

Abstract
Both types 1 and 2 diabetes mellitus (T1DM and T2DM) are associated with profound deterioration of calcium and bone metabolism, partly from impaired intestinal calcium absorption, leading to a reduction in calcium uptake into the body. T1DM is associated with low bone mineral density (BMD) and osteoporosis, whereas the skeletal changes in T2DM are variable, ranging from normal to increased and to decreased BMD. However, both types of DM eventually compromise bone quality through production of advanced glycation end products and misalignment of collagen fibrils (so-called matrix failure), thereby culminating in a reduction of bone strength. The underlying cellular mechanisms (cellular failure) are related to suppression of osteoblast-induced bone formation and bone calcium accretion, as well as to enhancement of osteoclast-induced bone resorption. Several other T2DM-related pathophysiological changes, e.g., osteoblast insulin resistance, impaired productions of osteogenic growth factors (particularly insulin-like growth factor 1 and bone morphogenetic proteins), overproduction of pro-inflammatory cytokines, hyperglycemia, and dyslipidemia, also aggravate diabetic osteopathy. In the kidney, DM and the resultant hyperglycemia lead to calciuresis and hypercalciuria in both humans and rodents. Furthermore, DM causes deranged functions of endocrine factors related to mineral metabolism, e.g., parathyroid hormone, 1,25-dihydroxyvitamin D3, and fibroblast growth factor-23. Despite the wealth of information regarding impaired bone remodeling in DM, the long-lasting effects of DM on calcium metabolism in young growing individuals, pregnant women, and neonates born to women with gestational DM have received scant attention, and their underlying mechanisms are almost unknown and worth exploring.
Keywords: Bone loss, Calcium wasting, Growth plate, T1DM, T2DM, Osteoporosis
Introduction
Diabetes mellitus (DM) is a worldwide metabolic disease characterized by chronic hyperglycemia, which results from insufficient insulin secretion and/or insulin resistance in target tissues or both [1, 2]. Chronic hyperglycemia leads to macrovascular and microvascular complications in various organs, e.g., heart, brain, kidney, eyes, and bone, the last of which culminates in the so-called diabetic osteopathy [1, 2]. In other words, DM not only disturbs normal functions of vital organs (for reviews, see [3–5]) but also impairs calcium metabolism by disrupting the functions of calcium-regulating organs, namely the intestine, bone, and kidney. The present article thus focuses on the underlying mechanisms of the negative effects of DM on calcium homeostasis as well as their long-term consequences.
Overview of DM and insulin resistance
Generally, DM is classified into 4 types based on defects in the mechanism of insulin action, that is, type 1 DM (T1DM), type 2 DM (T2DM), gestational DM (GDM) and other specific types of DM, the last of which includes genetic defects in insulin processing, insulin action, or β-cell function [1, 2]. T1DM, or insulin-dependent DM, accounts for 5–10 % of diabetic patients. It is caused by autoimmune destruction of β-cells in the islet of Langerhans, resulting in reduced β-cell mass and inadequate insulin secretion. Autoimmunity in T1DM comes in diverse forms, such as islet autoantibodies, insulin autoantibodies, autoantibodies to tyrosine phosphatase IA-2 and IA-2β and glutamic acid decarboxylase isoform-65 (GAD65), the last of which is usually detected in the early hyperglycemic stage of T1DM [1, 2]. T1DM commonly takes effect in childhood and adolescence, but it can occur at any age up to old age [1].
The more common type is T2DM or non-insulin-dependent DM, which accounts for 90–95 % of diabetic patients. It results from insulin resistance and relative insulin deficiency rather than absolute deficiency. Most T2DM patients have obesity or high body fat distribution that often occurs with insulin resistance in the target tissues, such as skeletal muscle and adipose tissue. This form of DM is more strongly correlated with genetic predisposition than immune-mediated T1DM. Because of insulin resistance, a compensatory increase in insulin secretion is required to maintain normal plasma glucose levels [6, 7]. A number of post-receptor defects have been proposed to explain the pathogenesis of insulin resistance, such as defects in insulin-regulated phosphorylation/dephosphorylation [7]. For example, DM causes a phosphatidylinositol-3-kinase (PI3K) signaling defect that may reduce translocation of glucose transporters (e.g., GLUT4) to the plasma membrane, thereby impairing glucose uptake [6, 8]. Other abnormalities include the accumulation of ectopic lipid metabolites that later impair mitochondrial oxidative phosphorylation and reduce insulin-stimulated mitochondrial ATP production, and also generate reactive oxygen species (ROS) [6–8]. Lipid metabolite-mediated insulin resistance results from the accumulation of diacylglycerol (DAG), which, in turn, activates protein kinase C isoform θ (PKCθ) and subsequently impairs the translocation of GLUT4-containing storage vesicles to the plasma membrane [6, 8]. Of note, inflammatory mediators secreted from adipocytes also contribute to insulin resistance, such as tumor necrosis factor (TNF)-α, and interleukin (IL)-6, and suppressor of cytokine signaling (SOC)-1 and -3 [6], some of which, particularly TNF-α, and IL-6, are also potent stimulators of osteoclastogenesis and bone resorption [9].
Regarding GDM, it is a condition in which women without previously diagnosed DM show hyperglycemia during pregnancy or persisting after delivery [1]. GDM women with hypertensive disorder, preterm delivery, and multiparity have increased risk of developing T2DM in their lifetime [10]. GDM is also more prevalent in women of certain ethnic groups, e.g., Hispanic, and Native American [11]. Interestingly, a clinical study investigating the risk factors associated with development of postpartum T2DM in 843 GDM women found that high pre-pregnancy body mass index (BMI) was an important risk factor for both early and late T2DM conversions [12]. Indeed, pregnancy places a stress on maternal calcium metabolism, especially in the third trimester, during which ~200 mg elemental calcium per day is transferred across the placenta for fetal osteogenesis (for reviews, see [13, 14]). However, bone changes in GDM mothers have received little attention.
As for other specific types of DM, the etiologies are diverse, but most of them are genetic diseases or consequences of exocrine pancreatic disorders and endocrinopathies [1]. The most common genetic defect of β-cell is maturity-onset diabetes of the young (MODY), which is characterized by the onset of hyperglycemia at an early age (generally before 25 years), and impairment of insulin secretion with minimal or no defect in insulin action [1]. MODY is a monogenic disorder that inherits in an autosomal dominant pattern. Approximately 1 % of MODY patients are misdiagnosed as T1DM or T2DM [1, 15]. The most common forms of MODY are associated with mutations of hepatocyte nuclear factor (HNF)-1α and glucokinase genes, which account for 80 % of all MODY cases [1, 15]. HNF-1α is a transcription factor directly involved in pancreatic development and β-cell differentiation [16]. Glucokinase serves as glucose detector for β-cells, and is responsible for the conversion of glucose to glucose-6-phosphate, which in turn stimulates insulin secretion [1]. The defect of the glucokinase gene thus directly affects β-cells secretion of insulin.
Body calcium homeostasis
Calcium is an important element for several biological processes, such as cell division and growth, blood coagulation, cardiovascular homeostasis, hormone responses, neural electrical activity, and bone formation [17, 18]. Ninety-nine percent of calcium is stored in bone, while the remaining one percent is distributed in the intracellular fluid (ICF) and extracellular fluid (ECF). The total plasma calcium and ionized calcium must be maintained within a narrow range (8.5–10.5 mg/dL and 4.65–5.25 mg/dL, respectively) [18]. Because a large change of plasma calcium concentration, either a decrease (hypocalcemia) or increase (hypercalcemia), is lethal, therefore, calcium level is tightly regulated by three classical calcium-regulating hormones, namely, parathyroid hormone (PTH), 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], and calcitonin, as well as other endocrine and paracrine factors [e.g., estrogen, androgens, adrenal glucocorticoids, prolactin, insulin-like growth factor (IGF)-1, and fibroblast growth factor (FGF)-23]. Not only the plasma calcium level, but the whole body calcium metabolism is also controlled by these calcium-regulating hormones to ensure adequate intestinal calcium absorption, proper storage of calcium in bone, and excretion of excess calcium via the kidney.
To maintain normal calcium homeostasis, two aspects of calcium metabolism have to be considered. The first aspect is the total amount of calcium in the body that depends on the balance between calcium absorbed by the intestine and that excreted by the kidney. In normal adults with daily calcium intake of 1000 mg, ~200 mg (~20 %) is absorbed by the intestine, while ~800 and ~200 mg are excreted in feces and urine, respectively [17]. Thus, calcium input balances total calcium output. The second aspect is the regulation of the plasma-ionized calcium within a normal range of 4.65–5.25 mg/dL. Specifically, hypocalcemia increases the circulating levels of parathyroid hormone (PTH) and 1,25(OH)2D3 levels. PTH stimulates calcium reabsorption and suppresses phosphate reabsorption in the renal tubules, and also enhances the conversion of 25(OH)D3 into 1,25(OH)2D3, which, in turn, increases the intestinal calcium absorption. PTH also stimulates bone resorption to release calcium into the circulation [17].
In contrast, when hypercalcemia occurs, negative feedback mechanisms limit calcium entry and increase calcium excretion and the normal level of plasma ionized calcium is restored [17]. The feedback mechanism involves calcium-sensing receptors (CaSR) in the parathyroid chief cells that sense elevated ionized calcium level, thus inhibiting PTH release and 1,25(OH)2D3 synthesis [17]. Recently, FGF-23 has been added to the list of calcium regulatory hormones as it can inhibit 1,25(OH)2D3-induced intestinal calcium absorption [19, 20]. Therefore, the intestinal calcium absorption is inhibited directly by reduction in 1,25(OH)2D3 production, and indirectly by increased FGF-23 production from the osteocytes in bone or from local production in the intestine [17, 19, 21].
In the diabetic condition, derangement of calcium homeostasis is multifactorial and, although it was reported in 1979 that diabetic patients exhibited normal calcium homeostasis, i.e., no change in plasma levels of total and ionized calcium, PTH, 25(OH)D3, and 1,25(OH)2D3 [22], new investigations in the 2000s mostly found impaired calcium homeostasis in DM and associations between calcium-regulating hormones and the development of DM [23, 24]. For example, individuals with hypovitaminosis D had a higher risk of developing insulin resistance, metabolic syndrome, and incidence of T2DM [23, 24]. Recently, there was a report that a high level of PTH was associated with DM, and this association varied according to ethnic groups—i.e., higher incidence in Caucasians [25]. Besides the calcium-regulating hormones, DM also affects the function of calcium-regulating organs, causing impaired intestinal calcium absorption, bone deterioration, decelerated bone elongation and renal calcium wasting (Fig. 1); the details are discussed below.
Fig. 1.
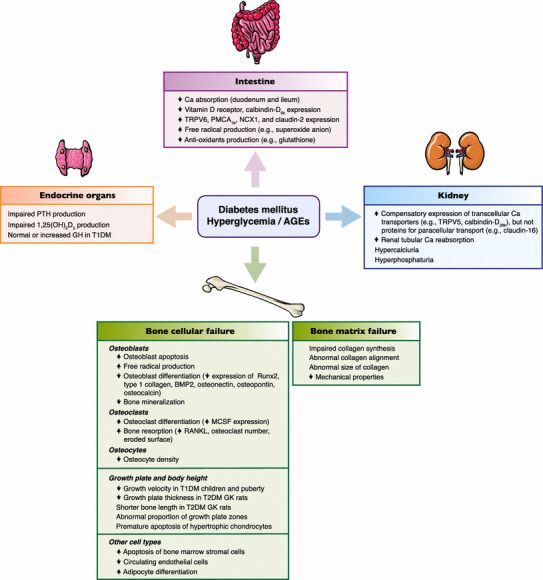
Possible deleterious effects of diabetes mellitus, hyperglycemia and advanced glycation end products (AGEs) on metabolism of calcium-regulating hormones and calcium-regulating organs, i.e., intestine, kidney, and bone
DM-associated impairment of intestinal calcium absorption
Unlike bone, intestinal calcium absorption, which is the only route for calcium entry into the body, is rarely investigated in DM. Most intestinal calcium absorption studies have been performed in alloxan- or streptozotocin (STZ)-induced T1DM animal models that showed lower rates in DM [26–29]. In alloxan-induced DM rats, the decrease in net calcium absorption was due to a decrease in the lumen-to-plasma calcium flux in the duodenum and ileum [27]. This reduction in calcium absorption could be restored to control level by insulin [28], confirming the association between DM and intestinal impaired calcium absorption.
At present, it is not known whether the reduction in intestinal calcium absorption results directly from insulin deficiency or from other hormonal disturbance. Several studies in diabetic rats reported that the reduction in intestinal calcium absorption occurred concurrently with decreases in the circulating 1,25(OH)2D3 level, intracellular vitamin D receptor number, and cytoplasmic calcium-binding protein calbindin-D9k in the enterocytes [29, 30]. However, other investigators have reported a compensatory upregulation of transcellular and paracellular calcium transporter expression in the intestinal epithelial cells of diabetic animals. As demonstrated by Rivoira and co-workers [26], the expressions of transient receptor potential vanilloid family calcium channel-6 (TRPV6), plasma membrane calcium-ATPase-1b (PMCA1b), sodium-calcium exchanger-1 (NCX1), and claudin-2 were significantly increased in the intestinal epithelium of STZ-induced T1DM rats. They also found that DM was accompanied by increased oxidative stress and production of free radicals (e.g., superoxide anion), which could alter duodenal permeability in STZ-induced diabetic rats [26]. However, whether T2DM affects intestinal calcium absorption and intestinal permeability in humans remains to be investigated.
Diabetic osteopathy
T1DM and T2DM have been reported to aggravate osteopenia and osteoporosis that increase fracture risk [31, 32], but pathological bone changes in T1DM and T2DM are markedly different in terms of cellular and molecular mechanisms [33–37]. Low bone mass is consistently observed in T1DM patients [33–35]. However, for T2DM, there have been reports of both high and low bone mineral density (BMD) [36–39]. Previously, it was thought that the high bone mass in T2DM might be an adaptation to sustain high body weight and might postpone osteoporosis [37, 39]. More advanced high-resolution, peripheral quantitative computed tomography (HR-pQCT) or microindentation techniques have supported the notion that T2DM patients indeed have deteriorated bone functions, such as impaired bone microstructure and mechanical properties, and abnormal function of bone cells [38, 40, 41]. Although evidence is scant, a few studies on GDM have reported greater reduction in maternal BMD and bone loss when compared with non-diabetic mothers [42].
Regardless of the types of DM, pathogenesis of DM-induced osteoporosis can result from pathophysiological changes in bone compositions, i.e., cellular failure and extracellular matrix failure, both of which are provoked by factors such as insulin resistance, advanced glycation end products (AGEs) and ROS. Regarding the cellular failure, DM is correlated with abnormal or diminished activities of bone cells (i.e., osteoblasts, osteoclasts and osteocytes) as well as neighboring cells in bone, such as adipocytes, mesenchymal cells, and endothelial progenitor cells [38, 43, 44]. Several in vitro investigations reported that high blood glucose induced ROS production in osteoblasts, which, in turn, promoted osteoblast apoptosis, and suppressed osteoblast differentiation and mineralization as indicated by decreased expressions of osteoblast differentiation markers, i.e., runt-related transcription factor-2 (Runx2), type I collagen, osteonectin and osteocalcin [43]. In addition, ROS enhanced transformation from osteoblast lineage to adipocyte lineage by increasing the expression of adipogenic markers, i.e., peroxisome proliferator-activated receptor (PPAR)-γ, adipocyte fatty acid binding protein (aP2), resistin and adipsin [45]. The final outcome is a reduction in the mature osteoblast numbers and thus bone formation [43, 45, 46]. Consistently, in vivo investigation in insulin-resistant high-fat diet-fed Zucker diabetic fatty (ZDF) rats also showed downregulation of osteoblast differentiation markers, namely Runx2, bone morphogenetic protein (BMP-2), osteocalcin, and osteopontin [47]. In Goto-Kakizaki (GK) T2DM rats, bone histomorphometric analysis confirmed that DM reduced osteoblast activities (e.g., osteoblast surface, mineralizing surface and bone formation rate), while increasing osteoclast bone resorption (e.g., osteoclast surface and eroded surface) [38]. This accelerated bone resorption resulted from an increase in osteoclast numbers and expression of osteoclastogenic mediators, e.g., TNF-α, macrophage colony-stimulating factor (MCSF), receptor activator of nuclear factor-κB ligand (RANKL) and vascular endothelial growth factor (VEGF)-A [48]. The DM-associated bone loss is consistent with increases in mRNA and circulating levels of osteoclastogenic cytokines, as summarized in Table 1.
Table 1.
Cytokines with osteoclastogenic activities in DM
| Cytokines | Major findings | Refs. |
|---|---|---|
| RANKL | Elevated in plasma of T1DM children | [101] |
| Elevated in bone marrow of polygenic TallyHo/JngJ T2DM mice | [102] | |
| Increased mRNA expression in STZ-treated T1DM mice | [48] | |
| IL-1 | Elevated in serum during early onset of T1DM in STZ-treated mice | [103] |
| IL-6 | Elevated in blood and bone marrow of TallyHo/JngJ T2DM mice | [102] |
| Elevated in serum during early onset of T1DM in STZ-treated mice | [103] | |
| TNF-α | Elevated in blood and bone marrow of TallyHo/JngJ T2DM mice | [102] |
| Increased mRNA expression in STZ-treated T1DM mice | [48] | |
| IFN-γ | Elevated in blood and bone marrow of TallyHo/JngJ T2DM mice | [102] |
| Elevated in serum during early onset of T1DM in STZ-treated mice | [103] | |
| MCSF | Increased mRNA expression in STZ-treated T1DM mice | [48] |
| MCP-1 | Elevated in serum during early onset of T1DM in STZ-treated mice | [103] |
RANKL receptor activator of nuclear factor-κB ligand, IL-1 interleukin-1, IL-6 interleukin-6, TNF-α tumor necrosis factor-α, IFN-γ interferon-γ, MCSF macrophage colony-stimulating factor, MCP-1 monocyte chemoattractant protein-1, STZ streptozotocin
Osteocytes—the most abundant bone cells entombed in the bone matrix—are also affected, as indicated by a reduction in osteocyte density (number of osteocyte-occupied lacunae per unit area) in diabetic rats [44]. Since osteocytes are mechanosensitive cells that play an important role in bone response to mechanical stimuli, DM may reduce the mechanical signal transduction for bone remodeling and may indirectly weaken the collagen alignment with increased fracture risk [44, 49, 50].
Besides bone cells, a number of other cell types in bone, such as bone marrow stromal cells (BMSCs) or endothelial progenitor cells, are also decreased in diabetic condition. The underlying mechanism—at least in rats—probably related to AGEs-induced apoptosis of BMSCs through TNF-α production, oxidative stress, and finally caspase activation [51]. The number of circulating endothelial cells are apparently decreased in diabetic mice, causing delay in angiogenesis, and eventually lead to poor bone quality, poor healing at fracture sites, and increased fracture risk [52].
Regarding the bone extracellular matrix failure, DM was recently reported to alter the structure of type I collagen causing impaired bone strength [53]. Normally, collagen synthesis takes place both intracellularly and extracellularly. Initially, it is synthesized as preprocollagen, which is exported from the cell in the form of procollagen. Upon secretion, procollagen undergoes three steps of transformation in the extracellular space, i.e., (1) enzymatic removal of the free endings by procollagen peptidase to give rise to tropocollagen, (2) cross-linking of tropocollagen molecules to form collagen fibrils by lysyl oxidase, and (3) side-by-side cross-linking of collagen fibrils to form collagen fibers [54]. Large amounts of AGEs and ROS commonly accumulated in diabetic condition contribute to bone deterioration by impairing collagen synthesis and collagen alignment [45, 50, 55]. AGEs are heterogeneous groups of molecules formed from the non-enzymatic reaction between reducing sugars and free amino groups of proteins, lipids, and nucleic acids. They can form crosslinks between proteins, and, in doing so, alter structure and function of those proteins in extracellular matrix, basement membrane, and blood vessel wall. AGEs are often found in the plasma proteins of patients with DM and renal failure [50, 56]. They can suppress the production of phosphoprotein-1 (the mature osteoblast marker) and lysyl oxidase, the latter being essential for collagen crosslink [50]. Recently, by using scanning electron microscopy (SEM) and transmission electron microscopy (TEM), abnormally large collagen fibrils and aberrant collagen fibril alignment were found in rats with AGEs accumulation induced by adenine treatment [50]. These animals exhibited low bone quality as evidenced by a reduction in mechanical bone strength, i.e., rigidity, yield moment, ultimate moment, and energy to fracture [57, 58]. In addition to AGEs, ROS produced by mitochondrial dysfunction (such as hydrogen peroxide), malondialdehyde, and cytosolic ROS also impaired osteoblastogenesis, thereby preventing type I collagen production and compromising bone quality [45, 55].
Growth retardation in DM
While DM-induced osteopathy has been extensively investigated, impaired longitudinal bone growth, especially during childhood and adolescence, another feature of bone defects associated with T1DM, T2DM, and other congenital DM, is largely unknown. Normally longitudinal bone growth is under the influence of both local and systemic factors, e.g., growth hormone (GH), insulin-like growth factor-1 (IGF-1) and insulin [59, 60]. GH from the anterior pituitary stimulates longitudinal bone growth specifically via indirect and direct actions. The indirect action is mediated by IGF-1 from the liver, which increases growth plate chondrocyte proliferation [59, 61]. GH and insulin also play important roles in longitudinal bone growth by increasing proliferation of the growth plate chondrocytes and expression of endochondral bone formation related genes [62, 63]. Thus, insulin deficiency in T1DM ultimately impairs growth plate development and also longitudinal bone growth. Again, there have been reports of both normal and impaired growth in DM subjects [64–66].
Growth retardation can occur after the onset of DM [64, 66]. Lebl et al. [64] reported longitudinal change of height in 587 diabetic children and teenagers (~0.3–20 years of age). They found that the heights of male and female T1DM in the first observation were higher than the mean standard, but subsequently declined. Consistently, Muñoz and co-workers [66] found that the heights of T1DM patients in different pubertal stages were within normal range, but the growth velocity was slightly below average. The reason why the height of T1DM patients was normal during diabetic onset but decreased later remains unclear, but it might link to synchronization between diabetic onset and pubertal growth spurt or partial GH resistance in T1DM patients [64, 66], since circulating GH was reportedly increased in T1DM adolescents [67].
Recent investigations by Lapmanee et al. [38] and Aiemlapa et al. [68] also showed aberrant growth plate morphology and abnormal chondrocyte function in diabetic animals. Generally, bone elongation is controlled by proliferation and differentiation of chondrocytes in the growth plate. The growth plate is histologically divided into 3 zones, i.e., resting zone (RZ), proliferative zone (PZ) and hypertrophic zone (HZ). The RZ contains small chondroblasts with low mitotic activity embedded in extracellular matrix. Chondroblasts gradually migrate to the PZ where cells become highly proliferative and align into parallel vertical columns. The proliferative chondrocytes enlarge and become hypertrophic cells in HZ [69]. Before undergoing apoptosis, they are replaced by osteoblasts that arrive with vascularization. The cartilaginous scaffold thus acts as a template for bone formation and bone elongation [54, 59, 69]. Apparently, strong stimuli that disrupt growth plate differentiation could have an effect on the overall growth plate morphology and the extent of longitudinal bone growth. Since MODY develops during childhood or adolescence, it could profoundly affect chondrocyte function and endochondral bone growth. Even though short stature is not an important clinical manifestation of MODY, its negative effect on the growth plate is likely to result in poor-quality bone tissues.
Laboratory investigations of the growth plate of diabetic animals mostly show growth arrest, reduction of growth plate height or abnormal proportion of growth plate zones [38, 68, 70, 71]. For example, STZ-induced T1DM rats showed growth arrest with a marked reduction of the growth plate height [70]. Consistent with T1DM, T2DM was found to produce similar manifestations. Histological study of femur and tibia of GK rats has revealed shorter femoral length with a reduction in total growth plate and HZ heights [38]. Furthermore, Aiemlapa et al. [68], by using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) technique, found that the shorter bone length in GK rats resulted from premature apoptosis of chondrocytes in HZ. Such a reduction in the number of mature chondrocytes induced compensatory overexpression of parathyroid hormone-related protein (PTHrP), vascular endothelial growth factor (VEGF) and Runx2, all of which induce chondrocytes to differentiate. Despite this body of evidence on T1DM and T2DM growth plate changes, more study is required to demonstrate the underlying cellular and molecular mechanisms that would help to understand the etiology of skeletal disorders in DM.
DM-related renal calcium wasting
Several lines of evidence have suggested that DM in both humans and rodents profoundly affects renal calcium handling, such as decreasing 1,25(OH)2D3 synthesis, as a consequence of prolonged hyperglycemia and dysregulation of calciotropic hormone metabolism [72–75]. An early study of glucose overload in rats showed that both oral and intravenous glucose administration induced hypercalciuria independently of urine flow rate and sodium excretion [76]. Tubular calcium reabsorption in STZ-induced diabetic rats was markedly reduced, which could be reversed by insulin administration [73]. GDM is also associated with severe hypercalciuria in rats [77]. Both urinary calcium and phosphate excretion were increased in female BioBreeding (BB) rats—an animal model of T1DM—and their neonates manifested fewer ossification centers and lower bone mineral content than neonates from healthy dams [78]. Hypercalciuria and hyperphosphaturia were also evident in T1DM children aged between 6 and 12 years [79]. However, the underlying cellular and molecular mechanisms are not well understood. Lee and co-workers [80] reported a paradoxical upregulation of transcellular calcium-transporting protein expression (e.g., TRPV5 and calbindin-D28k), but not the paracellular protein expression (i.e., claudin-16, normally required for calcium and magnesium reabsorption in the thick ascending limb of the loop of Henle) in the renal tubules of STZ-induced diabetic rats, suggesting that this is a compensatory response to long-term renal calcium wasting. Thus, calciuria in T1DM might have resulted from the DM-induced impairment of renal calcium transporter activities rather than their protein expression.
A decrease in insulin production and release in T1DM or insulin resistance in T2DM is believed to contribute to a reduction in renal calcium (and magnesium) reabsorption. A study in isolated perfused mouse thick ascending limb showed that insulin significantly increased the transepithelial potential difference—a driving force for passive calcium reabsorption—as well as the transepithelial calcium and magnesium transport [81]. Therefore, this driving force for calcium reabsorption is probably reduced under diabetic condition, thereby culminating in increased calcium excretion. On the other hand, amino acids that can induce insulin secretion are able to induce tubular calcium reabsorption [82]. Nevertheless, in parathyroidectomized female rats subjected to intravenous arginine infusion, the arginine-induced insulin secretion was associated with urinary calcium excretion [82]. This discrepancy can be explained by an increase in glomerular filtration rate secondary to amino acid loading, which leads to renal calcium excretion. Indeed, the stimuli or pattern of insulin secretion may also determine the final outcome of renal calcium handling.
In addition, renal calcium loss in DM can result from dysregulation of the calciotropic hormones, such as PTH and 1,25(OH)2D3. It has been reported that the PTH-induced stimulation of 1,25(OH)2D3 production was impaired in diabetic condition, whereas insulin treatment successfully rescued 1,25(OH)2D3 production [83]. Another evidence comes from the study of hyperinsulinemic ZDF rats (a T2DM model), in which a reduction in the renal expression of megalin and disabled (Dab)-2 proteins can cause an impaired receptor-mediated endocytosis of vitamin D-binding protein-bound 25(OH)D3, leading to urinary loss of 25(OH)D3 and a decrease in circulating 25(OH)D3 level [84]. It is of interest that the bone-derived phosphaturic hormone FGF-23 probably contributes to changes in renal calcium handling during diabetic condition. FGF-23 has recently been demonstrated to be a negative regulator of intestinal calcium absorption [19–21]. Meanwhile, this hormone is also postulated to be a counter-regulatory factor that alleviates vitamin D toxicity and hypercalcemia by directly suppressing PTH release and renal 1,25(OH)2D3 production [85]. Moreover, the TRPV5-mediated renal calcium reabsorption is dependent on FGF-23 and its co-receptor (soluble α-Klotho) [86, 87]. The release of soluble Klotho is insulin-dependent [88]. Thus, it is tempting to speculate that the FGF-23-regulated renal calcium reabsorption, if present, is probably impaired under diabetic condition. Fajol et al. [89] recently reported that transgenic mice expressing Akt/PKB/SGK-resistant glycogen synthase kinase (GSK)-3 is characterized by hypophosphatemia, phosphaturia, and calciuria with increases in serum FGF-23 levels. Since Akt, PKB, and SGK are known to mediate intracellular insulin signaling, this study suggested an interplay between insulin and FGF-23 in the regulation of renal calcium handling.
Furthermore, FGF-23 also has direct effects on osteoblasts by enhancing the expression of osteoblast differentiation markers, e.g., Runx2, osterix, alkaline phosphatase and osteopontin, in a dose-dependent manner [90]. They also express receptors for FGF-23 [90]. Therefore, abnormally high circulating FGF-23 in DM-associated chronic kidney disease may contribute to deterioration of the coupling process of bone remodeling and bone quality in T2DM [91].
Perspectives and conclusions
It is clear that DM, hyperglycemia and associated complications (e.g., AGEs production and microvascular damage) interfere with normal functions of the three calcium-regulating organs, namely the intestine, bone, and kidney. Moreover, they also lead to dysregulation of calciotropic hormone actions, thereby worsening the already impaired function of the calcium-regulating organs. As demonstrated in the small intestine and cultured intestinal cells, DM as well as elevated glucose concentration and AGEs diminish calcium absorption, presumably by downregulating nuclear vitamin D receptor expression and inducing oxidative stress [29, 30, 43, 45, 46]. However, how DM alters the expression of genes and proteins related to calcium transport in the small intestine, and whether calcium absorption in the large intestine is impaired, are not known. Moreover, since the proximal part of the large intestine, particularly the cecum, has the highest rate of calcium uptake compared to other intestinal segments, and since surgical removal of the cecum can cause osteopenia in rats [92–95], it is hypothesized that the DM-induced impairment of calcium transport across the large intestine would substantially contribute to impaired calcium metabolism and diabetic osteopathy.
In bone, DM induces both cellular and matrix failure in association with low BMD, especially in T1DM. Specifically, DM and hyperglycemia lead to uncoupling of bone remodeling, i.e., suppression of osteoblast function and enhancement of osteoclastogenesis and osteoclast function. These osteoblasts suffer from oxidative stress, downregulation of key protein expression (e.g., Runx2, type I collagen, and osteocalcin) and apoptosis, whereas osteoclasts become more active in resorbing bone. DM is also associated with a reduction in osteocyte density, while enhancing adipocyte differentiation. Meanwhile, hyperglycemia and overproduction of AGEs in both T1DM and T2DM impair collagen synthesis, produce abnormal collagen alignment and fibril size, and increase glycated collagen, thereby resulting in matrix failure and poor bone mechanical properties. Nevertheless, how other DM complications, such as microangiopathy in bone and dyslipidemia, affect bone remodeling are inconclusive. Furthermore, since the autonomic nervous system, especially the sympathetic activity through β-adrenergic receptors, has been reported to modulate bone remodeling [96–98], it is tempting to speculate that diabetic neuropathy and the resultant dysautonomia probably contribute to bone loss in DM patients.
Although the deleterious effects of DM on bone and calcium metabolism in adult and aging individuals have been widely investigated, the effects in young growing individuals, pregnant and lactating mothers, fetuses and neonates born to mothers with GDM have received much less attention. Available data suggest that DM in the young impairs longitudinal bone growth by inducing premature hypertrophic chondrocyte apoptosis, and early vascular invasion into the epiphyseal chondro-osseous junction [38, 68]. However, the circulating profiles of growth factors, which are important for the epiphyseal function and elongation of long bones, remain unknown. In addition, how DM alters local production of chondroregulatory factors, such as IGF-1, PTHrP, and Indian hedgehog (Ihh), is also not understood, and requires a systematic investigation. Other challenging questions include placental and mammary calcium transfer under diabetic conditions, long-term impact of GDM in both mothers and neonates, and interplay between classical calciotropic hormones and non-classical mineral-regulating hormones, e.g., prolactin, FGF-23, and insulin itself. In addition, because certain antidiabetic agents, including thiazolidinediones and FGF-21, can aggravate osteopenia [99, 100], it is necessary to develop a practice guideline for antidiabetic drug use in patients with diabetic osteopathy.
Acknowledgments
Our research was supported by grants from the Cluster and Program Management Office (CPMO), National Science and Technology Development Agency (P-11-00639 to NK and P-13-00100 to NC), the Thailand Research Fund (TRF)–Mahidol University through the TRF Senior Research Scholar Grant (RTA5780001 to NC), the Faculty of Allied Health Sciences, Burapha University and TRF through TRF Research Scholar Award Grant (RSA5780041 to KW), the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (6/2559 to KW).
Compliance with ethical standards
Conflict of interest
The authors declare that there is no conflict of interest.
References
- 1.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37(Suppl 1):S81–S90. doi: 10.2337/dc14-S081. [DOI] [PubMed] [Google Scholar]
- 2.American Diabetes Association 2. Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl):S8–S16. doi: 10.2337/dc15-S005. [DOI] [PubMed] [Google Scholar]
- 3.Isfort M, Stevens SC, Schaffer S, Jong CJ, Wold LE. Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail Rev. 2014;19:35–48. doi: 10.1007/s10741-013-9377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deli G, Bosnyak E, Pusch G, Komoly S, Feher G. Diabetic neuropathies: diagnosis and management. Neuroendocrinology. 2013;98:267–280. doi: 10.1159/000358728. [DOI] [PubMed] [Google Scholar]
- 5.Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–2340. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mlinar B, Marc J, Janez A, Pfeifer M. Molecular mechanisms of insulin resistance and associated diseases. Clin Chim Acta. 2007;375:20–35. doi: 10.1016/j.cca.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Powers AC. Diabetes mellitus. In: Jameson JL, editor. Harrison’s endocrinology. 3. New York: McGraw-Hill; 2013. pp. 261–307. [Google Scholar]
- 8.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hienz SA, Paliwal S, Ivanovski S. Mechanisms of bone resorption in periodontitis. J Immunol Res. 2015;2015:615486. doi: 10.1155/2015/615486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rayanagoudar G, Hashi AA, Zamora J, Khan KS, Hitman GA, Thangaratinam S. Quantification of the type 2 diabetes risk in women with gestational diabetes: a systematic review and meta-analysis of 95,750 women. Diabetologia. 2016;59:1403–1411. doi: 10.1007/s00125-016-3927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Committee on Practice Bulletins-Obstetrics Practice bulletin no. 137: gestational diabetes mellitus. Obstet Gynecol. 2013;122:406–416. doi: 10.1097/01.AOG.0000433006.09219.f1. [DOI] [PubMed] [Google Scholar]
- 12.Kwak SH, Choi SH, Jung HS, Cho YM, Lim S, Cho NH, Kim SY, Park KS, Jang HC. Clinical and genetic risk factors for type 2 diabetes at early or late post partum after gestational diabetes mellitus. J Clin Endocrinol Metab. 2013;98:E744–E752. doi: 10.1210/jc.2012-3324. [DOI] [PubMed] [Google Scholar]
- 13.Kovacs CS. Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiol Rev. 2016;96:449–547. doi: 10.1152/physrev.00027.2015. [DOI] [PubMed] [Google Scholar]
- 14.Charoenphandhu N, Wongdee K, Krishnamra N. Is prolactin the cardinal calciotropic maternal hormone? Trends Endocrinol Metab. 2010;21:395–401. doi: 10.1016/j.tem.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 15.McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50:403–415. doi: 10.1177/0004563213483458. [DOI] [PubMed] [Google Scholar]
- 16.Anık A, Çatlı G, Abacı A, Böber E. Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 2015;28:251–263. doi: 10.1515/jpem-2014-0384. [DOI] [PubMed] [Google Scholar]
- 17.White BA. Hormonal regulation of calcium and phosphate metabolism. In: Koeppen BM, Stanton BA, editors. Berne & Levy physiology. 6 (updated edition) St Louis: Mosby; 2010. pp. 696–705. [Google Scholar]
- 18.Favus MJ, Goltzman D. Regulation of cacium and magnesium. In: Rosen CJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. 7. Washington, D.C.: American Society for Bone and Mineral Research; 2008. pp. 104–108. [Google Scholar]
- 19.Khuituan P, Teerapornpuntakit J, Wongdee K, Suntornsaratoon P, Konthapakdee N, Sangsaksri J, Sripong C, Krishnamra N, Charoenphandhu N. Fibroblast growth factor-23 abolishes 1,25-dihydroxyvitamin D3-enhanced duodenal calcium transport in male mice. Am J Physiol Endocrinol Metab. 2012;302:E903–E913. doi: 10.1152/ajpendo.00620.2011. [DOI] [PubMed] [Google Scholar]
- 20.Khuituan P, Wongdee K, Jantarajit W, Suntornsaratoon P, Krishnamra N, Charoenphandhu N. Fibroblast growth factor-23 negates 1,25(OH)2D3-induced intestinal calcium transport by reducing the transcellular and paracellular calcium fluxes. Arch Biochem Biophys. 2013;536:46–52. doi: 10.1016/j.abb.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Wongdee K, Teerapornpuntakit J, Sripong C, Longkunan A, Chankamngoen W, Keadsai C, Kraidith K, Krishnamra N, Charoenphandhu N. Intestinal mucosal changes and upregulated calcium transporter and FGF-23 expression during lactation: contribution of lactogenic hormone prolactin. Arch Biochem Biophys. 2016;590:109–117. doi: 10.1016/j.abb.2015.11.038. [DOI] [PubMed] [Google Scholar]
- 22.Heath H, 3rd, Lambert PW, Service FJ, Arnaud SB. Calcium homeostasis in diabetes mellitus. J Clin Endocrinol Metab. 1979;49:462–466. doi: 10.1210/jcem-49-3-462. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Zhang X, Bao M, Liu L, Xian Y, Wu J, Li P. Effect of serum 25-hydroxyvitamin D3 on insulin resistance and β-cell function in newly diagnosed type 2 diabetes patients. J Diabetes Investig. 2016;7:226–232. doi: 10.1111/jdi.12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiu KC, Chu A, Go VL, Saad MF. Hypovitaminosis D is associated with insulin resistance and β cell dysfunction. Am J Clin Nutr. 2004;79:820–825. doi: 10.1093/ajcn/79.5.820. [DOI] [PubMed] [Google Scholar]
- 25.Reis JP, Selvin E, Pankow JS, Michos ED, Rebholz CM, Lutsey PL. Parathyroid hormone is associated with incident diabetes in white, but not black adults: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Metab. 2016;42:162–169. doi: 10.1016/j.diabet.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rivoira M, Rodríguez V, López MP, Tolosa de Talamoni N. Time dependent changes in the intestinal Ca2+ absorption in rats with type I diabetes mellitus are associated with alterations in the intestinal redox state. Biochim Biophys Acta. 2015;1852:386–394. doi: 10.1016/j.bbadis.2014.11.018. [DOI] [PubMed] [Google Scholar]
- 27.Schneider LE, Schedl HP. Diabetes and intestinal calcium absorption in the rat. Am J Physiol. 1972;223:1319–1323. doi: 10.1152/ajplegacy.1972.223.6.1319. [DOI] [PubMed] [Google Scholar]
- 28.Schneider LE, Nowosielski LM, Schedl HP. Insulin-treatment of diabetic rats: effects on duodenal calcium absorption. Endocrinology. 1977;100:67–73. doi: 10.1210/endo-100-1-67. [DOI] [PubMed] [Google Scholar]
- 29.Seino Y, Sierra RI, Sonn YM, Jafari A, Birge SJ, Avioli LV. The duodenal 1α,25-dihydroxyvitamin D3 receptor in rats with experimentally induced diabetes. Endocrinology. 1983;113:1721–1725. doi: 10.1210/endo-113-5-1721. [DOI] [PubMed] [Google Scholar]
- 30.Nyomba BL, Verhaeghe J, Thomasset M, Lissens W, Bouillon R. Bone mineral homeostasis in spontaneously diabetic BB rats. I. Abnormal vitamin D metabolism and impaired active intestinal calcium absorption. Endocrinology. 1989;124:565–572. doi: 10.1210/endo-124-2-565. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto M. Insights into bone fragility in diabetes: the crucial role of bone quality on skeletal strength. Endocr J. 2015;62:299–308. doi: 10.1507/endocrj.EJ15-0129. [DOI] [PubMed] [Google Scholar]
- 32.Sealand R, Razavi C, Adler RA. Diabetes mellitus and osteoporosis. Curr Diab Rep. 2013;13:411–418. doi: 10.1007/s11892-013-0376-x. [DOI] [PubMed] [Google Scholar]
- 33.Hamilton EJ, Rakic V, Davis WA, Chubb SA, Kamber N, Prince RL, Davis TM. Prevalence and predictors of osteopenia and osteoporosis in adults with type 1 diabetes. Diabet Med. 2009;26:45–52. doi: 10.1111/j.1464-5491.2008.02608.x. [DOI] [PubMed] [Google Scholar]
- 34.Saha MT, Sievänen H, Salo MK, Tulokas S, Saha HH. Bone mass and structure in adolescents with type 1 diabetes compared to healthy peers. Osteoporos Int. 2009;20:1401–1406. doi: 10.1007/s00198-008-0810-0. [DOI] [PubMed] [Google Scholar]
- 35.Lumachi F, Camozzi V, Tombolan V, Luisetto G. Bone mineral density, osteocalcin, and bone-specific alkaline phosphatase in patients with insulin-dependent diabetes mellitus. Ann N Y Acad Sci. 2009;1173(Suppl 1):E64–E67. doi: 10.1111/j.1749-6632.2009.04955.x. [DOI] [PubMed] [Google Scholar]
- 36.Yaturu S, Humphrey S, Landry C, Jain SK. Decreased bone mineral density in men with metabolic syndrome alone and with type 2 diabetes. Med Sci Monit. 2009;15:CR5–CR9. [PubMed] [Google Scholar]
- 37.Yamaguchi T, Kanazawa I, Yamamoto M, Kurioka S, Yamauchi M, Yano S, Sugimoto T. Associations between components of the metabolic syndrome versus bone mineral density and vertebral fractures in patients with type 2 diabetes. Bone. 2009;45:174–179. doi: 10.1016/j.bone.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Lapmanee S, Charoenphandhu N, Aeimlapa R, Suntornsaratoon P, Wongdee K, Tiyasatkulkovit W, Kengkoom K, Chaimongkolnukul K, Seriwatanachai D, Krishnamra N. High dietary cholesterol masks type 2 diabetes-induced osteopenia and changes in bone microstructure in rats. Lipids. 2014;49:975–986. doi: 10.1007/s11745-014-3950-3. [DOI] [PubMed] [Google Scholar]
- 39.Petit MA, Paudel ML, Taylor BC, Hughes JM, Strotmeyer ES, Schwartz AV, Cauley JA, Zmuda JM, Hoffman AR, Ensrud KE, Osteoporotic Fractures in Men Study Group Bone mass and strength in older men with type 2 diabetes: the osteoporotic fractures in men study. J Bone Miner Res. 2010;25:285–291. doi: 10.1359/jbmr.090725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farr JN, Drake MT, Amin S, Melton LJ, 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J Bone Miner Res. 2014;29:787–795. doi: 10.1002/jbmr.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patsch JM, Burghardt AJ, Yap SP, Baum T, Schwartz AV, Joseph GB, Link TM. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. J Bone Miner Res. 2013;28:313–324. doi: 10.1002/jbmr.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.To WW, Wong MW. Bone mineral density changes in gestational diabetic pregnancies-a longitudinal study using quantitative ultrasound measurements of the os calcis. Gynecol Endocrinol. 2008;24:519–525. doi: 10.1080/09513590802288184. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Zhang X, Zheng J, Yang J. High glucose stimulates adipogenic and inhibits osteogenic differentiation in MG-63 cells through cAMP/protein kinase A/extracellular signal-regulated kinase pathway. Mol Cell Biochem. 2010;338:115–122. doi: 10.1007/s11010-009-0344-6. [DOI] [PubMed] [Google Scholar]
- 44.Villarino ME, Sánchez LM, Bozal CB, Ubios AM. Influence of short-term diabetes on osteocytic lacunae of alveolar bone. A histomorphometric study. Acta Odontol Latinoam. 2006;19:23–28. [PubMed] [Google Scholar]
- 45.Zhang Y, Yang JH. Activation of the PI3 K/Akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J Cell Biochem. 2013;114:2595–2602. doi: 10.1002/jcb.24607. [DOI] [PubMed] [Google Scholar]
- 46.Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-γ2 expression in type I diabetic mice. Endocrinology. 2005;146:3622–3631. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamann C, Goettsch C, Mettelsiefen J, Henkenjohann V, Rauner M, Hempel U, Bernhardt R, Fratzl-Zelman N, Roschger P, Rammelt S, Günther KP, Hofbauer LC. Delayed bone regeneration and low bone mass in a rat model of insulin-resistant type 2 diabetes mellitus is due to impaired osteoblast function. Am J Physiol Endocrinol Metab. 2011;301:E1220–E1228. doi: 10.1152/ajpendo.00378.2011. [DOI] [PubMed] [Google Scholar]
- 48.Kayal RA, Tsatsas D, Bauer MA, Allen B, Al-Sebaei MO, Kakar S, Leone CW, Morgan EF, Gerstenfeld LC, Einhorn TA, Graves DT. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res. 2007;22:560–568. doi: 10.1359/jbmr.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burghardt AJ, Issever AS, Schwartz AV, Davis KA, Masharani U, Majumdar S, Link TM. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:5045–5055. doi: 10.1210/jc.2010-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aoki C, Uto K, Honda K, Kato Y, Oda H. Advanced glycation end products suppress lysyl oxidase and induce bone collagen degradation in a rat model of renal osteodystrophy. Lab Invest. 2013;93:1170–1183. doi: 10.1038/labinvest.2013.105. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg E, Maymon T, Weinreb M. AGEs induce caspase-mediated apoptosis of rat BMSCs via TNFα production and oxidative stress. J Mol Endocrinol. 2014;52:67–76. doi: 10.1530/JME-13-0229. [DOI] [PubMed] [Google Scholar]
- 52.Kang L, Chen Q, Wang L, Gao L, Meng K, Chen J, Ferro A, Xu B. Decreased mobilization of endothelial progenitor cells contributes to impaired neovascularization in diabetes. Clin Exp Pharmacol Physiol. 2009;36:e47–e56. doi: 10.1111/j.1440-1681.2009.05219.x. [DOI] [PubMed] [Google Scholar]
- 53.Hammond MA, Gallant MA, Burr DB, Wallace JM. Nanoscale changes in collagen are reflected in physical and mechanical properties of bone at the microscale in diabetic rats. Bone. 2014;60:26–32. doi: 10.1016/j.bone.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kierszenbaum AL, Tres LL. Connective tissue. In: Kierszenbaum AL, Tres LL, editors. Histology and cell biology: an introduction to pathology. 3. Philadelphia: Elsevier; 2012. pp. 111–149. [Google Scholar]
- 55.Feng YF, Wang L, Zhang Y, Li X, Ma ZS, Zou JW, Lei W, Zhang ZY. Effect of reactive oxygen species overproduction on osteogenesis of porous titanium implant in the present of diabetes mellitus. Biomaterials. 2013;34:2234–2243. doi: 10.1016/j.biomaterials.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 56.McCarthy AD, Molinuevo MS, Cortizo AM. Ages and bone ageing in diabetes mellitus. J Diabetes Metab. 2013;4:276. [Google Scholar]
- 57.Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21:195–214. doi: 10.1007/s00198-009-1066-z. [DOI] [PubMed] [Google Scholar]
- 58.Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, Wang X. Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res. 2009;24:1618–1627. doi: 10.1359/jbmr.090316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samsa WE, Zhou X, Zhou G. Signaling pathways regulating cartilage growth plate formation and activity. Semin Cell Dev Biol. 2016 doi: 10.1016/j.semcdb.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu S, Aguilar AL, Ostrow V, De Luca F. Insulin resistance secondary to a high-fat diet stimulates longitudinal bone growth and growth plate chondrogenesis in mice. Endocrinology. 2011;152:468–475. doi: 10.1210/en.2010-0803. [DOI] [PubMed] [Google Scholar]
- 61.Yakar S, Isaksson O. Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: lessons from mouse models. Growth Horm IGF Res. 2016;28:26–42. doi: 10.1016/j.ghir.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol. 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 63.Yao Y, Zhai Z, Wang Y. Evaluation of insulin medium or chondrogenic medium on proliferation and chondrogenesis of ATDC5 cells. Biomed Res Int. 2014;2014:569241. doi: 10.1155/2014/569241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lebl J, Schober E, Zidek T, Baldis S, Rami B, Pruhova S, Kolouskova S, Snajderova M, Frisch H. Growth data in large series of 587 children and adolescents with type 1 diabetes mellitus. Endocr Regul. 2003;37:153–161. [PubMed] [Google Scholar]
- 65.Armas LA, Akhter MP, Drincic A, Recker RR. trabe-cular bone histomorphometry in humans with type 1 diabetes mellitus. Bone. 2012;50:91–96. doi: 10.1016/j.bone.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muñoz MT, Barrios V, Pozo J, Argente J. Insulin-like growth factor I, its binding proteins 1 and 3, and growth hormone-binding protein in children and adolescents with insulin-dependent diabetes mellitus: clinical implications. Pediatr Res. 1996;39:992–998. doi: 10.1203/00006450-199606000-00011. [DOI] [PubMed] [Google Scholar]
- 67.Edge JA, Dunger DB, Matthews DR, Gilbert JP, Smith CP. Increased overnight growth hormone concentrations in diabetic compared with normal adolescents. J Clin Endocrinol Metab. 1990;71:1356–1362. doi: 10.1210/jcem-71-5-1356. [DOI] [PubMed] [Google Scholar]
- 68.Aeimlapa R, Wongdee K, Charoenphandhu N, Suntornsaratoon P, Krishnamra N. Premature chondrocyte apoptosis and compensatory upregulation of chondroregulatory protein expression in the growth plate of Goto-Kakizaki diabetic rats. Biochem Biophys Res Commun. 2014;452:395–401. doi: 10.1016/j.bbrc.2014.08.085. [DOI] [PubMed] [Google Scholar]
- 69.Wongdee K, Krishnamra N, Charoenphandhu N. Endochondral bone growth, bone calcium accretion, and bone mineral density: how are they related? J Physiol Sci. 2012;62:299–307. doi: 10.1007/s12576-012-0212-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hough S, Avioli LV, Bergfeld MA, Fallon MD, Slatopolsky E, Teitelbaum SL. Correction of abnormal bone and mineral metabolism in chronic streptozotocin-induced diabetes mellitus in the rat by insulin therapy. Endocrinology. 1981;108:2228–2234. doi: 10.1210/endo-108-6-2228. [DOI] [PubMed] [Google Scholar]
- 71.Bain S, Ramamurthy NS, Impeduglia T, Scolman S, Golub LM, Rubin C. Tetracycline prevents cancellous bone loss and maintains near-normal rates of bone formation in streptozotocin diabetic rats. Bone. 1997;21:147–153. doi: 10.1016/S8756-3282(97)00104-X. [DOI] [PubMed] [Google Scholar]
- 72.Raskin P, Stevenson MR, Barilla DE, Pak CY. The hypercalciuria of diabetes mellitus: its amelioration with insulin. Clin Endocrinol (Oxf) 1978;9:329–335. doi: 10.1111/j.1365-2265.1978.tb02218.x. [DOI] [PubMed] [Google Scholar]
- 73.Anwana AB, Garland HO. Renal calcium and magnesium handling in experimental diabetes mellitus in the rat. Acta Endocrinol (Copenh) 1990;122:479–486. doi: 10.1530/acta.0.1220479. [DOI] [PubMed] [Google Scholar]
- 74.Garland HO, Forshaw AG, Sibley CP. Dietary essential fatty acid supplementation, urinary calcium excretion and reproductive performance in the diabetic pregnant rat. J Endocrinol. 1997;153:357–363. doi: 10.1677/joe.0.1530357. [DOI] [PubMed] [Google Scholar]
- 75.Huang CQ, Ma GZ, Tao MD, Ma XL, Liu QX, Feng J. The relationship among renal injury, changed activity of renal 1-α hydroxylase and bone loss in elderly rats with insulin resistance or type 2 diabetes mellitus. J Endocrinol Invest. 2009;32:196–201. doi: 10.1007/BF03346452. [DOI] [PubMed] [Google Scholar]
- 76.Singh HJ, Garland HO. A comparison of the effects of oral and intravenous glucose administration on renal calcium excretion in the rat. Q J Exp Physiol. 1989;74:531–540. doi: 10.1113/expphysiol.1989.sp003300. [DOI] [PubMed] [Google Scholar]
- 77.Birdsey TJ, Husain SM, Garland HO, Sibley CP. The effect of diabetes mellitus on urinary calcium excretion in pregnant rats and their offspring. J Endocrinol. 1995;145:11–18. doi: 10.1677/joe.0.1450011. [DOI] [PubMed] [Google Scholar]
- 78.Verhaeghe J, Bouillon R, Nyomba BL, Lissens W, Van Assche FA. Vitamin D and bone mineral homeostasis during pregnancy in the diabetic BB rat. Endocrinology. 1986;118:1019–1025. doi: 10.1210/endo-118-3-1019. [DOI] [PubMed] [Google Scholar]
- 79.Kashef S, Karamizadeh Z. Hypercalciuria, hyperphosphaturia and growth retardation in children with diabetes mellitus. Iran J Med Sci. 2002;27:11–14. [Google Scholar]
- 80.Lee CT, Lien YH, Lai LW, Chen JB, Lin CR, Chen HC. Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney Int. 2006;69:1786–1791. doi: 10.1038/sj.ki.5000344. [DOI] [PubMed] [Google Scholar]
- 81.Mandon B, Siga E, Chabardes D, Firsov D, Roinel N, De Rouffignac C. Insulin stimulates Na+, Cl−, Ca2+, and Mg2+ transports in TAL of mouse nephron: cross-potentiation with AVP. Am J Physiol. 1993;265:F361–F369. doi: 10.1152/ajprenal.1993.265.3.F361. [DOI] [PubMed] [Google Scholar]
- 82.Gollaher CJ, Wood RJ, Holl M, Allen LH. A comparison of amino acid-induced hypercalciuria in sham-operated and parathyroidectomized rats. J Nutr. 1984;114:622–626. doi: 10.1093/jn/114.3.622. [DOI] [PubMed] [Google Scholar]
- 83.Wongsurawat N, Armbrecht HJ. Insulin modulates the stimulation of renal 1,25-dihydroxyvitamin D3 production by parathyroid hormone. Acta Endocrinol (Copenh) 1985;109:243–248. doi: 10.1530/acta.0.1090243. [DOI] [PubMed] [Google Scholar]
- 84.Anderson RL, Ternes SB, Strand KA, Rowling MJ. Vitamin D homeostasis is compromised due to increased urinary excretion of the 25-hydroxycholecalciferol-vitamin D-binding protein complex in the Zucker diabetic fatty rat. Am J Physiol Endocrinol Metab. 2010;299:E959–E967. doi: 10.1152/ajpendo.00218.2010. [DOI] [PubMed] [Google Scholar]
- 85.David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium regulates FGF-23 expression in bone. Endocrinology. 2013;154:4469–4482. doi: 10.1210/en.2013-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, Shalhoub V, Mohammadi M, Pohl EE, Lanske B, Erben RG. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014;33:229–246. doi: 10.1002/embj.201284188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wolf MT, An SW, Nie M, Bal MS, Huang CL. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms. J Biol Chem. 2014;289:35849–35857. doi: 10.1074/jbc.M114.616649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fajol A, Chen H, Umbach AT, Quarles LD, Lang F, Föller M. Enhanced FGF23 production in mice expressing PI3K-insensitive GSK3 is normalized by β-blocker treatment. FASEB J. 2016;30:994–1001. doi: 10.1096/fj.15-279943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teerapornpuntakit J, Wongdee K, Krishnamra N, Charoenphandhu N. Expression of osteoclastogenic factor transcripts in osteoblast-like UMR-106 cells after exposure to FGF-23 or FGF-23 combined with parathyroid hormone. Cell Biol Int. 2016;40:329–340. doi: 10.1002/cbin.10573. [DOI] [PubMed] [Google Scholar]
- 91.Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82:737–747. doi: 10.1038/ki.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nellans HN, Goldsmith RS. Transepithelial calcium transport by rat cecum: high-efficiency absorptive site. Am J Physiol. 1981;240:G424–G431. doi: 10.1152/ajpgi.1981.240.6.G424. [DOI] [PubMed] [Google Scholar]
- 93.Karbach U, Feldmeier H. The cecum is the site with the highest calcium absorption in rat intestine. Dig Dis Sci. 1993;38:1815–1824. doi: 10.1007/BF01296104. [DOI] [PubMed] [Google Scholar]
- 94.Charoenphandhu N, Suntornsaratoon P, Jongwattanapisan P, Wongdee K, Krishnamra N. Enhanced trabecular bone resorption and microstructural bone changes in rats after removal of the cecum. Am J Physiol Endocrinol Metab. 2012;303:E1069–E1075. doi: 10.1152/ajpendo.00242.2012. [DOI] [PubMed] [Google Scholar]
- 95.Jongwattanapisan P, Suntornsaratoon P, Wongdee K, Dorkkam N, Krishnamra N, Charoenphandhu N. Impaired body calcium metabolism with low bone density and compensatory colonic calcium absorption in cecectomized rats. Am J Physiol Endocrinol Metab. 2012;302:E852–E863. doi: 10.1152/ajpendo.00503.2011. [DOI] [PubMed] [Google Scholar]
- 96.Yirmiya R, Goshen I, Bajayo A, Kreisel T, Feldman S, Tam J, Trembovler V, Csernus V, Shohami E, Bab I. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci USA. 2006;103:16876–16881. doi: 10.1073/pnas.0604234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang HH, Brennan TC, Muir MM, Mason RS. Functional α1- and β2-adrenergic receptors in human osteoblasts. J Cell Physiol. 2009;220:267–275. doi: 10.1002/jcp.21761. [DOI] [PubMed] [Google Scholar]
- 98.Nuntapornsak A, Wongdee K, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Changes in the mRNA expression of osteoblast-related genes in response to β3-adrenergic agonist in UMR106 cells. Cell Biochem Funct. 2010;28:45–51. doi: 10.1002/cbf.1617. [DOI] [PubMed] [Google Scholar]
- 99.Charoenphandhu N, Suntornsaratoon P, Krishnamra N, Sa-Nguanmoo P, Tanajak P, Wang X, Liang G, Li X, Jiang C, Chattipakorn N, Chattipakorn S. Fibroblast growth factor-21 restores insulin sensitivity but induces aberrant bone microstructure in obese insulin-resistant rats. J Bone Miner Metab. 2016 doi: 10.1007/s00774-016-0745-z. [DOI] [PubMed] [Google Scholar]
- 100.Palermo A, D’Onofrio L, Eastell R, Schwartz AV, Pozzilli P, Napoli N. Oral anti-diabetic drugs and fracture risk, cut to the bone: safe or dangerous? A narrative review. Osteoporos Int. 2015;26:2073–2089. doi: 10.1007/s00198-015-3123-0. [DOI] [PubMed] [Google Scholar]
- 101.Tsentidis C, Gourgiotis D, Kossiva L, Doulgeraki A, Marmarinos A, Galli-Tsinopoulou A, Karavanaki K. Higher levels of s-RANKL and osteoprotegerin in children and adolescents with type 1 diabetes mellitus may indicate increased osteoclast signaling and predisposition to lower bone mass: a multivariate cross-sectional analysis. Osteoporos Int. 2016;27:1631–1643. doi: 10.1007/s00198-015-3422-5. [DOI] [PubMed] [Google Scholar]
- 102.Won HY, Lee JA, Park ZS, Song JS, Kim HY, Jang SM, Yoo SE, Rhee Y, Hwang ES, Bae MA. Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in TallyHo/JngJ mice. PLoS ONE. 2011;6:e18168. doi: 10.1371/journal.pone.0018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Motyl KJ, Botolin S, Irwin R, Appledorn DM, Kadakia T, Amalfitano A, Schwartz RC, McCabe LR. Bone inflammation and altered gene expression with type I diabetes early onset. J Cell Physiol. 2009;218:575–583. doi: 10.1002/jcp.21626. [DOI] [PubMed] [Google Scholar]