

Abstract
We examined the effect of the cytosolic Ca2+ concentration ([Ca2+]c) in marginal cells on the asphyxia- or furosemide-induced decrease in the endocochlear potential (EP) by perfusing the endolymph with or without a Ca2+ chelator or inhibitors of Ca2+-permeable channels or Ca2+-pump during transient asphyxia or intravenous administration of furosemide. We obtained the following results. (1) Endolymphatic administration of SKF96365 (an inhibitor of TRPC and L-type Ca2+ channels) or EGTA-acetoxymethyl ester (EGTA-AM) significantly inhibited both the transient asphyxia-induced decrease in EP (TAID) and the furosemide-induced decrease in EP (FUID). (2) Endolymphatic perfusion with nifedipine significantly inhibited the TAID but not the FUID. (3) The recovery from the FUID was significantly suppressed by perfusing the endolymph with EGTA-AM, nifedipine, or SKF96365. (4) Endolymphatic administration of thapsigargin inhibited both the FUID and TAID. (5) The recovery rate from the FUID was much slower than that from the TAID, indicating that furosemide may inhibit the Ca2+-pump. (6) A strong reaction in immunohistochemical staining for TRPC channels was observed in the luminal and basolateral membranes of marginal cells. (7) A positive staining reaction for the γ subunit of epithelial Na+ channels was observed in the luminal and basolateral membranes of marginal cells. (8) Positive EP was diminished toward 0 mV by the endolymphatic perfusion with 10 μM amiloride or 10 μM phenamil. Taken together, these findings suggest that [Ca2+]c regulated by endoplasmic Ca2+-pump and Ca2+-permeable channels in marginal cells may regulate the positive EP, which is partly produced by the diffusion potential of Na+ across the basolateral membrane in marginal cells.
Keywords: Endocochlear potential, Intracellular Ca2+, TRPC channels, ENaC
Introduction
Recent review articles have indicated that Ca2+-permeable channels, such as TRP channels, play an important role in Ca2+-signal transduction in many cells, and that malfunction of TRP channels, referred to as a channelopathy, is the direct cause of various diseases [1, 2]. Despite a few experiments with respect to the Ca2+-permeable channels in the marginal cells of the stria vascularis [3–7], the effect of these channels on the regulation of the endocochlear potential (EP) has not been precisely analyzed during anoxia or the administration of diuretics.
EP playing an essential role in the transduction of sound by hair cells has been considered to occur across the marginal cells in the cochlear stria vascularis [8, 9]. As regards the mechanisms for the generation of the EP, however, two hypotheses have been proposed over the past 20 years. The first hypothesis is that the basolateral membrane of marginal cells is primarily Na+ conductive and is the source for the positive EP [10]. The second one is that the K+ diffusion potential across the inner membrane of basal/intercalated cells produces EP; the intrastrial space, i.e., the interstitial space between marginal and basal cells, has a large positive potential with respect to the perilymph [11, 12].
In preceding reports, we provided evidence that nifedipine has the inhibitory effect on the transient asphyxia-induced decrease in the EP (TAID) [4, 6], that the L-type Ca2+ channels are expressed in the basolateral membrane in marginal cells, and that other Ca2+-permeable channels in marginal cells may also contribute to the regulation of the EP [6]. Studies from our laboratory also demonstrated that the endolymphatic application of EGTA-acetoxymethyl ester (EGTA-AM) almost completely inhibited the furosemide-induced decrease in the EP (FUID) but only partially inhibited the TAID [13]. These findings suggest that the FUID and TAID might be caused by the increased influx of Ca2+ into the endolymphatic surface cells (ESC) through different Ca2+-permeable channels, although a large increase in the Ca2+ concentration in the endolymph with a concomitant fall in the EP might be induced by a decrease in the shunt resistance in the ESC and/or a decrease in the EP produced by transient asphyxia or the intravenous administration of diuretics [6, 14].
On the other hand, it was earlier reported that Ca2+-ATPase is abundant in the marginal cells [15–18], and immuno-cytochemical and histochemical studies also demonstrated significantly greater expression of plasma membrane Ca2+-ATPase (PMCA) and endoplasmic reticulum Ca2+-ATPase (SERCA) in the stria vascularis than in the spiral ligament and the organ of Corti of the guinea pig [16, 17]. Therefore, we consider that cytosolic Ca2+ concentration ([Ca2+]c) regulated by the Ca2+-pump and Ca2+-permeable channels in the marginal cells plays an important role in maintenance of the EP.
The aim of the present study was to examine whether Ca2+-permeable channels, such as L-type Ca2+ channels or TRPC channels, and SERCA in ESC co-operatively regulate the EP. Moreover, we analyzed the function of epithelial Na+ channels (ENaC) in the cells of the stria vascularis because ENaC is known to be regulated by [Ca2+]c [19] and is considered to produce the positive EP [9, 20]. The present results indicate that maintaining the [Ca2+]c at a suitable level via the Ca2+-pump and Ca2+-permeable channels in the marginal cells may be a pivotal factor for maintaining the large positive EP, which is partly generated by Na+ diffusion potential across the basolateral membrane in the marginal cells.
Methods
Animal preparation and recording system
Experimental methods are similar to those described in preceding papers [6, 13, 14]. Guinea pigs with a normal Preyer reflex and weighing between 300 and 400 g were anesthetized by an intraperitoneal injection of pentobarbital sodium (28 mg/kg) and were artificially respired with room air through a tracheal cannula after the intramuscular injection of suxamethonium chloride (50 mg/kg). The ECG was monitored to check the circulatory condition of the experimental animals. The tympanic bulla was exposed, and a conventional microelectrode filled with 0.5 M KCl was inserted into the scala media at the second turn. Transient asphyxia was induced by interruption of the artificial ventilation for ~100 s, and long-term asphyxia was induced by stopping the artificial ventilation. Furosemide was administered via a jugular vein over a period of 60 s (total dose, 60 mg/kg). Tubocurarine (4 μM) was added to the animal to eliminate nerve-induced twitching of skeletal muscles and associated tissue movement. At the completion of all experimental protocols, the animals were given a lethal dose of pentobarbital sodium (i.v.).
The endolymphatic perfusion was conducted with solutions containing nifedipine (1 μg/ml, Sigma-Aldrich, St. Louis, MO), SKF96365 (100 μM, Calbiochem, San Diego, CA), 3,5-bistrifluoromethyl pyrazole derivative (BTP2, 100 μM, Calbiochem), thapsigargin (1 μM, Calbiochem), phenamil (10 μM, Calbiochem), amiloride (10 μM, Calbiochem), EGTA (Nacalai Tesque, Kyoto, Japan), or EGTA-AM (Calbiochem), while keeping the flow rate at 80–120 nl/min. The EGTA-containing solution was composed of 134 mM KCl, 25 mM KHCO3, 0.1 μM M Ca2+ (adjusted with 0.1 or 1 mM EGTA), and 5 mM HEPES bubbled with 5% CO2, 5% O2, and 90% N2. Although we discarded the data showing less than +70 mV of EP without endolymphatic perfusion as due to the damage to the stria vascularis, we collected some of the EP data with a value of less than +70 mV with endolymphatic perfusion because we found that endolymphatic perfusion with nifedipine or EGTA with EGTA-AM solution induced an increase (or recovery) of the EP to more than +70 mV.
The microelectrode for the measurement of the EP was connected to an electrometer (FD-223, WPI, Sarasota, FL) via a microelectrode holder (MEH1SF, WPI), and the output was recorded by a U-638 recorder (Nippon Denshi Kagaku, Tokyo, Japan) and MacLab 8 s (ADInstruments, New South Wales, Australia). The indifferent electrode was an Ag/AgCl wire connected via a KCl/agar bridge to a saline-soaked cotton wick placed on the exposed neck muscles. The rectal temperature of the animals was kept at 37°C. The results are expressed as the mean ± SD. Statistical significance was assessed by using the ANOVA test. P values <0.05 were considered to be significant. The care and use of animals in this study were approved by the Animal Care and Use Committee of Osaka Medical College.
Immunohistochemistry
Inner ear tissue from guinea pigs (n = 5, weighing 150–200 g) was used. The animals were deeply anesthetized by an intraperitoneal administration of sodium pentobarbital (0.5 mg/g body wt) and perfused via the heart with Ringer’s solution and then with 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate-buffered solution (PBS, pH = 7.4) [6, 21–23]. The inner ears were removed and postfixed for 2 h in the same fixative at 4°C. The specimens were then immersed in 10% EDTA-4Na (pH = 7.4) for 1 week at room temperature. After being briefly rinsed three times with PBS, the specimens were immersed in 30% sucrose in PBS overnight at 4°C. Sections (12-μm thick) were cut with a cryostat and air-dried at room temperature.
For immunohistochemical analysis of TRPC3/6/7 channels or ENaC, sections were incubated with Block Ace (Dainippon Pharmaceutical, Osaka, Japan) containing 0.1% Tritron X for 1 h at room temperature to control nonspecific reactions. They were then incubated with rabbit anti-TRPC3/6/7 channels (diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA), anti-α or β subunit of ENaC (diluted 1:100; Chemicon International, Temecula, CA) or anti-γ subunit of ENaC (diluted 1:300; Alomone Labs, Jerusalem, Israel) for 18 h at 4°C. The sections were then rinsed three times in PBS for 15 min at 4°C and incubated for 2 h with Alexa Fluor 488-conjugated goat anti-rabbit IgG (diluted 1:300; Molecular Probes, Eugene, OR) in 1.5% normal goat serum. The sections were finally examined with a confocal laser scanning microscope (LSM 510, Carl Zeiss, Oberkochen, Germany) equipped with 40× Plan-NEOFLUOAR (NA, 0.75) and 63× Plan-APOCHROMAT (NA, 1.40 oil) objectives. Immunoreactivity of each antibody was visualized as green fluorescence excited with a 488-nm argon laser, and nuclear staining with propidium iodide was visualized as red fluorescence obtained with a 543-nm HeNe laser. These two images taken from a single section (<~1.7 μm thickness) were merged to make one picture. As a negative control for immunostaining, other sections were incubated with IgG of nonimmune sera from the same species used for the primary antibody.
Results
Effect of endolymphatic perfusion with EGTA-AM, nifedipine, or SKF96365 on TAID or FUID
In a previous report, we indicated that endolymphatic perfusion with EGTA-AM inhibited the TAID or FUID [13]. Here, we again examined the effect of the endolymphatic application of EGTA-AM on the TAID and FUID. Application of EGTA-AM partially, but not completely, inhibited the TAID and FUID (Fig. 1a, b).
Fig. 1.
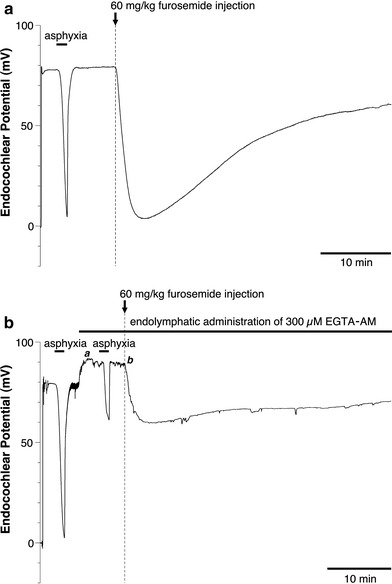
Typical recording of the EP after transient asphyxia or intravenous injection of 60 mg/kg furosemide with or without endolymphatic administration of 300 μM EGTA-AM. a Transient asphyxia for about 100 s or the intravenous administration of furosemide produces a rapid decrease in the EP. b After administration of EGTA-AM to the endolymph, the asphyxia- or the furosemide-induced decrease in the EP is significantly suppressed
The endolymphatic perfusion with nifedipine, an inhibitor of L-type Ca2+ channels, strongly inhibited the TAID and the recovery from the FUID, but only slightly reduced the FUID, which was not significantly different from that in the absence of nifedipine (Fig. 2a, see also Figs. 4, 5). By contrast, endolymphatic perfusion with SKF96365, an inhibitor of both L-type Ca2+ channels and TRPC channels, produced a large inhibition of both the TAID and FUID (Fig. 2b).
Fig. 2.
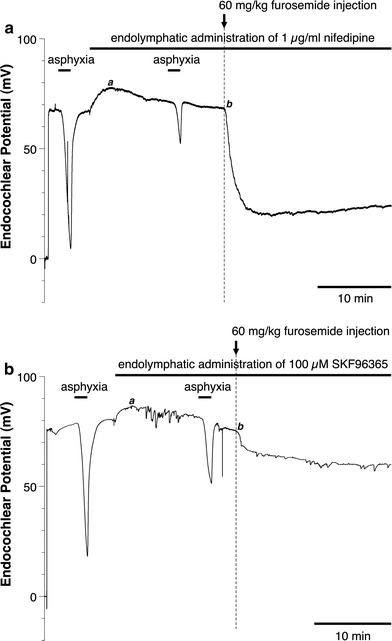
Recording of the EP after transient asphyxia or intravenous injection of 60 mg/kg furosemide with endolymphatic administration of 1 μg/ml nifedipine or 100 μM SKF96365. a Administration of nifedipine into the endolymph significantly suppresses the TAID, but only slightly reduces the FUID. b Administration of SKF96365 into the endolymph significantly suppresses both the TAID and FUID
Fig. 4.
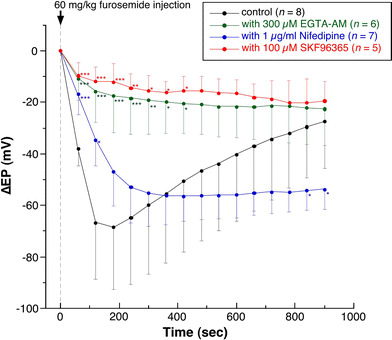
Summarized data on the changes in the decrease in EP induced by furosemide infusion with or without the endolymphatic administration of EGTA-AM, nifedipine, or SKF96365. Administration of EGTA-AM or SKF96365 into the endolymph significantly suppressed the FUID, but nifedipine did not. Moreover, in the presence of nifedipine, the recovery rate from the FUID was significantly retarded. *P > 0.05, **P > 0.01, ***P > 0.005 indicate significant differences from the corresponding data for the control experiment
Fig. 5.
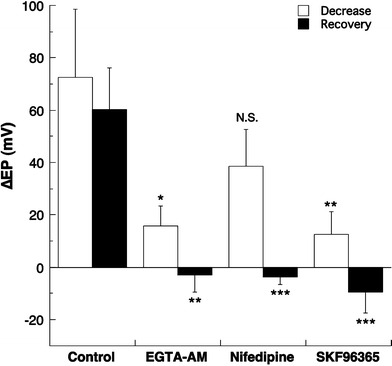
Summarized data on the maximum change in FUID and the recovery from it. The maximum change in FUID at 2.7 min after the furosemide injection was not significantly different from the recovery change in the FUID at 30 min after the maximum change in FUID. The recovery from FUID was significantly suppressed by the endolymphatic perfusion with EGTA-AM, nifedipine, or SKF96365, indicating that all of these drugs directly or indirectly suppress the Ca2+-pump in the ESC
These inhibitory effects of EGTA-AM, nifedipine, and SKF96365 on the TAID are summarized in Fig. 3, and those on the FUID in Fig. 4. The slight increase in the EP during the initial phase of endolymphatic perfusion throughout these experiments might have been due to the perfusion pressure, as reported previously [4, 6, 24, 25]. EGTA-AM, nifedipine, and SKF96365 significantly diminished the TAID, whereas the maximum value of FUID with nifedipine was significantly smaller than that with EGTA-AM or SKF96365. The inhibitory effect of EGTA-AM on the TAID was not different from that of nifedipine or SKF96365. Moreover, EGTA-AM and SKF96365 significantly diminished the FUID compared with the control value (only furosemide injection), and the mean diminution of the FUID was 74.6 or 82.4%, respectively, at 2.7 min after the furosemide injection. In contrast, nifedipine did not significantly affect the FUID at 2.7 min after the furosemide injection (see also Fig. 5). These results indicate that the FUID might be produced by the activation of SKF96365-sensitive channels (probably TRPC channels), but not nifedipine-sensitive channels. The maximum change in the FUID was observed at 2.7 min after the intravenous injection of furosemide (Fig. 4). No significant difference was observed between the value of maximum change in FUID and the recovery change in FUID at 30 min after the maximum change in FUID, indicating that the EP was almost recovered at ~30 min after the intravenous injection of furosemide.
Fig. 3.
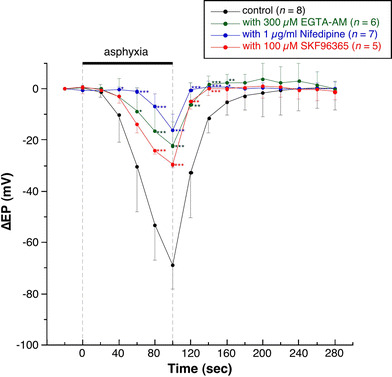
Summarized data on the changes in the decrease in EP induced by transient asphyxia with or without the endolymphatic administration of drugs. Administration of EGTA-AM, nifedipine, or SKF96365 into the endolymph significantly suppresses the asphyxia-induced decrease in EP. *P > 0.05, **P > 0.01, ***P > 0.005 indicate significant differences from the corresponding data for the control experiment
The changes in the FUID at 2.7 min and the recovery change in FUID at 30 min after the maximum change in FUID with the endolymphatic perfusion of EGTA-AM, nifedipine, or SKF96365 are also summarized in Fig. 5. As indicated there, in the presence of endolymphatic EGTA-AM, nifedipine, or SKF96365, the recovery from the FUID was suppressed, although the most inhibitory effect on the recovery change in the FUID was observed in the presence of nifedipine (P < 0.001, see also Figs. 2a, 4). These results suggest that in the presence of nifedipine, Ca2+-pump might be suppressed, or Ca2+ influx through the nifedipine-insensitive Ca2+-permeable channels was enhanced by furosemide administration in the ESC.
To assure the effects of furosemide on TRPC channels, we examined the effects of BTP2, which has a more specific inhibitory effect than SKF96365 on TRPC channels [26]. As shown in Fig. 6, BTP2 inhibited the FUID, indicating that furosemide may activate TRPC channels.
Fig. 6.
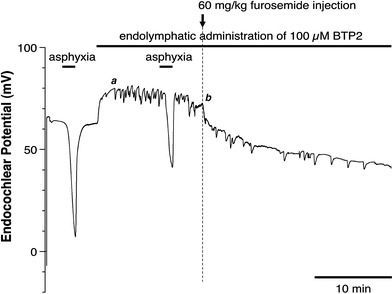
Effect of endolymphatic application of 100 μM BTP2 on TAID and FUID. Endolymphatic administration of BTP2 suppresses both the TAID and FUID and also suppresses the recovery from FUID
Effect of thapsigargin on TAID and FUID
It has been also reported that BTP2 is a useful tool to identify the function of store-operated Ca2+ entry [26]. In the next experiments, therefore, we examined the effect of thapsigargin on the TAID and FUID because BTP2 inhibited the TAID or FUID. If the TAID or FUID was caused by the store-operated Ca2+ influx through the activation of Ca2+-permeable channels in the ESC, application of thapsigargin may enhance the decrease in EP. After the initial increase in the EP induced by the endolymphatic perfusion, a small biphasic change in the EP (from a to d in Fig. 7) produced by an unknown mechanism was always observed in the endolymphatic perfusion with thapsigargin. However, the endolymphatic application of thapsigargin itself did not cause a decrease in EP after perfusion of 30 min (data are not shown). Furthermore, it partially inhibited the TAID by ~50% at the peak value and the FUID by ~30% at the peak value (Fig. 7). At ~30 min after the infusion of furosemide, the value for the EP with thapsigargin was +35 mV, which is significantly lower than that without thapsigargin (+60 mV). In the presence of thapsigargin, the FUID recovered quickly by ~15 mV, suggesting that PMCA or some other Ca2+ extrusion system in the ESC might have been activated under this condition. Furthermore, the gradual decrease in the EP (base line in the EP from c to d in Fig. 7, not TAID) as observed in Figs. 1b, 2a, b (from a to b) indicated that the Ca2+-pump in the ESC might have been suppressed. In any case, TAID, FUID, and recovery rate from the FUID were inhibited in the presence of thapsigargin, but we do not have any explanation for this presently. All of the above results indicate that keeping the [Ca2+]c at a suitable level in the ESC may be a pivotal factor for maintaining the large positive EP.
Fig. 7.
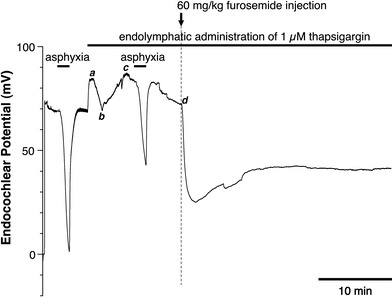
Effect of endolymphatic application of 1 μM thapsigargin on the TAID and FUID. Endolymphatic administration of thapsigargin suppresses both the TAID and FUID and also suppresses the recovery from FUID
Immunohistochemistry
In the next step, using an immunohistochemical method, we examined the existence of TRPC channels and ENaC in the cells of the stria vascularis.
TRPC3/6/7 channels
Strong immunoreactivity for TRPC channels was detected in the luminal side of the stria vascularis (Fig. 8). Furthermore, we also observed significant immunoreactivity for TRPC3/6/7 channels in the luminal and basolateral membrane of the marginal cells, whereas the intermediate cells and basal cells appeared to show faint immunoreactivity or failed to stain at all (Fig. 9). These results indicate that TRPC3/6/7 channels reside on the plasma membrane of marginal cells, but not on that of the intermediate or basal cells in the stria vascularis.
Fig. 8.
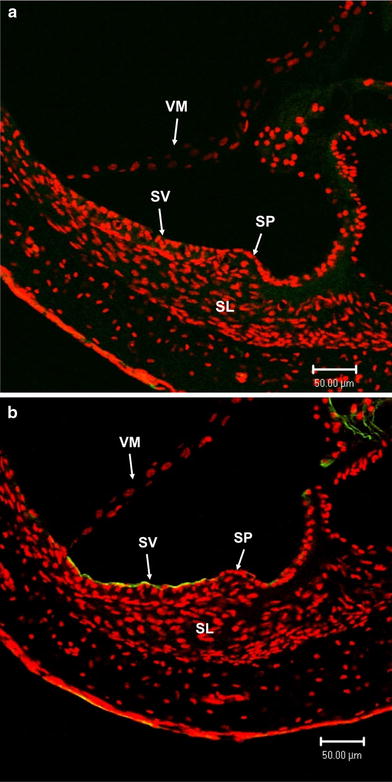
Expression of TRPC3/6/7 channels in the stria vascularis of cochlea. a Control. b Strong and significant immunoreactivity indicating TRPC3/6/7 channels (green) is seen in the luminal side in the stria vascularis
Fig. 9.
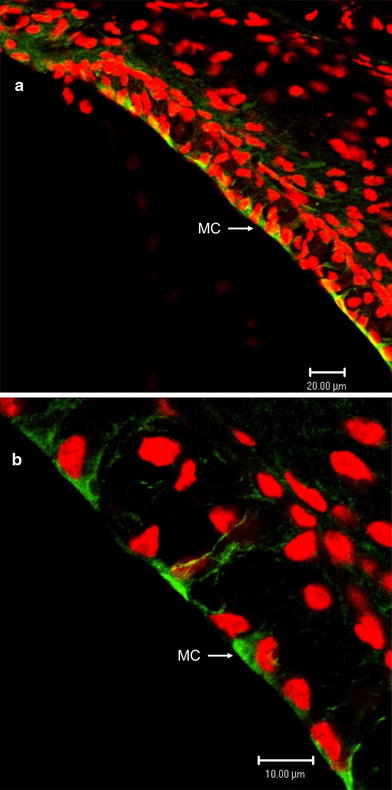
Expression of TRPC3/6/7 channels in the stria vascularis of cochlea. a Strong and significant immunoreactivity for TRPC3/6/7 channels (green) is seen in the marginal cells (MC) of the stria vascularis. b High magnification of the stria vascularis. Significant immunoreactivity from TRPC3/6/7 channels is evident in the luminal and basolateral membrane of marginal cells, but not in the intermediate or basal cells
ENaC
We also examined the existence of ENaC in the stria vascularis. As is shown in Fig. 10, significant immunoreactivity for the γ subunit of ENaC was seen in the luminal and basolateral membrane of marginal cells. However, the intermediate cells and basal cells appeared to show only faint immunoreactivity (Fig. 10). The α or β subunit of ENaCs failed to be specifically stained in the cells of the stria vascularis (data are not shown). These results indicate the presence of ENaC in the marginal cells.
Fig. 10.
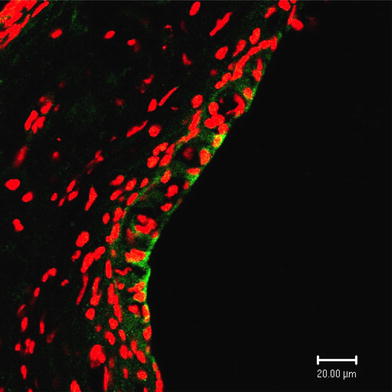
Expression of γENaC in the stria vascularis of the cochlea. High magnification of the stria vascularis. Significant and strong immunoreactivity indicating γENaC (green) is evident in the marginal cells, especially in their luminal and basolateral membranes
Effect of amiloride or phenamil on the EP
To confirm the presence of ENaC in the endolymphatic surface cells, especially in the marginal cells, we endolymphatically perfused amiloride or phenamil, an inhibitor of ENaC. As shown in Fig. 11, the EP was decreased toward 0 mV at ~5 min after the perfusion with amiloride or phenamil, indicating that the positive EP was generated by the ENaC in the basolateral membrane of the marginal cells. After the initial increase in the EP induced by the endolymphatic perfusion of amiloride or phenamil, a small biphasic change in the EP was always observed.
Fig. 11.
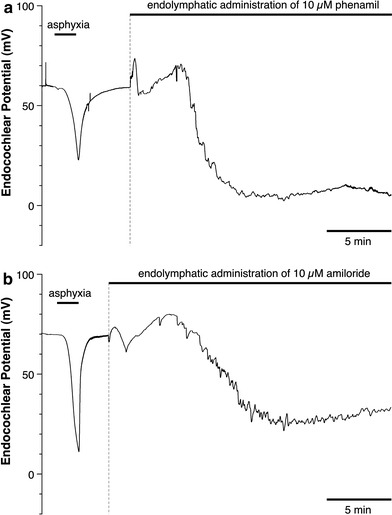
Effect of endolymphatic administration of 10 μM phenamil or 10 μM amiloride on the EP. a Effect of endolymphatic administration of phenamil on the EP. A few minutes after the administration of phenamil, the EP decreases to ~0 mV. b Effect of endolymphatic administration of amiloride on the EP. A few minutes after the administration of amiloride the EP drops to ~+20 mV. In both experiments, an initial biphasic change in the EP was observed, but this change cannot be explained at the present time
Discussion
The present study demonstrated that (1) endolymphatic perfusion with EGTA-AM, nifedipine, or SKF96365 inhibited both the TAID and FUID, (2) the recovery from the FUID was largely inhibited by endolymphatic application of nifedipine, and significantly inhibited by that of SKF96365 or EGTA-AM, (3) endolymphatic perfusion with thapsigargin inhibited both the TAID and FUID and suppressed the recovery rate from the FUID, (4) the recovery rate of the FUID was much slower than that of the TAID, indicating that furosemide itself may inhibit the Ca2+-pump in the cells of stria vascularis, and (5) TRPC3/6/7 channels and the γ subunit of ENaC were strongly stained immunohistochemically in luminal and basolateral membranes of marginal cells. These findings suggest that the cytosolic Ca2+ regulated by SERCA and Ca2+-permeable channels in marginal cells may play an important role in regulation of the large positive EP. Furthermore, γENaC was significantly stained in the marginal cells of the stria vascularis, and endolymphatic application of amiloride or phenamil decreased the EP to +20 to 0 mV. This result suggests that the positive EP may be generated partly by the Na+ diffusion potential across the basolateral membrane in the marginal cells.
Inhibitory effect of endolymphatic administration of nifedipine, SKF96365, BTP2, EGTA-AM, and thapsigargin on FUID
In a previous report, we demonstrated that endolymphatic administration of EGTA-AM almost completely inhibited the FUID. In the current study, we also demonstrated inhibition of the FUID by 75% in the presence of EGTA-AM and by 82% with SKF96365 at peak values. These results indicate that an increase in [Ca2+]c in marginal cells might be induced by furosemide, resulting in the decrease in EP, as suggested previously [6]. However, the recovery rate from the FUID with EGTA-AM, nifedipine, or SKF96365 was significantly different from that without these drugs, suggesting that the activity of the Ca2+-pump might be different between these two experimental conditions. In any case, we can consider four possible mechanisms for the furosemide-induced increase in [Ca2+]c in ESC: (1) The first possibility is that furosemide directly inhibits the Ca2+-pump. (2) The second one is that SKF96365- or BTP2-sensitive channels with Ca2+ permeability, such as TRPC channels, were activated directly by furosemide. (3) The third is that the intracellular Cl− concentration was decreased by the inhibition of Na–K–2Cl co-transporter in basolateral membrane, resulting in the release of Ca2+ from an intracellular store [27–29]. (4) The fourth possibility is the inhibition of ATP production [30, 31], resulting in the inhibition of Ca2+-pump.
The first possibility is probable because the recovery rate of the FUID was much slower than that of the TAID. Furthermore, a previous report suggested that the Ca2+-pump might not be inhibited but stimulated in the later phase of the TAID [6]. The second possibility is also probable because SKF96365 and BTP2 inhibited the FUID by 82% and about 70%, respectively, at 3 min after administration. Furthermore, TRPC3/6/7 channels were stained in the plasma membrane of the marginal cells. The third possible mechanism probably contributes little to the FUID because endolymphatic perfusion with thapsigargin inhibited only ~30% of the FUID (Fig. 6). Therefore, we consider that the FUID was not caused mainly by the release of Ca2+ from the intracellular Ca2+ store induced by the changes in intracellular Cl− concentration. Finally, the fourth possibility has little probability. It has been reported that a high concentration of furosemide or ethacrynic acid inhibited the electron transport system in mitochondria and suppressed the production of ATP [30, 31]. Recently, it has been reported that a decrease in the membrane potential of mitochondria produced the release of Ca2+ from mitochondria to cytosol [32, 33]. Histologically, abundant mitochondria were detected in marginal (intermediate) cells in the stria vascularis [9]. However, the ATP level in the cell might not change as quickly as the changes in the FUID.
In the current study, the most interesting phenomenon was the slower recovery rate from the FUID during the perfusion with the inhibitors of Ca2+-permeable channels or thapsigargin. In both cases, the recovery rate from the FUID was suppressed significantly from that without endolymphatic perfusion of these inhibitors (Figs. 4, 5). The suppression of the recovery rate from the FUID in the presence of inhibitors of Ca2+-permeable channels might also be explained by the compensatory increase in expression of TRPV5 and 6 during administration of furosemide in marginal cells, as has been already reported to occur in the renal distal tubule [34]. Furthermore, the suppression of the recovery rate from the FUID might be explained by the inhibition of Ca2+-pump after the administration of furosemide. In the current study, we did not study the effect of inhibitors of PMCA on the TAID and FUID because no specific inhibitors of PMCA are available at the present time.
If BTP2 is a useful tool to identify the function of store-operated Ca2+ entry [26], application of thapsigargin may enhance the TAID or the FUID because BTP2 inhibited the TAID or the FUID. Unexpectedly, thapsigargin suppressed both TAID and FUID (Fig. 7). Therefore, we considered that, at the normal extracellular Ca2+ concentration, store-operated Ca2+ entry does not work in the ESC. We propose that these phenomena could be explained by the functional linkage of Ca2+-permeable channels in plasma membrane and SERCA in the ESC. Finally, we should consider that endolymphatic perfusion with these drugs might suppress the Ca2+-pump and Ca2+-permeable channels in the ESC nonspecifically.
Taking all of the above into consideration, we conclude that [Ca2+]c regulated by SERCA and Ca2+-permeable channels in marginal cells may play an important role in regulating the large positive EP, although the mechanisms of the functional coupling of these systems are still unknown.
Mechanisms for the generation and regulation of the EP
Based on our previous and current studies, we would like to hypothesize mechanisms for the generation and the regulation of the EP, as shown in Fig. 12. The main mechanisms are as follows: (1) the EP is partly generated by Na+ diffusion potential across the basolateral membrane in the marginal cells, (2) this Na+ diffusion potential is regulated by [Ca2+]c in the marginal cells, which is regulated by the Ca2+-pump and Ca2+-permeable channels, and (3) cytosolic Ca2+ in the marginal cells regulates the shunt resistance.
Fig. 12.
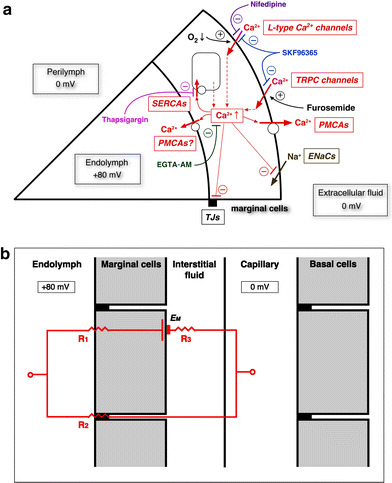
a Possible mechanisms of the regulation in ENaCs and tight junctions (TJs) by increase in [Ca2+]c in marginal cells. b Electrical circuit model in the stria vascularis. In this model, we propose the electrical shunt pathway through marginal cells, and we do not need to consider the negative potential of hair cells, as was proposed previously [9]. Negative EP should be analyzed under the consideration of shunt resistance in the ESC in the future. EP was almost determined by the shunt resistance, because EP ~ EM if the R 2 is much larger than R 1 or R 3. We considered that EM is mainly produced by Na+ diffusion potential across the basolateral membrane of marginal cells
As to the first mechanism, Offner et al. [10] proposed the hypothesis that the EP is generated by Na+ diffusion potential across the basolateral membrane in marginal cells. This hypothesis is supported by the experimental perfusion with a low Na+ solution [20]. However, it has been reported that amiloride perfusion does not affect the EP, although we found that the EP was decreased by the endolymphatic perfusion of 10 μM amiloride or 10 μM phenamil. Moreover, deafness has not been reported in knockout mice for the α subunit of ENaC, whereas the δ subunit was able to replace αENaC in forming functional Na+ channels [35–37]. The expression of this δ subunit overlaps with the expression of α, β, and γENaC in several tissues. Therefore, a difference in subunit composition might account for some variability in the sensitivity to amiloride [35].
In the present immunohistochemical study, we assured the presence of ENaC in the luminal and basolateral membranes of the marginal cells. Moreover, even when it is generally considered that there is no electrical potential in the intrastrial space because the claudin-11 knockout mouse is being used, ~+30 mV of EP still is present [38, 39]. Therefore, we conclude that the Na+ diffusion potential across the basolateral membrane in the marginal cells might partially contribute to the generation of the EP. Other types of TRP channels have been reported to be present in marginal cells [40]. These channels are nonselective cation channels. Therefore, it is possible that a cell-interior positive potential might be generated by Na+ diffusion through these channels.
As to the second mechanism, it is supported by previous immunohistochemical studies [2, 5–7, 15–17] and the current study, which demonstrate both Ca2+-ATPase and Ca2+-permeable channels in the marginal cells of the stria vascularis. In the present study, we demonstrated that the endolymphatic administration of EGTA-AM, nifedipine, SKF96365, BTP2, or thapsigargin inhibited the TAID or the FUID. Furthermore, ENaC is known to be inhibited by an increase in [Ca2+]c [18]. These findings suggest that the positive EP might be generated by Na+ diffusion potential at least in part in the marginal cells in which [Ca2+]c plays an important role for regulation of EP. In any case, further experiments are needed for the conclusion that the basolateral membrane potential of marginal cells is generated by the Na+ diffusion potential.
The third mechanism is supported by a previous experiment [6]. We calculated that Ca2+ flux during an asphyxia condition through tight junctions across the ESC was ~20 times larger than that during the nonasphyxia condition. As the TAID and FUID were inhibited by nifedipine, SKF96365, BTP2, and EGTA-AM, it is possible that either decrease of EP might be mainly caused by a decrease in the shunt resistance of ESC induced by the increase in [Ca2+]c in ESC, because Ca2+-permeable channels were strongly stained in the ESC (Figs. 8, 9 in the present study; and refs. [5, 6]), and several types of claudins constitute the tight junctions of ESC other than the organ of Corti [41]. It has been reviewed that these tight junctions are regulated by [Ca2+]c and that TRPC channels play an important role in regulating [Ca2+]c and signaling in epithelial cells [42]. Furthermore, it is reported that a decrease in the cochlear partition resistance (trans-stria vascularis resistance) was recorded along with a proportional decrease in the EP by endolymphatic perfusion with ATP [43]. This report suggests that the decrease in the shunt resistance through ESC would produce the decrease in EP, although there has been no discussion about the electrical shunt pathway through ESC except our previous report [6].
Thus, it is reasonable to conclude that the Ca2+ transport system in the ESC and ENaC in marginal cells may both contribute to the regulation and maintenance of the EP. In any case, further experiments are needed to clarify the role of Ca2+ and Na+ in the generation and regulation of the large positive EP in the guinea pig cochlea.
Acknowledgments
This study was supported by a grant-in aid for scientific research (No. 19590216 to T.K.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Prof. Yoshinori Marunaka, Kyoto Prefectural University of Medicine, for reading the manuscript.
References
- 1.Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- 2.Nilius B, Owsianik G, Votes T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke W, Choe Y, Marti-Renom MC, Bell AM, Denis CS, Sal A, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–535. doi: 10.1016/S0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nimura Y, Mori Y, Inui T, Takenaka H, Kubota T. Effects of CO2/HCO3 − in perilymph on endocochlear potential of guinea pigs. J Physiol Sci. 2007;57:15–22. doi: 10.2170/physiolsci.RP012006. [DOI] [PubMed] [Google Scholar]
- 5.Wangemann P, Nakaya K, Wu T, Maganti R, Erin MI, Sanneman J, Harbidge D, Billings S, Marcus DC. Loss of cochlear HCO3 − secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2007;292:F1345–F1353. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inui T, Mori Y, Watanabe M, Takamaki A, Yamaji J, Sohma Y, Yoshida R, Takenaka H, Kubota T. Physiological role of L-type Ca2+ channel in marginal cells in the stria vascularis of guinea pigs. J Physiol Sci. 2007;57:287–298. doi: 10.2170/physiolsci.RP006807. [DOI] [PubMed] [Google Scholar]
- 7.Nagata K, Zheng L, Madathany T, Castiglioni AJ, Bartles JR, Garcia-Anoveros J. The variant-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward-rectifying currents and causes cell degeneration. Proc Natl Acad Sci USA. 2008;105:353–358. doi: 10.1073/pnas.0707963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasaki I, Spyropoulos CS. Stria vascularis as source of endocochlear potential. J Neurophysiol. 1959;22:149–155. doi: 10.1152/jn.1959.22.2.149. [DOI] [PubMed] [Google Scholar]
- 9.Sterkers O, Ferrary E, Amiel C. Production of inner ear fluids. Physiol Rev. 1988;68:1083–1128. doi: 10.1152/physrev.1988.68.4.1083. [DOI] [PubMed] [Google Scholar]
- 10.Offner FF, Dallos P, Cheatham MA. Positive endocochlear potential: mechanism of production by marginal cells of stria vascularis. Hear Res. 1987;29:117–124. doi: 10.1016/0378-5955(87)90160-2. [DOI] [PubMed] [Google Scholar]
- 11.Wangemann P, Liu J, Marcus DC. Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear Res. 1995;84:19–29. doi: 10.1016/0378-5955(95)00009-S. [DOI] [PubMed] [Google Scholar]
- 12.Wangemann P. K+ cycling and endocochlear potential. Hear Res. 2002;165:1–9. doi: 10.1016/S0378-5955(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 13.Mineharu A, Mori Y, Nimura Y, Takamaki A, Araki M, Yamaji J, Yoshida R, Takenaka H, Kubota T. Endolymphatic perfusion with EGTA-acetoxymethyl ester inhibits asphyxia- and furosemide-induced decrease in endocochlear potential in guinea pigs. Jpn J Physiol. 2005;55:53–60. doi: 10.2170/jjphysiol.R2086. [DOI] [PubMed] [Google Scholar]
- 14.Takamaki A, Mori Y, Araki M, Mineharu A, Sohma Y, Tashiro J, Yoshida R, Takenaka H, Kubota T. Asphyxia and diuretic-induced changes in the Ca2+ concentration of endolymph. Jpn J Physiol. 2003;53:35–44. doi: 10.2170/jjphysiol.53.35. [DOI] [PubMed] [Google Scholar]
- 15.Crouch JJ, Sculte BA. Expression of plasma membrane Ca-ATPase in the adult and developing gerbil cochlea. Hear Res. 1995;92:112–119. doi: 10.1016/0378-5955(95)00201-4. [DOI] [PubMed] [Google Scholar]
- 16.Curtis LM, Garg LC, Rarey KE. Ca2+-ATPases in the cochlear duct. Acta Otolaryngol. 1997;117:553–558. doi: 10.3109/00016489709113436. [DOI] [PubMed] [Google Scholar]
- 17.Ågrup C, Bagger-Syoback D, Fryckstedt J. Presence of plasma membrane-bound Ca2+-ATPase in the secretory epithelia of the inner ear. Acta Otolaryngol. 1999;119:437–445. doi: 10.1080/00016489950180964. [DOI] [PubMed] [Google Scholar]
- 18.Wood JD, Muchinsky SJ, Filoteo AG, Penniston JT, Tempel BL. Low endolymph calcium concentrations in deafwaddler2J mice suggest that PMCA2 contributes to endolymph calcium maintenance. J Assoc Res Otolaryngol. 2004;5:99–110. doi: 10.1007/s10162-003-4022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa T, Marunaka Y, Rotin D. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+ . J Gen Physiol. 1998;111:825–846. doi: 10.1085/jgp.111.6.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shindo M, Miyamoto M, Abe N, Shida S, Murakami Y, Imai Y. Dependence of endocochlear potential on basolateral Na+ and Cl− concentrations: a study using vascular and perilymph perfusion. Jpn J Physiol. 1992;42:617–630. doi: 10.2170/jjphysiol.42.617. [DOI] [PubMed] [Google Scholar]
- 21.Carlton SM, Hayes ES. Light microscopic and ultrastructural analysis of GABA-immunoreactive profiles in the monkey spinal cord. J Comp Neurol. 1990;300:162–182. doi: 10.1002/cne.903000203. [DOI] [PubMed] [Google Scholar]
- 22.Abe H, Yanagawa Y, Kanbara K, Hayasaki H, Azuma H, Obata K, Katsuoka Y, Yabumoto M, Watanabe M. Epithelial localization of green fluorescent protein-positive cells in epididymis of the GAD67-GFP knock-in mouse. J Androl. 2005;26:16–25. doi: 10.2164/jandrol.04157. [DOI] [PubMed] [Google Scholar]
- 23.Hayasaki H, Sohma Y, Kanbara K, Maemura T, Kubota T, Watanabe M. A local GABAergic system within rat trigeminal ganglion cells. Eur J Neurosci. 2006;23:745–757. doi: 10.1111/j.1460-9568.2006.04602.x. [DOI] [PubMed] [Google Scholar]
- 24.Kakigi A, Takada T. Effect of artificial endolymph injection into the cochlear duct on the endocochlear potential. Hear Res. 1998;116:113–118. doi: 10.1016/S0378-5955(97)00209-8. [DOI] [PubMed] [Google Scholar]
- 25.Couloigner V, Fay M, Djelidi S, Farman N, Escoubet B, Runembert I, Sterkers O, Frielander G, Ferrary E. Location and function of the epithelial Na channel in cochlea. Am J Physiol Renal Physiol. 2001;280:F214–F222. doi: 10.1152/ajprenal.2001.280.2.F214. [DOI] [PubMed] [Google Scholar]
- 26.Li-P He, Hewavitharana T, Soboloff J, Spassova MA, Gill DL. A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrasole derivative, BPT2. J Biol Chem. 2005;280:10997–11006. doi: 10.1074/jbc.M411797200. [DOI] [PubMed] [Google Scholar]
- 27.Cooper GJ, Hunter M. Intracellular pH and calcium in frog early distal tubule: effects of transport inhibitors. J Physiol. 1997;498:49–59. doi: 10.1113/jphysiol.1997.sp021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper GJ, Hunter M. Membrane cross-talk in the early distal tubule segment of frog kidney: role of calcium stores and chloride. Pflugers Arch. 2001;442:243–247. doi: 10.1007/s004240100518. [DOI] [PubMed] [Google Scholar]
- 29.Shimamoto C, Umegaki E, Katsu K, Kato M, Fujiwara S, Kubota T, Nakahari T. [Cl]i modulation of Ca2+-regulated exocytosis in Ach-stimulated antral mucous cells of guinea pig. Am J Physiol Gastrointest Liver Physiol. 2007;293:G824–G837. doi: 10.1152/ajpgi.00125.2007. [DOI] [PubMed] [Google Scholar]
- 30.Manuel MA, Weiner MW. Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: inhibition of electron transport in the region of phosphorylation site II. J Pharmacol Exp Ther. 1976;198:209–221. [PubMed] [Google Scholar]
- 31.Manuel MA, Weiner MW. Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: inhibition of the adenine nucleotide translocase. Biochim Biophys Acta. 1977;460:445–454. doi: 10.1016/0005-2728(77)90083-4. [DOI] [PubMed] [Google Scholar]
- 32.Frienden M, Arnaudeau S, Castelbou C, Demaurex N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem. 2005;280:43198–43208. doi: 10.1074/jbc.M510279200. [DOI] [PubMed] [Google Scholar]
- 33.Gerasimenko O, Tepikin A. How to measure Ca2+ in cellular organelles? Cell Calcium. 2005;38:201–211. doi: 10.1016/j.ceca.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 34.Lee C-T, Chen H-C, Lai L-W, Yong K-C, Lien Y-H H. Effects of furosemide on renal calcium handling. Am J Physiol Renal Physiol. 2007;293:F1231–F1237. doi: 10.1152/ajprenal.00038.2007. [DOI] [PubMed] [Google Scholar]
- 35.Rüsch A, Hummler E. Mechano-electrical transduction in mice lacking the α-subunit of the epithelial sodium channel. Hear Res. 1999;131:170–176. doi: 10.1016/S0378-5955(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 36.Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem. 1995;270:27411–27414. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- 37.Fyfe GK, Quinn A, Canessa CM. Structure and function of the Mec-ENaC family of ion channels. Semin Nephrol. 1998;18:138–151. [PubMed] [Google Scholar]
- 38.Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, Yamauchi D, Marcus D, Kachar B. Deafness in claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci. 2004;24:7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitajiri S, Miyamoto T, Mineharu A, Sonoda N, Hata M, Furuse K, Sasaki H, Mori Y, Kubota T, Ito J, Furuse M, Tsukita S. Compartmentalization established by claudin-11-based tight junction in stria vascularis is required for hearing through generation of the endocochlear potential. J Cell Sci. 2004;117:5087–5096. doi: 10.1242/jcs.01393. [DOI] [PubMed] [Google Scholar]
- 40.Cuajungco MP, Grimm C, Heller S. TRP channels as candidates for hearing and balance abnormalities in vertebrates. Biochim Biophys Acta. 2007;1772:1022–1027. doi: 10.1016/j.bbadis.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitajiri S, Furuse M, Morita K, Kaishin-Kiuchi Y, Kido H, Ito J, Tsukita S. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear Res. 2004;187:25–34. doi: 10.1016/S0378-5955(03)00338-1. [DOI] [PubMed] [Google Scholar]
- 42.Vandenbrouke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 43.Thorne PR, Munoz DJB, Housley GD. Purinergic modulation of cochlear partition resistance and its effect on endocochlear potential in the guinea pig. JARO. 2003;5:58–65. doi: 10.1007/s10162-003-4003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]