

Abstract
We examined the role of nitric oxide (NO) in muscle repair and regeneration following repetitive eccentric contractions (ECC). A standardized exercise protocol was used to create eccentric contraction-induced injury to the left tibialis anterior muscle of 48 male Wistar rats (body wt 250–350 g), using a customized isokinetic test device and a bout of 40 ECCs under electrical stimulation. A nitric oxide synthase inhibitor, N(G)-nitro-l-arginine-methyl ester (l-NAME; 35 mg kg−1 day−1), was included in the diet for half the animals (n = 24) beginning 3 days prior to the ECC and continuing throughout the experiment, whereas the other half (n = 24) received a control diet. ECC/+l-NAME and ECC/−l-NAME were killed after the ECC protocol at 0, 1, 3 and 7 days (n = 6 on each day). An unexercised contralateral limb with and without l-NAME infusion served as a respective control muscle at each time point. Muscle NO content, skeletal muscle damage, leukocyte infiltration, calpain activity, and MyoD and myogenin expression were assessed. NO has both pro-inflammatory and anti-inflammatory properties, and several possible roles for NO in skeletal muscle damage have been postulated. NO content was greater in the ECC/−l-NAME group at all time points (p < 0.05) compared to ECC/+l-NAME. Additionally, significant differences in NO content were observed on day 0 (p < 0.05), and day 3 (p < 0.05), ECC/+l-NAME versus ECC/−l-NAME. One day following the bout of ECC, and NO levels were increased in the ECC/−l-NAME group. Three days following ECC, there was greater myofiber damage (measured by β-glucuronidase activity) and leukocyte invasion in the ECC/−l-NAME group as compared to the ECC/+l-NAME group. One day after ECC, calpain activity was significantly increased in ECC/−l-NAME compared with control muscles (p < 0.05). On days 3 and 7, Myo-D and myogenin gene expression was increased in both groups; however, the degree of regeneration was less in the ECC/+l-NAME-treated animals. These data suggest that NO dynamics have important implications in the regulation of various factors during skeletal muscle regeneration following damaging eccentric muscle contractions.
Keywords: Skeletal muscle, Nitric oxide, Eccentric muscle contraction, Regeneration
Introduction
Unaccustomed exercise involving eccentric contractions (ECC) induces skeletal muscle damage including decreased muscle strength, muscle edema, and loss of function. The sequence of events during skeletal muscle regeneration has been well characterized by remodeling of the extracellular and intracellular protein networks, which is maintained by a concerted regulation of the balance between protein synthesis and degradation.
It is well known that nitric oxide (NO) plays an important role in regulating blood flow and glucose metabolism [1], as well as the cellular redox state of skeletal muscle [2]. NO may also influence expression of antioxidant enzymes, as well as cellular inflammation in exercised skeletal muscles [3]. NO is produced in vivo through the conversion of l-arginine to l-citrulline by nitric oxide synthase (NOS). Skeletal muscle normally expresses the nNOS and eNOS isoforms of NOS. iNOS is also expressed in skeletal muscle in inflammatory conditions, and activation of iNOS can elicit NO in quantities sufficient to stimulate the inflammatory process [4–9]. Because NO has both pro-inflammatory and anti-inflammatory properties, several possible roles for NO in skeletal muscle damage have been postulated. NO produced endogenously by skeletal muscle and other cell types has the potential to inhibit calpain activity and cytoskeletal proteolysis [10]. The possibility that NO plays a role in skeletal myogenesis is suggested by observations that it participates in satellite cell activation [11, 12]. Moreover, iNOS is a muscle-injury inducer both in vitro and in vivo that triggers the loss of MyoD mRNA [13]. Thus, the precise role of NO in skeletal muscle damage and repair is not completely known.
Calpain proteins are a family of Ca2+-activated non-lysosomal proteases that have been reported to cleave muscle proteins. Three calpain family members are well characterized in skeletal muscle. μ-calpain and m-calpain are ubiquitously expressed and exhibit in vitro sensitivity to Ca2+ in the micro-molar and milli-molar range, whereas n-calpain exhibits sensitivity in the more physiologically feasible nano-molar Ca2+-range and is expressed predominantly, but not exclusively, in skeletal muscle [14]. NO modulates the mechanical behavior of skeletal muscle cells through a decrease in calpain-mediated cytoskeletal proteolysis [15]. Thus, NO inhibition of calpain may provide a potential therapeutic approach to protect from skeletal muscle injury [10].
In adult skeletal muscle, myogenic differentiation factor D (MyoD) and myogenin are both expressed at relatively low levels [16, 17]. However, in response to injury, expression of MyoD is related to activation and proliferation of satellite cells, whereas myogenin reflects terminal myoblast differentiation [18]. Several studies have aimed to study the expression of MyoD or myogenin in skeletal muscle, but, to the best of our knowledge, no study has shown their presence in damaged skeletal muscle specifically after sessions of ECC [19].
In this study, we observed the repair process for 7 days after mechanically damaging the tibial anterior (TA) muscles of rats treated with NO synthesis inhibitors (l-NAME). The purpose of the present investigation was to examine the role of NO in skeletal muscle regeneration following ECC. Specifically, we were interested in the time course for regeneration and the potential role for factors such as calpain and their effects on NO and muscle regeneration.
Methods
Animal care
Forty-eight male Wistar rats (body wt 250–350 g) were housed individually and fed food and water ad libitum in a temperature-controlled room on a 12:12 h light:dark cycle. All experiments on animals were performed following approval by the Tokyo University of Agriculture Research Animals and Resource Center Review Committee.
Experimental protocol
Following 1 week of acclimation in the laboratory, animals were randomly divided into two groups: one group (n = 24) received N(G)-nitro-l-arginine-methyl ester (l-NAME; NOS inhibitor, 35 mg kg−1 day−1) in drinking water starting 3 days before ECC and continuing until the end of the experiment. A second (n = 24) control group received tap drinking water during the experiment.
Eccentric contraction injury induction
The eccentric exercise was performed according to a condition of Kano et al. [20]. Briefly, during eccentric exercise and all surgical procedures, the rats were anesthetized with intraperitoneal injection of pentobarbital sodium (70 mg kg−1 i.p.) and supplemental doses of anesthesia were administered, as needed. The right TA muscle was stimulated electrically via a surface electrode (10 V stimulation, 100 Hz frequency, 700 ms stimulation period, i.e., 70 pulses). We confirmed that maximum muscle tension was achieved by electrical stimulation of a surface electrode (100 Hz, <10 V). In the resting condition before ECC, the right foot was attached to the clamp unit and the plate was connected to the electromotor system (Model RU-72; NEC medical systems). The right ankle joint was maintained at 50° as the initial angle and, during electro-stimulation of the TA muscle, the electromotor was rotated at an angular velocity of 260° s−1 to 180° of the ankle joint to lengthen the dorsiflexor muscle group. The muscle tension generated during ECC was monitored using a strain gauge that was incorporated into the plate fixing the foot. The strain gauge was calibrated using precision calibration weights that spanned the expected range of strains. The right TA muscle was subjected to 40 repeated ECCs. The TA muscle undergoing ECC treatment was isolated at 0, 1, 3, and 7 days. Animals were divided into two groups for each of the 4 time points: (1) ECC/−l-NAME, no l-NAME infusion (n = 6 on each day), and (2) ECC/+l-NAME (n = 6 on each day). An unexercised contralateral limb with and without l-NAME infusion served as a respective control muscle at each time point.
Histological evaluation for muscle damage
After the ECC protocols, the TA muscles of both legs were carefully dissected and the mid-belly region cut transversely to the long axis of the muscle. The tissue blocks were rapidly frozen in isopentane cooled by liquid nitrogen. Transverse 10-μm cross-sections were made with a cryostat (CM1510; Leica) at −20 °C and stained with hematoxylin–eosin (HE) to examine the histological features of muscle damage. To avoid sampling bias, each section was sub-sampled at three different regions (anterior, central, posterior) and each of these fields were analyzed in all muscles. Muscle fiber damage was determined by a point-counting method using a 24 × 33 mounted grid (i.e., 792 points total; one square = 30 × 30 μm) on microscopic fields. Damaged fibers were defined as those with infiltration of inflammatory cells, pale staining of the cytoplasm, swollen appearance. Muscle damage was expressed as a percentage of counted grid squares.
β-Glucuronidase activity assay
β-Glucuronidase activity was assayed in essence according to Koskinen et al. [21]. Briefly, 450 μl 0.1 M acetate buffer, pH 4.2, was added to 50 μl supernatant from muscle homogenate and incubated for 5 min at 37 °C. Next, 250 μl substrate (5 mM p-nitrophenyl-β-d-glucuronide; Sigma, St. Louis, MO, USA) was added and incubated 18 h at 37 °C. After overnight incubation, 1.5 ml 0.1 M glycine buffer (ice-cold), pH 10, was added. The tubes were centrifuged at 3,000g for 10 min at 4 °C. The absorbances were measured at the wavelength 420 nm. The results were calculated per soluble protein and incubation time. Protein concentration was measured using a commercial kit (Bio-Rad).
NO content
Samples were homogenized at 0–4 °C in ice-cold 0.1 M K2HPO4–KH2PO4 (pH 7.4) buffer (wt:vol, 1:10) with a motor-driven Potter-Elvehjem teflon glass homogenizer. Quantification of NO content was performed spectrophotometrically [22]. A range of standard solutions (concentration: 0–30 μM NO) was prepared using KNO3 as a standard. Because the Griess reagent measures only nitrite, and biological systems contain both nitrite and nitrate, addition of 0.01 U nitrate reductase (Sigma) was added to each well, followed by addition of NADH (0.02 mM). Samples were read at 540 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA) and the results were expressed as pmol/mg protein. Pilot assays established inter/intraassay variability of less than 3 % (data not shown).
Reverse-transcription polymerase chain reaction (RT-PCR) Analysis
Total RNA was prepared from the skeletal muscles using ISOGEN (NIPPON GENE, Tokyo, Japan). First-strand complementary DNA was obtained by incubating total RNA samples (2 μg) with reverse transcriptase (Superscript II; GIBCO BRL, Gaithersburg, MD, USA) in the reaction mixture (18 μl). The reverse transcription product (1 μl) was subjected to polymerase chain reaction (PCR) using Taq DNA polymerase (Perkin Elmer, Branchburg, NJ, USA). Twenty to thirty cycles of amplification were carried out using the following conditions for each cycle: denaturing at 94 °C for 1 min, annealing at 72 °C for 2 min, and extension at 72 °C for 3 min. The PCR products were electrophoresed in 1 % agarose gels containing ethidium bromide. The intensity of the bands was estimated by scanning with a Light-Capture Scanner (ATTO, Tokyo, Japan), and the optical density of each band was analyzed with the CS Analyzer (ATTO). The primers used are described as follows:
Myo D forward: 5′-CTACAGCGGCGACTCAGACG-3′
Myo D reverse: 5′-AGGATGTAGGCCGGGGTT-3′
Myogenin forward: 5′-ACTACCCACCGTCCATTCAC-3′
Myogenin reverse: 5′-TCTCTGTCACTCACGGGGCT-3′
GAPDH forward: 5′-CCAAAAGGGTCATCATCTCC-3′
GAPDH reverse: 5′-GGAGTTGCTGTTGAAGTCAC-3′
Solution calpain assay
Calpain activity was measured as described [23]. Briefly, 0.25 U m-calpain was solubilized in 0.4 ml 50 mM Tris–HCl, pH 7.4, containing 1 mM DTT, 5 mM CaCl2 or EDTA, and 0.5 %-casein. Samples were preincubated with varying concentrations of calpain inhibitor I for 15 min on ice. All samples were then incubated at room temperature for 30 min, and proteolysis was stopped by addition of 0.3 ml 10 % trichloroacetic acid. Samples were placed on ice for 15 min, centrifuged for 15 min at 12,000g, and the A280 of the supernatant was measured. Calpain activity was defined as the Ca2+-dependent increase in A280.
Data and statistical methods
All values are represented as mean ± SE. Statistical analysis was performed using two-way ANOVA and post hoc Scheffe test using the Prism (v.4.0). Statistical significance was accepted at p < 0.05.
Results
Muscle histology
In the control and 0 day muscles, myofibers demonstrated no visible evidence of muscle inflammation. In contrast, typical histological lesions appeared in the ECC muscle fiber around 1–3 days (Fig. 1). Area percentage of inflammation and regeneration muscle fibers was determined by microscopic fields (Table 1). On 1 day after ECC, some myofibers were swollen (ECC/−l-NAME: 5.2 ± 2.2 %, ECC/+l-NAME: 6.1 ± 1.5 % of total fiber area). On day 3, myofiber disruption was evident and the extent of damaged fiber area was significantly increased (ECC/−l-NAME: 43.1 ± 8.5 % of total fiber area, ECC/+l-NAME: 35.7 ± 7.4 % of total fiber area, p < 0.01 vs. day 0). Muscle regeneration was observed at 7 days following ECC, characterized by small diameter fibers with multiple central nuclei (ECC/−l-NAME: 6.5 ± 2.3 % of total fiber area). Animals infused with l-NAME had significant difference in regeneration at day 7 (ECC/+l-NAME: 3.7 ± 1.4 % of total fiber area, p < 0.05 vs. ECC/−l-NAME).
Fig. 1.
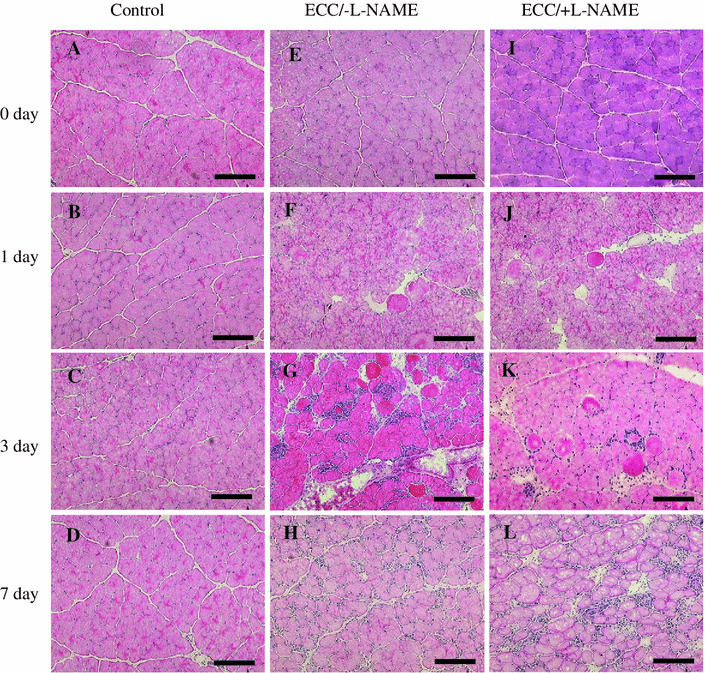
Histological analysis of muscle sections by H&E staining. Representative effects of ECC in tibialis anterior (TA) muscles at 0 day (e, i), 1 day (f, j), 3 days (g, k) and 7 days (h, l). Effects of ECC without l-NAME (e–h). Effects of ECC with l-NAME (i–l). Serial transverse sections from control [contralateral (left) no-ECC/−l-NAME muscle] (a–d). Bar 100 μm
Table 1.
Area percentage of inflammation and regeneration muscle fibers
| Control | 1 day | 3 days | 7 days | |
|---|---|---|---|---|
| Inflammation fibers area (%) | ||||
| ECC/−l-NAME | 0.0 ± 0.0 | 5.2 ± 2.3 | 43.1 ± 8.5** | 0.0 ± 0.0 |
| ECC/+l-NAME | 0.0 ± 0.0 | 6.1 ± 1.5 | 35.7 ± 7.4** | 0.0 ± 0.0 |
| Regeneration fibers area (%) | ||||
| ECC/−l-NAME | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 6.5 ± 2.3* |
| ECC/+l-NAME | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 3.7 ± 1.4 |
Control contralateral (left) no-ECC muscle. Values represent mean ± SEM
* p < 0.05, significantly different from ECC/+l-NAME; ** p < 0.01, significantly different from control
β-Glucuronidase activity
β-Glucuronidase activity was measured to provide us with an indirect assessment of muscle damage (Fig. 2). β-Glucuronidase activity was elevated 3 days after ECC in all groups, the increase was more pronounced in ECC/−l-NAME muscles than ECC/+l-NAME (p < 0.05). ECC/−l-NAME muscles showed increased β-glucuronidase activity at 3 days after ECC, as compared to 0, 1, and 7 days (p < 0.05). However, in ECC/+l-NAME muscles, β-glucuronidase activity increased continuously for 7 days.
Fig. 2.
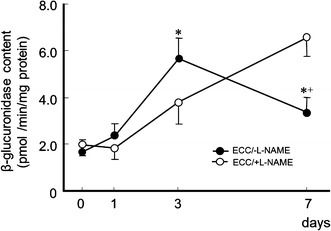
Time course for β-glucuronidase content at 0, 1, 3, and 7 days following ECC in rat TA muscle. Data are presented as the mean ± SEM for five animals at each time point. * Significantly different from ECC/+l-NAME. + Significantly different from 3 days from ECC/−l-NAME treatment
NO content
ECC/−l-NAME muscles showed decreased NO content at 1 day, as compared to muscles at 0, 3, and 7 days (p < 0.05) (Fig. 3). Baseline NO levels, in ECC/+l-NAME-treated animals were approximately 25 % of those of in ECC/−l-NAME group For all time points, ECC/−l-NAME values were significantly higher than ECC/+l-NAME (p < 0.05).
Fig. 3.
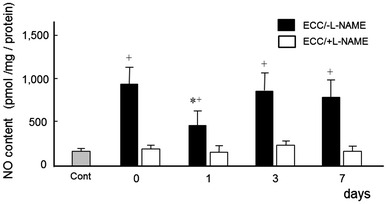
Time course for NO content at 0, 1, 3, and 7 days following ECC in rat TA muscle. NO data are presented as the mean ± SEM for five animals at each time point. Cont contralateral (left) no-ECC/−l-NAME muscle. * Significantly different from 0 day. + Significantly different from ECC/+l-NAME
Calpain activity
In ECC/+l-NAME muscles, calpain activity had no change with control muscles (Table 2). One day after ECC, calpain activity was significantly increased in ECC/−l-NAME compared with control muscles. But activity returned to the level of control muscles at 3 and 7 days.
Table 2.
Calpain activity
| Control | 0 day | 1 day | 3 days | 7 days | |
|---|---|---|---|---|---|
| ECC/−l-NAME | 13.4 ± 1.8 | 12.8 ± 1.9 | 26.3 ± 3.7* | 12.2 ± 4.4 | 14.2 ± 2.5 |
| ECC/+l-NAME | 12.8 ± 1.6 | 15.3 ± 3.2 | 16.6 ± 2.8 | 15.3 ± 3.5 | 17.9 ± 4.6 |
Expressed as nmol/min/g ww. Control contralateral (left) no-ECC muscle. Values represent mean ± SEM for five animals at each time point
* p < 0.05, significantly different from control values
Myogenin gene expression
Following ECC, myogenin mRNA levels in ECC/−l-NAME muscles significantly increased approximately fourfold after 3 and 7 days compared with the control condition (Fig. 4). In ECC/+l-NAME muscles; myogenin mRNA levels were increased only on day 7. Furthermore, l-NAME treatment resulted in a significant decrease in myogenin expression on days 0 and 3 (p < 0.05).
Fig. 4.
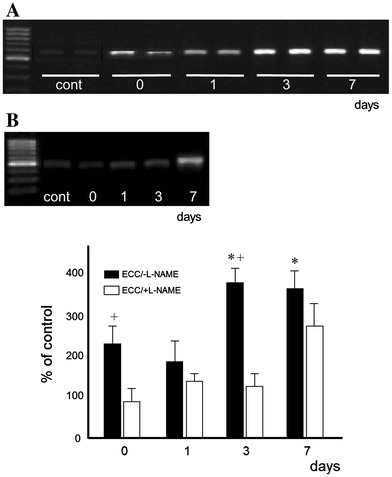
Myogenin mRNA expression. Samples were taken from rat immediately after ECC treatment muscles at times indicated. a ECC, b ECC/+l-NAME, cont contralateral (left) no-ECC/−l-NAME muscle. Values represent mean ± SEM for five animals at each time point, expressed as relative expression levels normalized by dividing by the GAPDH level. * Significantly different from 0 day. + Significantly different from ECC/+l-NAME
MyoD gene expression
In ECC/−l-NAME muscles, MyoD mRNA expression increased at day 1 (p < 0.05) and peaked at day 7 (~2.8-fold, p < 0.01) (Fig. 5). With ECC/+l-NAME treatment, MyoD mRNA level was higher on day 3, but significantly lower on day 7 compared to that in ECC/−l-NAME (p < 0.05).
Fig. 5.
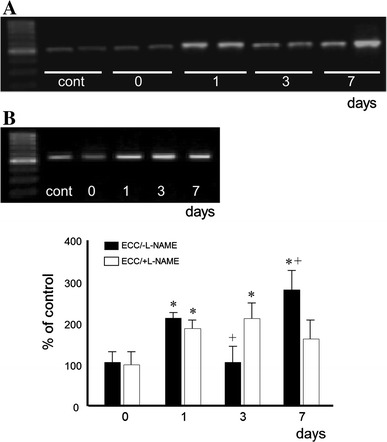
MyoD mRNA expression. Samples were taken from rat immediately after ECC treatment leg at times indicated. a ECC, b ECC/+l-NAME, cont contralateral (left) no-ECC/−l-NAME muscle. Values represent mean ± SEM for five animals at each time point, expressed as relative expression levels normalized by dividing by the GAPDH level. * Significantly different from 0 day. + Significantly different from ECC/+l-NAME
Discussion
NO production is known to increase dramatically in injured skeletal muscle [11, 24–26]. In addition, previous studies have shown that a decrease in NO accompanies inflammation and recovery after muscle injury [5, 6, 27]. However, there are many uncertainties regarding the role that NO plays in recovery after muscle injury. In this study, we examined the relationship between muscle NO levels resulting from altered NO synthesis and regeneration markers after eccentric contraction injury using a rat TA eccentric muscle injury model. The major finding in the current study was that NO dynamics have important implications in the regulation of various factors during skeletal muscle regeneration following damaging eccentric muscle contractions.
In our previous study, we reported that intramuscular NO concentration after injury exhibited a bimodal response [9]. In the current study, we reproduced this NO bimodality by showing a decreased NO concentration a day after ECC injury followed by an increase on day 3. On the other hand, the higher initial NO levels on day 0 compared with the control muscles may also have resulted from a general systemic blood-borne response from the injury as well as the surgery. The histological changes shown in Fig. 1, where muscle fiber damage caused by ECC on day 1 through day 3 began to recover by day 7 in animals with normal NO, but recovery was delayed in the NO-deprived animals, suggest to us that adequate NO exerts a crucial effect on muscle recovery after EEC-induced injury. Several studies have indicated that iNOS-derived NO is an important homeostatic regulator of leukocyte recruitment in the inflamed microcirculation, suggesting that one of its functions may be to act as an anti-inflammatory agent during inflammation [28].
Muscle injury was indirectly assessed by measuring β-glucronidase activity. This marker is a reliable indicator of necrosis caused by exercise-induced muscle injury [29–31]. In the ECC/−l-NAME group, ECC-triggered high β-glucronidase activity declined on day 7 in the presence of NO, whereas in ECC/+l-NAME rats, it continued to increase and almost doubled the level seen in ECC/−l-NAME rats. Hence, it may be that NO affected the balance between necrosis and apoptosis. This finding is in accordance with previous studies on the development of apoptosis due to ECC [32, 33].
The mRNA levels of MyoD, which are the activity and the proliferation marker of a satellite cell, and myogenin, which is the marker of myotube production, were both affected by the presence of NO. The expression of MyoD contributes to satellite cell proliferation [34]. When satellite cell proliferation enters the differentiation arrest step, the expression of myogenin is induced, and satellite cells differentiate into myotube cells. This stimulates maturation of myotube cells to become muscle fibers [35]. In this study, since myotube cells were found in large numbers in the tissue 3 days following ECC and the administration of l-NAME, it can be assumed that satellite cell proliferation and differentiation occurred. Under normal conditions, control of muscle cell differentiation that occurs after ECC injury is considered to be a cause of the significant increases in the expression of MyoD 1 day following injury and in the expression of myogenin 3 days after injury. An increase in NO suppresses MyoD [13]. Our results also showed that, as endogenous NO increased on day 3, MyoD was suppressed. These findings suggest that NO contributes to the time difference between the expression of MyoD and myogenin. Ulibarri et al. [36] demonstrated that myoblast proliferation is stimulated by sodium nitroprusside (SNP) and S-nitroso-N-acetyl-penicillamine (SNAP), but the addition of high concentrations of NO donor agents suppressed this stimulation. It was assumed that, since SNP is a calcium channel blocker, it had an effect on the expression of satellite cell proliferation markers according to the difference in calcium/calmodulin regulation. Myogenin and MyoD also contribute to regeneration in muscle fiber [37]. Myogenin plays an important role in the reconstruction of damaged neuromuscular connections [38] and the differentiation of terminal myoblasts [12]. There is a possibility that skeletal muscle damage accompanies regeneration, since this accounts for the fact that the expression of myogenin increased significantly from 3 days after injury.
Calpain plays an important role in the breakdown of proteins in skeletal muscle, inflammation, and induction during regeneration [39]. NO inhibits calpain activity. In addition, it has been reported that calpain reduces the expression of myogenic regulatory factors (MRFs), including myogenin [40]. The present study found further evidence to support these previous findings. The increase in calpain concentration 1 day after injury accelerates the disassembly of broken-down muscle protein, and that it is synchronized with satellite cell proliferation, as indicated by the expression of MyoD.
In summary, the results of the present study demonstrated the important role of NO in recovery after muscle injury. It was suggested that NO activation after injury exerts an effect on the expression of MRFs that occur during recovery, and that it makes a significant contribution to the reconstruction of tissue. Furthermore, there is a possibility that NO plays an important role in regulating both the destruction and the construction of tissue during recovery after skeletal muscle injury. Further research is required, since NO dynamics has important implications in the regulation of various factors during skeletal muscle regeneration.
Acknowledgments
The authors thank Dr. Mizuki Sudo (Central Research Institute for Physical Activity, Fukuoka University) for her expert technical assistance. This work was supported by Grant-in-Aid for Scientific Research (C) from the ministry of Education, Culture, Sports, Science and Technology of Japan (21500545). The work presented here was carried out in collaboration between all authors. Sakurai, Kashimura and Best defined the research theme. Sakurai and Izawa designed methods and experiments, carried out the laboratory experiments, analyzed the data, interpreted the results and wrote the paper. Kano and Ohno co-designed the dispersal and colonization experiments, and co-worked on associated data collection and their interpretation. Ji co-designed experiments, discussed analyses, interpretation, and presentation. All authors have contributed to, seen, and approved the manuscript.
Conflict of interest
The authors report no conflict of interest.
Reference
- 1.Kingwell BA. Nitric oxide as a metabolic regulator during exercise: effects of training in health and disease. Clin Exp Pharmacol Physiol. 2000;27:239–250. doi: 10.1046/j.1440-1681.2000.03232.x. [DOI] [PubMed] [Google Scholar]
- 2.Jackson MJ. Reactive oxygen species and redox-regulation of skeletal muscle adaptations to exercise. Philos Trans R Soc Lond B. 2005;360:2285–2291. doi: 10.1098/rstb.2005.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang CC, Lin TJ, Lu YF, Chen CC, Huang CY, Lin WT. Protective effects of l-arginine supplementation against exhaustive exercise-induced oxidative stress in young rat tissues. Chin J Physiol. 2009;52:306–315. doi: 10.4077/CJP.2009.AMH068. [DOI] [PubMed] [Google Scholar]
- 4.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filippin LI, Cuevas MJ, Lima E, Marroni NP, Gonzalez-Gallego J, Xavier RM. Nitric oxide regulates the repair of injured skeletal muscle. Nitric Oxide. 2011;24:43–49. doi: 10.1016/j.niox.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Filippin LI, Cuevas MJ, Lima E, Marroni NP, Gonzalez-Gallego J, Xavier RM. The role of nitric oxide during healing of trauma to the skeletal muscle. Inflamm Res. 2011;60:347–356. doi: 10.1007/s00011-010-0277-2. [DOI] [PubMed] [Google Scholar]
- 7.Soneja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol Rep. 2005;57:108–119. [PubMed] [Google Scholar]
- 8.Sriwijitkamol A, Christ-Roberts C, Berria R, Eagan P, Pratipanawatr T, DeFronzo RA, Mandarino LJ, Musi N. Reduced skeletal muscle inhibitor of kappaB beta content is associated with insulin resistance in subjects with type 2 diabetes: reversal by exercise training. Diabetes. 2006;55:760–767. doi: 10.2337/diabetes.55.03.06.db05-0677. [DOI] [PubMed] [Google Scholar]
- 9.Sakurai T, Hollander J, Brickson SL, Ohno H, Ji LL, Izawa T, Best TM. Changes in nitric oxide and inducible nitric oxide synthase following stretch-induced injury to the tibialis anterior muscle of rabbit. Jpn J Physiol. 2005;55:101–107. doi: 10.2170/jjphysiol.R614. [DOI] [PubMed] [Google Scholar]
- 10.Koh TJ, Tidball JG. Nitric oxide inhibits calpain-mediated proteolysis of talin in skeletal muscle cells. Am J Physiol Cell Physiol. 2000;279:C806–C812. doi: 10.1152/ajpcell.2000.279.3.C806. [DOI] [PubMed] [Google Scholar]
- 11.Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11:1859–1874. doi: 10.1091/mbc.11.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE. Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell. 2002;13:2909–2918. doi: 10.1091/mbc.E02-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, Marette A, Gallouzi IE. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol. 2005;25:6533–6545. doi: 10.1128/MCB.25.15.6533-6545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.König N, Raynaud F, Feane H, Durand M, Mestre-Francès N, Rossel M, Ouali A, Benyamin Y. Calpain 3 is expressed in astrocytes of rat and Microcebus brain. J Chem Neuroanat. 2003;25:129–136. doi: 10.1016/S0891-0618(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JS, Kraus WE, Truskey GA. Stretch-induced nitric oxide modulates mechanical properties of skeletal muscle cells. Am J Physiol Cell Physiol. 2004;287:C292–C299. doi: 10.1152/ajpcell.00018.2004. [DOI] [PubMed] [Google Scholar]
- 16.Costa A, Dalloul H, Hegyesi H, Apor P, Csende Z, Racz L, Vaczi M, Tihanyi J. Impact of repeated bouts of eccentric exercise on myogenic gene expression. Eur J Appl Physiol. 2007;101:427–436. doi: 10.1007/s00421-007-0510-z. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Capetanaki Y. Regulation of the mouse desmin gene: transactivated by MyoD, myogenin, MRF4 and Myf5. Nucleic Acids Res. 1993;21:335–343. doi: 10.1093/nar/21.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tajbakhsh S. Skeletal muscle stem cells in developmental versus regenerative myogenesis. J Intern Med. 2009;266:372–389. doi: 10.1111/j.1365-2796.2009.02158.x. [DOI] [PubMed] [Google Scholar]
- 19.Kadi F, Johansson F, Johansson R, Sjöström M, Henriksson J. Effects of one bout of endurance exercise on the expression of myogenin in human quadriceps muscle. Histochem Cell Biol. 2004;121:329–334. doi: 10.1007/s00418-004-0630-z. [DOI] [PubMed] [Google Scholar]
- 20.Kano Y, Sampei K, Matsudo H. Time course of capillary structure changes in rat skeletal muscle following strenuous eccentric exercise. Acta Physiol Scand. 2004;180:291–299. doi: 10.1111/j.0001-6772.2003.01250.x. [DOI] [PubMed] [Google Scholar]
- 21.Koskinen H, Järvisalo J, Huuskonen MS, Koivula T, Mutanen P, Pitkänen E. Serum lysosomal enzyme activities in silicosis and asbestosis. Eur J Respir Dis. 1983;64:182–188. [PubMed] [Google Scholar]
- 22.Nims RW, Cook JC, Krishna MC, Christodoulou D, Poore CM, Miles AM, Grisham MB, Wink DA. Colorimetric assays for nitric oxide and nitrogen oxide species formed from nitric oxide stock solutions and donor compounds. Methods Enzymol. 1996;268:93–105. doi: 10.1016/S0076-6879(96)68012-4. [DOI] [PubMed] [Google Scholar]
- 23.Graham-Siegenthaler K, Gauthier S, Davies PL, Elce JS. Active recombinant rat calpain II. Bacterially produced large and small subunits associate both in vivo and in vitro. J Biol Chem. 1994;269:30457–30460. [PubMed] [Google Scholar]
- 24.Kobzik L, Stringer B, Balligand JL, Reid MB, Stamler JS. Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships. Biochem Biophys Res Commun. 1995;211:375–381. doi: 10.1006/bbrc.1995.1824. [DOI] [PubMed] [Google Scholar]
- 25.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 26.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 27.Tidball JG. Inflammatory response to acute muscle stretch injury. Med Sci Sports Exerc. 1995;27:1022–1032. doi: 10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Hickey MJ, Sharkey KA, Sihota EG, Reinhardt PH, Macmicking JD, Nathan C, Kubes P. Inducible nitric oxide synthase-deficient mice have enhanced leukocyte–endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- 29.Komulainen J, Takala TE, Kuipers H, Hesselink MK. The disruption of myofibre structures in rat skeletal muscle after forced lengthening contractions. Pflugers Arch. 1998;436:735–741. doi: 10.1007/s004240050696. [DOI] [PubMed] [Google Scholar]
- 30.Komulainen J, Koskinen SO, Kalliokoski R, Takala TE, Vihko V. Gender differences in skeletal muscle fibre damage after eccentrically biased downhill running in rats. Acta Physiol Scand. 1999;165:57–63. doi: 10.1046/j.1365-201x.1999.00481.x. [DOI] [PubMed] [Google Scholar]
- 31.Salminen A. Lysosomal changes in skeletal muscles during the repair of exercise injuries in muscle fibers. Acta Physiol Scand Suppl. 1985;539:1–31. [PubMed] [Google Scholar]
- 32.Enns DL, Tiidus PM. Estrogen influences satellite cell activation and proliferation following downhill running in rats. J Appl Physiol. 2008;104:347–353. doi: 10.1152/japplphysiol.00128.2007. [DOI] [PubMed] [Google Scholar]
- 33.Sudo M, Kana Y. Myofibers apoptosis occur in the inflammation and regeneration phase following eccentric contractions in rats. J Physiol Sci. 2009;59:405–412. doi: 10.1007/s12576-009-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tidball JG, Wehling-Henricks M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J Physiol. 2007;578:327–336. doi: 10.1113/jphysiol.2006.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sassoon D, Lyons G, Wright WE, Lin V, Lassar A, Weintraub H, Buckingham M. Expression of two myogenic regulatory factors myogenin and MyoD1 during mouse embryogenesis. Nature. 1989;341:303–307. doi: 10.1038/341303a0. [DOI] [PubMed] [Google Scholar]
- 36.Ulibarri JA, Mozdziak PE, Schultz E, Cook C, Best TM. Nitric oxide donors, sodium nitroprusside and S-nitroso-N-acetylpencillamine, stimulate myoblast proliferation in vitro. In Vitro Cell Dev Biol Anim. 1999;35:215–218. doi: 10.1007/s11626-999-0029-1. [DOI] [PubMed] [Google Scholar]
- 37.Grounds MD, Garrett KL, Lai MC, Wright WE, Beilharz MW. Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 1992;267:99–104. doi: 10.1007/BF00318695. [DOI] [PubMed] [Google Scholar]
- 38.Sakuma K, Watanabe K, Sano M, Uramoto I, Sakamoto K, Totsuka T. The adaptive response of MyoD family proteins in overloaded, regenerating and denervated rat muscles. Biochim Biophys Acta. 1999;1428:284–292. doi: 10.1016/S0304-4165(99)00086-0. [DOI] [PubMed] [Google Scholar]
- 39.Koh TJ, Tidball JG. Nitric oxide inhibits calpain-mediated proteolysis of talin in skeletal muscle cells. Am J Physiol Cell Physiol. 2000;279:C806–C812. doi: 10.1152/ajpcell.2000.279.3.C806. [DOI] [PubMed] [Google Scholar]
- 40.Kook SH, Choi KC, Son YO, Lee KY, Hwang IH, Lee HJ, Chung WT, Lee CB, Park JS, Lee JC. Involvement of p38 MAPK-mediated signaling in the calpeptin-mediated suppression of myogenic differentiation and fusion in C2C12 cells. Mol Cell Biochem. 2008;310:85–92. doi: 10.1007/s11010-007-9668-2. [DOI] [PubMed] [Google Scholar]