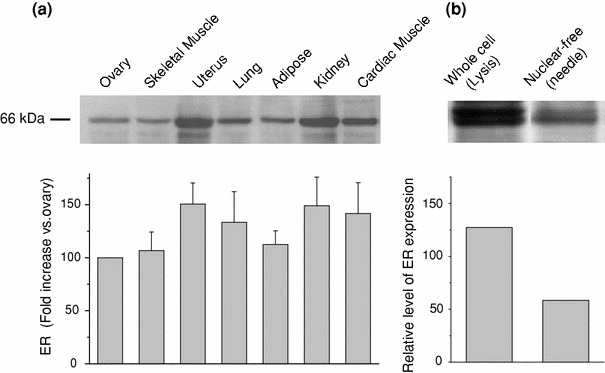
Distribution of estrogen receptors in various organs and tissues of adult mouse. a Western blot analysis of whole-cell fractions with a specific monoclonal estrogen receptor (ER) antibody. Upper panel immunoreactive bands corresponding to the ER protein (66 kDa). The histogram (lower) indicates the results of densitometric analysis for a 66-kDa band (mean ± SE, n = 4). Relative expression level with respect to the ovary is shown in %. From left to right: skeletal muscle, uterus, lung, adipose tissue, kidney, and myocardium. b Relative expression (to ovary) of whole cell and nonnuclear ER proteins obtained from C2C12 mouse myoblasts (n = 3)
In the following, for the convenience of experimentation and in order to explore a possible physiological impact of estrogen on ER in the skeletal muscle, we employed C2C12 myoblasts as a model cell.
Effects of 17β-estradiol (E2) on ER expression in C2C12 mouse myoblasts
To explore the possible effects of E2 treatment on ER expression in C2C12 myoblasts, we followed the time course of the ER mRNA level as well as the expression level of ER protein by using the real-time PCR and Western blot analyses, respectively. As summarized in Fig. 2a, the mRNA level started to increase only 30 min after addition of E2 (10−8 M) to the culture medium, reached a maximum plateau about 2.5-fold higher than control within 1 h, and then gradually declined. The expression of ER protein was also increased in both total (Fig. 3a) and non-nuclear fractions (Figs. 2b, 3b). The time course more thoroughly followed for nonnuclear ER fraction showed a rapid increase similar to that of ER mRNA, reaching the maximum (about two-fold higher than the control) within about 6 h and then declining to a level higher by about 20% than the control (Fig. 2b). The E2-induced increase in ER occurred dose-dependently in both whole-cell (10−12–10−5 M) and non-nuclear (10−10–10−6 M) fractions (Fig. 3a, b). These data were statistically significant.
Fig. 2.
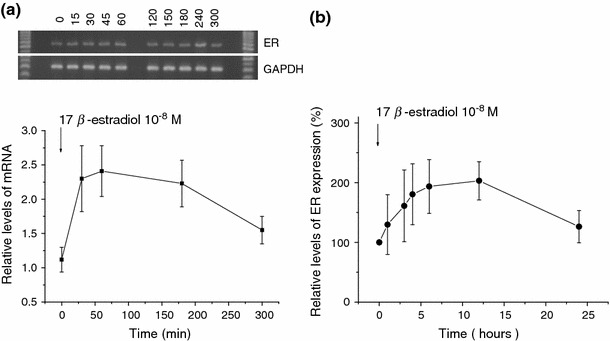
Time course of ER mRNA and protein expression during E2 exposure. a Representative of C2C12 mRNA transcripts for ER and GAPDH at several time points after exposure to E2 (10−8 M) (upper). The mRNA level of ER relative to GAPDH is averaged from five individual experiments and displayed as mean ± SE (statistically significant). At 0, 0.5, 1, 3, and 5 h (lower). b Proteins were extracted from nucleus-free C2C12 cells exposed to E2 (10−8 M) for varying periods of time (from left: 0, 1, 3, 4, 6, 12, and 24 h) and immunoblotted with the ER antibody. Immunodensity in a 66-kDa band is quantified by densitometry and presented as the % increase of control (time 0). Symbols and bars represent mean ± SE (n = 5); statistically significant
Fig. 3.
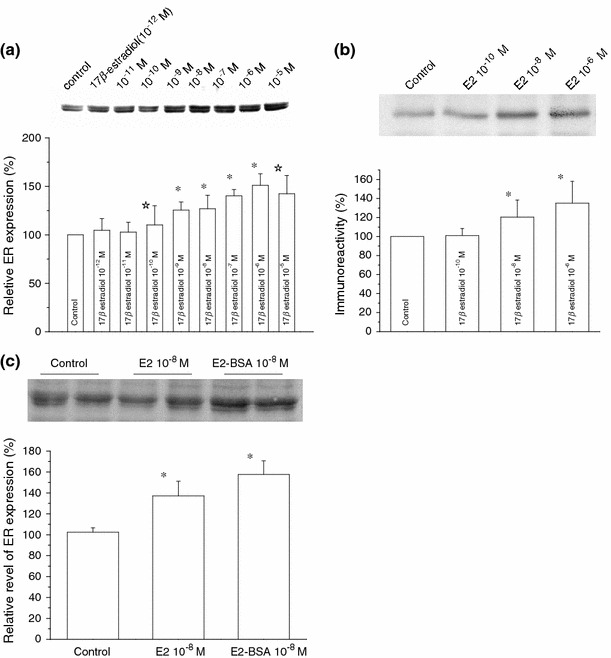
Dose–dependent effects of 17β-estradiol on the expression of total and nonnuclear ER. Representative immunoreactive bands for total ER in C2C12 myoblasts (upper panel) and the relative expression of total ER protein (a) or nonnuclear ER protein (b) quantified by densitometric analysis (lower panel); after treatment with either vehicle (control), different concentrations of 17β-estradiol (10−12–10−5 M) in a and (10−10–10−6 M) in b for 15 h. c Shows the relative expression of total ER protein after treatment with vehicle (control), 17β-estradiol (10−8 M), and BSA-conjugated E2 (10−8 M) for 15 h. Each bar is normalized to the control (far left) and expressed as %. Columns and bars indicate mean ± SE (n = 3 in a, n = 4 in b, and n = 5 in c). *P < 0.01, ☆ P < 0.05 compared with control values, determined using Student’s t test
A similar extent of the increase was also observed in total ER protein when a BSA-conjugated form of E2 (10−8 M) was used (Fig. 3c). Since this E2 form is incapable of penetrating the plasma membrane, it is the most likely that the increasing effect of E2 on ER expression occurred through cell-surface ER receptor(s).
Effects of 17β-estradiol on C2C12 myoblast proliferation
The observed increasing effect of E2 on nonnuclear ER expression may result from promoted cell proliferation, since it would increase the total protein content. To exclude this possibility, we assessed the effects of E2 on the proliferation rate of C2C12 myoblasts. C2C12 cells were serum-deprived for 48 h and subsequently subjected to various serum concentrations (1 and 10%) in the absence or presence of E2 (10−8 M). As summarized in Fig. 4, there was no significant difference between E2-free and -present conditions (open vs. filled symbols). We also examined the possible influence of culture medium by changing it from DMEM/F12 to DMEM or MEM, but we could not find significant differences (data not shown). These results suggest that increased E2 protein expression is not ascribable to accelerated cell proliferation and likely reflects the increased protein synthesis in each cell.
Fig. 4.
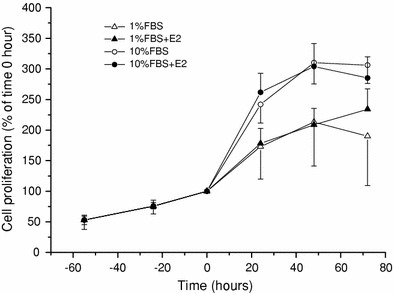
Influence of 17-βestradiol on C2C12 proliferation. C2C12 cells were grown in the serum-free medium for 48 h, which was then changed to ones with or without E2 (10−8 M) in 1% FBS (open triangle, closed triangle) or 10% FBS (open circle, closed circle). The rate of cell growth is shown as the cell number at every 24 h relative to 0 h. Symbols and bars represent the mean ± SE (n = 5)
Effects of estrogen antagonists, tamoxifen, and ICI 182,780 on ER expression
We next examined the effects of ER antagonists, tamoxifen, and ICI 182,780 on E2-inudced upregulation of ER. A nonsteroidal triphenylethlene compound, tamoxifen has widely been used because it demonstrates a strong anti-estrogenic activity. As summarized in Fig. 5, however, tamoxifen and 182,780 could not antagonize the E2-induced ER increase in total ER and nonnuclear ER (Fig. 5a, b). Similar agonistic effects were also observed for tamoxifen and ICI 182,780 (10−12 M–10−6 M, supplementary Figs. 2 and 3). These unexpected enhancing effects of ER antagonists on ER in C2C12 cells might reflect some structural difference of involved ER from the ‘classical’ type of ER, but we did not further pursue this subject in this study.
Fig. 5.
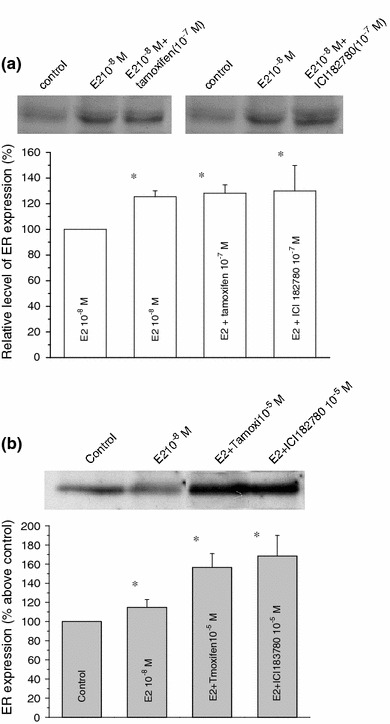
Effects of ER antagonists on E2-induced whole-cell and non-nuclear ER increase. C2C12 cells were treated with E2 (10−8 M) in the absence or presence of either tamoxifen or ICI 182,780 (10−7 M; a total ER) or (10−5 M; b non-nuclear fractions) for 15 h. Tamoxifen or ICI 182,780 was added to the culture medium 30 min prior to the administration of E2. Total and nonnuclear fractions of ER were prepared as described in “Materials and methods” and subjected to Western blotting. Histograms represent the extent of inhibition of E2-induced ER increase in total a and nonnuclear b fractions, which are expressed as the percentage of control (no stimulation), averaged from five separate experiments (mean ± SE). *P < 0.01, compared with control values, determined using Student’s t test
17β-Estradiol induced the new synthesis of ER protein
To gain further insight into the mechanism underlying E2-induced ER increase, we next performed immunoprecipitation of ER combined with the [35S]-methionine incorporation method to selectively detect a newly synthesized ER protein. As shown in Fig. 6, basal 35S-radioactivity was detected under unstimulated conditions (three far left lanes and columns), suggesting the presence of constitutive ER synthesis. When 17β-estradiol (E2: 10−8 M) was applied, the rate of 35S incorporation was significantly increased in hours and reached to more than a four-fold higher level than that at rest, at 5 h later. This rapid time course of 35S-coopertation into ER or new ER synthesis was similar to that observed for nonnuclear ER expression increase induced by the same concentration of E2 (10−8 M; see above). The increased rate of ER synthesis induced by E2 was further enhanced by the presence of TPA 10 μM. These results suggest that E2 treatment greatly facilitates the de novo synthesis of ER, which may be positively upregulated by simultaneous activation of PKC.
Fig. 6.
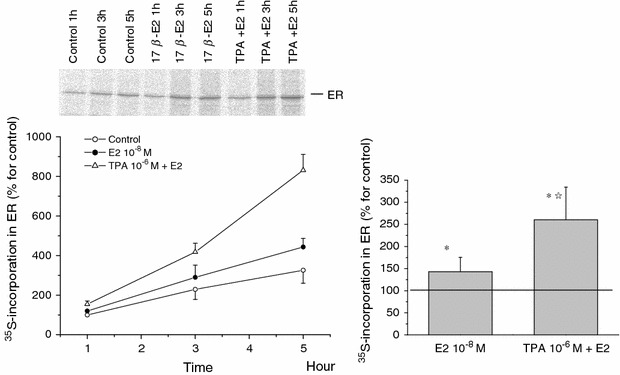
Effects of 17β-estradiol and/or TPA on ER de novo synthesis. In the absence (control) or presence of 17β-estradiol (10−8 M) and/or TPA (10−6 M), C2C12 cells were labeled with [35S] methionine. At the time points indicated in the figure, the total ER fractions were prepared and subjected to selective immunoprecipitation for ER. The immunoprecipitates were analyzed by SDS-PAGE and image scanning. Data are the mean ± SE from three separate experiments. Left panel time course of 35methionine incorporation in ER. Right panel rates of the increase at 5 h by the relative ratio of control. *P < 0.01, compared with control values, determined using Student’s t test. ☆ P < 0.01, compared with E2 values, determined using Student’s t test
The activation of the expression of estrogen receptor induced by 17β-estradiol involves ERK1/2
In the final step of the present study, we explored the possible intracellular signaling pathway(s) involved in the above-described rapid E2-induced ER increase. It was previously reported that the MAPK cascade plays a pivotal role in a rapid E2/ER signaling in different types of cells [21]. Therefore, the monolayer of C2C12 myoblasts was pre-incubated for 30 min with the specific ERK1/2 inhibitor PD98059 (10 μM) prior to addition of E2 (10−8 M). As shown in Fig. 7a, treatment with PD98059 reduced the expression level of ER not only after E2 treatment (by 60%), but also in basal conditions (by 50%). To confirm that this inhibition indeed involves ERK phosphorylation by activation of its upstream signaling cascade, we performed immunoblot analysis with a phospho-epitope-specific antibody recognizing a phosphorylated form of ERK. Treatment with E2 (10−8 M) increased the phosphorylation of ERK time-dependently, which peaked at 30 min and then gradually declined (Fig. 7b), the time course being similar to that of ER mRNA increase (Fig. 2a). These results together suggest that ERK phosphorylation would be an essential downstream event of cell membrane ER activation by E2, which mediates the transcription of the ER gene.
Fig. 7.
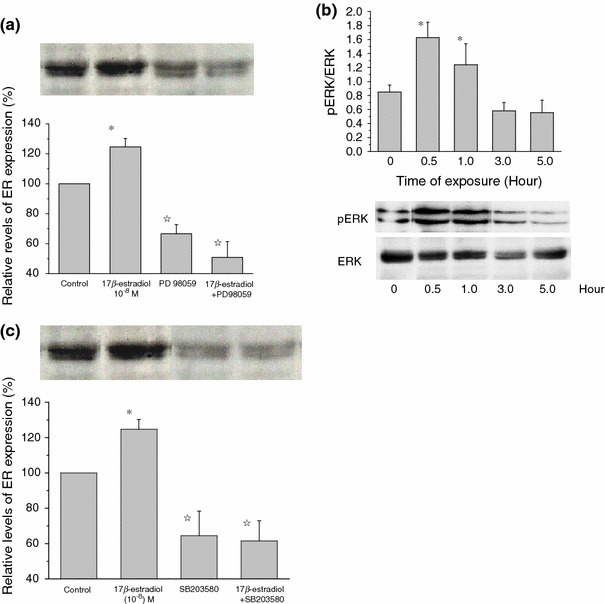
E2-induced ER increase involves ERK1/2 and p38 pathways. Histograms indicate the relative density of ER immunoblots normalized to control (basal level). a From left to right, vehicle, E2 10−8, PD9809 alone, and E2 + PD98059. b Time course of ERK phosphorylation induced by E2. ERK phosphorylation assessed by pERK-specific antibody. c From left to right, vehicle, E2 (10−8 M), SB203580 (10 μM) alone, and E2 + SB203580. In a and c, PD98059 (10 μM) or SB203580 (10 μM) was added into the culture medium 30 min before addition of E2 (10−8 M). The bars show means ± SE of three independent experiments performed in duplicate. *P < 0.01, compared with control values, determined using Student’s t test. ☆ P < 0.01, compared with E2 values, determined using Student’s t test
In another series of experiments, we also investigated the possible involvement of the p38-MAPK pathway. The specific p38 inhibitor SB203580 (10 μM), which was applied prior to the addition of 10−8 M E2, significantly inhibited the E2-induced ER increase less than the level in unstimulated conditions (Fig. 7c).
Discussion
In this study, we have obtained evidence that C2C12 myoblasts derived from mouse skeletal muscle constitutively express the estrogen receptor (ER) at a high level comparable to the major target organs of estrogen, such as uterus and ovary, and a considerable part of the receptor is distributed outside the nucleus. Consistent with our finding, the expression of ER in the same cell line has recently been found confined largely to extracellular regions, such as mitochondria [22]. In addition to this, we also found that the expression level of ER in C2C12 cells is positively regulated by 17β-estradiol (E2) in both total and nonnuclear fractions, at least part of which seems mediated via increased de novo synthesis.
The observed E2-induced upregulation of ER seems mediated by a signaling pathway initiated from a cell membrane ER, since a similar extent of ER increase could be induced by a membrane-impermeable, BSA-conjugated E2. Furthermore, the rapid activation of MAPKs following E2 stimulation and ‘agonistic’ actions of ER antagonists (tamoxifen and ICI1182,780) strongly indicate that the E2-induced ER increase clearly differs from the genomic actions of estrogen and would occur through a non-genomic, cytosolic pathway via a receptor pharmacologically different from classical ERs [23]. Although clear evidence is still lacking, these results support the previously proposed idea that an ER isoform that is distinct from classical ERs exists in the cell membrane and mediates rapid nongenomic actions.
The observed time course of E2-induced ER increase (requiring several hours to become overt) is considerably slower than other reported non-genomic effects of estrogen, such as Ca2+ release via stimulated turnover of phosphatidylinositides. This may reflect multiple processes involved in E2-induced ER protein synthesis, e.g., transcriptional activation of genes after MAPKs activation (Fig. 7). In contrast, our previous experiment in frog skeletal muscle revealed the presence of a much faster signaling initiated by the cell membrane ER, where administration of E2 quickly (in minutes) enhanced the twitch tension, presumably because of increased myoplasmic Ca2+ concentration [18]. These findings may suggest, as in other cell types, the presence of distinct non-genomic signalings linked to the cell membrane ERs, which operate in both fast and slow fashions via PLC- and MAPK-mediated pathways, respectively.
The physiological significance of E2-induced membrane-bound ER upregulation in skeletal myoblasts remains to be determined. It could, however, be speculated that this upregulation may serve as positive feedback to boost the actions of estrogen on skeletal muscle function effectively. In fact, similar estrogen-dependent upregulation of estrogen receptor (ERα) has been reported in hyppocampal dendric spines, where the expression level of ERα was much higher in proesterus female than diesterus female or male rats [24]. Furthermore, in many distinct types of neurons and neuronal cell lines, it has been reported that estrogen activates ERK1/2 and/or Akt/CREB phosphorylation-mediated signaling cascades to exert neuroprotective actions in an ERE-independent manner, which may underlie the well-known beneficial effects of estrogen on the progression and severity of neurological disorders such as Parkinson’s and Alzheimer’s disease [7]. Thus, simply extrapolating the present results, an estrogen-induced non-genomic signaling may be involved in protecting the skeletal muscle tissues from a variety of cell-invasive stresses that cause degenerative and atrophic disorders and diseases. Alternatively, as has been suggested by a recent study, the extranuclear ER receptor, which is expressed in mitochondria, may contribute to the inhibition of the apoptotic process in skeletal muscle [25]. Further study will clearly be needed to elucidate how and to what extent such beneficial effects of estrogen could be carried out through non-genomic signaling in intact skeletal muscle.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgment
We thank Dr. Yukisato Ishida (Bunkyo Gakuin University) for the helpful discussions.
References
- 1.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 2.Nadal A, Diaz M, Valverde M. The estrogen trinity: membrane, cytosolic, and nuclear effects. News Physiol Sci. 2001;16:251–255. doi: 10.1152/physiologyonline.2001.16.6.251. [DOI] [PubMed] [Google Scholar]
- 3.Ho KJ, Liao JK. Nonnuclear action of estrogen. Arterioscler Thromb Vasc Biol. 2002;22:1952–1961. doi: 10.1161/01.ATV.0000041200.85946.4A. [DOI] [PubMed] [Google Scholar]
- 4.Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-α functions. BBRC. 2004;316:878–883. doi: 10.1016/j.bbrc.2004.02.129. [DOI] [PubMed] [Google Scholar]
- 5.Boonyaratanakonkit V, Edwards DP. Receptor mechanisms mediating non-genomic actions of sex steroids. Semim Reprod Med. 2007;25(3):139–153. doi: 10.1055/s-2007-973427. [DOI] [PubMed] [Google Scholar]
- 6.Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148(7):3236–3245. doi: 10.1210/en.2006-1605. [DOI] [PubMed] [Google Scholar]
- 7.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–153. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- 8.Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- 9.Norfleet AM, Clarke CH, Gametchu B, Watson CS. Antibodies to the estrogen receptor-α modulate rapid prolactin release from rat pituitary tumor cells through plasma membrane estrogen receptors. FASEB J. 2000;14:157–165. doi: 10.1096/fasebj.14.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson CS, Norfleet AM, Pappas TC, Gametchu B. Rapid actions of estrogens in GH3/B6 pituitary tumor cells via a plasma membrane version of estrogen receptor-α. Steroids. 1999;64:5–13. doi: 10.1016/S0039-128X(98)00107-X. [DOI] [PubMed] [Google Scholar]
- 11.Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singth M, Connolly ES, Jr, Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and ischemic brain injury. J Neurosci. 2002;22:8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 13.Revankar CM, Cimino DF, Sklar LA, Arterburm JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 14.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segars J, Driggers P. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab. 2002;13:349–354. doi: 10.1016/S1043-2760(02)00633-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERα expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999;13:307–319. doi: 10.1210/me.13.2.307. [DOI] [PubMed] [Google Scholar]
- 17.Canderelli R, Leccesse LA, Miller NL, Unruh DJ. Benefits of hormone replacement therapy in postmenopausal women. J Am Acad Nurse Pract. 2007;19:635–641. doi: 10.1111/j.1745-7599.2007.00269.x. [DOI] [PubMed] [Google Scholar]
- 18.Hatae J. Effects of 17-βestradiol on tension responses and fatigue in the skeletal twitch muscle fibers of frog. Jpn J Physiol. 2001;51:753–759. doi: 10.2170/jjphysiol.51.753. [DOI] [PubMed] [Google Scholar]
- 19.Hatae J, Takami N, Hai L, Honda A, Inoue R. Genomic and non-genomic action of estrogen in the mouse skeletal muscle cells. J Physiol Sci. 2008;58(Suppl.):S69. [Google Scholar]
- 20.Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 21.Nethrapalli IS, Tinnikov AA, Krishnan V, Lei CD, Toran-Allerand CD. Estrogen activates mitogen-activated protein kinase in native, nontransfected CHO-K1, COS-7, and RAT2 fibroblast cell lines. Endocrinology. 2005;146(1):56–63. doi: 10.1210/en.2004-1106. [DOI] [PubMed] [Google Scholar]
- 22.Milanesi L, de Boland AR, Boland R. Expression and localization of estrogen receptor alpha in the C2C12 murine skeletal muscle cell line. J Cell Biochem. 2008;104(4):1254–1273. doi: 10.1002/jcb.21706. [DOI] [PubMed] [Google Scholar]
- 23.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 24.Romeo RD, McCarthy JB, Wang A, Milner TA, McEwen BS. Sex differences in hippocampal estradiol-induced N-methyl-d-aspartic acid binding and ultrastructural localization of estrogen receptor-alpha. Neuroendocrinology. 2005;81(6):391–399. doi: 10.1159/000089557. [DOI] [PubMed] [Google Scholar]
- 25.Vasconsuelo A, Milanesi L, Boland R. 17β-Estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatilinositol 3-kinase/Akt pathway. J Endocrinol. 2008;196:385–397. doi: 10.1677/JOE-07-0250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.