

Abstract
This study was designed to clarify whether nitric oxide (NO) participates in the regulation of local cerebral blood flow (CBF) during hypoxia (inhalation of 15% O2 in N2). The CBF response to hind-paw stimulation (evoked CBF) of Sprague-Dawley (SD) rats was measured by laser-Doppler flowmetry. Physiological variables, such as heart rate, mean blood pressure, and PaCO2 during hypoxia, were identical to those under normoxic conditions. Hypoxia increased the baseline CBF (17.5 ± 14.3%) and the normalized peak amplitude of evoked CBF (31.1 ± 18.5%) relative to those during normoxia. When an NOS inhibitor was infused intravenously, these differences were abolished in both the baseline CBF or evoked CBF between normoxic and hypoxic conditions, whereas the heart rate decreased and the mean blood pressure increased during hypoxia in comparison with these during normoxia. The field potential was constant under all experimental conditions. These results suggest that NO plays a major role in the regulation of baseline and evoked CBF during hypoxia.
Keywords: Cerebral blood flow, Functional activation, Laser-Doppler flowmetry, Somatosensory stimulation, Rat, Nitric oxide
Introduction
Changes in local cerebral blood flow (CBF) are closely related to neural activity; therefore, the cerebral hemodynamic response has been used extensively to map brain function in humans and animals. One of the major roles of CBF is to supply oxygen to the brain tissues. The relationship between oxygen metabolism and CBF is of considerable interest to many researchers.
In the case of baseline CBF, it has been reported that an increased arterial oxygen supply (hyperoxia) results in a decrease in the baseline level of CBF in rats [1], whereas a decreased arterial oxygen supply (hypoxia) increases the baseline level of CBF in humans [2] and in the somatosensory cortex of rats [3–5]. These observations indicate that the baseline CBF is affected by the O2 level in the supplying blood in brain tissue.
On the other hand, in the case of an increase in local CBF (evoked CBF), induced by neuronal activation, the relationship between evoked CBF and blood oxygenation in the activated brain area is still a disputable and controversial issue. In our previous study, we reported that hyperoxia provokes the enhancement of evoked CBF in the V1 area of the human brain [6, 7] and in the somatosensory cortex of rats [1]. In a human study, using positron emission tomography (PET), Mintun et al. [2] reported that hypoxia does not enhance evoked CBF, suggesting that evoked CBF is independent of the oxygen supply or metabolic demand. In contrast, several studies in rats, using functional magnetic resonance imaging (fMRI) and optical spectroscopy, showed that hyperoxia provokes a decrease in evoked CBF, while hypoxia induces its enhancement [3, 4, 8]. These studies suggest that evoked CBF depends on the oxygen metabolic demand.
The dynamics of the evoked CBF is highly complicated and many biochemical mediators could be considered in the regulation system, such as nitric oxide (NO), cyclooxygenase-1 (COX-1), COX-2, and adenosine [9–11]. NO, produced by endothelial and neuronal nitric oxide synthase (NOS), acts as a neurotransmitter and is involved in the regulation of cerebral circulation. In particular, it is also an important mediator of the cerebral vasodilatation in response to changes in the physiological parameters during hypercapnia [12] and hyperoxia [1]; however, the role of NO in evoked CBF regulation remains to be elucidated. In our previous study, we reported that the NOS inhibition was accompanied by a decrease in baseline CBF during normoxia and suppression of evoked CBF during hyperoxia [1]. We speculated that NOS inhibition would also suppress the effect of hypoxia on cerebral circulation.
In the present study, we investigated the effect of hypoxia on the baseline levels of CBF and evoked CBF using laser-Doppler flowmetry (LDF) to clarify the regulation mechanism of CBF in relation to the metabolic oxygen demand. The effect of NOS inhibition on CBF regulation under hypoxia was also demonstrated using N ω-nitro-l-arginine (LNA) as a pharmacological tool.
Materials and methods
Animal preparation and control of physiological conditions
All experiments were conducted in accordance with the guidelines of the Physiological Society of Japan and were approved by the Animal Care and Use Committee of the National Institute of Radiological Sciences, Chiba, Japan.
Sprague-Dawley rats (370–420 g) were anesthetized with isoflurane (4% for induction and 1.5% during surgery) in 30% O2 and 70% N2 using a face mask. Subcutaneous 2% lidocaine was used before incision to prevent vasospasm during catheter insertion. The tail artery and left femoral vein were cannulated for blood pressure monitoring, blood gas sampling, and intravenous drug administration. After tracheotomy, α-chloralose (75 mg/kg body weight, i.v.) was administered and isoflurane administration was discontinued. Anesthesia was maintained with α-chloralose (44 mg/kg/h, i.v.), and muscle relaxation was maintained with pancuronium bromide (0.7 mg/kg/h, i.v.) during the experimental period. Body temperature was monitored with a rectal probe and maintained at approximately 37.0°C using a heating pad (ATC-210; Unique Medical, Japan). The rat was fixed in a stereotactic frame, and the parietal bone was thinned to translucency at the left somatosensory cortex using a dental drill (an area 3 × 3 mm, centered 2.5 mm caudal and 2.5 mm lateral to the bregma). To ensure a stable physiological condition of the animal, the measurements were performed 3 h after the preparation of the parietal bone (Fig. 1).
Fig. 1.
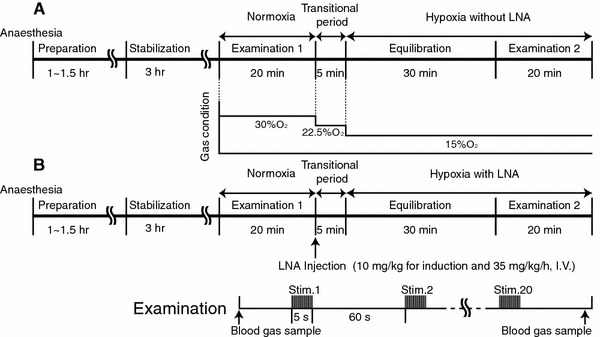
Experimental protocol of hypoxia a without LNA (experiment A) and b hypoxia with LNA (experiment B). The experiment was carried out approximately 3 h after the preparation of rats. The evoked CBF and field potential were measured under normoxia, hypoxia without LNA, and hypoxia with LNA. In each examination, 20 successive stimuli (5 Hz and 5 s) were applied at 60 s intervals. The animals were ventilated with a 30% O2 and 70% N2 mixture during normoxia (control). To avoid a change in arterial blood pressure, the O2 concentration was gradually decreased in a stepwise manner from 30 to 22.5 and 15% during hypoxia
The rat was paralyzed by injection of pancuronium bromide and artificially ventilated using a respirator (SN-480-7; Shinano, Japan) with a mixture of N2 and O2 to achieve physiological arterial blood levels of O2 and CO2 tension (PaO2 and PaCO2, respectively). During the experiments, the changes in respiratory O2 and CO2 concentrations were monitored with a capnometer (SurgiVet V9004, Smiths Medical, USA). Arterial blood pressure from the tail artery was measured with a pressure sensor (TP400T; Nihon Kohden, Japan) and recorded continuously using MacLab data acquisition software (AD Instruments, Australia) during the experiments; the mean arterial blood pressure (MABP) was calculated as the average at three time points (i.e., before, during, and immediately after each stimulation period). Arterial blood samples were serially collected before and immediately after each step of the experiment and analyzed for gas values.
Baseline and evoked CBF measurements
The evoked CBF was measured by LDF (FLO-C1; OMEGA FLO) equipped with a probe with a tip diameter of 0.55 mm (Probe NS; OMEGA FLO). The sampling volume for the LDF measurement was approximately 1 mm3 [13]. A time constant of 0.1 s was used to detect the LDF signal. The LDF probe was positioned over the thinned skull (over the somatosensory area of the hind paw). Electrical hind paw stimulation was performed with two needle electrodes inserted subdermally into the right hind paw contralateral to the LDF probe. A current stimulus of 1.5 mA (0.1 ms pulse) was applied at 5 Hz frequency and 5 s duration. In each experiment, 20 successive stimuli were applied at 60 s intervals and accumulated using MacLab data acquisition software. These stimulus parameters did not cause any change in the systemic arterial blood pressure and heart rate during stimulation.
The evoked CBF response to somatosensory stimulation was investigated under three experimental conditions as follows: normoxia (17 rats), hypoxia without LNA (9 rats), and hypoxia with LNA (8 rats) (Fig. 1).
Hypoxia and application of NOS inhibitor
Under normoxia (control), the animals were ventilated with a 30% O2 and 70% N2 mixture. Hypoxia was induced by decreasing the concentration of inspired O2 to 15%. The acute change from 30% O2 to 15% O2 often caused mild hypotension, so the O2 concentration was gradually decreased in a stepwise manner from 30 to 22.5% and 15% (Fig. 1). This stepwise hypoxia had no effect on arterial blood pressure (Table 1). In the “hypoxia without LNA” experiment (Fig. 1a), we first examined the evoked CBF under normoxia. The oxygen concentration was then decreased in a stepwise manner from 30 to 22.5% and 15%. After an equilibration time of 30 min, the evoked CBF was measured during hypoxia without LNA. In the “hypoxia with LNA” experiment (Fig. 1b), we also examined the evoked CBF under normoxia. LNA was then applied intravenously (10 mg kg−1 for induction and 35 mg kg−1 h−1 during experiments), and the O2 concentration was decreased in a stepwise manner from 30 to 22.5% and 15%. After an equilibration time of 30 min, evoked CBF responses were measured under hypoxia with LNA administration.
Table 1.
Physiological variables

aNumber of rats; mean ± SD
* P < 0.01
Measurement of neuronal activity
To estimate the correlation between the change in evoked CBF and neuronal activation, field potentials were measured in both experiments: “hypoxia without LNA” (6 rats) and “hypoxia with LNA” (6 rats). A tungsten microelectrode (12 MΩ) was inserted into the somatosensory area of the hind paw through the thinned portion of the skull and fixed using dental cement. The tip of the electrode was set at a depth of approximately 0.5 mm from the surface of the cortex. An Ag-AgCl indifferent electrode was placed between the skull bone and scalp. Field potentials under normoxia, hypoxia without LNA, and hypoxia with LNA were recorded using the same time schedule as evoked CBF measurements (Fig. 1). Twenty successive signals of the field potential recordings were also accumulated using MacLab data acquisition software and were digitized at 100 Hz, which was the same frequency as that in our previous study [1, 11, 14]. The mean amplitude of the field potentials was calculated as the average of the negative components of each spike to evaluate the neuronal activity during stimulation [1, 14].
Data analysis
The LDF signal was normalized towards the baseline level as percent changes from the baseline (normalized evoked CBF). The rise time of the evoked CBF was defined as the time at the intersection of the extrapolated lines, which was drawn with the baseline on the response curve from 90 to 10% of the peak [15, 16]. The peak amplitude indicated the percent change from the baseline to the maximum evoked CBF. Values were statistically analyzed by Student’s t test and presented as the mean ± SD.
Results
Physiological variables during hypoxia and LNA administration
The physiological variables (i.e., heart rate, MABP, and PaCO2) during hypoxia without LNA were almost identical to those during normoxia (control) (Table 1). Only the PaO2 value (49.8 ± 3.0 mmHg) was significantly different from that measured during normoxia (PaO2 = 93.3 ± 11.0 mmHg) (P < 0.01).
The systemic administration of the NOS inhibitor LNA during hypoxia (hypoxia with LNA) significantly increased MABP and decreased the heart rate relative to those parameters during normoxia (P < 0.01). PaO2 was significantly lower during hypoxia with LNA (48.1 ± 1.4 mmHg) than during normoxia (101.8 ± 11.9 mmHg) (P < 0.01). There was no clear change in the respiratory CO2 concentration between normoxia and hypoxia (data not shown).
Effects of hypoxia on CBF
During hypoxia (in the absence of LNA), the baseline level of CBF was significantly higher than that during normoxia (P < 0.05) (Fig. 2). As shown in Fig. 3a, hypoxia enhanced the normalized evoked CBF in comparison with normoxia. There was a significant difference in the peak amplitude of the normalized evoked CBF between the hypoxic condition (20.4 ± 7.4%) and normoxic condition (15.3 ± 5.4%) (P < 0.05) (Fig. 3b). The rise time of evoked CBF was 0.67 ± 0.22 s during hypoxia and 0.51 ± 0.30 s during normoxia, although no significant difference in the rise time between the two conditions was detected. Presumably, some mediators involved in vasodilatation enhance baseline and evoked CBF during hypoxia. In the subsequent experiments, we focused on NO as one of the possible candidates responsible for vasodilatation.
Fig. 2.
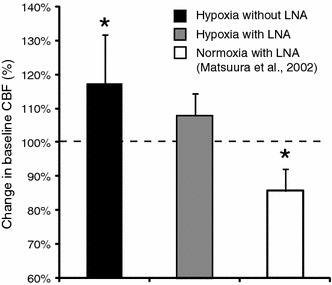
Change in baseline CBF. The values of baseline CBF, obtained during normoxia, were considered to be 100%, indicated by a dashed line. Black and gray bars indicate the baseline CBF during hypoxia without LNA and hypoxia with LNA, respectively. The white bar indicates the baseline CBF during normoxia with LNA, which was obtained in the previous study [1]. The data were normalized to those under normoxia without LNA after statistical analysis. To evaluate the changes in baseline CBF, the absolute value of the baseline CBF was statistically compared for each animal group by paired t test. Note that the baseline CBF during hypoxia with LNA was not significantly different from that during normoxia, whereas the baseline CBF was higher during hypoxia without LNA than during normoxia. Asterisks indicate significant differences in levels of baseline CBF between hypoxia without LNA and normoxia without LNA, and normoxia with LNA and normoxia without LNA (P < 0.05). Error bars are SD
Fig. 3.
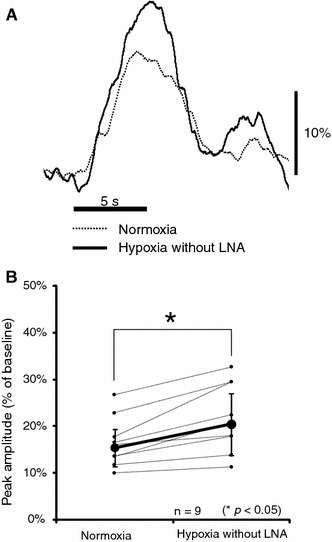
Change in normalized evoked CBF under normoxia and hypoxia without LNA. a Normalized evoked CBF with somatosensory stimulation from one representative animal. Curves were normalized to the baseline level. Horizontal and vertical bars indicate the stimulation period and 10% increase from the baseline level, respectively. b Peak amplitude of normalized evoked CBF under normoxia and hypoxia without LNA. Each line corresponds to the data obtained from one rat. Bold lines and error bars indicate the mean value and SD, respectively (n = 9). Note that the normalized evoked CBF was significantly higher during hypoxia than during normoxia (*P < 0.05)
Application of NOS inhibitor during hypoxia
The injection of NOS inhibitors (LNA) in rats subjected to hypoxia resulted in the same baseline CBF as that during normoxia (Fig. 2). The normalized evoked CBF during hypoxia with LNA did not increase in comparison with that during normoxia (Fig. 4b). The peak amplitude of the normalized evoked CBF was 22.8 ± 10.8% during normoxia and 19.5 ± 7.6% during hypoxia with LNA (Fig. 4c). A slight (but not significant) decrease in the evoked CBF was detected in rats subjected to hypoxia and with LNA. There was no significant difference in the rise time of evoked CBF between normoxia and hypoxia with LNA: 0.60 ± 0.23 s during hypoxia with LNA and 0.57 ± 0.29 s during normoxia. The results suggest that NO is involved in the enhancement of baseline and evoked CBF during hypoxia. The NO inhibitor abolished the effect of hypoxia completely at the baseline level and evoked CBF; therefore, NO could be considered as a major mediator of the relationship between hypoxia and evoked CBF response.
Fig. 4.
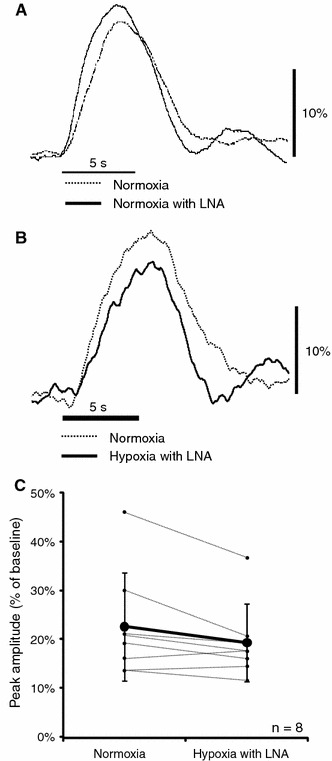
Change in normalized evoked CBF during normoxia, normoxia with LNA, and hypoxia with LNA. a Normalized evoked CBF during normoxia without LNA and normoxia with LNA (cited from reference [1]). b Normalized evoked CBF during normoxia without LNA and hypoxia with LNA. Curves were normalized to the baseline level and shown for one representative animal. Horizontal and vertical bars indicate the stimulation period and 10% increase from baseline level, respectively. c Peak amplitude of normalized evoked CBF under normoxia and hypoxia with LNA. Each line corresponds to the data obtained from one rat. Bold lines and error bars indicate the mean value and SD, respectively (n = 8). No significant difference in the peak amplitude of normalized evoked CBF between normoxia and hypoxia with LNA was observed
Neural activities during experiments
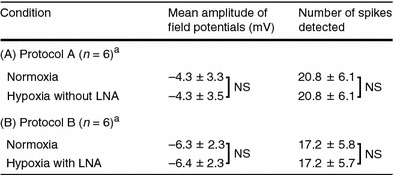
Figure 5 shows one of the representative field potentials from the somatosensory cortex during normoxia, hypoxia without LNA, and hypoxia with LNA. The field potential during hypoxia without LNA was almost the same as that during normoxia (Fig. 5a). There were no significant differences in the mean amplitude and number of spikes of field potentials between hypoxia without LNA and normoxia (Table 2). The results indicate that the enhancement of evoked CBF is not caused by an increase in neuronal activity during hind paw stimulation.
Fig. 5.
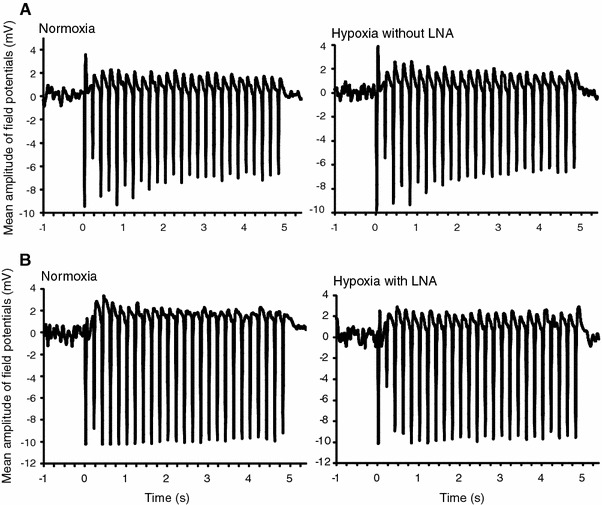
Representative field potential recordings from the somatosensory cortex during hind paw stimulation. a Hypoxia without LNA. Neuronal activity during hypoxia without LNA was almost the same as that during normoxia. b Hypoxia with LNA. Field potential during hypoxia with LNA was almost identical to that during normoxia. In both experiments, there were no significant differences in the mean amplitude and number of spikes of field potentials between hypoxia and normoxia (see Table 2)
Table 2.
Parameters of neuronal activity during each examination
Hind-paw stimulation (5 Hz and 5 s)
NS not significant
aNumber of rats; mean ± SD
The field potential during hypoxia with LNA was also almost the same as that during normoxia (Fig. 5b). There were no significant differences in the mean amplitude and number of spikes of field potentials between hypoxia with LNA and normoxia (Table 2), indicating that the application of LNA did not affect neuronal activity.
Discussion
In the present study, we investigated the effects of hypoxia on the regulation of baseline and evoked CBF in rats, as well as the role of NO in this relationship. NO is synthesized mainly by endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS) in the brain tissue. To inhibit the activities of all these NOS isoenzymes, a nonselective NOS inhibitor, LNA, was used. A previous study showed that the intracortical administration of the NOS inhibitor affected the CBF during hypoxia [17]. We also found that the NOS inhibitor LNA infused intravenously attenuated the CBF response to hyperoxic conditions [1]. The intravenous infusion of LNA may cause excess accumulations of the drug leading to unfavorable effects on the evoked CBF. However, we revealed that the systemic administration of LNA under normoxic condition (normoxia with LNA) did not affect the neuronal activity and normalized evoked CBF (Fig. 4a), although it caused a decline in the level of the baseline CBF (Fig. 2) [1]. The experimental protocol used in this study should not cause any side effects of the drug leading to the normalized evoked CBF.
It has been reported that hypoxia causes mild hypotension [4, 5]. In our experiment, we avoided a significant reduction of MABP during hypoxia by applying a stepwise decrease in the inspired O2 concentration and mild hypoxia (15% O2) (Fig. 1). The other physiological parameters (i.e., heart rate and PaCO2) during hypoxia were identical to those during normoxia (Table 1), which is in agreement with the previous report [5]. It is known that hypoxia activates distinct cellular and molecular processes in the brain depending on both its severity and time course (e.g., rate of decrease in oxygen tension and duration). The protocol in the hypoxia experiment was targeting PaO2 within 40–50 mmHg, which was consistent with previous studies [3–5], but kept the PaCO2, MABP, and heart rate within the normal range. The stepwise method of hypoxia used in this study is one of the best ways to achieve this and did not cause any serious damage to the brain tissue. Moreover, the inhalation of 15% O2 in N2 did not evoke a hypoxic ventilatory response (i.e., increases in minute ventilation) despite the fact that PaO2 was significantly lowered (Table 1). In this study, rats were anesthetized and relaxed with continuous infusion of α-chloralose and pancuronium bromide during the experiment. Muscle relaxation with pancuronium bromide does not cause hypoxic ventilatory responses.
We reported that the baseline CBF was ~5.0% lower during hyperoxia than during normoxia [1], and we found in the present study that it was ~17.5% higher during hypoxia than during normoxia (Fig. 2). These results indicate that the baseline CBF under resting conditions is regulated to maintain a constant oxygen supply. The infusion of LNA in rats during hypoxia abolished the effect of hypoxia and returned the baseline to the level obtained during normoxia (Fig. 2). A decrease in the baseline CBF under normoxia with LNA has been reported (Fig. 2) [1]. Their results suggest that the systemic administration of LNA suppresses the NO activity and the decline in the baseline CBF. We considered that the increase in the baseline CBF during hypoxia was caused by an increase in the level of NO activity.
Mintun et al. [2] reported following a PET study that there is no significant difference in the normalized regional CBF response to visual stimulation between normoxia and hypoxia and that the baseline level of CBF is higher during hypoxia than during normoxia. The discrepancy between our findings and the results of the PET study might be a result of the differences in experimental protocols and measurement techniques. For example, the time resolution of LDF in this study is much higher than that of PET. A higher time resolution of LDF is feasible in detecting the detailed time course of evoked CBF during hypoxia. Thus, it is possible that an increase in evoked CBF during hypoxia could be detected in our present LDF study that might have been smoothed out in the PET study.
The peak amplitude of normalized evoked CBF is higher under hyperoxia than under normoxia [1, 7]. The normalized evoked CBF during hypoxia was also enhanced in comparison with that during normoxia (Fig. 3). These observations suggest that the evoked CBF is enhanced by abnormal oxygen supply, such as high and low levels of oxygen in the brain tissue. The peak amplitude of normalized evoked CBF during hypoxia with LNA was also the same as that during normoxia (Fig. 4b). We previously reported that LNA administration during hyperoxia decreased not only evoked CBF but also the neural activity. Therefore, the increase in the evoked CBF during hyperoxia was not simply accounted for by the up-regulated NOS expression. On the other hand, there was no significant difference in neuronal activity between normoxia and hypoxia with LNA (Fig. 5; Table 2). These observations suggest that hypoxia is accompanied by an increase in NOS activity, and that LNA administration during hypoxia suppresses this process.
It has been reported that although the baseline CBF is higher during hypercapnia than that during normocapnia, there was no significant difference in normalized evoked CBF between the hypercapnic and normocapnic conditions [18]. These results indicated that the absolute value of evoked CBF increases according to the baseline CBF during hypercapnia; that is, the evoked CBF under hypercapnia is proportional to the baseline CBF. In contrast, we established that the normalized evoked CBF is higher during hypoxia than during normoxia despite the increase in baseline CBF (Fig. 3). This suggests that there are some differences in the mechanism of NOS activity between hypercapnia and hypoxia. Since hypoxia is accompanied by free radical generation in the hypoxic area [19], it may be speculated that nitric oxide radicals and their intermediate products are involved in the regulation of evoked CBF response during hypoxia, while hypercapnia is not accompanied by such free radical mechanism.
A previous study showed that the increase in neuronal activity induced by stimulation in rat was unchanged during acute hypoxia relative to that under normoxia [3]. As mentioned above, in the present study, hypoxia was caused by a stepwise mild decrease in the respiratory O2 concentration. The neuronal activity was not significantly different between hypoxia and normoxia (Fig. 5; Table 2), suggesting that the neural activity induced by stimulation is stable during the experiment. The enhancement of evoked CBF during hypoxia in this study may be caused by a low oxygen supply level within the activated brain area.
It is well known that NO is a key mediator of cerebrovascular responses to carbon dioxide and involved in neurovascular coupling. However, the effects of NOS inhibitors, such as LNA, on the neurovascular coupling differ with each experimental condition. LNA by itself reduced the evoked CBF slightly but not completely. This is because the CBF response to neuronal activation is mediated by various mediators, e.g., NO, prostaglandins, adenosine, and ions [11, 16]. LNA blocks only NO synthesis, but not the effects of other mediators of neurovascular coupling. In our study, we observed that LNA abolished completely the effect of hypoxia on the evoked CBF response. Moreover, the evoked CBF response is slightly lower in the presence of LNA than under normoxia (Fig. 4). This means that a part of the evoked CBF response to normoxia, but not the total response, could be explained by the effect of NO.
Acknowledgments
The present work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and KAKENHI from the Japan Society for the Promotion of Science.
Contributor Information
Hiroyuki Takuwa, Phone: +81-43-2063425, FAX: +81-43-2063425, Email: takuwa@nirs.go.jp.
Tetsuya Matsuura, Phone: +81-19-6216376, FAX: +81-19-6216376, Email: matsuura@iwate-u.ac.jp.
References
- 1.Matsuura T, Kanno I. Effect of nitric oxide synthase inhibitor on the local cerebral blood flow evoked by rat somatosensory stimulation under hyperoxia. Comp Biochem Physiol A. 2002;131:267–274. doi: 10.1016/S1095-6433(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 2.Mintun MA, Lundstrom BN, Snyder AZ, Vlassenko AG, Shulman GL, Raichle ME. Blood flow and oxygen delivery to human brain during functional activity: theoretical modeling and experimental data. Proc Natl Acad Sci USA. 2001;98:6859–6864. doi: 10.1073/pnas.111164398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindauer U, Gethmann J, Kühl M, Kohl-Bareis M, Dirnagl U. Neuronal activity-induced changes of local cerebral microvascular blood oxygenation in the rat: effect of systemic hyperoxia or hypoxia. Brain Res. 2003;975:135–140. doi: 10.1016/S0006-8993(03)02602-7. [DOI] [PubMed] [Google Scholar]
- 4.Sicard KM, Duong TQ. Effects of hypoxia, hyperoxia, and hypercapnia on baseline and stimulus-evoked BOLD, CBF, and CMRO2 in spontaneously breathing animals. Neuroimage. 2005;25:850–858. doi: 10.1016/j.neuroimage.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukhotinsky I, Dilekoz E, Moskowitz MA, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab. 2008;28:1369–1376. doi: 10.1038/jcbfm.2008.35. [DOI] [PubMed] [Google Scholar]
- 6.Kanno I, Fujita H, Hatazawa J. Enhancement of CBF response for V1 stimuli during hyperoxia: behavior of oxygen in neuronal activation revisited. J Cereb Blood Flow Metab. 1996;17:S646. [Google Scholar]
- 7.Kashikura K, Kershaw J, Kashikura A, Matsuura T, Kanno I. Hyperoxia-enhanced activation-induced hemodynamic response in human VI: an fMRI study. Neuroreport. 2000;11:903–906. doi: 10.1097/00001756-200004070-00001. [DOI] [PubMed] [Google Scholar]
- 8.Duong TQ. Cerebral blood flow and BOLD fMRI responses to hypoxia in awake and anesthetized rats. Brain Res. 2007;1135:186–194. doi: 10.1016/j.brainres.2006.11.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy demand. Science. 1999;22:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 10.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 11.Matsuura T, Takuwa H, Bakalova R, Obata T, Kanno I. Effect of cyclooxygenase-2 on the regulation of cerebral blood flow during neuronal activation in the rat. Neurosci Res. 2009;65:64–70. doi: 10.1016/j.neures.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Iadecola C. Does nitric oxide mediate the increases in cerebral blood flow elicited by hypercapnia? Proc Natl Acad Sci USA. 1992;89:3913–3916. doi: 10.1073/pnas.89.9.3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solfrizzi V, D’Introno A, Colacicco AM, Capurso C, Del Parigi A, Capurso S, Gadaleta A, Capurso A, Panza F. Dietary fatty acids intake: possible role in cognitive decline and dementia. Exp Gerontol. 2005;40:257–270. doi: 10.1016/j.exger.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Matsuura T, Kanno I. Quantitative and temporal relationship between local cerebral blood flow and neuronal activation induced by somatosensory stimulation in rats. Neurosci Res. 2001;40:281–290. doi: 10.1016/S0168-0102(01)00236-X. [DOI] [PubMed] [Google Scholar]
- 15.Matsuura T, Fujita H, Seki C, Kashikura K, Kanno I. CBF change by somatosensory activation measured by laser-Doppler flowmetry: independent evaluation of RBC velocity and RBC concentration. Jpn J Physiol. 1999;49:289–296. doi: 10.2170/jjphysiol.49.289. [DOI] [PubMed] [Google Scholar]
- 16.Bakalova R, Matsuura T, Kanno I. The cyclooxygenase inhibitors indomethacin and rofecoxib reduce regional cerebral blood flow evoked by somatosensory stimulation in rats. Exp Biol Med (Maywood) 2002;227:465–473. doi: 10.1177/153537020222700710. [DOI] [PubMed] [Google Scholar]
- 17.Pelligrino DA, Wang Q, Koenig HM, Albrecht RF. Role of nitric oxide, adenosine, N-methyl-d-aspartate receptors, neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res. 1995;704:61–70. doi: 10.1016/0006-8993(95)01105-6. [DOI] [PubMed] [Google Scholar]
- 18.Matsuura T, Fujita H, Kashikura K, Kanno I. Evoked local cerebral blood flow induced by somatosensory stimulation is proportional to the baseline flow. Neurosci Res. 2000;38:341–348. doi: 10.1016/S0168-0102(00)00175-9. [DOI] [PubMed] [Google Scholar]
- 19.Packer L, Prillipko L, Christen Y, editors. Free radicals in the brain. Berlin: Springer; 1992. [Google Scholar]