

Abstract
It has been confirmed that exercise preconditioning (EP) has a protective effect on acute cardiovascular stress. However, how Hsp70 participates in EP-induced cardioprotection is unknown. EP may involve Hsp70 to repair unfolded proteins or may also stabilize the function of the endoplasmic reticulum via Hsp70-related autophagy to work on a protective formation. Our EP protocol involves four periods of 10 min running with 10 min recovery intervals. We added a period of exhaustive running to test this protective effect, using histology and molecular biotechnology methods to detect related markers. EP provided cardioprotection at its early and late phases against exhaustive exercise-induced ischemic myocardial injury. Results showed that Hsp70 co-chaperone protein BAG3, ubiquitin adaptor p62 and critical autophagy protein LC3 were significantly upregulated at the early phase. Meanwhile, Hsp70, Hsp70/BAG3 co-localization extent, LC31 and LC3II were significantly upregulated at the late phase. Hsp70 mRNA levels and LC3II/I ratios were also consistent with the extent of myocardial injury following exhaustive exercise. Hsp70 increase was delayed relative to BAG3 and p62 after EP, indicating a pre-synthesized phenomenon of BAG3 and p62 for chaperone-assisted selective autophagy (CASA). The decreased Hsp70, BAG3 and p62 levels and increased Hsp70/BAG3 co-localization extent and LC3 levels induced by exhaustive exercise after EP suggest that EP-induced cardioprotection might associate with CASA. Hsp70 has a cardioprotective role and has a closer link with CASA in LEP. Additionally, EP may not cause exhaustion-dependent excessive autophagy regulation. Collectively, during early and late EP, CASA potentially plays different roles in cardioprotection.
Keywords: Exercise preconditioning, Cardioprotection, Molecular chaperone, Macroautophagy, Hsp70
Introduction
As exercise is an intense and workload stimulus factor, it greatly enhances myocardial oxygen consumption, which can result in absolute or relative myocardial hypoxia [1]. Repeated short-term exercise can cause repeated transient absolute or relative myocardial ischemia, similar to the process of ischemic preconditioning (IP). Studies have shown that one-off high-intensity intermittent aerobic exercise can induce endogenous cardioprotection in organisms, allowing the myocardium to be protected during subsequent sustained ischemia [2]. This method of endogenous myocardial protection, induced by exercise, is known as exercise preconditioning (EP) [3, 4]. Therefore, the cardioprotective mechanisms of EP, IP and exercise are strongly associated. Also similar to IP, two windows of cardioprotection for EP exist: early exercise preconditioning (EEP), which occurs immediately after EP until 2–3 h, and late exercise preconditioning (LEP), which occurs at 12 h for 24–36 h after EP; they provide cardioprotection, involving different mechanisms between two windows, associating with the PKC family, natriuretic peptide family, ATP-sensitive potassium channels and NADPH oxidase, as previously reported [3, 5–8]. At the early and late phases of IP, increased Hsp70 participates in the formation of cardioprotection [9, 10]. Recently, Hsp70's participation in EP-induced cardioprotection has also been evaluated [11]. According to Sheng et al., IP provides cardioprotection by inhibiting signal enhancement of caspase 12, mediated by the unfolded protein response (UPR) [12]. Also, Hsp70 upregulation encourages cardioprotection immediately and 24 h after exercise as well [13–16]. Recent studies have found that Hsp70 is an anti-apoptotic protein that participates in short-term exercise, protecting against ischemia-reperfusion (I/R) injury [13]. As a molecular chaperone, it also plays a critical role in cardioprotection induced by exercise [17]. However, for the purpose of our research, whether changes of Hsp70 in EP can have a cardioprotective role needs to be confirmed.
According to our previous studies, intermittent high-intensity EP accompanied with enhanced protein synthesis protects the myocardium from acute cardiovascular stress, induced by exhaustive exercise [3, 5, 6]. Hsp70 may play a key role in the stabilization of endoplasmic reticulum (ER) function in EP. In acute cardiovascular stress, increased ATP consumption, ROS and other factors induced by myocardial hypoxia and ischemia give rise to a decline of processing and transport proteins in the ER. This results in ER calcium uptake and release disruption as well as misfolded and unfolded protein accumulation [18], ultimately causing endoplasmic reticulum stress (ERS). The ER is not able to transport and fold newly synthesized proteins, which leads to UPR, also inducing programmed cell death due to caspase 12 activation [19]. Timely removal of misfolded proteins is a way to reduce UPR and to prevent further exacerbated ERS. It has been reported that protein elimination of the Hsp70s family when acting as a chaperone does not only occur through the ubiquitin-proteasome system (UPS) [20], but also through mediated autophagy. Chaperone-mediated autophagy (CMA) induced by Hsc70, which is an Hsp70 homologous protein, is a supplement to macroautophagy and plays a leading role in selective degradation of soluble proteins [21]. It was recently found that Hsc70-dependent UPS and CMA were unable to affect bigger protein aggregates, but macroautophagy was effectively affected [20]. In this way, we hypothesized that Hsp70 and related CASA may be important factors for EP-induced cardioprotection.
It should be noted that cellular macroautophagy is also induced directly by oxidative stress, lack of energy, calcium overload and other factors [22]. However, activated autophagy aims to facilitate harmful effects of various stresses [23, 24]. Recently, EP providing cardioprotection through mechanisms regulating calcium homeostasis and antioxidants has been reported [8, 25, 26]. Although a broad range of autophagy exists, indicating the possibilities of EP, whether autophagy can be truly induced by EP still needs to be confirmed. Also, macroautophagy is essential for generating cardioprotection in IP and exercise, being as important as Hsp70 [27–29]. Studies found that BAG3 and p62 may be the link between Hsp70 and macroautophagy [30, 31]. To evaluate the extent of CASA, as our research purposes, the interaction between Hsp70 and macroautophagy should be explicit, involving the expression of BAG3, p62 and the critical autophagy membrane protein, LC3. Furthermore, autophagy characters may switch between cell damage and protection when affected by stress [32]. Also, the autophagy characteristics also need to be evaluated at different stress levels.
Materials and methods
Experimental animals
Healthy 8-week-old male Sprague-Dawley rats (n = 120 rats, Siper BK, Shanghai, China), weighing about 256 ± 13 g, were housed at five rats per cage. Rats were fed with standard rodent mash and water ad libitum and maintained at constant temperature and humidity in a 12 h light/dark cycle. All animal procedures were in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee (NIH Publication no. 85–23, revised 1996) and approved the Ethics Committee for Science Research at Shanghai University of Sport.
Experimental protocol
All animals performed 5 days of adaptive treadmill running (10–20 min at 15 m/min, 0% grade) following 1 day of rest. On the 7th day, rats were randomly distributed into six groups (n = 20 rats per group) and underwent the insertion procedures described in the following sessions. All exercise groups underwent prior warm-up at an initial velocity of 15 m/min, 0% grade for 5 min, and with 5-min symmetrical velocity increase until 30 m/min. The exercise preconditioning protocol was the same as previously reported [3, 7, 33]. Following the previous classic EP research, time points at 0.5 h and 24 h after EP were used in this research to examine the EP-induced cardioprotective effect of exhaustive exercise. The exhaustive exercise group (EE group) performed consecutive running till exhaustion, which was defined as when the rat was unable to right itself when placed on its back; the rats were killed at 30 min after exhaustion. Both the early exercise preconditioning group (EEP group) and late exercise preconditioning group (LEP group) sessions consisted of the above-described warm-up and an EP scenario (approximately 75%VO2max) [34]. The EP protocol was four periods of 10-min running with 10-min intervallic recovery and terminated gradually via a 10-min cool-down after the last 10 min of running. While the EEP group was killed at 30 min after exercise, the LEP group was killed at 24 h after exercise. The early exercise preconditioning plus exhaustive exercise group (EEP + E group) and late exercise preconditioning plus exhaustive exercise group (LEP + E group) respectively underwent an exhaustive exercise period at 30 min or 24 h after the EP period and were killed at 30 min after exhaustion.
All rats were anesthetized by intraperitoneal injection of 10% trichloroacetaldehyde monohydrate at 400 mg/kg body weight. The abdomen was opened along its midline to take a 5-ml blood sample from the inferior vena cava. An infusion needle was injected into the left ventricle from the apex cordis to perfuse heparin sodium for anticoagulation followed by 0.85% saline; after that, the the inferior vena cava was cut to let the perfusate flow out. For myocardial detection by Western blot and RT-PCR, at this time the heart was quickly removed and the tissue close to the apex was clipped from the left ventricle wall. Tissue pieces were quick frozen in liquid nitrogen and stored at −80 °C. For histological analysis, supplementary 4% paraformaldehyde was perfused after saline, and then the heart was excised and kept in 4% paraformaldehyde for 24 h fixation.
Detection of cTnI in serum
The rat blood samples were kept for 30 min at room temperature until serum precipitation and then centrifuged for 15 min at 3000 r/min to collect the serum. By using automated immunochemiluminescence on an Access 2 immunoassay system (Beckman Coulter, USA) to measure the serum cTnI, the linear range was between 0.01–99.99 ng/ml; the cTnI antibodies were anti-human and had about 92.8% amino acid sequence homology with rats.
Histological handling
After 24 h fixation, heart tissue was cut to 1 mm3, washed and immersed in PBS for 24 h (0.01 M, pH = 7.4). Gradient alcohol dehydration was carried out as well as wax dips with different melted paraffins at 60 °C and then the samples were embedded. The paraffin slices were dewaxed in water and washed three times in PBS after washing in distilled water. For visible light microscopic observation, the nuclei of the slices were stained with hematoxylin, and then 1% hydrochloric acid alcohol differentiation was added. For each experimental group, five slices were randomly selected from different samples, and five non-overlapping regions were randomly selected from each slice at 400× magnification. Consequently, 25 regions were collected from each group. Image acquisition and processing were done using an image analysis system.
HBFP (hematoxylin-basic fuchsin-picric acid) staining
After the nucleus had been stained by hematoxylin, the slices were put in 0.1% basic fuchsin for 3 min and then washed in distilled water three times, followed by pure acetone for 5 s, staining with 0.1% picric acid for 15 s and another 5-s wash in acetone. Image acquisition was obtained by using an optical photographic microscope (Olympus, Tokyo, Japan). The integrated optical density (IOD) and positive area were measured using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA).
Immunofluorescence staining
After dewaxing in water, slices were washed three times in PBS for digestion by pepsin complex at room temperature and PBS immersion. Goat serum was used for blocking. For double-labeled immunofluorescence staining, anti-HSP70 mouse antibody (non-cross-reactive with HSC 70, anti-rat, 1:200, Santa Cruz, CA, USA) and anti-BAG3 rabbit antibody (anti-rat, 1:500, Abcam, CBG, UK) were mixed, added and incubated for 24 h at 4 °C; the negative control used PBS instead of primary antibody. In the same way as for single-labeled immunofluorescence staining, p62 antibody (anti-rat, 1:200, Novus, CO, USA) and LC3 antibody (anti-rat, 1:200, Sigma, CA, USA) were respectively added to different tissue slices. After overnight incubation, PBST was used for washing.
After combination with a fluorescein-labeled secondary antibody mixture of FITC (anti-rabbit, 1:200, Beyotime Biotech, Jiangsu, China) and CY3 (anti-mouse, 1:200, Beyotime Biotech, Jiangsu, China), or separated FITC and CY3 (anti-rabbit, 1:200, Beyotime Biotech, Jiangsu, China), slices were conjugated in a dark reaction at 30 °C. Nuclei were labeled with Dapi. Images were acquired by laser scanning confocal microscopy (Zeiss, Jena, German) or fluorescence microscopy (Olympus, Tokyo, Japan). Immunofluorescence images of Hsp70 (red) and BAG3 (green) were analyzed using Zen 2012 (black version) software (Zeiss, Jena, German). The co-localization extent between Hsp70 and BAG3 was defined as the R value (Pearson’s correlation coefficient) [35]. Immunofluorescence images of p62 and LC3 were directly merged with the Dapi image using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA). The average integral optical density (AIOD) value was evaluated by ImageJ software (NIH, Bethesda, MD, USA).
Western blotting analysis
Cardiac tissues were homogenized to collect the supernatant liquor. Standard curves were obtained with the BCA method for quantitative measurement of the sample's protein concentration. An SDS-PAGE electrophoresis system was used; the samples were distributed at 50 μg per well for gel electrophoresis at 4 °C in an electrophoresis buffer. At 4 °C the samples were transferred to PVDF membranes. The transfer membrane was incubated in 5% BSA solution at room temperature for 1 h to block nonspecific binding. After blocking, Hsp70 (Santa Cruz, USA), p62 (Novus, USA), LC3 (Sigma, USA) and BAG3 (Abcam, UK) antibody at 1:3,000 dilution were added, as was GAPDH antibody at 1:10,000 dilution, overnight at 4 °C for antigen-antibody binding. After washing the membrane in TBST, HRP-labeled secondary antibody was added at 1:5,000 dilution to bind the primary antibody, and then the membranes were incubated at room temperature for 1 h. KC™ chemiluminescent reaction was carried out for incubation at room temperature for 3 min. The membrane was exposed to X-ray film. Images were scanned and measured for the gray value of specific bands via ImageJ software (NIH, Bethesda, MD, USA).
RT-PCR analysis
Total RNA was isolated from the left ventricle tissue using Trizol. After centrifugation, RT reactive solution [5 × First-Strand Buffer, 4 μl; 0.1 M DTT, 1 μl; RNase inhibitor, 1 μl; SuperScript III RT, 1 μl (Invitrogen, Carland, CA, USA)] was added to the tubes. After mixing at 37 °C for 1 min, 50 °C incubation for 60 min and 70 °C incubation for 15 min were carried out to inactivate the enzyme.
HSP70 primer sequences were as follows:
F: 5′GCAACAGCCACAGACACCA3′, R: 5′GGCGTCATTCCGTTCCTT3′.
GADPH primer sequences were:
F: 5′GGAAAGCTGTGGCGTGAT3′, R: 5′TGCAGGTCCAGCCAGAACT3′.
The reaction system included NTP (2.5 mMeach), 1.0 μl; 10 × PCR buffer, 1.0 μl; MgCl2 solution, 0.6 μl; Taq polymerase, 0.5 units; SybergreenI at a final concentration of 0.25×; 10 μM primer F, 0.4 μl; 10 μM primer R, 0.4 μl; cDNA 2.0, 0.4 μl, adding water to a total volume of 100.4 μl. The real-time PCR reaction program was carried out at 95 °C for 3 min with 40 PCR cycles (95 °C, 15 s; 59 °C, 20 s; 72 °C, 20 s; 82.5 °C, 20 s), and then the fluorescence was collected. After completion of the reaction amplification, it was set to 95 °C, 15 s; 59 °C, 20 s; 72 °C, 20 s; 99 °C, 15 s. Then, the sample was slowly heated from 72 to 99 °C to establish a PCR product melting curve, and a standard curve was established using GAPDH.
Statistical analysis
Data were analyzed by SPSS 20.0 statistical software (Chicago, IL, USA). One-way ANOVA was used on the basis of significant group differences in pairwise comparisons (SNK). The results are shown as mean ± SD (P < 0.05) and demonstrated statistically significant differences.
Results
EP provides cardioprotection against exhaustive exercise at the early and late phases
To accurately evaluate the extent of myocardial injury induced by exercise, we measured serum cardiac troponin I (cTnI) levels (Fig. 1a). The exhaustive exercise group (EE group) had a significant increase (EE vs. C, 3.87 ± 5.04 vs. 0.02 ± 0.01, P < 0.05). By using an intermittent high-intensity EP protocol for 90 min, the EEP group (0.5 h after EP) and LEP group (24 h after EP) did not have significant serum cTnI level elevation. We therefore presumed that the EP for the myocardium was safe. To address the question whether EP provides protection at early and late phases, an exhaustive running exercise was added after the EEP and LEP. The results demonstrated that serum cTnI levels in the EEP + E and LEP + E group compared with the EE group were significantly decreased (EEP + E; LEP + E vs. EE, 1.09 ± 0.83; 0.83 ± 0.63 vs. 3.87 ± 5.04, P < 0.05).
Fig. 1.
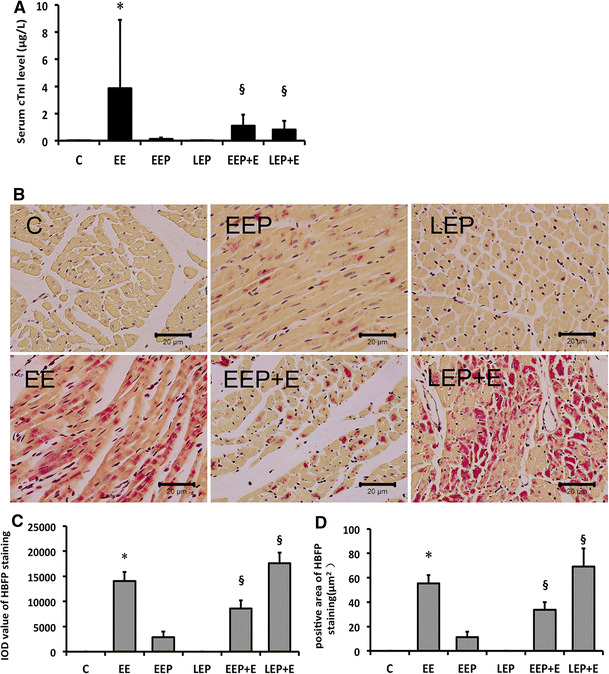
Exercise preconditioning protects against myocardial injury induced by exhaustive exercise at early and late phases. a Serum cTnI level changes in EP and EP-induced cardioprotection in the EE group compared with the C group were significantly different, *P < 0.05. The EEP + E group and LEP + E group compared with the EE group were significantly different, § P < 0.05. b HBFP staining shows myocardial hypoxia-ischemia. Non-ischemic myocardial cells with stained cytoplasm in brown. Hypoxic-ischemic area is bright crimson. The EE group, EEP group, EEP + E group and LEP + E group present different degrees of myocardial hypoxia-ischemia. Group C and the LEP group without a significant hypoxic-ischemic region stained in brown. In the EE, EEP + E group and especially LEP + E group, crimson hypoxic-ischemic regions are significant. Original magnification ×400; bar 20 μm. c, d HBFP positively stained area and integral optical density (IOD) changes in EP and EP-induced cardioprotection. The EE group compared with C group. The difference was significant, *P < 0.05. The EEP + E and LEP + E groups compared with the EE group were significantly different, § P < 0.05. C control, EE exhaustive exercise, EEP early exercise preconditioning, LEP late exercise preconditioning, EEP + E early exercise preconditioning plus exhaustive exercise, LEP + E late exercise preconditioning plus exhaustive exercise (color figure online)
Using hematoxylin basic fuchsin picric acid staining (HBFP staining) (Fig. 1b), rats’ cardiomyocyte nuclei were stained blue, and the cytoplasm was light brown without ischemic crimson staining in the C group. There was a small number of red stains in the EEP group, and non-ischemic staining in the LEP group could be observed. After exhaustive exercise, in the EE, EEP + E and LEP + E groups, crimson hypoxic-ischemic regions were significant. The results of images analysis show (Fig. 1c, d) the EE group had a significant increase in the positive area and integrated optical density (IOD) value (EE vs. C, 55.26 ± 6.91 vs. 0 ± 0; 14092.39 ± 1761.04 vs. 0 ± 0, P < 0.05). A serum cTnI-like consistency was also detected in the EEP + E group because of the protective effect induced by EP (EEP + E vs. EE, 33.77 ± 6.24 vs. 55.26 ± 6.91; 8610.24 ± 1590.79 vs.14092.39 ± 1761.04, P < 0.05). However, two HBFP staining data in the LEP + E group were significantly higher than in the EE group (LEP + E vs. EE, 69.09 ± 14.79 vs. 55.26 ± 6.91; 17617.76 ± 2107.04 vs.14092.39 ± 1761.04, P < 0.05), perhaps due to the increased running ability (Table 1). EP facilitated exhaustive exercise-induced hypoxia and ischemia at the early phase, but EP still prevented myocardial injury, which was indicated by serum cTnI elevation.
Table 1.
Average exhaustive running distance and volume of rats
| Groups | n | Distance of exhaustive exercise (m) | Volume of exhaustive exercise (min) |
|---|---|---|---|
| EE | 19 | 2657.37 ± 975.57 | 88.58 ± 32.52 |
| EEP + E | 20 | 2058.00 ± 852.28 | 69.26 ± 29.03 |
| LEP + E | 20 | 4590.00 ± 1582.82*,§ | 155.74 ± 52.73*,§ |
* P < 0.05 vs. EE
§ P < 0.05 vs. EEP + E
Hsp70 and BAG3 expression and interaction changes in EP-induced cardioprotection
The immunofluorescence images of Hsp70 show red immunoreactive products appearing as granules that are relatively evenly distributed within the myocardium. The BAG3 images show green immunoreactive products distributed as aggregates in the myocardium. The merged images show more dispersed distribution of red and green fluorescence in groups C, EE and EEP (Fig. 2a). The fluorescence intensity of Hsp70 indicated that the LEP group had a significant increase (LEP vs. C, 159.48 ± 24.03 vs. 139 ± 20.61, P < 0.05); the LEP + E group had a significant decrease when compared with LEP (LEP + E vs. LEP, 118.77 ± 29.68 vs. 159.48 ± 24.03, P < 0.05) (Fig. 2b). The fluorescence intensity of BAG (Fig. 2c) showed the BAG3 expression in EEP was markedly increased (EEP vs. C, 186.1 ± 16.78 vs. 150.77 ± 16.51, P < 0.05), and increased BAG3 consumption was accompanied by exhaustion after EEP (EEP + E vs. EEP, 146.46 ± 23.65 vs. 186.1 ± 16.78, P < 0.05). The R value of Hsp70 and BAG3 co-localization (Fig. 2d) was significantly higher in the LEP group than in C (LEP vs. C, 0.24 ± 0.1 vs. 0.08 ± 0.06, P < 0.05). It was was significantly higher in the EEP + E group than in C, EE and EEP (EEP + E vs. C; EE; EEP, 0.28 ± 0.11 vs. 0.08 ± 0.06; 0.11 ± 0.04; 0.08 ± 0.04, P < 0.05) and significantly higher in the LEP + E group than in C and EE (LEP + E vs. C; EE, 0.31 ± 0.11 vs. 0.08 ± 0.06; 0.11 ± 0.04, P < 0.05).
Fig. 2.
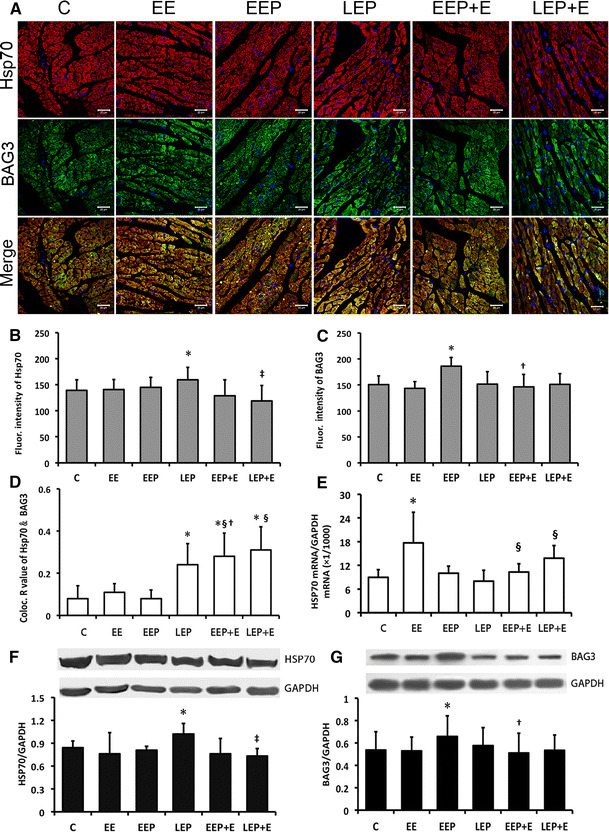
HSP70 and BAG3 expression and interaction changes in rat myocardium. a, b Laser scanning images. Hsp70 shows immunoreactive products (red) appeared as relatively evenly distributed granules within the myocardium. The BAG3 images show green immunoreactive products distributed as aggregates in the myocardium. The merged images show more dispersed distribution of red and green fluorescence in groups C, EE and EEP. Original magnification ×400; bar 20 μm. b Average integral optical density (AIOD) value of HSP70 immunoreactive image analysis in cardiomyocytes indicates the LEP group compared with the C group. The difference was significant, *P < 0.05. The LEP + E group compared with the LEP group was significantly different, ‡ P < 0.05. c AIOD value of BAG3 immunoreactive image analysis in cardiomyocytes indicates that in the EEP group compared with the C group, the difference was significant, *P < 0.05. The EEP + E group compared with the EEP group was significantly different, † P < 0.05. d The co-localization R values of Hsp70/BAG3 in the LEP, EEP + E and LEP + E groups compared with the C group were significantly different, *P < 0.05. The EEP + E and LEP + E groups compared with EE showed a significant difference, § P < 0.05. The EEP + E group compared with the EEP group was significantly different, † P < 0.05. e For HSP70 mRNA level changes in EP and EP-induced cardioprotection, the EE group compared with group C was significantly different, *P < 0.05. The EEP + E group and LEP + E group compared with the EE group were significantly different, § P < 0.05. f HSP70 protein expression changes in EP and EP-induced cardioprotection detected by immune blotting. In the LEP group compared with the C group, the difference was significant, *P < 0.05. The LEP + E group compared with the LEP group was significantly different, ‡ P < 0.05. g BAG3 protein expression changes in EP and EP-induced cardioprotection detected by immune blotting. In the EEP group compared with the C group, the difference was significant, *P < 0.05. The EEP + E group compared with the EEP group was significantly different, † P < 0.05. C control, EE exhaustive exercise, EEP early exercise preconditioning, LEP late exercise preconditioning, EEP + E early exercise preconditioning plus exhaustive exercise, LEP + E late exercise preconditioning plus exhaustive exercise (color figure online)
The levels of Hsp70 mRNA were detected via RT-PCR (Fig. 2e). A phenomenon of Hsp70 mRNA upregulation could be defined as exhaustive exercise dependency (EE vs. C, 17.71 ± 7.73 vs. 8.97 ± 1.97, P < 0.05). Moreover, compared with the EE group, the levels in the EEP + E and LEP + E groups were significantly downregulated (EEP + E; LEP + E vs. EE, 0.35 ± 2.04; 13.85 ± 3.19 vs. 17.71 ± 7.73, P < 0.05). We determined Hsp70 and BAG3 levels by Western blotting, and the results demonstrated that Hsp70 expression (Fig. 2f) was highly upregulated by LEP (LEP vs. C, 1.02 ± 0.14 vs. 0.84 ± 0.09, P < 0.05), and consumption was accompanied by exhaustion after LEP (LEP + E vs. LEP, 0.72 ± 0.1 vs. 1.02 ± 0.14, P < 0.05). The levels of BAG3 expression indicated (Fig. 2g) the development of BAG3 was significant in the EEP group compared with C (EEP vs. C, 0.66 ± 0.18 vs. 0.53 ± 0.16, P < 0.05), and it had a significant decline in the EEP + E group relative to the EEP group (EEP + E vs. EE, 0.51 ± 0.18 vs. 0.66 ± 0.18, P < 0.05).
P62 expression changes in EP-induced cardioprotection
The immunofluorescence images of p62 show immunoreactive products appeared as relatively evenly distributed granules within the myocardium in the C, EE, EEP and EEP + E groups. The average integral optical density (AIOD) value of p62 (Fig. 3a) indicated that the EEP group was brighter than groups C, LEP, EEP + E and LEP + E (EEP vs. C; LEP; EEP + E; LEP + E, 1.17 ± 0.38 vs.0.89 ± 0.42; 0.82 ± 0.26; 0.69 ± 0.32; 0.76 ± 0.41, P < 0.05). The EEP + E group had a deepening relative to EE as well (EEP + E vs. EE, 0.68 ± 0.25 vs. 1.1 ± 0.42, P < 0.05) (Fig. 3b). Based on our supplemental Western blotting results (Fig. 4c), it was further found that the EEP group had significantly higher levels of p62 compared with C (EEP vs. C, 0.32 ± 0.14 vs. 0.21 ± 0.10, P < 0.05), and the EEP + E and LEP + E groups were significantly lower than the EEP group (EEP + E; LEP + E vs. EEP, 0.20 ± 0.14; 0.19 ± 0.13 vs. 0.32 ± 0.14, P < 0.05).
Fig. 3.
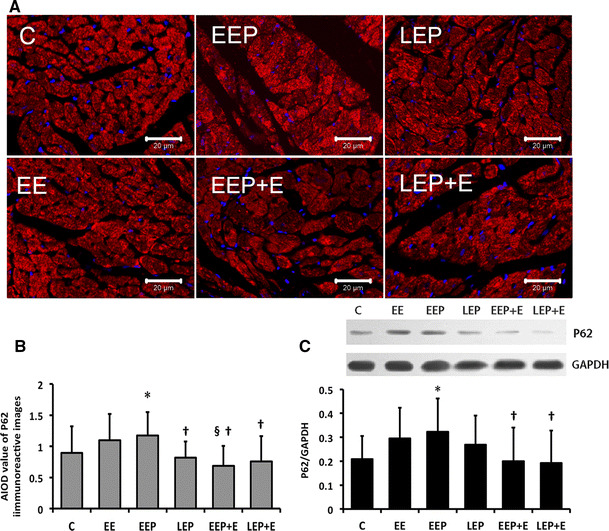
p62 expression changes in rat myocardium. a, b p62 immunofluorescence expression in cardiomyocytes. Immunoreactive regions of each group were stained bright red. Few immunoreactivity differences among groups can be observed except that the EEP group showed significantly more than the C group. Original magnification ×400; bar 20 μm. b The average integral optical density (AIOD) value of p62 immunoreactive image analysis in cardiomyocytes indicates that the difference in the EEP group was significantly different compared with the C group, *P < 0.05. The EEP + E group compared with the EE group was significantly different, § P < 0.05. The LEP, EEP + E and LEP + E groups were significantly different compared with the EEP group, † P < 0.05. c p62 protein expression changes in EP and EP-induced cardioprotection, detected by immune blotting. In the EEP group compared with the C group, the difference was significant, *P < 0.05. In the EEP + E group and LEP group compared with the EEP group, the difference was significant, † P < 0.05. C control, EE exhaustive exercise, EEP early exercise preconditioning, LEP late exercise preconditioning, EEP + E early exercise preconditioning plus exhaustive exercise, LEP + E late exercise preconditioning plus exhaustive exercise (color figure online)
Fig. 4.
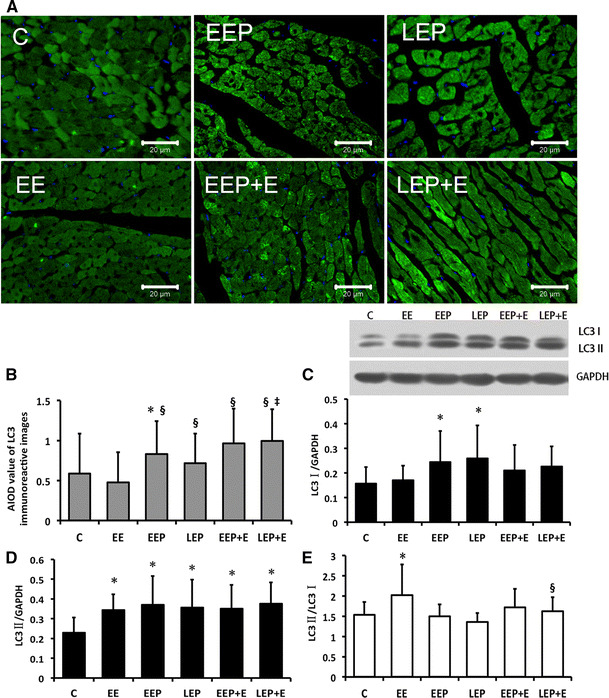
LC3 expression changes in rat myocardium. a, b LC3 immunofluorescence expression in cardiomyocytes. Immunoreactive regions of each group were stained bright green. As observed, positive products in the C group were significantly less than in other groups except the EE group. Original magnification, ×400; bar 20 μm. b Average integral optical density (AIOD) value of LC3 immunoreactive image analysis in cardiomyocytes indicates the level in the EEP group was significantly higher than in the C group, *P < 0.05. All groups were significantly higher than the EE group except for the C group, § P < 0.05. Compared with the LEP group, the LEP + E group was significantly different, ‡ P < 0.05. c, d Presented LC3 protein expression changes in EP and EP-induced cardioprotection detected by immune blotting. c LC3I protein expression: the difference between the LEP group and C group was significant, *P < 0.05. d LC3II protein expression: the EE, EEP, LEP, EEP + E and LEP + E groups were significantly different compared with the C group, *P < 0.05. e LC3II/I ratio changes in EP and EP-induced cardioprotection: the EE group was significantly different compared with the group C, *P < 0.05. The LEP + E group was significantly different compared with the EE group, § P < 0.05. C control, EE exhaustive exercise, EEP early exercise preconditioning, LEP late exercise preconditioning, EEP + E early exercise preconditioning plus exhaustive exercise, LEP + E late exercise preconditioning plus exhaustive exercise (color figure online)
LC3 expression changes in EP-induced cardioprotection
The immunofluorescence images of LC3 show immunoreactive products distributed as aggregates in the myocardium as the bright green color in the C group (Fig. 4a). There were differences in brightness among the cells and evenly within the cytoplasm. A similar phenotypic distribution was observed in all groups; nevertheless, there was a relatively enlarged positive region in the EEP, LEP, EEP + E and LEP + E groups. According to the AIOD value of total LC3 (Fig. 4b), the EEP group had a significant increase (EEP vs. C, 0.83 ± 0.41 vs. 0.59 ± 0.50, P < 0.05). An EP-induced LC3 elevation could be identified when compared with exhaustive exercise (EEP; LEP vs. EE, 0.83 ± 0.41; 0.71 ± 0.37 vs. 0.48 ± 0.37, P < 0.05). After the supplemental exhaustion period, fluorescence reactions in the EEP + E and LEP + E groups were higher compared with the EE group (EEP + E; LEP + E vs. EE, 0.96 ± 0.43; 1.00 ± 0.39 vs. 0.48 ± 0.37, P < 0.05). Especially the LEP + E group was significantly higher than the LEP group (LEP + E vs. LEP, 1.00 ± 0.39 vs. 0.71 ± 0.37, P < 0.05).
In the immune blotting analysis of LC3 (Fig. 4b, c), an Hsp70-like feature trend (Fig. 2c) was detected on LC3I as a significant increase in the EEP and LEP group compared with C (EEP; LEP vs. C, 0.24 ± 0.13; 0.26 ± 0.13 vs. 0.16 ± 0.07, P < 0.05). LC3II in the C group was significantly different from the other groups (C vs. EE; EEP; LEP; EEP + E; LEP + E, 0.23 ± 0.08 vs. 0.34 ± 0.08; 0.37 ± 0.15; 0.36 ± 0.14; 0.35 ± 0.12; 0.38 ± 0.11, P < 0.05). Additional calculation of the LC3II/I ratio, which mirrored the autophagy levels (Fig. 4d), showed that the EE group had a significant increase compared with C (EE vs. C, 2.02 ± 0.76 vs. 1.54 ± 0.32, P < 0.05), and the LEP + E group was significantly lower than the EE group (0.32 ± 0.14 vs. 0.21 ± 0.10, P < 0.05). This was also confirmed by the Hsp70 mRNA or serum cTnI level (Figs. 1a, 2d).
Discussion
Hsp70 synthesis is mainly regulated by the heat shock factor (HSF), and Hsp70 transcription is promoted by binding between the DNA heat shock element and HSF, which can transport it into the nucleus, where it is subsequently activated by intracellular ERK1/2 [36, 37]. Thus, the expression and synthesis of Hsp70 are susceptible to stress. In this study, Hsp70 mRNA levels were upregulated by stress levels, consistent with extent of myocardial injury, in agreement with the finding that exhaustive exercise-induced cardiovascular stress may result in an excessive elevation of Hsp70 gene transcription [37]. The unchanged serum cTnI level indicated EEP and LEP were safe and also associated with an unchanged Hsp70 mRNA level. EEP and LEP markedly blocked the Hsp70 mRNA increases caused by exhaustion, while exhaustive exercise-induced Hsp70 mRNA upregulation could not be attributed to Hsp70 protein expression. Instead, increased Hsp70 in LEP may substantively participate in the cardioprotective effect. We speculated that exhaustive exercise-induced Hsp70 transcriptional and protein expressive changes may be associated with a higher ERS level. Because the amount of damaged protein produced by ERS may competitively bind with Hsp70, this will stimulate HSF-induced gene transcription [15].
Exercise-induced heatstroke associated with Hsp70 overexpression acting in cardioprotection has been reported [38]. However, the myocardial Hsp70 level shows a sharp decline at 60–100 min after continuous heat stress in vivo [39]. The downregulation of Hsp70 may be partly due to substrate degradation of the CASA pathway [40]. In this case, an inverted level of increasing Hsp70 may only be detected at 24 h or longer following high-intensity exhaustive exercise and EP [41]. Myocardial Hsp70 exists as a cumulative process perhaps due to adaptive development after EP, and delayed Hsp70 is a decisive factor for the late protective window. Rats may suffer more serious cardiac hypoxia-ischemia from exhaustion after LEP, which can be considered a stronger running ability caused by EP pretreatment and 24-h recovery.
The hydrophobic surface of misfolded proteins could easily lead to protein aggregation and encumber normal proteins, as protein aggregates cannot be degraded by UPS and CMA pathways, but they can be degraded by CASA. BAG3 is E3 enzyme-like with its co-chaperone protein CHIP, Hsp70 and HspB8. They combine together into larger misfolded proteins, assisting in p62 recruitment for p62 body formation, which induces selective wrapping of autophagy membranes [20]. Hence, molecular chaperone Hsp70 greatly promotes the ubiquitination of E3 ubiquitin ligase BAG3, which contributes to selective autophagy for protein aggregates. In addition, when misfolded proteins are not repaired in a timely fashion, BAG3 mediates microtubule transportation, encouraging larger aggregates and concentrations degraded by CASA, and this process does not depend on substrate ubiquitination [30]. Therefore, BAG3 may actually induce inhibition of monomeric protein-degraded UPS and CMA. Our study also suggests that the BAG3/Hsp70 complex generated in groups LEP, EEP + E and LEP + E may inhibit proteasome-mediated intracellular Akt degradation [42]. Also, Akt itself plays a role in IP and exercise-induced cardioprotection [43]. Another study demonstrated that BAG3 and Hsp70 could promote stabilization of myocardial fibers and Z-disc binding, participating in mechanical stress-induced injury repair and cardioprotective formation, accompanied by translocation [44]. As observed, exhaustive exercise influenced the distributive centralization of BAG3, likely confirming this finding. One study indicated BAG3-dependent CASA is affected by mechanical tension in muscles [31]. We hypothesized the cardiovascular functions of EP also act on the anti-injury mechanism of BAG3, which is similar to other exercise protocols [45].
Chaperone-associated macroautophagy acts as a key factor to inhibit ERS, and BAG3 is a marker of CASA. We found BAG3 and p62 to be significantly elevated at the early phase and downregulated in EEP + E. Both of the change trends suggested a closer collaborative relationship between BAG3 and p62. This phenomenon has been reported in a cardiac study of a spontaneous hypertensive rat protocol by Bloemberg et al. [46]. However, our data indicated that the generated BAG3 will not largely conjugate to Hsp70 in EEP unless Hsp70 is evaluated in LEP. As such, the groups EE and EEP may not relate to Hsp70/BAG3 complex generation. The R value and p62 level in the LEP + E group show no change when compared with LEP; therefore, the amount of Hsp70/BAG3 complex in the LEP + E group greatly depends on the original level in LEP rather than the effect of EE. In other words, it demonstrates that, through the accumulation of Hsp70/BAG3 complex, LEP is a key factor in stably inducing CASA, generating and extending the late protective window, and enhancing long-term adaptability. The R values of HSP70 to BAG3 were greatly elevated in LEP, EEP + E and LEP + E compared to the others, without big differences in the fluorescence intensities and the protein expression levels of Hsp70 and BAG3 between groups. The results strongly suggest that both proteins were translocated or co-localized to specific regions to participate in EP-induced cardioprotection.
Combined with p62 and LC3II changes, an exercise-induced autophagy activity can be identified in groups EE, EEP, LEP, EEP + E and LEP + E. We demonstrated that Hsp70-induced selective macroautophagy plays a potential role in late cardioprotection of EP, which depends on the Hsp70 level and extent of co-localization being consistent with LC3I and LCII levels in LEP. Mostly, increased LC3II and decreased p62 levels demonstrate autophagy activity [47–49]. However, a more intense autophagy can also be composed of both increases in the heart and may be associated with a fresh balance of the p62 level between generation and degradation [50]. According to the AIOD value of p62 in LEP, p62 actually can be consumed by autophagy during 24-h recovery since EEP induced p62 generation. Also the p62 protein in the EEP + E group originally came from EEP, but not for the LEP + E group. In IP, autophagy provides protection against myocardial ischemia by regulating ERS [12]. LC3 is a key protein for forming autophagosomes, and degraded Akt phosphorylation in rat hearts will activate autophagy via the Akt/TSC/mTOR pathway. It was the factor affecting increased LC3 mRNA transcription [51, 52]. BAG3 induces an Akt increas, which may delay the peak appearance time of LC3 after EP [36, 42]. We also investigated the LC3I, LC3II and total LC3 level, which could be elevated by EEP, indicating the enlarged LC3 product surpassing its consumption or inhibition. This may be due to other types of macroautophagy induced by EEP. Because of unchanged the Hsp70 levels and Hsp70/BAG3 co-localization, autophagy in groups EEP or EE is possibly not related to CASA. However, the elevated levels of BAG3 and p62 in EEP, which rapidly decreased in EEP + E, will provide an immediately increased CASA to protect against EE. As a limitation, more time points following EP may need to be selected to capture the peak timing of CASA generation. This would deepen the understanding of CASA's participation in cardioprotection. In conclusion, CASA is another key factor affecting the qualitative difference between the early and late windows of EP.
Atg4 participates in LC3 recycling, but LC3, which is attached to the inner membrane of the autophagosome and its ubiquitin-binding protein p62, will be degraded by lysosomes as substrates are irreversible [53]. Our data showed significantly elevated LC3I in EEP and LEP, which may result from higher Atg4 activity. Atg4 converts partial LC3II to LC3I and further causes an LC3I cumulative effect [49]. On the other hand, the p62 level in LEP had a significant decrease relative to EEP. We hypothesized that EP associates with autophagy-induced p62 consumption whose peak may lie somewhere between the early and late phase of EP; increased CASA partially removed p62 bodies generated by EEP during this period. However, according to HBFP staining, a low level of p62 in LEP will affect p62 body generation in LEP + E and may reduce the endurance of ischemic-hypoxia during exhaustion after LEP partly.
Restricted to several factors such as the efficiency of scavenging autophagosomes via lysosomes and the activity of Atg4, which is susceptible to oxidative stress, LC3II accumulation is likely to result in an increased LC3II/I ratio during acute stress [54, 55]. In this study, the LC3II/I ratio presented the same trend as the extent of stress level, similar to the Hsp70 mRNA levels. Due to the dual modality characteristics of autophagy, moderate autophagy expedites damage repair, and excessive autophagy participates in cell death [32, 56]. ERS-induced autophagy elevation results in cell apoptosis and has a link with inhibition of the Akt/TSC/mTOR pathway; within this chaperone Hsp70 may play a regulatory role leading to autophagy’s protective role [57]. Exhaustive exercise led to an increase in the LC3II/I ratio, which may be accompanied by apoptotic protein upregulation, which mediates myocardial apoptosis [58]. This further suggests that high Hsp70 expression could inhibit phospholipid binding of LC3, which may be the reason for the significant decrease in the LC3II/I in the LEP + E group relative to the EE group [52]. When rats were forced to exercise to exhaustion after LEP, LC3I was upregulated by LEP but did not display an increase in the LC3II/I ratio during subsequent exhaustive running, indicating that factors such as oxidative stress, ER stress and oxidosis were a result of hypoxia-ischemia-induced damaged autophagy [32].
However, increased Hsp70 in LEP may provide assistance to damage repair and stress adaptation and act on the associated autophagy. The characteristics of Hsp70 may possibly be reparative as well. For late cardioprotection induced by EP, upregulated LC3I reflects an endogenous protective cardiac mechanism advancement and leads to increased synergy with Hsp70, contributing to protein aggregate scavenging. This helps to further obtain a protective effect on exhaustive exercise after LEP. Further evidence demonstrated that when exhaustion was added after LEP, decreased Hsp70 and p62 levels in the LEP + E group were associated with an increased LC3 level. These indicated that Hsp70 is more evident concurrently with autophagy and participates in EP-induced late cardioprotection.
Conclusion
EP provided cardioprotection at its early and late phases. Hsp70 and associated CASA can be induced by LEP, but not by EE or EEP. Hsp70 plays a cardioprotective role in LEP. The Hsp70 increase was delayed relative to BAG3 and p62 after EP, indicating a pre-synthesized phenomenon of BAG3 and p62 for CASA. CASA participates in EP-induced early cardioprotection via BAG3 and p62 accumulation in EEP. CASA participates in EP-induced late cardioprotection via Hsp70/BAG3 complex accumulation in LEP. Hsp70 has a closer link with CASA in LEP and LEP-induced cardioprotection.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 31471136).
Abbreviations
- EP
Exercise preconditioning
- EEP
Early exercise preconditioning
- LEP
Late exercise preconditioning
- Hsp70
Heat shock protein 70
- BAG3
Bcl-2 associated athanogene 3
- LC3
Microtubule-associated protein 1A/1B light chain 3
- UPR
Unfolded protein response
- ERS
Endoplasmic reticulum stress
- UPS
Ubiquitin-proteasome system
- CMA
Chaperone-mediated autophagy
- Hsc70
70 kDa heat shock cognate protein
- CASA
Chaperone-assisted selective autophagy
- EE
Exhaustive exercise
- cTnI
Cardiac troponin I
- HSF
Heat shock factor
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Hoshimoto-Iwamoto M, Koike A, Nagayama O, Tajima A, Suzuki T, Uejima T, Sawada H, Aizawa T. Prognostic value of end-tidal CO2 pressure during exercise in patients with left ventricular dysfunction. J Physiol Sci. 2009;59(1):49–55. doi: 10.1007/s12576-008-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jimenez SK, Jassal DS, Kardami E, Cattini PA. A single bout of exercise promotes sustained left ventricular function improvement after isoproterenol-induced injury in mice. J Physiol Sci. 2011;61(4):331–336. doi: 10.1007/s12576-011-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hao Z, Pan SS, Shen YJ, Ge J. Exercise preconditioning-induced early and late phase of cardioprotection is associated with protein kinase C epsilon translocation. Circ J. 2014;78(7):1636–1645. doi: 10.1253/circj.CJ-13-1525. [DOI] [PubMed] [Google Scholar]
- 4.Marongiu E, Crisafulli A. Cardioprotection acquired through exercise: the role of ischemic preconditioning. Curr Cardiol Rev. 2014;10(4):336–348. doi: 10.2174/1573403X10666140404110229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen YJ, Pan SS, Ge J, Hao Z. Exercise preconditioning provides early cardioprotection against exhaustive exercise in rats: potential involvement of protein kinase C delta translocation. Mol Cell Biochem. 2012;368(1–2):89–102. doi: 10.1007/s11010-012-1346-3. [DOI] [PubMed] [Google Scholar]
- 6.Lu J, Pan SS. Elevated C-type natriuretic peptide elicits exercise preconditioning-induced cardioprotection against myocardial injury probably via the up-regulation of NPR-B. J Physiol Sci. 2016 doi: 10.1007/s12576-016-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domenech R, Macho P, Schwarze H, Sánchez G. Exercise induces early and late myocardial preconditioning in dogs. Cardiovasc Res. 2002;55(3):561–566. doi: 10.1016/S0008-6363(02)00334-6. [DOI] [PubMed] [Google Scholar]
- 8.Parra VM, Macho P, Sánchez G, Donoso P, Domenech RJ. Exercise preconditioning of myocardial infarct size in dogs is triggered by calcium. J Cardiovasc Pharmacol. 2015;65(3):276–281. doi: 10.1097/FJC.0000000000000191. [DOI] [PubMed] [Google Scholar]
- 9.Yin C, Salloum FN, Kukreja RC. A novel role of microRNA in late preconditioning: upregulation of endothelial nitric oxide synthase and heat shock protein 70. Circ Res. 2009;104(5):572–575. doi: 10.1161/CIRCRESAHA.108.193250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang JK, Kim JM, Kim YK, Kim SD, Park SC, Kim JI, Nam HW, Kim J, Moon IS. The early protective effect of glutamine pretreatment and ischemia preconditioning in renal ischemia-reperfusion injury of rat. Transpl Proc. 2013;45(9):3203–3208. doi: 10.1016/j.transproceed.2013.08.028. [DOI] [PubMed] [Google Scholar]
- 11.Madden LA, Sandstrom ME, Lovell RJ, McNaughton L. Inducible heat shock protein 70 and its role in preconditioning and exercise. Amino Acids. 2008;34(4):511–516. doi: 10.1007/s00726-007-0004-7. [DOI] [PubMed] [Google Scholar]
- 12.Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, Zhang XY, Qin ZH. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8(3):310–325. doi: 10.4161/auto.18673. [DOI] [PubMed] [Google Scholar]
- 13.Melling CW, Thorp DB, Milne KJ, Noble EG. Myocardial Hsp70 phosphorylation and PKC-mediated cardioprotection following exercise. Cell Stress Chaperones. 2009;14(2):141–150. doi: 10.1007/s12192-008-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powers SK, Demirel HA, Vincent HK, Coombes JS, Naito H, Hamilton KL, Shanely RA, Jessup J. Exercise training improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. Am J Physiol. 1998;275(5 Pt 2):R1468–R1477. doi: 10.1152/ajpregu.1998.275.5.R1468. [DOI] [PubMed] [Google Scholar]
- 15.Xu T, Zhang B, Yang F, Cai C, Wang G, Han Q, Zou L. HSF1 and NF-kappaB p65 participate in the process of exercise preconditioning attenuating pressure overload-induced pathological cardiac hypertrophy. Biochem Biophys Res Commun. 2015;460(3):622–627. doi: 10.1016/j.bbrc.2015.03.079. [DOI] [PubMed] [Google Scholar]
- 16.Lee BJ, Emery-Sinclair EL, Mackenzie RW, Hussain A, Taylor L, James RS, Thake CD. The impact of submaximal exercise during heat and/or hypoxia on the cardiovascular and monocyte HSP72 responses to subsequent (post 24 h) exercise in hypoxia. Extreme Physiol Med. 2014;3:15. doi: 10.1186/2046-7648-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis MS, Min JN, Wang S, McDonough H, Lockyer P, Wadosky KM, Patterson C. Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise. Cell Biochem Funct. 2013;31(8):724–735. doi: 10.1002/cbf.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang XL, Hu XL, Zhang YA, Qin FZ, Zhang WF. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim Biophys Acta. 2015;1852(5):805–815. doi: 10.1016/j.bbadis.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 20.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS One. 2012;7(10):e46834. doi: 10.1371/journal.pone.0046834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013;5(5):427–433. doi: 10.1159/000351979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurusamy N, Das DK. Autophagy, redox signaling, and ventricular remodeling. Antioxid Redox Signal. 2009;11(8):1975–1988. doi: 10.1089/ars.2009.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omatsu-Kanbe M, Matsuura H. Ischemic survival and constitutively active autophagy in self-beating atypically-shaped cardiomyocytes (ACMs): characterization of a new subpopulation of heart cells. J Physiol Sci. 2013;63(1):17–29. doi: 10.1007/s12576-012-0236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarrett CL, D’Lugos AC, Mahmood TN, Gonzales RJ, Hale TM, Carroll CC, Dickinson JM, Angadi SS (2016) Effect of high intensity exercise preconditioning and training on antioxidant enzymes in cardiomyocytes during doxorubicin treatment. FASEB J 30(1 Supplement):lb601–lb601
- 26.Wang L, Deng W, Yuan Q, Yang H. Exercise preconditioning reduces ischemia reperfusion-induced focal cerebral infarct volume through up-regulating the expression of HIF-1alpha. Pak J Pharm Sci. 2015;28(2 Suppl):791–798. [PubMed] [Google Scholar]
- 27.Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr, Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res. 2010;3(4):365–373. doi: 10.1007/s12265-010-9189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Park S, Kim WK. Exercise preconditioning reduces acute ischemic renal injury in Hsp70.1 knockout mouse. Histol Histopathol. 2013;28(9):1223–1233. doi: 10.14670/HH-28.1223. [DOI] [PubMed] [Google Scholar]
- 30.Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12(2):149–156. doi: 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulbricht A, Hohfeld J. Tension-induced autophagy: may the chaperone be with you. Autophagy. 2013;9(6):920–922. doi: 10.4161/auto.24213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793(9):1485–1495. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Shen YJ, Pan SS, Zhuang T, Wang FJ. Exercise preconditioning initiates late cardioprotection against isoproterenol-induced myocardial injury in rats independent of protein kinase C. J Physiol Sci. 2011;61(1):13–21. doi: 10.1007/s12576-010-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lennon SL, Quindry JC, French JP, Kim S, Mehta JL, Powers SK. Exercise and myocardial tolerance to ischaemia-reperfusion. Acta Physiol Scand. 2004;182(2):161–169. doi: 10.1111/j.1365-201X.2004.01346.x. [DOI] [PubMed] [Google Scholar]
- 35.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300(4):C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy-cooperation and control. Autophagy. 2015;11(2):200–213. doi: 10.1080/15548627.2015.1009776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melling CW, Thorp DB, Milne KJ, Krause MP, Noble EG. Exercise-mediated regulation of Hsp70 expression following aerobic exercise training. Am J Physiol Heart Circ Physiol. 2007;293(6):H3692–H3698. doi: 10.1152/ajpheart.00827.2007. [DOI] [PubMed] [Google Scholar]
- 38.Hung CH, Chang NC, Cheng BC, Lin MT. Progressive exercise preconditioning protects against circulatory shock during experimental heatstroke. Shock. 2005;23(5):426–433. doi: 10.1097/01.shk.0000159557.95285.96. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Adam A, Cheng Y, Tang S, Hartung J, Bao E. Localization and expression of heat shock protein 70 with rat myocardial cell damage induced by heat stress in vitro and in vivo. Mol Med Rep. 2015;11(3):2276–2284. doi: 10.3892/mmr.2014.2986. [DOI] [PubMed] [Google Scholar]
- 40.Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W, Hohfeld J. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11(3):538–546. doi: 10.1080/15548627.2015.1017186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Periard JD, Ruell PA, Thompson MW, Caillaud C. Moderate- and high-intensity exhaustive exercise in the heat induce a similar increase in monocyte Hsp72. Cell Stress Chaperones. 2015;20(6):1037–1042. doi: 10.1007/s12192-015-0631-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doong H, Rizzo K, Fang S, Kulpa V, Weissman AM, Kohn EC. CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: accumulation of poly-ubiquitinated Hsp90 client proteins. J Biol Chem. 2003;278(31):28490–28500. doi: 10.1074/jbc.M209682200. [DOI] [PubMed] [Google Scholar]
- 43.Mozaffari MS, Liu JY, Abebe W, Baban B. Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am J Cardiovasc Dis. 2013;3(4):180. [PMC free article] [PubMed] [Google Scholar]
- 44.Hishiya A, Kitazawa T, Takayama S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ Res. 2010;107(10):1220–1231. doi: 10.1161/CIRCRESAHA.110.225649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson NM, Greufe SE, Hydock DS, Hayward R. Doxorubicin-induced vascular dysfunction and its attenuation by exercise preconditioning. J Cardiovasc Pharmacol. 2013;62(4):355–360. doi: 10.1097/FJC.0b013e31829c9993. [DOI] [PubMed] [Google Scholar]
- 46.Bloemberg D, McDonald E, Dulay D, Quadrilatero J. Autophagy is altered in skeletal and cardiac muscle of spontaneously hypertensive rats. Acta Physiologica (Oxford, England) 2014;210(2):381–391. doi: 10.1111/apha.12178. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109(3):296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park S, Choi SG, Yoo SM, Son JH, Jung YK. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy. 2014;10(11):1906–1920. doi: 10.4161/auto.32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang P, Mizushima N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods. 2015;75:13–18. doi: 10.1016/j.ymeth.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 50.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paula-Gomes S, Goncalves DA, Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Insulin suppresses atrophy- and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res. 2013;45(12):849–855. doi: 10.1055/s-0033-1347209. [DOI] [PubMed] [Google Scholar]
- 52.Dokladny K, Zuhl MN, Mandell M, Bhattacharya D, Schneider S, Deretic V, Moseley PL. Regulatory coordination between two major intracellular homeostatic systems: heat shock response and autophagy. J Biol Chem. 2013;288(21):14959–14972. doi: 10.1074/jbc.M113.462408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 54.Perez-Perez ME, Zaffagnini M, Marchand CH, Crespo JL, Lemaire SD. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy. 2014;10(11):1953–1964. doi: 10.4161/auto.34396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharifi MN, Mowers EE, Drake LE, Macleod KF. Measuring autophagy in stressed cells. Methods Mol Biol. 2015;1292:129–150. doi: 10.1007/978-1-4939-2522-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gurusamy N, Das DK. Is autophagy a double-edged sword for the heart? Acta Physiol Hung. 2009;96(3):267–276. doi: 10.1556/APhysiol.96.2009.3.2. [DOI] [PubMed] [Google Scholar]
- 57.Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–247. doi: 10.4161/auto.6.2.11062. [DOI] [PubMed] [Google Scholar]
- 58.Smuder AJ, Kavazis AN, Min K, Powers SK. Doxorubicin-induced markers of myocardial autophagic signaling in sedentary and exercise trained animals. J Appl Physiol (Bethesda, Md: 1985) 2013;115(2):176–185. doi: 10.1152/japplphysiol.00924.2012. [DOI] [PubMed] [Google Scholar]