

Abstract
Atypically-shaped cardiomyocytes (ACMs) are a new subpopulation of spontaneously beating heart cells with a peculiar morphology identified within a culture of cardiac myocyte-depleted fraction (CMDF) cells obtained from adult mouse heart. ACMs originate from small cells in CMDF and grow in size and start beating within ~3 days culture without appreciable proliferation or express stem cell marker proteins, but stay in the heart until elderly stages. However, the characteristics of ACMs are largely unclear. The present study examined whether pre-exposure of CMDF cells to severe ischemia abolished the ability of ACMs to develop into beating cells. Of ACMs that underwent ischemia, ~50 % grew in size, changed the morphology, and started beating during the subsequent culture under normoxia. ACMs displayed constitutively active autophagy during the culture. The results suggest the possibility that the development of beating ACMs could occur in injured heart, even if the surviving cell population is small.
Keywords: Atypically-shaped cardiomyocytes, Beating, Spontaneous Ca2+ transients, Ischemia, Autophagy
Introduction
The adult mammalian heart comprises heterogeneous cells, such as cardiomyocytes, fibroblasts, endothelial cells, and vascular smooth muscle cells. In addition, the heart contains several cardiac stem or progenitor cell populations with the ability of long-term self-renewal and differentiation into cardiomyocytes, characterized based on the expression of stem cell marker proteins [1]. These cells proliferate in culture and can be converted into functional cardiomyocytes not only by treating with chemicals [21, 27, 33] but also by in vivo transplantation into injured heart by intravenous [27, 32] or intramyocardial [18] injections. More recently, transplantation of cardiac progenitor cell sheet is demonstrated to improve the delivery of these cells into injured myocardium [44]. Therefore, it is now generally accepted that the heart is not a rigidly terminally differentiated organ, but instead contains heterogeneous cell lineages of cardiac stem cell-like cells, thus ensuring the ability to achieve the lifelong renewal of cardiomyocytes [15].
There is a novel population of heart cells in adult mouse cardiac ventricles that spontaneously develop into beating cardiomyocytes with a peculiar morphology, defined as atypically-shaped cardiomyocytes (ACMs) [28, 30]. Beating ACMs were identified in cultures of cardiac myocyte-depleted fraction (CMDF) cells from ~3 days after plating. ACMs do not appreciably proliferate or express stem cell surface marker proteins, such as stem cells antigen 1 (Sca-1), stem cell factor receptor (c-kit), CD45 (leukocyte common antigen), CD31 (platelet-endothelial adhesion molecule), CD34 (muscle stem cell marker), and Flk-1 (vascular endothelial cell growth factor receptor 2) [28]. ACMs survive the long-term post-natal development of cardiac ventricles while preserving the expression of the fetal cardiac gene products, such as atrial natriuretic peptide (ANP) and α1H voltage-gated T-type Ca2+ channel CaV3.2 [28]. However, many of the characteristics of ACMs remain unclear. The aim of the present study was to examine whether pre-exposure of the isolated CMDF cells to the severe ischemia abolish the ability of ACMs to develop into beating cells. The results indicate that approximately half of the ACMs that undergo ischemia grow in size, change the morphology, and start beating during the subsequent culture under normoxia. We also demonstrate that the ultrastructure of ACMs appears to be quite different from that of ventricular myocytes, showing the constitutively active autophagy during the culture.
Materials and methods
Animals
Adult male C57BL/6 J mice were purchased from Charles River Japan. All animal experiments were performed in accordance with the guidelines of the institution’s Animal Care and Use Committee (No. 2010-12-6).
Solutions
The cell isolation buffer contained (in mM): 130 NaCl, 5.4 KCl, 0.5 MgCl2, 0.33 NaH2PO4, 22 glucose, 50 µU/mL bovine insulin, and 25 Hepes (pH adjusted to 7.4 with NaOH). Tyrode solution contained (in mM) 140 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.33 NaH2PO4, 5.5 glucose, and 5.0 Hepes (pH adjusted to 7.4 with NaOH). The cell suspension buffer contained Tyrode solution supplemented with 0.2 mg/mL bovine serum albumin (BSA), 100 µg/mL penicillin, 0.1 mg/mL streptomycin, and 15 µg/mL phenol red.
Preparation and culture of CMDF cells
The animals (8–16 weeks of age, body weight of 20–30 g) were killed by the i.p. injection of sodium pentobarbital overdose (>300 mg/kg) with heparin (8,000 U/kg). CMDF cells were isolated according to the method previously described [28]. Briefly, the heart was quickly excised, placed in an ice-chilled Tyrode solution, cannulated via the ascending aorta, and retrogradely perfused with an enzyme solution containing collagenase, trypsin, and protease in cell isolation buffer at 37 °C [38]. Both ventricles were then cut out at the level of 2/3 from the ventricular apex. These atrial tissue-free ventricular tissues were further digested with enzymes with intermittent pipetting, and washed by centrifugations. The ventricular myocyte fraction was precipitated at 14g (300 rpm) for 3 min and the supernatant was filtered through a 40-µm mesh filter and further centrifuged at 150g (1,000 rpm) for 5 min. The pelleted cells of the final centrifugation were obtained as CMDF cells. The CMDF cells resuspended in the cell suspension buffer were added into the semisolid culture medium at a dilution of 1:10 and vortexed for 2 s to disperse the cells. The semisolid culture medium contained an 80:20 mixture of methylcellulose-based medium MethoCult® M3231 (StemCell Technologies) and Iscove’s-modified Dulbecco’s medium (IMDM) in the absence of antibiotics. The total composition of the medium was 1 % methylcellulose, 30 % fetal bovine serum (FBS), 1 % BSA, 2 mM l-glutamine and 0.1 mM 2-mercaptoethanol in IMDM. CMDF cells were then plated in ∅35-mm plastic culture dishes (Falcon-BD) or high quality plastic dishes µ-Dishes (ibidi) with a cell density of ~1,700 cells/∅35-mm dish and maintained at 37 °C in a humidified atmosphere of 95 % air and 5 % CO2.
Confocal [Ca2+]i measurement
The cells cultured on the glass cover slips pre-coated with gelatin/fibronectin [4] were loaded with 5 µM of membrane-permeant Ca2+ indicator fluo 4-AM (Dojin Chemicals) in the cell suspension buffer in the dark for 30 min at 37 °C and washed twice. Fluo 4-loaded cells were then incubated in dye-free solution for 30 min at 37 °C and were transferred to the recording chamber mounted on the stage of an Axiovert 200M inverted microscope (Zeiss) with a ×20 objective equipped with a confocal laser scanning system LSM 510 META (Zeiss). The recording chamber was maintained at 36 °C and was perfused continuously at a rate of 1.5 mL/min with Tyrode or test solution.
Fluo 4-loaded cells were excited with the 488-nm line of a 30-mW argon laser, and the emitted light was filtered with a 505- to 530-nm band-pass filter before detection. The pinhole size was set to 1 Airy unit, resulting in an optical slice thickness of <4.1 µm. The pixel width was between 0.03 and 0.05 µm. Line-scan images were acquired by repeatedly scanning the laser beam at 10-ms intervals along with a single line, averaging four pixels in each scan line. Sequentially, the scanned lines were stacked over time and analyzed as 2.5D line-scan images using the included software package. The intensity values of the line-scan image (F) were normalized to the average fluorescence intensity at rest (F0).
Simulated lethal ischemia
A simulated lethal ischemia assay was performed according to the previously described method [7]. The CMDF cells obtained from the 2/3 size of the cardiac ventricles were suspended in 1.5 mL of cell suspension buffer. A 500-µL aliquot of the CMDF cell suspension was centrifuged at 150g (1,000 rpm) for 5 min and the supernatant was removed except for a volume equivalent to the pellet thickness. The cell pellet and supernatant were covered by ~5-mm-thick mineral oil layer and incubated for 90 min at 37 °C without agitation. The cells were then resuspended in the fresh solution, mixed with the semisolid culture medium in a 1:10 proportion and cultured for 6 days. The spontaneously beating ACMs identified in the culture were counted under a microscope.
Microscopy
Phase contrast images of ACMs were acquired using an inverted microscope DIAPHOT 300 (Nikon) and a charged-coupled device camera RETIGA 2000R (QImaging), and then analyzed using the Image Pro Plus software program (MediaCybernetics). The sample size of each acquired image was 1,600 × 1,200 pixels (588 × 442 µm with a ×20 objective).
Transmission electron microscopy (TEM)
ACMs cultured on µ-Dishes were fixed with 2.5 % glutaraldehyde in PBS for 30 min at 4 °C and washed twice with PBS for 3 min, and then secondarily fixed with 2 % osmium tetroxide for 30 min at 4 °C. The fixed cells were washed twice with PBS for 3 min at room temperature. The cells were then dehydrated by immersion in a series of ethanol dilutions, exchanged by immersion in a series of propylene oxide, and flat embedded on siliconized glass slides in epoxy resin (EPON812 resin embedding kit, TAAB). The resin polymerized for 6 h at 60 °C. Areas of interest were cut off and 50-nm ultra-thin sections were cut with an Ultracut E ultramicrotome (Reichert-Jung). The ultra-thin sections were mounted on 200-mesh nickel thin bar grids (Gilder Grid). The grids were stained with 2 % uranyl acetate and lead staining solution (Sigma-Aldrich), and then observed with an H-7500 electron microscope (Hitachi).
Scanning electron microscopy (SEM)
ACMs cultured on µ-Dishes were firstly fixed with 2.5 % glutaraldehyde in PBS for 30 min at 4 °C and washed twice with PBS for 3 min. The cells were secondarily fixed with 1 % osmium tetroxide in PBS for 30 min at 4 °C and washed twice with PBS for 3 min. The cells were then dehydrated by immersion in a series of ethanol dilutions, dried, and covered with 20 nm of gold. The final samples were observed with a field emission scanning electron microscope JSM-7505FA (JEOL Ltd).
Immunostaining
Cells cultured on µ-Dishes were fixed and immunostained with antibodies. The dilutions of antibodies used were: mouse monoclonal anti α-actinin (1:400, A7811, clone EA-53, Sigma) and rabbit polyclonal anti-microtubule-associated protein 1 light chain 3 (anti-LC3, 1:500, Medical and Biological Laboratories) and anti-Atg12 (1:100, Cell Signaling Technology) antibodies. Nuclei were stained with 4′-6-diamino-2-phenylindole (DAPI). Fluorescent signals and differential interference contrast (DIC) images were analyzed using a confocal laser scanning system C1si (Nikon) on an Eclipse TE2000-E inverted microscope (Nikon). Each image acquired by C1si was 1,024 × 1,024 pixels (636.5 × 636.5 µm with a ×20 objective) in size.
The anti-LC3 antibody used for the present study react with both cytosolic LC3-I and autophagosome-associated membrane-bound LC3-II. Therefore, dotted fluorescent signals (intensity ≥55 of 255 gradation, I m) were counted for autophagosome-associated membrane-bound LC3, and uniformly distributed fluorescent signals (intensity <55 of 255 gradation, I c) were estimated as cytosolic LC3 [29]. The area of dotted signals, I m (µm2), divided by the area of whole cell signals, I m + I c (µm2), calculated by computer using the Image-Pro Plus software program (MediaCybernetics) was estimated as the immunofluorescent signals for membrane-bound LC3.
Visualization of lysosomes
Lysosomes in living ACMs were visualized by fluorescent acidotropic probes for labeling and tracking acidic organelles in live cells LysoTracker®Red (Molecular Probes-Invitrogen). ACMs cultured on µ-Dishes were incubated with 50 nM LysoTracker for 20 min at 37 °C and analyzed using a confocal laser scanning system C1si (excitation/emission, 577/590 nm).
Statistical analysis and image data
All data are represented as the means ± SEM and n is the number of different cell preparations. Student’s t test or one-way ANOVA followed by Tukey’s test were used where applicable. Values of p < 0.05 were considered to be significant. Images are representative of 3 different cell preparations.
Results
Spontaneously beating ACMs identified in culture
The isolated CMDF cells contained heterogeneous small cells (Fig. 1a, 0 day). Some of the rounded CMDF cells that tightly adhered to the bottom of the dish changed the cell shape and started beating within ~3 days of culture, identified as ACMs (Fig. 1a, 3 days). These beating cells underwent a morphological maturation process over the period of several days, while growing in size and forming a peculiar morphology of highly branched and elongated shapes with many projections and/or bulges (Fig. 1a, 6 days). During the prolonged culture, the edges of the projections of ACMs sometimes adhered to the surface membrane of the neighboring proliferating cells and stretched rhythmically with those cells at a spontaneous beating rate (Fig. 1a, 9 days).
Fig. 1.
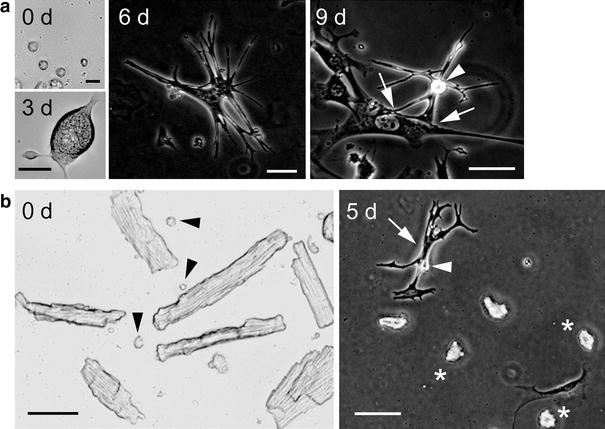
Spontaneously beating ACMs identified in culture. a Light microscopy of isolated CMDF cells (0 days; bar 15 µm) and beating ACM identified at 3 days culture (bar 25 µm), and phase contrast microscopy of beating ACMs cultured for 6 and 9 days (bar 50 µm). Arrowhead indicates surface bulge and arrows the edges of the projections of ACMs that adhered to the surface membrane of neighboring fibroblast-like cell. b Left panel the light microscopy of the ventricular myocyte fraction. Arrowheads indicate the small cells co-precipitated with the cardiomyocytes. Right panel the phase contrast image of ventricular myocyte fraction cultured in the same culture condition of CMDF cells for 5 days. Arrow, arrowhead, and asterisks indicate the beating ACM, surface bulge, and dead cardiomyocytes, respectively. Bar 50 µm
The isolated ventricular myocyte fraction contained heterogeneous small cells (∅ ~5–20 µm) co-precipitated with cardiomyocytes (Fig. 1b, 0 day, arrowheads). To examine the cell lineage of ACMs more precisely, we cultured the ventricular myocyte fraction in the same culture condition of CMDF cells. The small cells co-existing with cardiomyocytes slowly adhered to the bottom of the culture dish and changed the cells shapes depending on each cell type. Some of these small cells developed into beating ACMs at ~4 days after plating (Fig. 1b, 5 days, arrow), whereas most of the ventricular myocytes died during the culture (Fig. 1b, 5 days, asterisks).
Spontaneous Ca2+ transients recorded from beating ACMs
Cardiac contraction is regulated by rhythmic changes in the cytoplasmic Ca2+ concentration ([Ca2+]i), due to the cycling of Ca2+ between the cytoplasm and sarcoplasmic reticulum. The β-adrenergic stimulation of spontaneous action potentials and contractions is one of the characteristic responses in cardiac sino-atrial (SA) nodal cells [8]. We examined the effects of β-adrenergic agonist isoproterenol on the frequency of rhythmic spontaneous Ca2+ transients in fluorescent Ca2+ indicator fluo 4-loaded beating ACMs with confocal line-scan image analyses (Fig. 2a). The average peak intervals of spontaneous Ca2+ transients in the fluo 4-loaded ACMs perfused with normal Tyrode solution was 535 ± 56 ms (n = 5; Fig. 2b). This value is similar to the spontaneous cycle length of ACMs, 207–698 ms, obtained by the electrophysiological experiments as previously described [28]. The subsequent bath application of 10−6 M isoproterenol significantly reduced the peak intervals to 265 ± 40 ms (n = 5), a ~2-fold increase in the frequency of spontaneous Ca2+ transients (Fig. 2b). These results indicate that the ACMs respond to β-adrenergic stimulation on the spontaneous Ca2+ transients similar to that on the spontaneous electrical activities well known in cardiac SA nodal pacemaker cells [10].
Fig. 2.
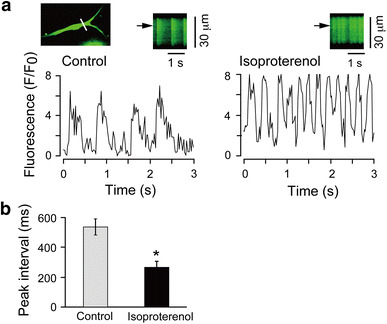
Spontaneous Ca2+ transients recorded from beating ACMs. a Changes in [Ca2+]i in the fluo 4-loaded beating ACMs recorded by confocal line-scan imaging. Cultured for 6 days. White line on the cell image indicates the scanned line (upper left). Representative traces show the fluorescent time course at the site marked by the arrows on the line-scan images (upper right of each trace). The cells were perfused with normal Tyrode solution (control) and then were subsequently exposed to 10−6 M isoproterenol. b Peak intervals of the spontaneous Ca2+ transients detected in ACMs before (control) and after exposure to isoproterenol (n = 5 cells, paired experiments). *p < 0.05
Culture of CMDF cells pre-exposed to ischemia
To examine whether pre-exposure of CMDF cells to severe ischemia abolishes the ability of ACMs to develop into beating cells during the subsequent culture under normoxia, we exposed the isolated CMDF cells to simulated lethal ischemic conditions prior to the plating. After 6 days of culture, the cell number of the beating ACMs observed in culture of 500 µL of the CMDF cell suspension with or without (control) ischemic pre-exposure was 92.1 ± 4.3 cells or 196.3 ± 20.3 cells, respectively (n = 8; Fig. 3a). These observations indicate that ~50 % of the ACMs that survived severe ischemia preserve the ability to develop into beating cells during the subsequent culture under normoxic conditions. There were no appreciable differences between the phase contrast images of the control and ischemia-surviving ACMs (Fig. 3a, right panels). We further confirmed the resistance of the ACMs to ischemia by exposing the isolated CMDF cell pellet to 100 % nitrogen gas in a humidified atmosphere for 90 min at 37 °C prior to the culture. The cell number of beating ACMs identified in 500 µL of CMDF cell suspension in normoxic conditions was 223 ± 20.8 cells and that from an equal volume of CMDF cell suspension exposed to nitrogen gas was 119.3 ± 17.4 cells (n = 3, data not shown). Therefore, the beating ACMs exposed to nitrogen gas represent ~50 % of those in the normoxic cell preparations. To examine the effects of pre-exposure to severe ischemic conditions on the spontaneous contractile activity, the rhythmic spontaneous Ca2+ transients were recorded from ACMs surviving ischemia (Fig. 3b). The time course of the line-scan imaging indicated that there were no statistical differences between the average values of the peak intervals of spontaneous Ca2+ transients obtained in ACMs of control, 542 ± 38 ms (n = 23), and ischemia-surviving, 485 ± 42 ms (n = 15), cell preparations (Fig. 3c).
Fig. 3.
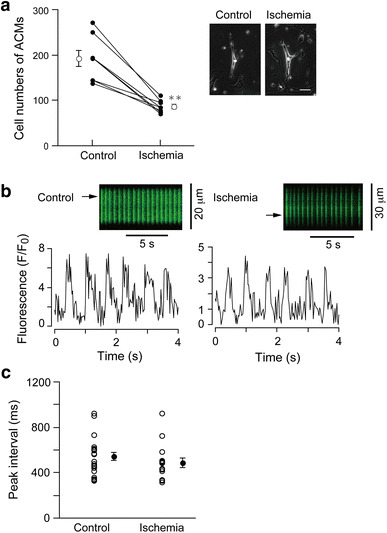
Beating ACMs detected in the culture of CMDF cells pre-exposed to ischemia. a The cell number of beating ACMs identified in culture of CMDF cells with or without (control) pre-exposure to ischemia. Closed symbols indicate individual values and open symbols the mean values (n = 8 cell preparations). **p < 0.001. Right panels indicate phase contrast microscopy of beating ACMs cultured for 6 days. Bar 50 µm. b Spontaneous Ca2+ transients recorded from fluo 4-loaded ACMs identified within culture of CMDF cells that were or were not (control) pre-exposed to ischemia. Representative traces show the fluorescent time course at the site marked by the arrow on the line-scan images. c The peak intervals of the spontaneous Ca2+ transients. Open circles indicate the individual values and closed circles the mean ± SEM (n = 22 cells for control, n = 15 cells for ischemia obtained from 3 mice). No statistically significant differences were noted
Ultrastructures of cardiac ventricle and ACMs
One of the survival mechanisms in the cells under stresses, such as ischemia and starvation, is the activation of autophagy. Autophagy is an evolutionally conserved cell process for the degradation and recycling of long-lived proteins and organelles, which is activated in nutrient-deprived cells [13, 16, 19]. Autophagy remains at a low level in the heart under physiological conditions and plays a role in the maintenance of the cells [26], but is up-regulated in response to various types of stress, including ischemia and starvation, essentially for reducing damage or protecting cells from death [6, 9, 14, 20, 35, 37]. The process of autophagy sequesters cellular components within an isolation membrane to form autophagic vacuoles, termed autophagosomes, which subsequently fuse with lysosomes for the bulk degradation and recycling. To examine whether the ultrastructure of ACMs is different from that of ventricular myocytes, we conducted the transmission electron microscopic (TEM) analyses of those cells.
The TEM image of the mouse cardiac ventricle demonstrated well-organized myofibers and a large number of mitochondria (Fig. 4a). The TEM images of ACMs also displayed the presence of myofibers in the cytoplasm (Fig. 4b–d), consisting of the immunostaining of α-actinin demonstrated in the previous studies [28, 30]. However, the amount and the distribution pattern of myofibers in ACMs are similar to those observed in SA nodal cells rather than ventricular or atrial myocytes [2, 3, 11, 29, 31].
Fig. 4.
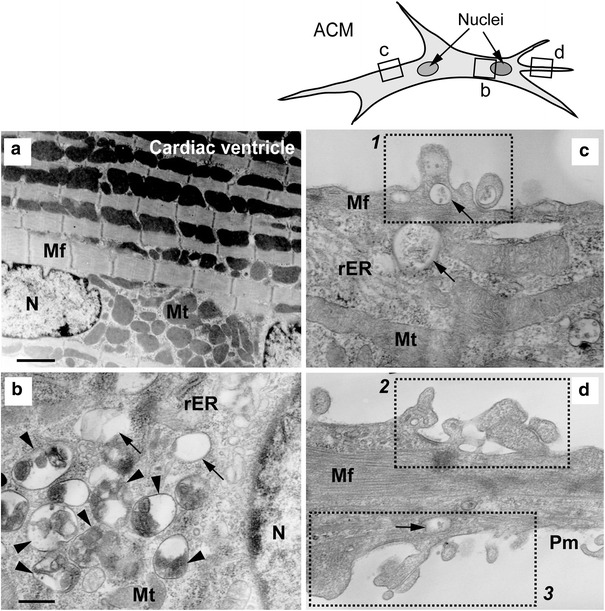
Ultrastructures of cardiac ventricle and ACMs. TEM images of mouse cardiac ventricle (a, ×6,000; bar 1.7 µm) and various parts of ACM cultured for 5 days (b–d; ×25,000; bar 400 nm). b–d the enlargement of the small areas of corresponding squares on a line drawing of the cell (upper right). N nucleus, Mt mitochondria, Mf myofiber, rER rough endoplasmic reticulum, Pm plasma membrane. Arrowheads and arrows indicate autophagic bodies containing degraded multiple organelles and almost empty vacuoles, respectively. Dotted square (1–3) the plasma membrane ruffles
The most noticeable characteristic of the ultrastructure of ACMs was the existence of numerous autophagosomes in both the perinuclear and the peripheral area of the cells (Fig. 4b, c). The autophagosomes detected in ACMs display various stages of lysosomal degradation, such as vacuoles containing several degraded organelles (Fig. 4b, arrowheads) and a small amount of indigestible remnants (Fig. 4b–d, arrows). During autophagy, intracellular organelles and/or cytoplasm sequestered within autophagosomes are finally delivered to lysosomes for bulk degradation [13, 16, 19]. The observations in TEM analyses indicate that the autophagy is indeed up-regulated in ACMs. Degraded cristae membrane structures, residual bodies with typical myelin-like figures, were observed in the majority of the autophagosomes (Fig. 4, arrowheads), thus indicating that the mitochondria are one of the major target organelles for the autophagy in these cells.
Surface structure of ACMs
In addition to the presence of numerous autophagosomes, the TEM images demonstrate unusual morphology of the plasma membrane, such as membrane extrusion holding vacuoles just beneath the membrane (Fig. 4c, square 1) and membrane invagination (Fig. 4d, squares 2 and 3). To investigate the surface membrane structure of ACMs, scanning electron microscopic (SEM) analyses of these cells were conducted. The SEM images of ACMs with lower magnifications demonstrate the rough appearance of the cell surface (Fig. 5a, b). The images with higher magnification of the area of the rough plasma membrane revealed the accumulation of a large number of papillae (Fig. 5c, d) and several papillae are stacked on the others (Fig. 5e, arrowheads). Some of the papillae were observed to have rupture (Fig. 5e, f, asterisks), resulting in holes in the plasma membrane. An SEM image of ACMs with ×100,000 magnification demonstrates the inner structure of the upper papillae of the stacking, showing several ruptured papillae in the bottom of the hole of the plasma membrane (Fig. 5f, arrow). These results indicate that the “plasma membrane disorders” observed in the TEM images (Fig. 4c, d) could be the images of the perpendicular sections of the stacked papillae on the cell surface with or without membrane rupture demonstrated in the SEM images (Fig. 5).
Fig. 5.
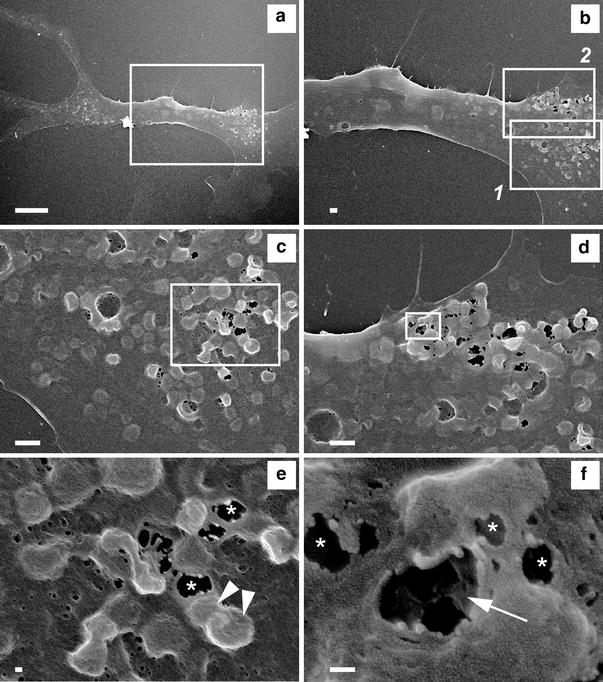
Surface structure of ACMs. SEM images of ACM cultured for 5 days. a Whole cell image (×1,300; bar 10 µm. b Enlargement of the area marked in (a) (×3,000; bar 1 µm. c Enlargement of the area marked in (b1) (×10,000; bar 1 µm). d Enlargement of the area marked in (b2) (×10,000; bar 1 µm). e Enlargement of the area marked in (c). Asterisks and arrowheads indicate ruptured and stacked papillae, respectively (×30,000; bar 100 nm). f Enlargement of the area marked in (d). Arrow indicates the inner structure of the upper papillae of the stacking. Several ruptured papillae were observed in the bottom of the hole of the plasma membrane (×100,000; bar 100 nm)
Immunostaining of LC3 and Atg12 in ACMs
Microtubule-associated protein 1 light chain 3 (LC3), a homologue of yeast Apg8p, is essential for autophagy, and two forms of LC3, cytosolic LC3-I and autophagosome-associating membrane bound LC3-II, are post-translationally produced in various cells [12]; therefore, the presence of LC3-II is recognized as a marker protein for autophagosomes. Immunocytochemical analyses confirmed that punctate staining signals for membrane-bound LC3 [14, 20] were detected in ACMs cultured for both 3 (Fig. 6a) and 6 (Fig. 6b) days, displaying an accumulation of the signals at the perinuclear area and a scatter of dotted signals in both bulge and cell body including the peripheral area. Formation of the autophagosome involves the conjugation system in which autophagy-related gene 12 (Atg12) is covalently bound to Atg5 and targeted for autophagosome vesicles [13, 16, 19, 23]; therefore, the detection of membrane-bound Atg12 is another marker for autophagosomes. The immunocytochemical analyses also confirmed that punctate fluorescent signals for membrane-bound Atg12 were detected in ACMs cultured for both 3 (Fig. 6c) and 6 (Fig. 6d) days.
Fig. 6.
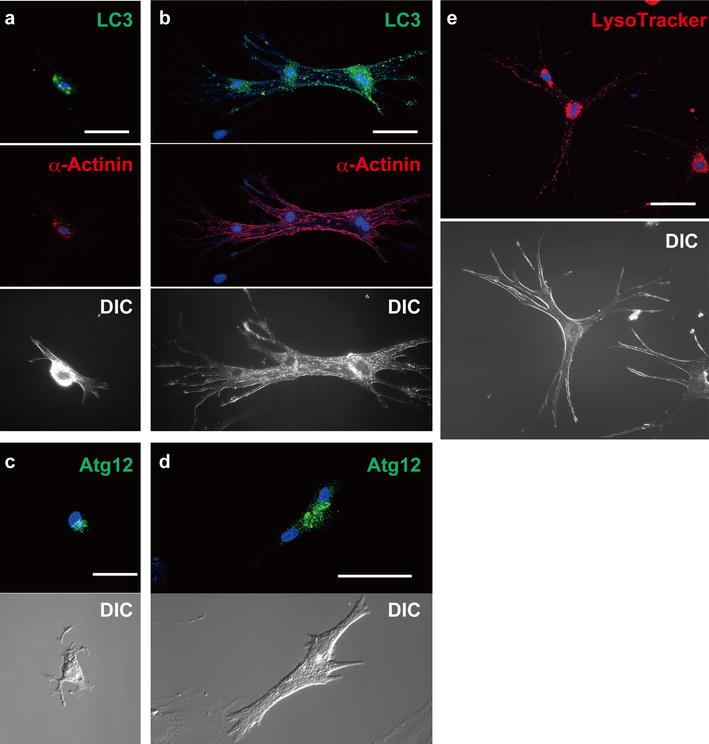
Immunostaining of LC3 and Atg12, and visualization of lysosomes. a, b Confocal laser scanning microscopy of double-immunostaining of LC3 (green) and α-actinin (red), DAPI staining (blue) and DIC images of ACMs cultured for 3 (a) and 6 (b) days. c, d Immunostaining of Atg12 of ACMs cultured for 3 (c) and 6 (d) days. e Fluorescent staining of lysosomes with LysoTracker (red) and nuclei with Hoechst 33342 (blue) in beating ACMs cultured for 6 days. Bars (a, c) 25 µm, (b, d, e) 50 µm
The autophagosomes detected in ACMs display various stages of the degradation of the trapped organelles (Fig. 4b–d), suggesting that the majority of autophagosomes are fused with lysosomes for bulk degradation [13, 16, 19]. To track the lysosomes in living ACMs, we labeled lysosomes with fluorescent acidotropic probe LysoTracker Red. The visualization of lysosomes in beating ACMs cultured for 6 days demonstrates that the lysosomes were concentrated in perinuclear area and scattered among whole cells including the periphery (Fig. 6e), similar to the distribution pattern of the immunosignals for membrane-bound LC3 (Fig. 6a, b). These results further suggest that autophagy is activated in the ACMs.
Effects of ischemia on the ventricular myocytes
It is expected that the ventricular myocytes are sensitive to ischemia. As demonstrated in Fig. 7a, the exposure of ventricular myocytes to the simulated lethal ischemia reduced the percentage of viable cells from 77.0 ± 2.5 % (control) to 15.0 ± 1.1 % (n = 8). No viable myocytes were detected in the dishes after 6 days culture (data not shown).
Fig. 7.
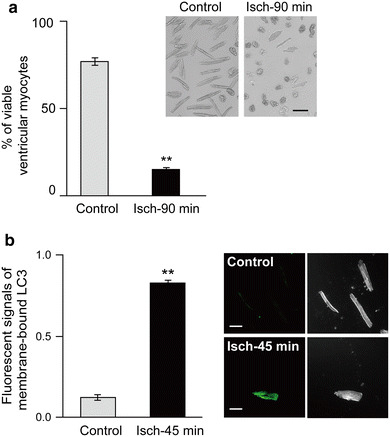
Effects of ischemia on the isolated ventricular myocytes. a Viability of ventricular myocytes pre-exposed to ischemia for 90 min. The percentage of the cell number of viable ventricular myocytes with or without (control) exposure to ischemia for 90 min (Isch-90 min, n = 8, paired experiments). Rounded cells (defined as cells in which the ratio between the length and width as <2) were considered to be irreversibly injured or dead [36]. **p < 0.001. Upper right panels show the representative light microscopy of ventricular myocytes. Bar 50 µm. b Immunofluorescent signals for the estimated membrane-bound LC3 in isolated ventricular myocytes with or without (control) ischemia for 45 min (Isch-45 min). n = 5 cells obtained from 3 mice. **p < 0.001. Right panels show the representative confocal laser scanning microscopy of immunostaining of LC3 (left) and DIC images (right) of ventricular myocytes (bars 50 µm)
To examine the ischemia-induced autophagy of ventricular myocytes, the ventricular myocytes were exposed to ischemia for 45 min, the conditions in which more cardiomyocytes can survive. The immunocytochemical analysis confirmed that the exposure of the isolated ventricular myocytes to ischemia significantly increased the immunofluorescent signals of membrane-bound LC3 from 0.12 ± 0.02 (control) to 0.83 ± 0.02 (n = 5, Isch-45 min; Fig. 7b). These observations indicate that the hypoxic stress induces the autophagy in isolated ventricular myocytes, consistent with the previous reports [6, 9, 20, 37].
Requirement of bafilomycin A1-sensitive autophagy for the development and ischemic tolerance of ACMs
Vacuole H+-ATPase inhibitor bafilomycin A1 reversibly inhibits the fusion between autophagosomes and lysosomes and accumulates autophagosomes in cytoplasmic space, resulting in the inhibition of the degradation of the sequestered organelles and/or cytoplasm [42]. To examine whether the inhibition of autophagy affects the development and ischemic tolerance of ACMs, each 250-µL CMDF cell suspension was exposed to normoxia or ischemia in the presence or absence of 100 nM bafilomycin A1 (Calbiochem) and subsequently cultured in the absence of the inhibitor for 5 days.
Under normoxic conditions, pre-treatment of CMDF cells with bafilomycin A1 significantly reduced the cell number of the beating ACMs from 112.3 ± 8.5 to 47.3 ± 6.1 cells (n = 3; Fig. 8a). These observations indicate that bafilomycin A1-sensitive autophagy in the early period plays an important role in the development of ACMs. Bafilomycin A1 also reduced the cell number of surviving ACMs underwent ischemia from 57.0 ± 4.9 to 21.0 ± 0.7 (n = 3; Fig. 8a). The bafilomycin A1-induced decrease in the cell number of ischemia-surviving ACMs may contain, at least partly, the inhibitory effects of this inhibitor on the development of ACMs. There were no appreciable differences between the phase contrast images of ACMs in those four groups (Fig. 8a, right). Since it is not currently possible to identify small ACMs among CMDF cells, we cultured CMDF cells in the continued presence of bafilomycin A1. As demonstrated in Fig. 8b, ACMs were not detected during 4 days culture in the presence of bafilomycin A1. Although the effect of bafilomycin A1 is reversible [42], very few beating ACMs were observed at ~5 days after removal of the inhibitor (data not shown).
Fig. 8.
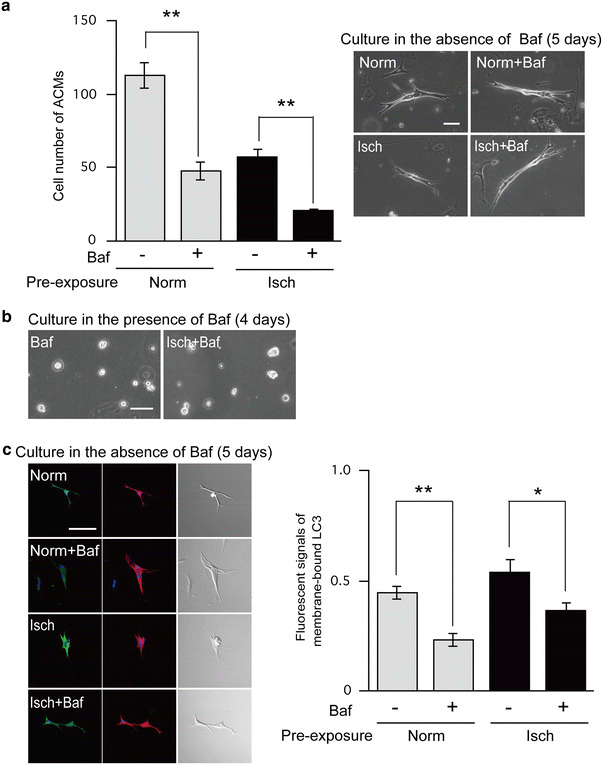
Effect of bafilomycin A1 on the development and ischemic tolerance of ACMs. 1 mL of CMDF cell suspension was divided into four groups (250 µL each). The CMDF cells were pre-exposed to normoxia (Norm) or ischemia (Isch) in the presence or absence of 100 nM bafilomycin A1 (Baf) for 90 min at 37 °C, washed out with centrifugation to remove Baf and subsequently cultured in the absence of Baf. The cells without Baf treatment were given the same volume of DMSO. a Left panel shows the cell number of beating ACMs cultured for 5 days. Data are mean ± SEM (n = 3 cell preparations). **p < 0.001. Right panels show the phase contrast microscopy of beating ACMs (bars 50 µm). b Phase contrast images of CMDF cells with or without pre-exposure to ischemia in the presence of Baf and cultured in the continued presence of Baf for 4 days (bar 50 µm). c Left panels show the confocal laser scanning microscopy of double-immunostaining of LC3 (green) and α-actinin (red), DAPI staining (blue) and DIC images of ACMs cultured in the absence of Baf for 5 days. The conditions of pre-treatment of CMDF cells are the same as (a) (bar 50 µm). Right panel shows the immunofluorescent signals for the estimated membrane-bound LC3. Data are mean ± SEM. (n = 8 cells obtained from 3 mice). **p < 0.001, *p < 0.005
Pre-incubation of CMDF cells with bafilomycin A1 under normoxia reduced the immunofluorescent signals for membrane-bound LC3 in ACMs cultured for 5 days (in the absence of the inhibitor) from 0.45 ± 0.03 to 0.23 ± 0.03, and under ischemia also decreased those in surviving ACMs from 0.54 ± 0.03 to 0.36 ± 0.04 (n = 8; Fig. 8c). These results suggest that the inhibition of autophagy in the early period of the development has a long-term effect on the cellular function of ACMs.
Discussion
The present study demonstrates that approximately half of the ACMs that underwent severe ischemia preserve the ability to develop into beating cells during the subsequent culture under normoxia. We also show the constitutively active autophagy in ACMs during culture.
The quiescent muscle precursor cells in adult skeletal muscle satellite cells are located under the basal lamina of the muscle fiber [22] and become activated and proliferated in response to damaged fibers, in order to repair the damaged muscle fibers. The regenerated muscle fibers then develop into new satellite cells [24]. On the other hand, the dedifferentiated cardiomyocytes exhibiting the characteristics of cardiac stem cell-like cells and/or fetal cardiomyocytes re-enter the cell cycle and can be re-differentiated into cardiomyocytes during long-term culture [34, 43, 45]. The dedifferentiation may be a metabolic link to the return to the fetal gene program in response to pathophysiological conditions in the heart [40]. The facts that ACMs start beating within ~3 days culture without appreciable proliferation (Fig. 1a) and originate from the small cells different from the ventricular myocytes (Fig. 1b) indicate that ACMs are a distinct cell lineage from cardiac satellite cells or dedifferentiated cardiomyocytes [28, 30].
ACMs that have undergone severe ischemia would be divided into three cell types, that is, dead cells, living cells that lost the ability to become beating cells (silent cells), and living cells that develop into beating cells (active cells). We do not know the cell number of dead or silent cells, but estimate the active cells at ~50 % of ACMs pre-exposed to the severe ischemic conditions (Fig. 3). In the adult mouse heart, Limana et al. [17] estimated the number of cardiomyocytes to be ~3 × 106 while we previously estimated the number of ACMs in 2/3 of the ventricles to be ~600 [30], thus indicating that the proportion of ACM to cardiomyocyte would be 1:~3,000. In the normal heart, ACMs are expected to be located in the extracellular space among the cardiomyocytes as small interstitial cells, and thus have been demonstrated to be non-myocytes [25, 39]. Since ACMs are likely to be more resistant to the severe ischemic condition compared to the ventricular myocytes, it is possible that some of the ACMs resident in the cardiac ventricles become active cells to start beating when the adjacent ventricular myocytes are lost in the injured heart [5, 41].
Notably, a number of autophagic vacuoles are present beneath the plasma membrane and the papillae with or without surface rupture are accumulated and/or stacked on the plasma membrane (Figs. 4, 5). The observations presumably indicate the high level of autophagy in ACMs, resulting in the continuous discharge of indigestible content to the extracellular space mediated through exocytosis-like pathways. These surface papillae may explain the rough appearance of the cell surface of ACMs observed in the bright-field images with low magnifications (Figs. 1, 3, 8).
Under physiological conditions, the autophagy is up-regulated in various tissues in mice, including cardiac muscles, during a specific early neonatal period and returns to the low basal levels, which allows for the maintenance of energy homeostasis to survive the neonatal starvation period by the autophagic degradation of self-proteins [14]. The evidence that the isolated small ACMs (∅ ~10 µm) rapidly develop into large beating cells with many branching within several days of culture (Fig. 1) indicates that ACMs need excess energy and source materials in comparison to other cell types in CMDF cells. The absence of the surface papillae on other CMDF cells cultured in the same dish of ACMs, such as fibroblast-like cells (data not shown), also suggests that the high level of autophagy is likely specific to ACMs. Although the precise role of the constitutively active autophagy in ACMs is not clear, the results indicate that ACMs require the bafilomycin A1-sensitive autophagy, especially in the early period, to develop into beating cells and obtain an ischemic tolerance (Fig. 8). It is likely that the constitutively active autophagy plays a role in both cell development and functions of ACMs, similar to that observed in the heart of early neonatal starvation period [14].
In the present study, it was difficult to perform a quantitative measurement of the autophagic marker proteins, such as western blotting analyses of LC3-II and Atg5-conjugated Atg12, because it is not currently possible to harvest ACMs scattered in the culture dish and collect sufficient cell numbers to conduct quantitative analyses. Therefore, we are now seeking to find appropriate cell surface marker proteins for ACMs to identify the original localization of these cells in the heart and also to harvest these cells for extracting the mRNA and proteins.
The previous studies showed that the cell number of ACMs in the neonatal heart is highest in the postnatal life [30], suggesting that ACMs are likely to be one of the residual fetal heart cells. However, it is still unclear whether functional ACMs play a beneficial role, such as providing a source of replacement cardiomyocytes, or a harmful role, such as a cause of arrhythmia, under physiological/pathophysiological conditions. Future research to improve the methods to convert ACMs into SA nodal cells and/or ventricular myocytes would therefore positively contribute to the development of new approaches for cell therapy, while also providing a new insight into the pathogenesis of arrhythmia in the injured heart.
Acknowledgments
We thank Takefumi Yamamoto for the technical assistance. We appreciate the helpful suggestions from Professor Hiroyuki Sugihara, Department of Pathology, Shiga University of Medical Science. This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (22590204 to M.O.K. and 22590205 to H.M.).
References
- 1.Barile L, Messina E, Giacomello A, Marban E. Endogenous cardiac stem cells. Prog Cardiovasc Dis. 2007;50:31–48. doi: 10.1016/j.pcad.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Bleeker WK, Mackaay AJ, Masson-Pevet M, Bouman LN, Becker AE. Functional and morphological organization of the rabbit sinus node. Circ Res. 1980;46:11–22. doi: 10.1161/01.RES.46.1.11. [DOI] [PubMed] [Google Scholar]
- 3.Bouman LN, Jongsma HJ. Structure and function of the sino-atrial node: a review. Eur Heart J. 1986;7:94–104. doi: 10.1093/oxfordjournals.eurheartj.a062047. [DOI] [PubMed] [Google Scholar]
- 4.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colucci WS. Molecular and cellular mechanisms of myocardial failure. Am J Cardiol. 1997;80:15L–25L. doi: 10.1016/S0002-9149(97)00845-X. [DOI] [PubMed] [Google Scholar]
- 6.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz RJ, Losito VA, Mao GD, Ford MK, Backx PH, Wilson GJ. Chloride channel inhibition blocks the protection of ischemic preconditioning and hypo-osmotic stress in rabbit ventricular myocardium. Circ Res. 1999;84:763–775. doi: 10.1161/01.RES.84.7.763. [DOI] [PubMed] [Google Scholar]
- 8.Dukes ID, Vaughan Williams EM. Effects of selective α1-, α2-, β1-and β2-adrenoceptor stimulation on potentials and contractions in the rabbit heart. J Physiol. 1984;355:523–546. doi: 10.1113/jphysiol.1984.sp015436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 10.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993;73:197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- 11.James TN, Sherf L, Fine G, Morales AR. Comparative ultrastructure of the sinus node in man and dog. Circulation. 1966;34:139–163. doi: 10.1161/01.CIR.34.1.139. [DOI] [PubMed] [Google Scholar]
- 12.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 15.Leri A, Kajstura J, Anversa P. Cardiac stem cells and mechanisms of myocardial regeneration. Physiol Rev. 2005;85:1373–1416. doi: 10.1152/physrev.00013.2005. [DOI] [PubMed] [Google Scholar]
- 16.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 17.Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, Nadal-Ginard B, Izumo S, Anversa P. bcl-2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci USA. 2002;99:6257–6262. doi: 10.1073/pnas.092672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lutz M, Rosenberg M, Kiessling F, Eckstein V, Heger T, Krebs J, Ho AD, Katus HA, Frey N. Local injection of stem cell factor (SCF) improves myocardial homing of systemically delivered c-kit+ bone marrow-derived stem cells. Cardiovasc Res. 2008;77:143–150. doi: 10.1093/cvr/cvm027. [DOI] [PubMed] [Google Scholar]
- 19.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 20.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 21.Matsuura K, Nagai T, Nishigaki N, Oyama T, Nishi J, Wada H, Sano M, Toko H, Akazawa H, Sato T, Nakaya H, Kasanuki H, Komuro I. Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem. 2004;279:11384–11391. doi: 10.1074/jbc.M310822200. [DOI] [PubMed] [Google Scholar]
- 22.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 24.Morgan JE, Partridge TA. Muscle satellite cells. Int J Biochem Cell Biol. 2003;35:1151–1156. doi: 10.1016/S1357-2725(03)00042-6. [DOI] [PubMed] [Google Scholar]
- 25.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 26.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 27.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, Entman ML, Schneider MD. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omatsu-Kanbe M, Matsuura H. A novel type of self-beating cardiomyocytes in adult mouse ventricles. Biochem Biophys Res Commun. 2009;381:361–366. doi: 10.1016/j.bbrc.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 29.Omatsu-Kanbe M, Yamamoto T, Matsuura H. Autophagy is constitutively active in normal mouse sino-atrial nodal cells. Acta Histochem Cytochem. 2011;44:223–231. doi: 10.1267/ahc.11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Omatsu-Kanbe M, Yamamoto T, Mori Y, Matsuura H. Self-beating atypically shaped cardiomyocytes survive a long-term postnatal development while preserving the expression of fetal cardiac genes in mice. J Histochem Cytochem. 2010;58:543–551. doi: 10.1369/jhc.2010.955245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Opthof T, de Jonge B, Mackaay AJ, Bleeker WK, Masson-Pevet M, Jongsma HJ, Bouman LN. Functional and morphological organization of the guinea-pig sinoatrial node compared with the rabbit sinoatrial node. J Mol Cell Cardiol. 1985;17:549–564. doi: 10.1016/S0022-2828(85)80024-9. [DOI] [PubMed] [Google Scholar]
- 32.Oyama T, Nagai T, Wada H, Naito AT, Matsuura K, Iwanaga K, Takahashi T, Goto M, Mikami Y, Yasuda N, Akazawa H, Uezumi A, Takeda S, Komuro I. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol. 2007;176:329–341. doi: 10.1083/jcb.200603014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker KK, Ingber DE. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philos Trans R Soc Lond B. 2007;362:1267–1279. doi: 10.1098/rstb.2007.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poindexter BJ, Smith JR, Buja LM, Bick RJ. Calcium signaling mechanisms in dedifferentiated cardiac myocytes: comparison with neonatal and adult cardiomyocytes. Cell Calcium. 2001;30:373–382. doi: 10.1054/ceca.2001.0249. [DOI] [PubMed] [Google Scholar]
- 35.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol. 2008;294:C833–C841. doi: 10.1152/ajpcell.00313.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J. 2001;65:965–968. doi: 10.1253/jcj.65.965. [DOI] [PubMed] [Google Scholar]
- 38.Shioya T. A simple technique for isolating healthy heart cells from mouse models. J Physiol Sci. 2007;57:327–335. doi: 10.2170/physiolsci.RP010107. [DOI] [PubMed] [Google Scholar]
- 39.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.RES.83.1.15. [DOI] [PubMed] [Google Scholar]
- 40.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann NY Acad Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 43.Zaglia T, Dedja A, Candiotto C, Cozzi E, Schiaffino S, Ausoni S. Cardiac interstitial cells express GATA4 and control dedifferentiation and cell cycle re-entry of adult cardiomyocytes. J Mol Cell Cardiol. 2009;46:653–662. doi: 10.1016/j.yjmcc.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Zakharova L, Mastroeni D, Mutlu N, Molina M, Goldman S, Diethrich E, Gaballa MA. Transplantation of cardiac progenitor cell sheet onto infarcted heart promotes cardiogenesis and improves function. Cardiovasc Res. 2010;87:40–49. doi: 10.1093/cvr/cvq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Li TS, Lee ST, Wawrowsky KA, Cheng K, Galang G, Malliaras K, Abraham MR, Wang C, Marban E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE. 2010;5:e12559. doi: 10.1371/journal.pone.0012559. [DOI] [PMC free article] [PubMed] [Google Scholar]